
Abstract
Hypoxic-ischemic encephalopathy (HIE) is an important cause of permanent damage to central nervous system (CNS) that may result in neonatal death or manifest later as mental retardation, epilepsy, cerebral palsy, or developmental delay. The primary cause of this condition is systemic hypoxemia and/or reduced cerebral blood flow with long-lasting neurological disabilities and neurodevelopmental impairment in neonates. About 20 to 25% of infants with HIE die in the neonatal period, and 25-30% of survivors are left with permanent neurodevelopmental abnormalities. The mechanisms of hypoxia-ischemia (HI) include activation and/or stimulation of myriad of cascades such as increased excitotoxicity, oxidative stress, N-methyl-d-aspartic acid (NMDA) receptor hyperexcitability, mitochondrial collapse, inflammation, cell swelling, impaired maturation, and loss of trophic support. Different therapeutic modalities have been implicated in managing neonatal HIE, though translation of most of these regimens into clinical practices is still limited. Therapeutic hypothermia, for instance, is the most widely used standard treatment in neonates with HIE as studies have shown that it can inhibit many steps in the excito-oxidative cascade including secondary energy failure, increases in brain lactic acid, glutamate, and nitric oxide concentration. Granulocyte-colony stimulating factor (G-CSF) is a glycoprotein that has been implicated in stimulation of cell survival, proliferation, and function of neutrophil precursors and mature neutrophils. Extensive studies both in vivo and ex vivo have shown the neuroprotective effect of G-CSF in neurodegenerative diseases and neonatal brain damage via inhibition of apoptosis and inflammation. Yet, there are still few experimentation models of neonatal HIE and G-CSF’s effectiveness, and extrapolation of adult stroke models is challenging because of the evolving brain. Here, we review current studies and/or researches of G-CSF’s crucial role in regulating these cytokines and apoptotic mediators triggered following neonatal brain injury, as well as driving neurogenesis and angiogenesis post-HI insults.
Similar content being viewed by others

Background
Numerous studies have shown that the most common contributor to early neonatal mortality is birth asphyxia with prematurity, infections, and low birth weight being other major contributors [1]. Birth asphyxia leads to significant brain injury with about 20-25% of asphyxiated newborns with hypoxic ischemic encephalopathy (HIE) dying within the newborn period and another 25% developing long-term sequelae, most commonly, cerebral palsy, epilepsy, autism spectrum disorder, and sensory deficits [2]. These neurological insults result in significant short-term and long-term physical, emotional, and financial cost [3]. HIE has a myriad of etiologies commonly categorized into prenatal and perinatal [4]. The pathophysiological presentation of HIE has been extensively explored, including but not limited to activation of inflammatory agents, apoptotic cascades, excitotoxicity, activation of microglia and astrocytes, N-methyl-d-aspartic acid (NMDA) receptor hyperexcitability, mitochondrial impairment and oxidative stress, and delayed cell death post-HI [5,6,7,8], categorized into three phases outlined in Fig. 1 [1, 9].

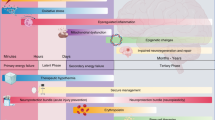
Schematic illustration of pathogenic mechanisms of HIE following HI brain injury. Primary energy failure occurs immediately after the hypoxic–ischemic insult. After reperfusion, there is a secondary energy failure, which can extend in duration from 6 to 48 h. Brain injury (tertiary phase) continues to occur months to years after the injury resulting in decreased plasticity and reduced number of neurons. Latent period following resuscitation is ideal for interventions to decrease the impact of secondary energy failure. However, strategies are developed to attenuate tertiary brain damage which will expand the therapeutic window, substantially increasing the beneficial effects of neuroprotection in these infants and hence its impact on long-term outcome. The up arrows represent an increase while the down arrows show a decrease in the corresponding metabolite/process
The clinical manifestations of neonatal HIE are insidious because the immature brain is more resistant to injury from hypoxia-ischemia (HI) events due to lower cerebral metabolic rate, plasticity of immature central nervous system (CNS), and immaturity in the development of balance in the functional neurotransmitters [10]. Thus, neonates suffering from HIE will go unnoticed during the early stages of HI, rendering them more susceptible to secondary injury occurring 6 to 72 h after the initial insults (Fig. 1). Similarly, clinical assessments are sometimes inconclusive or vague due to the nature of disease status and presentation in these infants. Diagnostic guidelines have been set out in neonates with HIE by the American Academy of Pediatrics (AAP) and the American College of Obstetrics and Gynecology (ACOG) [4] for initial assessment and appropriate management strategies. Routine serum biomarkers, magnetic resonance imaging (MRI), and electroencephalogram (EEG) [11,12,13,14,15] are the most commonly used diagnostic tools in recognizing brain injury in neonate that helps guide timely intervention and assessment of treatment outcome and prognostication.
Potential neuroprotective strategies targeting different pathways leading to neuronal cell death in response to hypoxic-ischemic insult have been investigated, including hypothermia, erythropoietin, magnesium, allopurinol, xenon, melatonin, growth factors (G-CSF, SCF, Epo), barbiturates, statins, and stem cells in various animal models of neonatal HIE [2, 16,17,18]. Therapeutic hypothermia (TH) is the most widely used standard treatment in neonatal HIE [19, 20], by inhibiting inflammatory cascades, reduced production of reactive oxygen species, inhibited apoptosis, an endogenous neuroprotective effect, reduced concentrations of free radicals, and neurotransmitters such as glutamate, glutamine, GABA, and aspartate [17, 21, 22], while others report ineffectiveness and/or adverse effects [23,24,25]. For instance, Arteaga et al. [9] reported that about 50% of infants treated with TH had adverse outcomes, such as cognitive impairment. A randomized control trial (RCT) by Shankaran et al. [26] concluded that cooling for 120 h or to 32.0 °C, or both, may be deleterious. Subsequently, TH did not improve EEG recovery when cooling was extended from 72 to 120 h and that it further impaired neuronal survival [27]. Thus, other neuroprotective agents are being explored that offer promise either as a monotherapy or in combination with therapeutic hypothermia.
Granulocyte-colony stimulating factor (G-CSF) is a 20-kDa protein that readily crosses the blood-brain barrier (BBB), which is a member of the hematopoietic growth factor family, that promote the proliferation, differentiation, and survival of hematopoietic stem cells with the obvious protective effect on neurons in peripheral and central nervous system [28,29,30]. The past few decades have explored its role from neutropenia-induced chemotherapy to neurological diseases and traumatic brain injury through regulation of both inflammatory and apoptotic mediators and enhancing neurogenesis and angiogenesis (Fig. 2) [29, 31,32,33,34]. G-CSF and its receptors are expressed by neurons, and their expression is regulated by ischemia, which points to an autocrine protective signaling mechanism [35]. Extensive studies in both in vivo and ex vivo have shown the neuroprotective effect of G-CSF in neurodegenerative diseases (Table1) such as Parkinson’s disease [53, 54], Alzheimer’s disease [55], and stroke [56,57,58], and clinical trials are ongoing to determine its efficacy and safety in these neurological diseases [59, 60]. Recent studies have focused on its neuroprotective effect in neonatal brain injury by regulating inflammatory and apoptotic mediators, thus attenuating neuroinflammation and neuronal apoptosis as well as enhancing neurogenesis and angiogenesis. Indeed, there are current reviews on HIE that have detailed its pathogenesis [4, 27], diagnostic modalities [13, 15], treatment interventions [2, 17], and emerging therapeutic agents [1], including G-CSF. In addition to these reviews, ours mainly focused on G-CSF role in modulating inflammatory and apoptotic mediators, driving of neurogenesis and angiogenesis, and signalling pathways mediated by G-CSF in attenuating neonatal ischemic brain damage. We therefore review current studies and/or researches of G-CSF’s crucial role in regulating these cytokines and apoptotic cascades triggered following neonatal hypoxia-ischemia injury and subsequently its role in promoting neurogenesis and angiogenesis, thus shedding more light on the current understanding of G-CSF’s potential protective mechanism(s) in neonatal brain injury.

Schematic illustration of the potential mechanism of G-CSF action in hypoxia ischemia injury. In the acute phase of cerebral ischemia, G-CSF can protect the brain by inhibiting glutamate release, anti-inflammatory, anti-apoptotic, and inhibit edema formation. In subacute phase, GCSFR can stimulate endogenous neuronal regeneration, mobilization of bone marrow stem cells, driving neuronal regeneration and functional repair, and promote neovascularization (angiogenesis and neurogenesis) during the chronic phase
Regulation of cytokines and apoptosis by G-CSF in neonatal HIE
There are numerous studies that have shown the neuroprotective effect of G-CSF via inhibition of apoptosis and inflammation as well as by stimulating angiogenesis and neurogenesis both in adults and neonatal animal HI models [61,62,63,64]. Indeed, G-CSF is implicated in the regulation of cytokines that are being disrupted following HI insults by decreasing pro-inflammatory and increasing anti-inflammatory cytokines. Similarly, G-CSF inhibits pro-apoptotic factors and increases anti-apoptotic factors.
Regulation of cytokines by G-CSF in neonatal HIE
Prior studies have shown that interleukin-1 beta (IL-1ß) and tumor necrosis factor alpha (TNF-α) are early response cytokines in neuronal injury [65]. Increased expression of TNF-α induces neutrophil infiltration that increases endotheliocyte permeability and activates the matrix metalloproteinases (MMPs), which damage the blood-brain barrier (BBB) leading to swelling and degeneration of neurons and glial cells [66], while IL-1ß acts primarily through transcriptional activation of inducible nitric oxide synthase (iNOS) gene and nitric oxide (NO) generation thereby exacerbating brain injury [42]. These cytokines’ inflammatory response even persist in school-age children with neonatal encephalopathy (NE) showing poor neurodevelopmental outcome [48, 67]. Increased TNF-α expression also results in increase of caspase-3 cleavage and causes neuronal apoptosis [68]. Thus, TNF-α not only participates in neuronal inflammation but also in inducing apoptosis in neonatal HI injury. The imbalance between pro- and anti-inflammatory cytokines caused by HI damage also favors oligodendrocyte precursor proliferation into astrocytes instead of oligodendrocytes, and as a result, there is subsequent impairment of myelination, increased production of pro-inflammatory cytokines (PICs), and suppression of anti-inflammatory cytokine levels [69]. Activated astrocyte (astrogliosis) has both positive and negative roles in cytokine regulation following brain injury [70]; activated microglia have similar effects by expressing both pro-inflammatory and anti-inflammatory mediators depending on the degree and duration of insult being inflicted [4, 5, 71, 72].
