
Abstract
Background
Low back pain has become a serious social and economic burden and the leading cause of disability worldwide. Among a variety of pathophysiological triggers, intervertebral disc (IVD) degeneration plays a primary underlying role in causing such pain. Specifically, multiple independent endplate changes have been implicated in the initiation and progression of IVD degeneration.
Methods
In this study, we built a signaling network comprising both well-characterized IVD pathology-associated proteins as well as some potentially correlated proteins that have been associated with one or more of the currently known pathology-associated proteins. We then screened for the potential IVD degeneration-associated proteins using patients’ normal and degenerative endplate specimens. Short hairpin RNAs for receptor interacting serine/threonine kinase 1 (RIPK1) were constructed to examine the effects of RIPK1 knockdown in primary chondrocyte cells and in animal models of caudal vertebra intervertebral disc degeneration in vivo.
Results
RIPK1 was identified as a potential IVD degeneration-associated protein based on IVD pathology-associated signaling networks and the patients’ degenerated endplate specimens. Construction of the short hairpin RNAs was successful, with short-term RIPK1 knockdown triggering inflammation in the primary chondrocytes, while long-term knockdown triggered apoptosis through cleavage of the caspase 3 pathway, down-regulated NF-κB and mitogen-activating protein kinase (MAPK)s cascades, and decreased cell survival and inflammation. Animal models of caudal vertebra intervertebral disc degeneration further demonstrated that apoptosis was induced by up-regulation of tumor necrosis factor (TNF) accompanied by down-regulation of NF-κB and MAPKs cascades that are dependent on caspase and RIPK1.
Conclusions
These results provide proof-of-concept for developing novel therapies to combat IVD degeneration through interfering with RIPK1-mediated apoptosis signaling pathways especially in patients with RIPK1 abnormality.
Similar content being viewed by others


Background
The social and economic burden of low back pain continues to escalate due mainly to changes in lifestyle and demographic profile, with such pain now the leading cause of disability worldwide [1]. Although many different pathophysiological causes might trigger low back pain, it has been demonstrated that intervertebral disc (IVD) degeneration plays a primary underlying role. By connecting the neighboring vertebrae of the spinal column and allowing slight movement flexibility of the spine, IVDs act to absorb axial compressive forces and facilitate load transmission. IVDs comprise an annulus fibrosus surrounding the central nucleus pulposus, the fibrocartilage, and endplates.
Decades of research into IVDs have revealed many secrets of structure, function, and molecular mechanisms; however, how these large structures can survive and function even under the most difficult physiological conditions remains an enigma. The lumbar spine carries considerable forces and has no dedicated blood supply, thus the vertebral endplate plays an important role in balancing the contradictory functions of permeability for nutrients to diffuse between disc cells and capillaries in the adjacent vertebra and vascularized tissues with sufficient strength to prevent damage or fracture. In addition, endplates function to absorb and separate significant pressure from the spine’s mechanical burden and prevent the nucleus pulposus from bulging into the adjacent vertebrae and are essential for disc metabolism.
Some studies have suggested that IVD degeneration is closely correlated with the state of the vertebral endplates [2, 3], which often show significant morphological changes with aging-related IVD degenerations [4]. The biochemical changes in endplates have been reported extensively, from normal to different degenerative conditions [4]. Ariga et al. showed that increased apoptosis in the cartilaginous endplate with age resulted in markedly decreased cell density and destruction of the cartilaginous endplate [5], followed by the structure of the cartilaginous endplate beginning to disappear. Multiple independent factors can cause the initiation and progression of degeneration through endplate changes (reviewed in [6,7,8]). The process is a chain of biochemical, cellular, structural, and functional changes in the endplates, with mechanical stress, nutrient supply, osmotic and ionic environments, hormones, cytokines, growth factors, and matrix molecules all reported to affect disc cell degeneration, and many other pathological causes still to be explored.
Receptor interacting serine/threonine kinase 1 (RIPK1) is involved in Toll-like receptor (TLR), tumor necrosis factor (TNF), interferon, and interleukin (IL)1α signaling pathways [9,10,11,12,13,14]. Several studies found that activated RIPK1 can associate with RIPK3 to induce mixed lineage kinase domain like pseudokinase (MLKL)-dependent necroptosis and production of inflammatory cytokines or recruit Fas-associated protein with death domain (FADD) and activate caspase-8 to induce apoptosis following DNA damage or TLR signaling. In addition, it participates in the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) activation independent of its kinase activity. Abnormal activities of RIPK1 have been indicated in several disease processes, such as ischemic injuries, chronic and acute inflammatory diseases, axonal degeneration, neutrophilic dermatosis, autoinflammatory and autoimmune pathology, and cancers [14]. Due to RIPK1′s role in regulating necroptosis and apoptosis, it has gained interests as a treatment target for the RIPK1-dependent diseases already mentioned. In addition, the cumulative effects of various functions performed by RIPK1 may collaboratively contribute to molecular pathologies of autoimmune, degenerative, and inflammatory diseases.
This study sought to identify potential molecular pathogenic markers of IVD degeneration through building signaling networks based on known pathways important in IVD degeneration and to screen against the network using clinical specimens to reveal the underlying mechanisms of such molecules.