The effect of G-CSF is demonstrated by preventing neuronal and glial pro-inflammatory cytokine expression. This is evidenced by preventing an overactivation of monocytes and lymphocytes by reducing the release of pro-inflammatory mediators while simultaneously activating the anti-inflammatory defense neutrophils [73]. Our previous study has shown that TNF-α and IL-1ß were upregulated after HI in neonatal rat model and were further elevated by rapamycin treatment with increased immunoreactivity of neuronal cell bodies. Their expression levels were subsequently decreased by G-CSF treatment [74]. Treatment with IL-10 is thought to decrease the level of TNF-α and IL-1ß after traumatic brain injury (TBI) [38]. Similarly, our previous study showed a decreased level of IL-10 expression after HI injury, while G-CSF administration was associated with its increased expression level thereby reducing further surges of TNF-α and IL-1ß and thus improving neurologic outcomes [74]. Xiao et al. [40] demonstrated that G-CSF treatment increased the mobilization of circulating CD34+ cells, polarizes T cell differentiation from Th1 to Th2 cells, and induces Th2 responses with the production of IL-4 and IL-10, accompanied by a decrease in production of IFN-ɣ and IL-2, thereby suppressing T cell proliferative responses to allogeneic stimulation that are anti-inflammatory and decreasing HI-induced injury, especially in the dentate gyrus by generating new neurons. G-CSF is said to significantly elevate the CD4 + CD25+ regulatory T cell subset in microglia-mediated reactive T cells as well as to inhibit MHC-II expression of microglia after lipopolysaccharide (LPS) activation or in the interactions of microglia and reactive T cells [75]. Other studies have shown that G-CSF effectively mobilized CD34-positive hematopoietic stem cells (HSC) in the preterm sheep thereby promoting proliferation of endogenous neural stem cells [76]. G-CSF can also stabilize the BBB and modulate neuroinflammation, and G-CSF is more neuroprotective in neonatal HI injury compared to adults [77].
Certain other studies have reported mixed results about the effectiveness of G-CSF treatment in ischemic stroke in terms of improving neurodegenerative and neurobehavioral outcomes, especially during the hyperacute and acute stage of HI injury [64, 78,79,80]. One study reported that G-CSF exacerbate brain damage as G-CSF increases the availability of neutrophils, which in turn enhances the inflammatory response in the brains of newborns [81]. Kallmunzer et al. [36] also reported a lack of neuroprotective or neuroregenerative effects of G-CSF in a rodent model of intracerebral hemorrhage. In a meta-analysis, England et al. [80] reported that G-CSF did not improve stroke outcome in individual patient with ischemic stroke when assessed by the National Institute of Health Stroke Scale (NIHSS) or Barthel Index (BI) while Huang et al. [64] analyzed 14 trials of G-CSF therapy in stroke and did not identify adequate evidence for the beneficial effects of this treatment modality in patients. Specifically, no favorable effects were noted on stroke outcomes including NIHSS score, the incidence of severe adverse events (SAEs), and mortality in patients treated with G-CSF versus control or placebo-treated patients. In another trial, the efficacy and tolerability of G-CSF were examined in patients with amyotrophic lateral sclerosis (ALS), a neurodegenerative disease, and the authors concluded that it has no clinical benefit in subjects with ALS [41], in contrast to previous studies [82, 83].
As some of these results and findings are mainly focused on adult ischemic stroke, it should be noted that differences do exist between the adult and neonate reaction to HI-induced brain insults, possibly due to immaturity of neonatal neurons. Specifically, there are differences in functional BBB response to acute experimental stroke between neonates and adults, as well as in gene expression of cerebral endothelial cells [25]. Other striking factors include initiation of G-CSF treatment, that is, the timing of G-CSF treatment might influence its neuroprotective effect; different dosages have been used both in ischemic stroke and neonatal HIE animal models, ranging from 10 to 250 μg/kg for subcutaneous injection and 5 to 60 mg/kg for intravenous injection [43, 61, 78]; route of administration varies according to each study; and duration of treatment ranges from 3 to 10 days [64, 80, 84]. Intranasal administration of G-CSF has also been examined in ischemic brain injury as potentially more effective and feasible [57]. All these factors influence the effectiveness of G-CSF across treatment modalities both in vivo and ex vivo. Therefore, such results should be judiciously interpreted and translated into neonates with HIE. Hence, there are still some loopholes and more studies are needed to examine the neuroprotective effect of G-CSF in neonatal brain injury at different times, doses, and duration in relation to regulation of neuroinflammatory and apoptotic cascades. Similarly, long-term neurofunctional assessment of G-CSF in neonatal hypoxia-ischemia brain injury should be warranted.
Regulation of apoptosis by G-CSF in neonatal HIE
Apoptosis can be triggered via the extrinsic pathway, which involves activation of cell surface death receptors, or the intrinsic pathway, which requires mitochondrial outer membrane permeabilization (MOMP) [85], which leads to loss of mitochondrial transmembrane potential, formation of transition pores, and production of reactive oxygen species, subsequently leading to release of cytochrome c from mitochondria resulting in activation of caspases and other effectors of DNA fragmentation and cell death [7, 86, 87]. Increased expression of pro-apoptotic mediators causes translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus, where it interacts with DNA and stimulates chromatin condensation. Overexpression of AIF can aggravate neonatal brain injury after HI that further increases its translocation [39].
Studies have shown that neonatal HI injury is associated with Bax translocation to mitochondria with a concomitant decrease in BCL-2, resulting in activation of caspase-3 leading to apoptotic cell death that peaks from 24 to 72 h post HI brain injury [33]. BAD is involved in apoptotic and nonapoptotic processes, and these dual activities are regulated by post-transcriptional modifications. Activation of BAX and BAD also promote MOMP [88]. The BCL-2 family proteins are key regulators of MOMP and play critical role in the intrinsic apoptotic pathway, classified into anti-apoptotic (Bcl-2, Bcl-xl, Bcl-w) and pro-apoptotic (Bax, Bak, Bim, Bid, Bad) [45]. The increased expression of pro-apoptotic markers is also influenced by the hormones involved in the pituitary–adrenal response [89], while in the extrinsic pathway, binding ligands to death receptors (TNF-α, Fas, TRAIL, etc.) leads to activation of caspase-8 [49]. In addition, BCL-2 has been demonstrated to play a critical role in preventing apoptosis induced by rapamycin derivatives that have been approved for the treatment of patients with various malignancies, thus suggesting that overexpression of antiapoptotic proteins such as BCL-2 might serve as a surrogate marker for resistance to rapalogues [90]. GSK-3ß is highly expressed in brain regions including the cerebral cortex, hippocampus, and cerebellum, and its overactivation is involved in neuronal pro-apoptosis, and dysregulation of this kinase has a devastating effect on neurodevelopment [91]. Its overexpression can increase caspase-3 activity [92], which in turn activates apoptosis.
G-CSF administration downregulates GSK-3ß activity, resulting in reduced neuronal cell death, apoptosis, and infarct volume, as well as upregulating anti-apoptotic protein Bcl-2 expression levels [45, 77]. Our previous study has demonstrated that increased Bax expression levels and cleaved caspase-3 (CC3) activation were attenuated significantly by G-CSF treatment and simultaneously increasing BCL-2 expression levels that were decreased following HI-induced injury. We further stated that the effect of G-CSF in modulating these apoptotic factors was abolished by rapamycin, an inhibitor of mTOR [74]. Other studies have reported similar neuroprotective effects of G-CSF by inhibiting apoptosis [63]. The neuronal anti-apoptotic action of G-CSF may also be mediated in part by the anti-apoptotic protein cIAP2 [93]. G-CSF also inhibits the mitochondrial-dependent activation of caspase-3, an apoptotic activator during HI injury [94]. Thus, G-CSF’s underlying mechanism(s) in the neuronal anti-apoptotic effect can be indirect through suppression of TNF-α, which increases caspase-3 cleavage or directly inhibiting caspase-3 and other pro-apoptotic mediators.
G-CSF in neurogenesis and angiogenesis
Neurogenesis is an important process for the reconstruction of neural networks and recovery of functional outcomes that are believed to continue throughout adulthood. It mainly occurs in the subventricular zone (SVZ) and subgranular layer of the hippocampal dentate gyrus, where the local environment tightly regulates neurogenesis [2]. Nonetheless, endogenous neurogenesis, stimulated by cerebral ischemia is not sufficient for the recovery of neurological functions [2]. The SVZ is more susceptible to neonatal HI brain injury located in the hippocampal dentate gyrus. Previous studies have shown that G-CSF facilitates bone marrow cell mobilization to the brain and drives neurogenesis and synaptic efficacy recovery thereby improving long-term functional outcome, which is mainly observed in the SVZ of the injured neonatal brain post-HI insult [61, 95, 96]. G-CSF also enhances concentrations of neurotrophic factors (GDNF and BDNF) that stimulate hippocampal neurogenesis as well as neuroplasticity by altering synaptic activity and possesses anti-apoptotic properties augmenting the neurogenic response to brain injury [97]. Endogenous role of G-CSF in the brain neuroprotective mechanism has also been demonstrated in ischemic model, where mice deficient of G-CSF showed overwhelming upregulation of matrix metalloproteinase 9 (MMP-9), a key factor in the activation of microglia and astrocytes, while treatment of G-CSF suppresses its expression [2, 98].
Angiogenesis is a physiological process by which new blood vessels are formed from pre-existing blood vessels. It is a complex and highly ordered process that relies upon extensive signaling networks both among and within endothelial cells (ECs) and their associated cells such as vascular endothelial growth factor (VEGF) proteins and angiopoietin-1 (Ang-1), which are required for angiogenesis [99]. Hypoxia-inducing factor-1 alpha (HIF-1α) is involved in early brain development and proliferation of neuronal progenitor cells. HIF also modulates cerebral hypoxic stress responses and activates endogenous neuroprotective systems during acute and late stages of HI damage of the developing brain [100]. HIF-1α induces VEGF expression and its receptors FMS-like tyrosine kinase (FLK-1) and fetal liver kinase-1 (Flk-1) in neurons facilitating blood reperfusion recovery, correlating with angiogenesis [25]. Late-stage of HIF-1α induction increases VEGF production that improves functional recovery and brain repair [101].
G-CSF administration increases local VEGF expression, which is necessary for vascular angiogenesis [102], thus corroborating its role in promoting angiogenesis through upregulation of VEGF expression via signaling pathways. Angiopeitin-1 (Ang-1), which is upregulated by G-CSF, is thought to reduce vascular solute permeability and contributes to vascular maturation and BBB stabilization by increasing the expression of BBB-related tight junction proteins (occludin, claudin-5, and zonula occludens protein 1) [103]. G-CSF treatment regulates the expression of VEGF and early growth response-1 (Erg-1) thereby ameliorating acute ischemic cerebral injury [29]. Mobilization of monocyte into blood vessels by G-CSF also stimulate angiogenesis [40]. Other reports argued that VEGF and MMP are involved in the initial opening of BBB within hours of an HI insult, which disrupt the basilar membrane and cause further damage to the BBB, especially VEGF-A which increases vascular permeability by uncoupling endothelial cell-cell junctions, resulting in BBB leakage and worsened outcomes [2, 103, 104]. Zhang et al. [25] stated that acute increases of VEGF results in the BBB leakage, whereas delayed upregulation of VEGF around the ischemic boundary area may prompt angiogenesis and reconstruction of the neurovascular unit (NVU). Thus, further research is needed to elucidate the pertinent role of VEGF after neonatal HI injury and its subsequent regulation by G-CSF.
Combinational therapy with other agents
Recent reports have advocated for combination strategies in neonatal HI brain injury and that it is more effective than G-CSF monotherapy. For instance, Yu and colleagues [105] demonstrated that both erythropoietin (Epo) and G-CSF combined produce functional recovery in a mouse model of hypoxic-ischemic brain injury in a time-dependent manner, and the underlying mechanisms may be the induction of HIF-1α activity in both cytosol and nucleus, and an early change in the cell fate determination from astrogliosis toward neurogenesis. Erythropoietin (EPO) a glycoprotein that controls erythropoiesis is expressed in neural progenitor cells, mature neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells. Epo has anti-apoptotic and anti-inflammatory effects and supports tissue remodeling by promoting neurogenesis, oligodendrogenesis, and angiogenesis [106]. Both G-CSF and EPO can readily cross the BBB, making them a possible candidate in neonates with HI insults. Liu et al. [107] reported that combination of G-CSF and EPO could synergistically promote proliferation of neural progenitor cells residing in the hippocampus and subventricular region of the brain. Another study examined the repetitive and long-term use of G-CSF+ EPO in stroke patients and reported similar neuroprotective effect with good tolerability and no associated adverse effects observed [108]. But in Yu and colleagues’ study, they reported that combined G-CSF and EPO does not show improvement during the chronic phase of HI injury [105]. Thus, the neuroprotective effects of combinational therapy of G-CSF and EPO in neonatal HIE need to be elucidated further to understand the underlying mechanisms.
Doycheva et al. [109] also concluded that G-CSF + SCF (stem cell factor) improved body weight, reduce brain-tissue atrophy, and improved neurological outcome following HI in the neonatal rat pup. Specifically, G-CSF + SCF induced more stable and long-lasting functional improvement in chronic strokes by increasing angiogenesis and neurogenesis through bone marrow-derived cells and the direct effects on stimulating neurons to form new neuronal networks [110, 111]. Hence, both G-CSF and SCF may work synergistically in improving the overall outcome of neonatal HI brain damage. Caspase-3 activation is reduced when G-CSF and SCF treatment are combined resulting in decreased apoptotic cell death, though other studies further reported that G-CSF/SCF and FL treatment did not affect apoptosis-inducing factor-dependent apoptosis or cell proliferation, and thus does not convey neuroprotection in neonatal HIE [112, 113]. Indeed, there are mixed reports about the neuroprotective effect of combinational G-CSF and SCF treatment in neonatal HI-induced brain injury. One study on HI mice demonstrated that G-CSF and SCF, given separately or in combination, have no neuroprotective effect, but rather a deleterious impact on neonatal excitotoxic brain damage [81].
G-CSF + hUCB can also decrease the number of MHCII+ cells not only in the corpus callosum and fornix but also in the cerebral peduncle. As G-CSF crosses the BBB, it acts upon neurons and glial cells through the G-CSF receptor. Indeed, glial cell activation has been demonstrated to downregulate expression of proinflammatory cytokines and to enhance neurogenesis [114]. One study demonstrated that intranasal administration of umbilical cord mesenchymal stem cells (UC-MSCs) significantly reduces neuroinflammation and protects hippocampal neurons, as well as increased concentration of the anti-inflammatory cytokine IL-10 in serum, thus contributing to neuroprotection [23]; similarly, UCB, especially its subtype EPCs (endothelial progenitor cells), has the ability to modulate neuroinflammation and reduce brain injury and behavioral deficits in perinatal HI brain injury [115]. Mesenchymal stem cells (MSCs) derived from human umbilical cord blood (hUCB) conferred neuroprotective and neuroregenerative benefits by improving angiogenesis and vasculogenesis [116]. MSCs plus G-CSF also decreased oxidative stress factors aggravated by HI brain injury [117]. G-CSF in combination with taurine is protective in primary cortical neurons against excitotoxicity induced by glutamate as well as suppresses endoplasmic reticulum (ER) stress [118]. Thus, synergistic therapies may exert and offer better functional improvements in neonatal HI brain injury, though more research is needed to explore the underlying mechanism(s).
Recently, Griva et al. [119] reported that the combination of neuroprotective treatments of G-CSF and enriched environment (EE) may enhance neuroprotection and it might be a more effective strategy for the treatment of neonatal hypoxic-ischemic brain injury by altering synaptic plasticity reflected by increased synaptophysin expression levels thus further enhancing cognitive function. Doycheva et al. [30] demonstrated that G-CSF + Ab improved body weight, reduced brain tissue damage, and improved long-term neurological function when assessed at 96 h and 5 weeks post HI in the neonatal rat pups, as well as conferring greater neuroprotection by depleting neutrophil accumulation, while G-CSF + metyrapone treatment not only lower caspase-3 expression level but also reduce corticosterone levels in neonates after HI injury [89]. Surprisingly, there has not been any study evaluating combinational therapy of G-CSF and therapeutic hypothermia (TH), as other modalities have been tried and underwent and/or are undergoing clinical trials in neonatal HIE, such as with epo and TH [120,121,122], xenon and TH [51, 123, 124], melatonin and TH [46, 47], and allopurinol and TH [125]. Thus, more study is needed to explore and evaluate the neuroprotective effect of G-CSF in combination with therapeutic hypothermia as well as other emerging agents.
G-CSF-mediated signaling pathways in neonatal HIE
Stimulation of G-CSF by its receptor activates many downstream signaling pathways such as the Janus kinase (JAK)/signal transducer and activator of transcription (STAT), the Ras/mitogen-activated protein kinase (MAPK), and the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathways [50] thereby exerting its neuroprotective effect.
The JAK-STAT signaling pathway is a chain of interactions between protein cells and is involved in many cellular processes that communicate information from chemical signals outside of a cell to the cell nucleus, resulting in the activation of genes through transcriptional processes [126]. The neuroprotective effect of G-CSF is manifested by activating the anti-apoptotic pathway via the JAK/STAT3 signaling, and it does so by suppressing the pro-apoptotic mediators and upregulating anti-apoptotic mediators via binding to its receptors (G-CSFR) in neurons. Moreover, G-CSF increases the activation of STAT3 pathway in glial cells together with increased cIAP2 expression which is a member of the inhibitor of apoptosis protein (IAP) family, subsequently regulating the activity of both initiator (caspase-9) and effector caspases (caspase-3 and -7) in ischemic mouse models [40, 94]. It can directly activate the JAK/STAT3 pathway as well [33, 93, 127], thereby promoting neurogenesis. Pim-1 increases cell survival through the regulation of bcl-2 proteins and that its upregulation after HI is enhanced by G-CSF treatment. Its expression is said to be paralleled to STAT3 expression, suggesting an association between the two in ischemic pathology [33, 94]. G-CSF also increased expression of STAT3 in the penumbra mediated by G-CSFR [128]. In short, these substrates and/or proteins are regulated by G-CSF and its receptor along the JAK/STATS signaling pathway following HI injury, which in turn favors neuroprotection.
The PI3K/AKT pathway is a key signaling pathway that participates in various cellular processes such as cell proliferation, survival, differentiation, and apoptosis [129, 130]. G-CSF enhances neurogenesis and neuroblast migration after stroke by regulating the PI3K/Akt pathway as well as modulates the NMDA receptor of glial cells exposed to PLS-induced brain damage [68]. The nuclear factor-kappa B (NF-κB) is a central transcriptional factor that is regulated and activated by Akt accompanied by activation of the inhibitor of κB (IKB) kinase (IKK) [131]. Activation of NF-κB, especially the canonical pathway, triggers the production of pro-inflammatory cytokines (PICs) and nitric oxide (NO) [132]. Phosphorylated Akt by G-CSF inactivates the canonical NF-κB pathway and inhibit the production of PICs and NO, further ameliorating neuroinflammation. Moreover, G-CSF phosphorylates Akt leading to downstream inactivation of GSK-3ß that will ultimately decrease apoptosis. G-CSF as well reduces adherens (VCAM-1 and ICAM-1) and increases tight junction (claudin 3 and 5) protein expression levels via the G-CSFR/PI3K/Akt/GSK-3ß signaling pathway [50].
The mammalian target of rapamycin/p70 ribosomal S6 protein kinase (mTOR/p70S6K) pathway has been implicated in neurogenesis as well, by decreasing the expression of PICs [133], as well as increasing IL-10 expression [134]. mTOR acts as a molecular system integrator to support organismal and cellular interactions with the environment [135], which regulates cellular metabolism, growth, and proliferation through two protein complexes, mTORC1 and mTORC2. mTOR activity is also thought to upregulate the translation of synaptic mRNAs via 4E-BP and S6K [136] by facilitating neuronal plasticity and activity [137]. Previous studies have demonstrated a decreased activation of S6K by rapamycin, an mTOR inhibitor, as well as loss of S6K function leads to increased astrocyte death in ischemic models, while G-CSF treatment increases S6K expression levels [138, 139], driving cellular functions as S6K phosphorylates and activates several substrates that promote mRNA translation initiation and other cellular processes [140]. Our previous study has shown that treatment with G-CSF decreases inflammatory mediators and apoptotic factors, attenuating neuroinflammation and neuronal apoptosis via the mTOR/p70S6K signaling pathway, which represents a potential target for treating HI induced brain damage in neonatal HIE [74].
The PI3K/AKT/mTOR pathway is an intracellular signaling pathway important in regulating the cell cycle. Therefore, it is directly related to cellular quiescence, proliferation, cancer, and longevity. PI3K activation phosphorylates and activates AKT, localizing it in the plasma membrane [52, 141]. G-CSF can upregulate brain-derived neurotrophic factor (BDNF) which in turn induces autophagy through the PI3K/Akt/mTOR pathway [2]. The neurotrophic nuclear transcription factor phosphorylated cAMP-responsive element-binding protein at serine 133 (pCREBSer-133) serves an important role in neurological regulation of ion channel function, neuronal differentiation and maturation, and the processes of learning and memory. This transcription factor is decreased during HI insult, while treatment of G-CSF was shown to significantly increase the expression of pRaf-pERK1/2-pCREBSer-133 pathway in neonates exposed to perinatal hypoxia [61]. One study has demonstrated that G-CSF treatment inhibits steroidogenesis through activation of the JAK2/PI3K/PDE3B signaling pathway by reducing the levels of cAMP expression in HI-induced brain injury [142]. Activation of ERK has been shown to be neuroprotective, both in adults and neonatal brain injury, while MAPK p38 is best known for transduction of stress-related signals, regulation of inflammatory gene production, and NF- κB recruitment to selected targets, and both ERK and MARK p38 are regulated by G-CSF [37]. G-CSF downregulates the activation of the phosphorylated JNK and c-jun pathway in the cerebral ischemia-reperfusion rats model [44].
Thus, G-CSF plays diverse roles in neonatal HIE through diverse signaling pathways. Modi et al. [45] summarizes the steps and effect of G-CSF across the signaling pathways that have been implicated in neonatal HI injury either through inhibition or upregulation and phosphorylation of their substrates thereby eliciting neuroprotection.
Conclusion
There are numerous studies that have shown the important role G-CSF plays in neurodegenerative diseases, ischemic stroke, and traumatic brain injury both using in vivo and ex vivo models. Recent research has specifically focused on its neuroprotective effect in neonatal HIE with both positive and mixed results. Indeed, G-CSF exerts a pivotal role in the control of immune response and acts as an anti-inflammatory cytokine, preventing an overactivation of monocytes and lymphocytes by reducing the release of pro-inflammatory cytokines as well as stabilizing the BBB and modulating neuroinflammation; inhibits pro-apoptotic mediators; enhances concentration of neurotrophic factors and facilitates bone marrow cell mobilization thereby driving neurogenesis; increases local VEGF expression necessary for vascular angiogenesis; upregulates Ang-1 that reduces vascular solute permeability; and contributes to vascular maturation and BBB stabilization. Despite these progresses being made, there are still few experimentation models of neonatal HIE and G-CSF’s neuroprotectiveness either directly or through signaling pathways, and extrapolation of adults’ stroke models is challenging due to the evolving neonatal brain. Also, there are mixed results about the effectiveness of G-CSF in improving HI-induced brain injury in neonates, specifically in the area of initiation of treatment, dosage, route of administration, and duration of treatment; this calls for more in-depth research to elucidate its underlying mechanisms and pertinent role in neuroprotection, as well as its long-term effect in neurological and behavioral outcomes.
Recent studies have focused on combinational treatment in neonatal HI brain injury and that it is more effective than G-CSF monotherapy. Therapeutic hypothermia has also been advocated for with other agents such as EPO, melanin, xenon, stem cells, and anticonvulsants with promising results. As most of these agents have controversial effectiveness in improving HI-induced brain injury and most of them are under clinical trials for efficacy and safety, at present, it appears that combination of these therapeutic agents with TH could be the promising intervention strategies to treat newborns suffering from HIE, while awaiting the outcomes of both preclinical and clinical trials that are under investigation.
In this review, we highlighted recent researches in the role of G-CSF in regulating cytokines and inhibition of apoptotic mediators, as well as promoting neurogenesis and angiogenesis thereby enhancing cell survival and proliferation, and modulation of inflammatory responses in the injured neonatal brain through activation of multistep signaling pathways.
Availability of data and materials
Not applicable.
Abbreviations
- AIP:
-
Apoptosis-inducing factor
- Akt:
-
Protein kinase B
- Ang-1:
-
Angiopoietin-1
- BAD:
-
Bcl-2-antagonist of cell death
- BAX:
-
Bcl-2-associated X protein
- BBB:
-
Blood-brain barrier
- BLC-2:
-
B cell lymphoma 2
- BDNF:
-
Brain-derived neurotrophic factor
- cAMP:
-
Cyclic adenosine monophosphate
- CC3:
-
Cleaved caspase-3
- CNS:
-
Central nervous system
- EE:
-
Enriched environment
- EEG:
-
Electroencephalogram
- EPCs:
-
Endothelial progenitor cells
- EPO:
-
Erythropoietin
- FL:
-
Fms-related tyrosine kinase 3 ligand
- FLK-1:
-
Fms-like tyrosine kinase 1
- GABA:
-
Gamma-aminobutyric acid
- G-CSF:
-
Granulocyte-colony stimulating factor
- G-CSFR:
-
Granulocyte-colony stimulating factor receptor
- GDNF:
-
Growth-derived neurotrophic factor
- GSK-3β:
-
Glycogen synthase kinase-3 beta
- HI:
-
Hypoxia ischemia
- HIE:
-
Hypoxic ischemic encephalopathy
- HIF-1α:
-
Hypoxia inducing factor-1 alpha
- HSCs:
-
Hematopoietic stem cells
- hUCB:
-
Human umbilical cord blood
- IAP:
-
Inhibitor of apoptosis protein
- ICAM1:
-
Intracellular adhesion molecule 1
- IL-1β:
-
Interleukin-1 beta
- iNOS:
-
Inducible nitric oxide synthase
- JAK:
-
Janus kinase
- MAPK:
-
Mitogen-associated protein kinase
- MHC:
-
Major histocompatibility complex
- MMPs:
-
Matrix metalloproteinases
- MMP-9:
-
Matrix metalloproteinase-9
- MOMP:
-
Mitochondrial outer membrane permeabilization
- mTOR:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor-kappa B
- NMDA:
-
N-methyl-d-aspartic acid
- NO:
-
Nitric oxide
- NVU:
-
Neurovascular unit
- PI3K:
-
Phosphatidylinositol 3 kinase
- PICs:
-
Pro-inflammatory cytokines
- PDKI:
-
Pyruvate dehydrogenase isoenzyme
- P70S6K:
-
P70 ribosomal S6 protein kinase
- RCT:
-
Randomized control trial
- SAEs:
-
Severe adverse effects
- SCF:
-
Stem cell factor
- STAT:
-
Signal transducer and activator of transcription
- SVZ:
-
Subventricular zone
- TBI:
-
Traumatic brain injury
- TH:
-
Therapeutic hypothermia
- TNF-α:
-
Tumor necrosis factor alpha
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- UCB:
-
Umbilical cord blood
- UC-MSCs:
-
Umbilical cord mesenchymal stem cells
- VCAM1:
-
Vascular cell adhesion molecule 1
- VEGF:
-
Vascular endothelial growth factor
References
Nair J, Kumar VHS. Current and emerging therapies in the management of hypoxic ischemic encephalopathy in neonates. Children. 2018;5(7):99 Published 2018 Jul 19. https://doi.org/10.3390/children5070099.
Dixon BJ, Reis C, Ho WM, Tang J, Zhang JH. Neuroprotective strategies after neonatal hypoxic ischemic encephalopathy. Int J Mol Sci. 2015;16(9):22368–401 Published 2015 Sep 15. https://doi.org/10.3390/ijms160922368.
Jelin AC, Salmeen K, Gano D, Burd I, Thiet MP. Perinatal neuroprotection update. F1000Res. 2016;5:F1000 Faculty Rev–1939 Published 2016 Aug 9. https://doi.org/10.12688/f1000research.8546.1.
Riljak V, Kraf J, Daryanani A, Jiruška P, Otáhal J. Pathophysiology of perinatal hypoxic-ischemic encephalopathy - biomarkers, animal models and treatment perspectives. Physiol Res. 2016;65(Suppl 5):S533–45 https://doi.org/10.33549/physiolres.933541.
Qin X, Cheng J, Zhong Y, et al. Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front Mol Neurosci. 2019;12(88) Published 2019 Apr 11. https://doi.org/10.3389/fnmol.2019.00088.
Zhao M, Zhu P, Fujino M, Zhuang J, Guo H, Sheikh I, Zhao L, Li XK. Oxidative stress in hypoxic-ischemic encephalopathy: molecular mechanisms and therapeutic strategies. Int J Mol Sci. 2016;17(12):2078 https://doi.org/10.3390/ijms17122078 PMID: 27973415; PMCID: PMC5187878.
Kochanek PM, Jackson TC, Ferguson NM, Carlson SW, Simon DW, Brockman EC, Ji J, Bayır H, Poloyac SM, Wagner AK, Kline AE, Empey PE, Clark RS, Jackson EK, Dixon CE. Emerging therapies in traumatic brain injury. Semin Neurol. 2015;35(1):83–100 https://doi.org/10.1055/s-0035-1544237 Epub 2015 Feb 25. PMID: 25714870; PMCID: PMC4356170.
Johnston MV, Fatemi A, Wilson MA, Northington F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011;10(4):372–82 https://doi.org/10.1016/S1474-4422(11)70016-3 PMID: 21435600; PMCID: PMC3757153.
Arteaga O, Álvarez A, Revuelta M, Santaolalla F, Urtasun A, Hilario E. Role of antioxidants in neonatal hypoxic-ischemic brain injury: new therapeutic approaches. Int J Mol Sci. 2017;18(2):265 Published 2017 Jan 28. https://doi.org/10.3390/ijms18020265.
James A, Patel V. Hypoxic ischaemic encephalopathy. Paediatr Child Health. 2014;24:9.
Chalak LF, Sánchez PJ, Adams-Huet B, Laptook AR, Heyne RJ, Rosenfeld CR. Biomarkers for severity of neonatal hypoxic-ischemic encephalopathy and outcomes in newborns receiving hypothermia therapy. J Pediatr. 2014;164(3):468–74.e1 https://doi.org/10.1016/j.jpeds.2013.10.067.
Graham EM, Everett AD, Delpech JC, Northington FJ. Blood biomarkers for evaluation of perinatal encephalopathy: state of the art. Curr Opin Pediatr. 2018;30(2):199–203 https://doi.org/10.1097/MOP.0000000000000591.
Mathur AM, Neil JJ, Inder TE. Understanding brain injury and neurodevelopmental disabilities in the preterm infant: the evolving role of advanced magnetic resonance imaging. Semin Perinatol. 2010;34(1):57–66 https://doi.org/10.1053/j.semperi.2009.10.006.
Horn AR, Swingler GH, Myer L, et al. Early clinical signs in neonates with hypoxic ischemic encephalopathy predict an abnormal amplitude-integrated electroencephalogram at age 6 hours. BMC Pediatr. 2013;13:52 Published 2013 Apr 10. https://doi.org/10.1186/1471-2431-13-52.
Abbasi H, Unsworth CP. Electroencephalogram studies of hypoxic ischemia in fetal and neonatal animal models. Neural Regen Res. 2020;15(5):828–37 https://doi.org/10.4103/1673-5374.268892.
Buonocore G, Perrone S, Turrisi G, Kramer BW, Balduini W. New pharmacological approaches in infants with hypoxic-ischemic encephalopathy. Curr Pharm Design. 2012;18:3086 https://doi.org/10.2174/1381612811209023086.
Singhi S, Johnston M. Recent advances in perinatal neuroprotection. F1000Res. 2019;8:F1000 Faculty Rev–2031 Published 2019 Nov 29. https://doi.org/10.12688/f1000research.20722.1.
Koehler RC, Yang ZJ, Lee JK, Martin LJ. Perinatal hypoxic-ischemic brain injury in large animal models: relevance to human neonatal encephalopathy. J Cereb Blood Flow Metab. 2018;38(12):2092–111 https://doi.org/10.1177/0271678X18797328.
Dereymaeker A, Matic V, Vervisch J, et al. Automated EEG background analysis to identify neonates with hypoxic-ischemic encephalopathy treated with hypothermia at risk for adverse outcome: a pilot study. Pediatr Neonatol. 2019;60(1):50–8 https://doi.org/10.1016/j.pedneo.2018.03.010.
Romeo DM, Bompard S, Serrao F, et al. Early neurological assessment in infants with hypoxic ischemic encephalopathy treated with therapeutic hypothermia. J Clin Med. 2019;8(8):1247 Published 2019 Aug 18. https://doi.org/10.3390/jcm8081247.
Lee CYZ, Chakranon P, Lee SWH. Comparative efficacy and safety of neuroprotective therapies for neonates with hypoxic ischemic encephalopathy: a network meta-analysis. Front Pharmacol. 2019;10:1221 Published 2019 Oct 25. https://doi.org/10.3389/fphar.2019.01221.
Silveira RC, Procianoy RS. Hypothermia therapy for newborns with hypoxic ischemic encephalopathy. J Pediatr. 2015;91(6 Suppl 1):S78–83 https://doi.org/10.1016/j.jped.2015.07.004.
McDonald CA, Djuliannisaa Z, Petraki M, et al. Intranasal delivery of mesenchymal stromal cells protects against neonatal hypoxic ischemic brain injury. Int J Mol Sci. 2019;20(10):2449 Published 2019 May 17. https://doi.org/10.3390/ijms20102449.
Reiss J, Sinha M, Gold J, Bykowski J, Lawrence SM. Outcomes of infants with mild hypoxic ischemic encephalopathy who did not receive therapeutic hypothermia. Biomed Hub. 2019;4(3):1–9 Published 2019 Oct 10. https://doi.org/10.1159/000502936.
Zhang W, Zhu L, An C, et al. The blood brain barrier in cerebral ischemic injury – disruption and repair. Brain Hemorrhages. 2020;1:34–53 https://doi.org/10.1016/j.hest.2019.12.004.
Shankaran S, Laptook AR, Pappas A, et al. Effect of depth and duration of cooling on deaths in the NICU among neonates with hypoxic ischemic encephalopathy: a randomized clinical trial. JAMA. 2014;312(24):2629–39 https://doi.org/10.1001/jama.2014.16058.
Wassink G, Davidson JO, Lear CA, et al. A working model for hypothermic neuroprotection. J Physiol. 2018;596(23):5641–54 https://doi.org/10.1113/JP274928.
Calipari ES, Godino A, Peck EG, et al. Granulocyte-colony stimulating factor controls neural and behavioral plasticity in response to cocaine. Nat Commun. 2018;9(1):9 Published 2018 Jan 16. https://doi.org/10.1038/s41467-017-01881-x.
Zhou DG, Shi YH, Cui YQ. Impact of G-CSF on expressions of Egr-1 and VEGF in acute ischemic cerebral injury. Exp Ther Med. 2018;16(3):2313–8 https://doi.org/10.3892/etm.2018.6486.
Doycheva DM, Hadley T, Li L, Applegate RL 2nd, Zhang JH, Tang J. Anti-neutrophil antibody enhances the neuroprotective effects of G-CSF by decreasing number of neutrophils in hypoxic ischemic neonatal rat model. Neurobiol Dis. 2014;69:192–9 https://doi.org/10.1016/j.nbd.2014.05.024.
D’Souza A, Jaiyesimi I, Trainor L, Venuturumili P. Granulocyte colony-stimulating factor administration: adverse events. Transfus Med Rev. 2008;22(4):280–90 https://doi.org/10.1016/j.tmrv.2008.05.005.
Molineux G. Granulocyte colony-stimulating factors. Cancer Treat Res. 2011;157:33–53 https://doi.org/10.1007/978-1-4419-7073-2_3.
Yata K, Matchett GA, Tsubokawa T, Tang J, Kanamaru K, Zhang JH. Granulocyte-colony stimulating factor inhibits apoptotic neuron loss after neonatal hypoxia-ischemia in rats. Brain Res. 2007;1145:227–38 https://doi.org/10.1016/j.brainres.2007.01.144.
Huang H, Zhang Q, Liu J, Hao H, Jiang C, Han W. Granulocyte-colony stimulating factor (G-CSF) accelerates wound healing in hemorrhagic shock rats by enhancing angiogenesis and attenuating apoptosis. Med Sci Monit. 2017;23:2644–53 https://doi.org/10.12659/msm.904988 PMID: 28559534; PMCID: PMC5461887.
Wallner S, Peters S, Pitzer C, Resch H, Bogdahn U, Schneider A. The granulocyte-colony stimulating factor has a dual role in neuronal and vascular plasticity. Front Cell Dev Biol. 2015;3(48) https://doi.org/10.3389/fcell.2015.00048 PMID: 26301221; PMCID: PMC4528279.
Kallmünzer B, Tauchi M, Schlachetzki JC, et al. Granulocyte colony-stimulating factor does not promote neurogenesis after experimental intracerebral haemorrhage. Int J Stroke. 2014;9(6):783–8 https://doi.org/10.1111/ijs.12217.
Vexler ZS, Yenari MA. Does inflammation after stroke affect the developing brain differently than adult brain? Dev Neurosci. 2009;31(5):378–93 https://doi.org/10.1159/000232556.
Kamm K, Vanderkolk W, Lawrence C, Jonker M, Davis AT. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J Trauma. 2006;60(1):152–7 https://doi.org/10.1097/01.ta.0000196345.81169.a1.
Li T, Li K, Zhang S, et al. Overexpression of apoptosis inducing factor aggravates hypoxic-ischemic brain injury in neonatal mice. Cell Death Dis. 2020;11(1):77 Published 2020 Jan 30. https://doi.org/10.1038/s41419-020-2280-z.
Xiao BG, Lu CZ, Link H. Cell biology and clinical promise of G-CSF: immunomodulation and neuroprotection. J Cell Mol Med. 2007;11(6):1272–90 https://doi.org/10.1111/j.1582-4934.2007.00101.x.
Amirzagar N, Nafissi S, Tafakhori A, Modabbernia A, Amirzargar A, Ghaffarpour M, Siroos B, Harirchian MH. Granulocyte colony-stimulating factor for amyotrophic lateral sclerosis: a randomized, double-blind, placebo-controlled study of Iranian patients. J Clin Neurol. 2015;11(2):164–71 https://doi.org/10.3988/jcn.2015.11.2.164 PMID: 25851895; PMCID: PMC4387482.
Ziemka-Nalecz M, Jaworska J, Zalewska T. Insights into the neuroinflammatory responses after neonatal hypoxia-ischemia. J Neuropathol Exp Neurol. 2017;76(8):644–54 https://doi.org/10.1093/jnen/nlx046.
Pastuszko P, Schears GJ, Kubin J, Wilson DF, Pastuszko A. Granulocyte colony-stimulating factor significantly decreases density of hippocampal caspase 3-positive nuclei, thus ameliorating apoptosis-mediated damage, in a model of ischaemic neonatal brain injury. Interact Cardiovasc Thorac Surg. 2017;25(4):600–5 https://doi.org/10.1093/icvts/ivx047.
Li YG, Liu XL, Zheng CB. Granulocyte colony-stimulating factor regulates JNK pathway to alleviate damage after cerebral ischemia reperfusion. Chin Med J. 2013;126(21):4088–92.
Modi J, Menzie-Suderam J, Xu H, et al. Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J Biomed Sci. 2020;27(1):19 Published 2020 Jan 6. https://doi.org/10.1186/s12929-019-0597-7.
Paprocka J, Kijonka M, Rzepka B, Sokół M. Melatonin in hypoxic-ischemic brain injury in term and preterm babies. Int J Endocrinol. 2019;2019:9626715 Published 2019 Feb 20. https://doi.org/10.1155/2019/9626715.
Robertson NJ, Martinello K, Lingam I, et al. Melatonin as an adjunct to therapeutic hypothermia in a piglet model of neonatal encephalopathy: a translational study. Neurobiol Dis. 2019;121:240–51 https://doi.org/10.1016/j.nbd.2018.10.004.
Zareen Z, Strickland T, Eneaney VM, et al. Cytokine dysregulation persists in childhood post neonatal encephalopathy. BMC Neurol. 2020;20(1):115 Published 2020 Mar 30. https://doi.org/10.1186/s12883-020-01656-w.
Thornton C, Leaw B, Mallard C, Nair S, Jinnai M, Hagberg H. Cell death in the developing brain after hypoxia-ischemia. Front Cell Neurosci. 2017;11:248 Published 2017 Aug 23. https://doi.org/10.3389/fncel.2017.00248.
Li L, McBride DW, Doycheva D, et al. G-CSF attenuates neuroinflammation and stabilizes the blood-brain barrier via the PI3K/Akt/GSK-3β signaling pathway following neonatal hypoxia-ischemia in rats. Exp Neurol. 2015;272:135–44 https://doi.org/10.1016/j.expneurol.2014.12.020.
Sabir H, Osredkar D, Maes E, Wood T, Thoresen M. Xenon combined with therapeutic hypothermia is not neuroprotective after severe hypoxia-ischemia in neonatal rats. PLoS One. 2016;11(6):e0156759 Published 2016 Jun 2. https://doi.org/10.1371/journal.pone.0156759.
King D, Yeomanson D, Bryant HE. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015;37(4):245–51 https://doi.org/10.1097/MPH.0000000000000329.
McCollum M, Ma Z, Cohen E, et al. Post-MPTP treatment with granulocyte colony-stimulating factor improves nigrostriatal function in the mouse model of Parkinson’s disease. Mol Neurobiol. 2010;41(2-3):410–9 https://doi.org/10.1007/s12035-010-8118-4.
Meuer K, Pitzer C, Teismann P, et al. Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson’s disease. J Neurochem. 2006;97(3):675–86 https://doi.org/10.1111/j.1471-4159.2006.03727.x.
Wu J-Y, Modi J, Menzie J, Chou HY, Tao R, Morrell A, Trujillo P, Medley K, Altamimi A, Jasica Shen J, Prentice H. Granulocyte colony stimulating factor (GCSF) gene therapy in stroke and Alzheimer’s disease model. J Neurol Exp Neurosci. 2018;4(Supplement 1):S17.
Lee ST, Chu K, Jung KH, Ko SY, Kim EH, Sinn DI, Lee YS, Lo EH, Kim M, Roh JK. Granulocyte colony-stimulating factor enhances angiogenesis after focal cerebral ischemia. Brain Res. 2005;1058(1-2):120–8 https://doi.org/10.1016/j.brainres.2005.07.076 Epub 2005 Sep 8. PMID: 16150422.
Sun BL, He MQ, Han XY, Sun JY, Yang MF, Yuan H, Fan CD, Zhang S, Mao LL, Li DW, Zhang ZY, Zheng CB, Yang XY, Li YV, Stetler RA, Chen J, Zhang F. Intranasal delivery of granulocyte colony-stimulating factor enhances its neuroprotective effects against ischemic brain injury in rats. Mol Neurobiol. 2016;53(1):320–30 https://doi.org/10.1007/s12035-014-8984-2 Epub 2014 Nov 29. PMID: 25432887.
Hasselblatt M, Jeibmann A, Riesmeier B, Maintz D, Schäbitz WR. Granulocyte-colony stimulating factor (G-CSF) and G-CSF receptor expression in human ischemic stroke. Acta Neuropathol. 2007;113(1):45–51 https://doi.org/10.1007/s00401-006-0152-y Epub 2006 Oct 18. PMID: 17047971.
Schäbitz WR, Laage R, Vogt G, et al. AXIS: a trial of intravenous granulocyte colony-stimulating factor in acute ischemic stroke. Stroke. 2010;41(11):2545–51 https://doi.org/10.1161/STROKEAHA.110.579508.
Ringelstein EB, Thijs V, Norrving B, et al. Granulocyte colony-stimulating factor in patients with acute ischemic stroke: results of the AX200 for Ischemic Stroke trial. Stroke. 2013;44(10):2681–7 https://doi.org/10.1161/STROKEAHA.113.001531.
Chen WF, Hsu JH, Lin CS, et al. Granulocyte-colony stimulating factor alleviates perinatal hypoxia-induced decreases in hippocampal synaptic efficacy and neurogenesis in the neonatal rat brain. Pediatr Res. 2011;70(6):589–95 https://doi.org/10.1203/PDR.0b013e3182324424.
Popa-Wagner A, Stöcker K, Balseanu AT, Rogalewski A, Diederich K, Minnerup J, Margaritescu C, Schäbitz WR. Effects of granulocyte-colony stimulating factor after stroke in aged rats. Stroke. 2010;41(5):1027–31 https://doi.org/10.1161/STROKEAHA.109.575621 Epub 2010 Apr 1. PMID: 20360546.
Kim BR, Shim JW, Sung DK, et al. Granulocyte stimulating factor attenuates hypoxic-ischemic brain injury by inhibiting apoptosis in neonatal rats. Yonsei Med J. 2008;49(5):836–42 https://doi.org/10.3349/ymj.2008.49.5.836.
Huang X, Liu Y, Bai S, Peng L, Zhang B, Lu H. Granulocyte colony stimulating factor therapy for stroke: a pairwise meta-analysis of randomized controlled trial. PLoS One. 2017;12(4):e0175774 Published 2017 Apr 13. https://doi.org/10.1371/journal.pone.0175774.
Sun Y, Calvert JW, Zhang JH. Neonatal hypoxia/ischemia is associated with decreased inflammatory mediators after erythropoietin administration. Stroke. 2005;36(8):1672–8 https://doi.org/10.1161/01.STR.0000173406.04891.8c.
Borjini N, Sivilia S, Giuliani A, et al. Potential biomarkers for neuroinflammation and neurodegeneration at short and long term after neonatal hypoxic-ischemic insult in rat. J Neuroinflammation. 2019;16(1):194 Published 2019 Oct 28. https://doi.org/10.1186/s12974-019-1595-0.
Aslam S, Molloy EJ. Biomarkers of multiorgan injury in neonatal encephalopathy. Biomark Med. 2015;9(3):267–75 https://doi.org/10.2217/bmm.14.116.
Yang YN, Su YT, Wu PL, et al. Granulocyte colony-stimulating factor alleviates bacterial-induced neuronal apoptotic damage in the neonatal rat brain through epigenetic histone modification. Oxid Med Cell Longev. 2018;2018:9797146 Published 2018 Feb 1. https://doi.org/10.1155/2018/9797146.
Sisa C, Agha-Shah Q, Sanghera B, Carno A, Stover C, Hristova M. Properdin: a novel target for neuroprotection in neonatal hypoxic-ischemic brain injury. Front Immunol. 2019;10:2610 Published 2019 Nov 29. https://doi.org/10.3389/fimmu.2019.02610.
Welcome MO. Neuroinflammation in CNS diseases: molecular mechanisms and the therapeutic potential of plant derived bioactive molecules. Pharma Nutr. 2020;11:100176 https://doi.org/10.1016/j.phanu.2020.100176.
Liu F, McCullough LD. Inflammatory responses in hypoxic ischemic encephalopathy. Acta Pharmacol Sin. 2013;34(9):1121–30 https://doi.org/10.1038/aps.2013.89.
Chaparro-Huerta V, Flores-Soto ME, Merin Sigala ME, et al. Proinflammatory cytokines, enolase and S-100 as early biochemical indicators of hypoxic-ischemic encephalopathy following perinatal asphyxia in newborns. Pediatr Neonatol. 2017;58(1):70–6 https://doi.org/10.1016/j.pedneo.2016.05.001.
Boneberg EM, Hartung T. Molecular aspects of anti-inflammatory action of G-CSF. Inflamm Res. 2002;51(3):119–28 https://doi.org/10.1007/pl00000283.
Dumbuya JS, Chen L, Shu SY, et al. G-CSF attenuates neuroinflammation and neuronal apoptosis via the mTOR/p70SK6 signaling pathway in neonatal hypoxia-ischemia rat model. Brain Res. 2020;1739:146817 https://doi.org/10.1016/j.brainres.2020.146817.
Peng W. Neuroprotective effects of G-CSF administration in microglia-mediated reactive T cell activation in vitro. Immunol Res. 2017;65(4):888–902 https://doi.org/10.1007/s12026-017-8928-9 PMID: 28646409.
Jellema RK, Lima Passos V, Zwanenburg A, et al. Cerebral inflammation and mobilization of the peripheral immune system following global hypoxia-ischemia in preterm sheep. J Neuroinflammation. 2013;10:13 Published 2013 Jan 24. https://doi.org/10.1186/1742-2094-10-13.
Li L, Klebe D, Doycheva D, et al. G-CSF ameliorates neuronal apoptosis through GSK-3β inhibition in neonatal hypoxia-ischemia in rats. Exp Neurol. 2015;263:141–9 https://doi.org/10.1016/j.expneurol.2014.10.004.
Schlager GW, Griesmaier E, Wegleiter K, et al. Systemic G-CSF treatment does not improve long-term outcomes after neonatal hypoxic-ischaemic brain injury. Exp Neurol. 2011;230(1):67–74 https://doi.org/10.1016/j.expneurol.2010.11.021.
Prasad K, Kumar A, Sahu JK, et al. Mobilization of stem cells using G-CSF for acute ischemic stroke: a randomized controlled, pilot study. Stroke Res Treat. 2011;2011:283473 https://doi.org/10.4061/2011/283473.
England TJ, Sprigg N, Alasheev AM, et al. Granulocyte-colony stimulating factor (G-CSF) for stroke: an individual patient data meta-analysis. Sci Rep. 2016;6:36567 Published 2016 Nov 15. https://doi.org/10.1038/srep36567.
Keller M, Simbruner G, Górna A, et al. Systemic application of granulocyte-colony stimulating factor and stem cell factor exacerbates excitotoxic brain injury in newborn mice. Pediatr Res. 2006;59(4 Pt 1):549–53 https://doi.org/10.1203/01.pdr.0000205152.38692.81.
Zhang Y, Wang L, Fu Y, Song H, Zhao H, Deng M, Zhang J, Fan D. Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2009;10(5-6):430–1 https://doi.org/10.3109/17482960802588059 PMID: 19922135.
Duning T, Schiffbauer H, Warnecke T, Mohammadi S, Floel A, Kolpatzik K, Kugel H, Schneider A, Knecht S, Deppe M, Schäbitz WR. G-CSF prevents the progression of structural disintegration of white matter tracts in amyotrophic lateral sclerosis: a pilot trial. PLoS One. 2011;6(3):e17770 https://doi.org/10.1371/journal.pone.0017770 PMID: 21423758; PMCID: PMC3056779.
Fathali N, Lekic T, Zhang JH, Tang J. Long-term evaluation of granulocyte-colony stimulating factor on hypoxic-ischemic brain damage in infant rats. Intensive Care Med. 2010;36(9):1602–8 https://doi.org/10.1007/s00134-010-1913-6.
Sarosiek KA, Chi X, Bachman JA, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51(6):751–65 https://doi.org/10.1016/j.molcel.2013.08.048.
Lilly M, Sandholm J, Cooper JJ, Koskinen PJ, Kraft A. The PIM-1 serine kinase prolongs survival and inhibits apoptosis-related mitochondrial dysfunction in part through a bcl-2-dependent pathway. Oncogene. 1999;18(27):4022–31 https://doi.org/10.1038/sj.onc.1202741.
Thornton C, Hagberg H. Role of mitochondria in apoptotic and necroptotic cell death in the developing brain. Clin Chim Acta. 2015;451(Pt A):35–8 https://doi.org/10.1016/j.cca.2015.01.026 Epub 2015 Feb 4. PMID: 25661091; PMCID: PMC4661434.
Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37(3):299–310 https://doi.org/10.1016/j.molcel.2010.01.025.
Charles MS, Ostrowski RP, Manaenko A, Duris K, Zhang JH, Tang J. Role of the pituitary–adrenal axis in granulocyte-colony stimulating factor-induced neuroprotection against hypoxia–ischemia in neonatal rats. Neurobiol Dis. 2012;47(1):29–37 https://doi.org/10.1016/j.nbd.2012.03.021 PMID: 22779090; PMCID: PMC3606051.
Faivre S, Decaens T. Raymond E. mTOR and cancer therapy: clinical development and novel prospects. In: Polunovsky VA, Houghton PJ, editors. mTOR Pathway and mTOR inhibitors in Cancer therapy, Cancer drug discovery and development; 2010. p. 133–48.
Huang S, Wang H, Turlova E, et al. GSK-3β inhibitor TDZD-8 reduces neonatal hypoxic-ischemic brain injury in mice. CNS Neurosci Ther. 2017;23(5):405–15 https://doi.org/10.1111/cns.12683.
King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3beta and attenuated by lithium. Brain Res. 2001;919(1):106–14 https://doi.org/10.1016/s0006-8993(01)03005-0.
Solaroglu I, Cahill J, Tsubokawa T, Beskonakli E, Zhang JH. Granulocyte colony-stimulating factor protects the brain against experimental stroke via inhibition of apoptosis and inflammation. Neurol Res. 2009;31(2):167–72 https://doi.org/10.1179/174313209X393582.
Solaroglu I, Tsubokawa T, Cahill J, Zhang JH. Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience. 2006;143(4):965–74 https://doi.org/10.1016/j.neuroscience.2006.09.014.
Yang YN, Lin CS, Yang CH, Lai YH, Wu PL, Yang SN. Neurogenesis recovery induced by granulocyte-colony stimulating factor in neonatal rat brain after perinatal hypoxia. Pediatr Neonatol. 2013;54(6):380–8 https://doi.org/10.1016/j.pedneo.2013.04.011.
Yang DY, Chen YJ, Wang MF, Pan HC, Chen SY, Cheng FC. Granulocyte colony-stimulating factor enhances cellular proliferation and motor function recovery on rats subjected to traumatic brain injury. Neurol Res. 2010;32(10):1041–9 https://doi.org/10.1179/016164110X12807570510013.
Song S, Kong X, Acosta S, Sava V, Borlongan C, Sanchez-Ramos J. Granulocyte colony-stimulating factor promotes behavioral recovery in a mouse model of traumatic brain injury. J Neurosci Res. 2016;94(5):409–23 https://doi.org/10.1002/jnr.23714.
Sevimli S, Diederich K, Strecker JK, Schilling M, Klocke R, Nikol S, Kirsch F, Schneider A, Schäbitz WR. Endogenous brain protection by granulocyte-colony stimulating factor after ischemic stroke. Exp Neurol. 2009;217(2):328–35 https://doi.org/10.1016/j.expneurol.2009.03.018 Epub 2009 Mar 28. PMID: 19332060.
Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–78 https://doi.org/10.1038/nrm2183 PMID: 17522591.
Trollmann R, Gassmann M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. 2009;31(7):503–9 https://doi.org/10.1016/j.braindev.2009.03.007.
Liang X, Liu X, Lu F, Zhang Y, Jiang X, Ferriero DM. HIF1α signaling in the endogenous protective responses after neonatal brain hypoxia-ischemia. Dev Neurosci. 2019:1–10 https://doi.org/10.1159/000495879 [published online ahead of print, 2019 Mar 5].
Hong Y, Deng C, Zhang J, Zhu J, Li Q. Neuroprotective effect of granulocyte colony-stimulating factor in a focal cerebral ischemic rat model with hyperlipidemia. J Huazhong Univ Sci Technolog Med Sci. 2012;32(6):872–8 https://doi.org/10.1007/s11596-012-1050-2.
Zhou S, Yin DP, Wang Y, Tian Y, Wang ZG, Zhang JN. Dynamic changes in growth factor levels over a 7-day period predict the functional outcomes of traumatic brain injury. Neural Regen Res. 2018;13(12):2134–40 https://doi.org/10.4103/1673-5374.241462.
Larpthaveesarp A, Ferriero DM, Gonzalez FF. Growth factors for the treatment of ischemic brain injury (growth factor treatment). Brain Sci. 2015;5(2):165–77 Published 2015 Apr 30. https://doi.org/10.3390/brainsci5020165.
Yu JH, Seo JH, Lee JE, Heo JH, Cho SR. Time-dependent effect of combination therapy with erythropoietin and granulocyte colony-stimulating factor in a mouse model of hypoxic-ischemic brain injury. Neurosci Bull. 2014;30(1):107–17 https://doi.org/10.1007/s12264-013-1397-9.
Nonomura M, Harada S, Asada Y, et al. Combination therapy with erythropoietin, magnesium sulfate and hypothermia for hypoxic-ischemic encephalopathy: an open-label pilot study to assess the safety and feasibility. BMC Pediatr. 2019;19(1):13 Published 2019 Jan 8. https://doi.org/10.1186/s12887-018-1389-z.
Liu SP, Lee SD, Lee HT, et al. Granulocyte colony-stimulating factor activating HIF-1alpha acts synergistically with erythropoietin to promote tissue plasticity. PLoS One. 2010;5(4):e10093 https://doi.org/10.1371/journal.pone.0010093 Published 2010 Apr 9. [published correction appears in PLoS One. 2010;5(10). doi:10.1371/annotation/433064f4-e30a-4000-8e5a-9e8d1775d820. Su, Ching-Yuan [added]; Li, Hung [added]].
Shin YK, Cho SR. Exploring erythropoietin and G-CSF combination therapy in chronic stroke patients. Int J Mol Sci. 2016;17(4):463 https://doi.org/10.3390/ijms17040463 PMID: 27043535; PMCID: PMC4848919.
Doycheva D, Shih G, Chen H, Applegate R, Zhang JH, Tang J. Granulocyte-colony stimulating factor in combination with stem cell factor confers greater neuroprotection after hypoxic-ischemic brain damage in the neonatal rats than a solitary treatment. Transl Stroke Res. 2013;4(2):171–8 https://doi.org/10.1007/s12975-012-0225-2.
Zhao LR, Piao CS, Murikinati SR, Gonzalez-Toledo ME. The role of stem cell factor and granulocyte-colony stimulating factor in treatment of stroke. Recent Pat CNS Drug Discov. 2013;8(1):2–12 https://doi.org/10.2174/1574889811308010002 PMID: 23173646; PMCID: PMC3740536.
De La Peña I, Sanberg PR, Acosta S, Lin SZ, Borlongan CV. G-CSF as an adjunctive therapy with umbilical cord blood cell transplantation for traumatic brain injury. Cell Transpl. 2015;24(3):447–57 https://doi.org/10.3727/096368915X686913.
Neubauer V, Wegleiter K, Posod A, et al. Delayed application of the haematopoietic growth factors G-CSF/SCF and FL reduces neonatal excitotoxic brain injury. Brain Res. 2016;1634:94–103 https://doi.org/10.1016/j.brainres.2015.12.058.
Posod A, Wegleiter K, Neubauer V, et al. Short, mid, and long-term effect of granulocyte colony-stimulating factor/stem cell factor and Fms-related tyrosine kinase 3 ligand evaluated in an in vivo model of hypoxic-hyperoxic ischemic neonatal brain injury. Biomed Res Int. 2019;2019:5935279 Published 2019 Mar 13. https://doi.org/10.1155/2019/5935279.
Dela Peña I, Sanberg PR, Acosta S, Tajiri N, Lin SZ, Borlongan CV. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: aging as a comorbidity factor. J Neurosurg Sci. 2014;58(3):145–9.
McDonald CA, Penny TR, Paton MCB, et al. Effects of umbilical cord blood cells, and subtypes, to reduce neuroinflammation following perinatal hypoxic-ischemic brain injury. J Neuroinflammation. 2018;15(1):47 Published 2018 Feb 17. https://doi.org/10.1186/s12974-018-1089-5.
Liska MG, Dela PI. Granulocyte-colony stimulating factor and umbilical cord blood cell transplantation: synergistic therapies for the treatment of traumatic brain injury. Brain Circ. 2017;3(3):143–51 https://doi.org/10.4103/bc.bc_19_17.
Ghahari L, Safari M, Rahimi Jaberi K, Jafari B, Safari K, Madadian M. Mesenchymal stem cells with granulocyte colony-stimulating factor reduce stress oxidative factors in Parkinson’s disease. Iran Biomed J. 2020;24(2):89–98 https://doi.org/10.29252/ibj.24.2.89 Epub 2019 Nov 2. PMID: 31677610; PMCID: PMC6984711.
Pan C, Gupta A, Prentice H, Wu JY. Protection of taurine and granulocyte colony-stimulating factor against excitotoxicity induced by glutamate in primary cortical neurons. J Biomed Sci. 2010;17(Suppl 1):S18 https://doi.org/10.1186/1423-0127-17-S1-S18 PMID: 20804592; PMCID: PMC2994407.
Griva M, Lagoudaki R, Touloumi O, et al. Long-term effects of enriched environment following neonatal hypoxia-ischemia on behavior, BDNF and synaptophysin levels in rat hippocampus: effect of combined treatment with G-CSF. Brain Res. 2017;1667:55–67 https://doi.org/10.1016/j.brainres.2017.05.004.
Oorschot DE, Sizemore RJ, Amer AR. Treatment of neonatal hypoxic-ischemic encephalopathy with erythropoietin alone, and erythropoietin combined with hypothermia: history, current status, and future research. Int J Mol Sci. 2020;21(4):1487 Published 2020 Feb 21. https://doi.org/10.3390/ijms21041487.
Traudt CM, McPherson RJ, Bauer LA, et al. Concurrent erythropoietin and hypothermia treatment improve outcomes in a term nonhuman primate model of perinatal asphyxia. Dev Neurosci. 2013;35(6):491–503 https://doi.org/10.1159/000355460.
Fang AY, Gonzalez FF, Sheldon RA, Ferriero DM. Effects of combination therapy using hypothermia and erythropoietin in a rat model of neonatal hypoxia-ischemia. Pediatr Res. 2013;73(1):12–7 https://doi.org/10.1038/pr.2012.138.
Lobo N, Yang B, Rizvi M, Ma D. Hypothermia and xenon: novel noble guardians in hypoxic-ischemic encephalopathy? J Neurosci Res. 2013;91(4):473–8 https://doi.org/10.1002/jnr.23178.
Rüegger CM, Davis PG, Cheong JL. Xenon as an adjuvant to therapeutic hypothermia in near-term and term newborns with hypoxic-ischaemic encephalopathy. Cochrane Database Syst Rev. 2018;8(8):CD012753 Published 2018 Aug 20. https://doi.org/10.1002/14651858.CD012753.pub2.
Maiwald CA, Annink KV, Rüdiger M, et al. Effect of allopurinol in addition to hypothermia treatment in neonates for hypoxic-ischemic brain injury on neurocognitive outcome (ALBINO): study protocol of a blinded randomized placebo-controlled parallel group multicenter trial for superiority (phase III). BMC Pediatr. 2019;19(1):210 Published 2019 Jun 27. https://doi.org/10.1186/s12887-019-1566-8.
Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002;296(5573):1653–5 https://doi.org/10.1126/science.1071545.
Schneider A, Kuhn HG, Schäbitz WR. A role for G-CSF (granulocyte-colony stimulating factor) in the central nervous system. Cell Cycle. 2005;4(12):1753–7 https://doi.org/10.4161/cc.4.12.2213.
Schäbitz WR, Kollmar R, Schwaninger M, Juettler E, Bardutzky J, Schölzke MN, Sommer C, Schwab S. Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischemia. Stroke. 2003;34(3):745–51 https://doi.org/10.1161/01.STR.0000057814.70180.17 Epub 2003 Feb 13. PMID: 12624302.
Si W, Wang J, Li M, et al. Erythropoietin protects neurons from apoptosis via activating PI3K/AKT and inhibiting Erk1/2 signaling pathway. 3 Biotech. 2019;9(4):131 https://doi.org/10.1007/s13205-019-1667-y.
Zeng SS, Bai JJ, Jiang H, et al. Treatment with liraglutide exerts neuroprotection after hypoxic-ischemic brain injury in neonatal rats via the PI3K/AKT/GSK3β pathway. Front Cell Neurosci. 2020;13:585 Published 2020 Jan 30. https://doi.org/10.3389/fncel.2019.00585.
Xi JS, Wang YF, Long XX, Ma Y. Mangiferin potentiates neuroprotection by isoflurane in neonatal hypoxic brain injury by reducing oxidative stress and activation of phosphatidylinositol-3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling. Med Sci Monit. 2018;24:7459–68 Published 2018 Oct 19. https://doi.org/10.12659/MSM.908142.
Simmons LJ, Surles-Zeigler MC, Li Y, Ford GD, Newman GD, Ford BD. Regulation of inflammatory responses by neuregulin-1 in brain ischemia and microglial cells in vitro involves the NF-kappa B pathway. J Neuroinflammation. 2016;13(1):237 Published 2016 Sep 6. https://doi.org/10.1186/s12974-016-0703-7.
Nandipati KC, Subramanian S, Agrawal DK. Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem. 2017;426(1-2):27–45 https://doi.org/10.1007/s11010-016-2878-8.
Cheng P, Zuo X, Ren Y, et al. Adenosine A1-receptors modulate mTOR signaling to regulate white matter inflammatory lesions induced by chronic cerebral hypoperfusion. Neurochem Res. 2016;41(12):3272–7 https://doi.org/10.1007/s11064-016-2056-0.
Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–91 https://doi.org/10.1016/j.neuron.2014.09.034.
Switon K, Kotulska K, Janusz-Kaminska A, Zmorzynska J, Jaworski J. Molecular neurobiology of mTOR. Neuroscience. 2017;341:112–53 https://doi.org/10.1016/j.neuroscience.2016.11.017.
Russo E, Citraro R, Constanti A, De Sarro G. The mTOR signaling pathway in the brain: focus on epilepsy and epileptogenesis. Mol Neurobiol. 2012;46(3):662–81 https://doi.org/10.1007/s12035-012-8314-5.
Chi OZ, Barsoum S, Vega-Cotto NM, Jacinto E, Liu X, Mellender SJ, Weiss HR. Effects of rapamycin on cerebral oxygen supply and consumption during reperfusion after cerebral ischemia. Neuroscience. 2016;316:321–7 https://doi.org/10.1016/j.neuroscience.2015.12.045 Epub 2015 Dec 29. PMID: 26742793; PMCID: PMC4881736.
Pastor MD, García-Yébenes I, Fradejas N, Pérez-Ortiz JM, Mora-Lee S, Tranque P, Moro MA, Pende M, Calvo S. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem. 2009;284(33):22067–78 https://doi.org/10.1074/jbc.M109.033100 Epub 2009 Jun 17. PMID: 19535330; PMCID: PMC2755931.
Saxton RA, Sabatini DM. mTOR Signaling in growth, metabolism, and disease [published correction appears in Cell. 2017 Apr 6;169(2):361-371]. Cell. 2017;168(6):960–76 https://doi.org/10.1016/j.cell.2017.02.004.
Peltier J, O’Neill A, Schaffer DV. PI3K/Akt and CREB regulate adult neural hippocampal progenitor proliferation and differentiation. Dev Neurobiol. 2007;67(10):1348–61 https://doi.org/10.1002/dneu.20506.
Charles MS, Drunalini Perera PN, Doycheva DM, Tang J. Granulocyte-colony stimulating factor activates JAK2/PI3K/PDE3B pathway to inhibit corticosterone synthesis in a neonatal hypoxic-ischemic brain injury rat model. Exp Neurol. 2015;272:152–9 https://doi.org/10.1016/j.expneurol.2015.03.019.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dumbuya, J.S., Chen, L., Wu, JY. et al. The role of G-CSF neuroprotective effects in neonatal hypoxic-ischemic encephalopathy (HIE): current status. J Neuroinflammation 18, 55 (2021). https://doi.org/10.1186/s12974-021-02084-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-021-02084-4