Materials and methods
Antibodies and other reagents
The following antibodies were used in this study: RIPK1 (Cell Signaling Technology, Danvers, MA), p-IKKα/βSer176/Ser180 (Thermo Fisher Scientific, Waltham, MA), p-JNKThr183 (Abcam, Cambridge, MA), p-IKKα/βSer176/180, p-p38Thr180/Tyr182, β-actin, and cleaved caspase 3 (Cell Signaling Technology). Other reagents purchased are as follows: 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (X-Gal), zVAD (N-benzyloxycarbonyl-Val-Ala-Asp (O-Me) fluoromethyl ketone), and necrostatin-1 (Nec1) from Sigma-Aldrich (St. Louis, MO), cycloheximide (CHX) from Santa Cruz Biotech (Dallas, Texas), collagenase II from Thermo Fisher Scientific, BCA Protein Assay Kit from Beyotime Biotechnology (Shanghai, China), HiScript Q Select RT SuperMix for qPCR and AceQ qPCR Probe Master Mix from Vazyme Biotech (Nanjing, China), and all remaining reagents from Sigma-Aldrich unless otherwise specified.
Building a signaling network representing intervertebral disc diseases with pathology-associated proteins
Reported pathology-associated proteins in IVD diseases were used as signaling nodes [1, 6,7,8]. These molecules were input into the meta-search engine of protein–protein interaction database String [15], organized, and analyzed as previously described [16]. Interactions identified by experiment, database, neighborhood, gene fusion, co-expression, and co-occurrence were included in the search. A confidence score of 0.15 was used and only direct protein–protein interactions were counted. Due to the limited amounts of mRNA extracted from patient specimens, 70 genes with the highest confidence and interaction scores were maintained in the signaling network for later experiments.
Quantification of mRNA expression in patients’ specimens and primary chondrocyte cells using quantitative real-time PCR (qRT-PCR)
Conditions of normal and degenerative endplates were confirmed using nuclear magnetic resonance (NMR) analysis, the Modic scoring system, and the Pfriimann disc degeneration grading system. These endplates were removed from a patient’s spine during surgery then rapidly frozen using liquid nitrogen. Total RNAs were extracted using a RNeasy Plus Micro Kit (Qiagen) according to the manufacturer’s instructions, and fold changes were calculated based on mRNA expression levels of the degenerated versus normal endplates, with change thresholds set at 0.8 and 1.2 to pick hits. Primary chondrocyte cells were plated in 6-well dishes, treated, and then harvested. Total RNAs were extracted in TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions for qRT-PCR experiments, carried out as previously described [17] using the LightCycler 96 (Roche Diagnostics, Rotkreuz, Switzerland). Primers used are listed in Additional file 1: Table S1. The cycling conditions were: 95 °C for 5 min, 40 cycles of 95 °C for 10 s and 60 °C for 30 s, and then 95 °C for 15 s, 60 °C for 60 s, and 95 °C for 15 s. The fluorescence was measured during 40 cycles of the 60 °C step. Relative mRNA expression was analyzed using the 2−ΔΔCt method.
shRIPK1-mediated silencing in primary chondrocyte cells
RIPK1 knockdown was achieved through viral transduction in primary chondrocyte cells using lentiviral transduction particles for shRNAs. The sequences for the short hairpin RNAs for RIPK1 (shRIPK1) are listed in Additional file 2: Table S2. The shRIPK1s were cloned into the vector pTripz, characterized, and then sequenced. Lentiviral vector packaging and lentiviral transduction were carried out as described previously [18], and shRNA expression was inducted in the presence of doxycycline.
Overexpression of RIPK1 in primary chondrocyte cells
Full-length cDNA encoding RIPK1 (NM_001359997.1) was amplified from the Mus musculus fibroblast cell line NIH/3T3 (ATCC, USA) using the following primers: 5′-GCTCTAGAGCCACCATGCAACCAGACATGTCCTTGGACA-3′ (Xbal) and 5′-TAGGATCCGCTCTGGCTGGCACGAATCAAGTGG-3′ (BamHI). These cDNAs were then cloned into the vector pCDH-EF1-MCS-T2A-Puro (Addgene, Cambridge, MA), characterized, and sequenced. Lentiviral vector packaging and lentiviral transduction were carried out as described previously [18].
Isolation, culture, and identification of primary chondrocyte cells
Cartilage was obtained from the hind knees of 6- to 10-day-old ICR mice or from IVD degeneration models at the indicated times. After incision of the fiber annulus and removal of the nucleus pulposus with a blade under a dissecting microscope, the translucent endplate cartilage was exposed. The cartilages are shallow, dish-like structures, thin in the center and thick at the boundary. They were minced into 1 mm3 pieces with ophthalmic scissors, washed three times with phosphate-buffered saline (PBS) containing 1000 U/mL penicillin and streptomycin 1 mg/L, and then collected aseptically on a sterile bench. Chondrocytes were obtained by digestion with collagenase II, then the cells were washed with PBS and cultured in petri dishes in a humidified incubator containing 5% CO2 and 10% O2 at 37 °C. Chondrocytes were maintained in Dulbecco’s modified Eagle’s medium (DMEM)/F-12 medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and streptomycin 0.1 mg/L). The cells were trypsinized and replated for a few times to purify chondrocytes. Primary chondrocyte cells were identified using Toluidine blue staining. Cells were fixed in 4% formaldehyde and stained with Toluidine blue for microscope observation.
Cellular senescence assays
Senescence β-galactosidase Staining Kits (Cell Signaling Technology) were used for SA-β-gal staining according to the manufacturer’s instructions. Cells were cultured in petri dishes at 37 °C in a humidifier incubator containing 5% CO2 and 10% O2. Briefly, primary chondrocyte cells were fixed with 2% formaldehyde and 0.2% glutaraldehyde, and then incubated with X-gal staining solution. Cells were visualized and imaged with a Nikon Eclipse Ni-U microscope (Nikon, Tokyo, Japan). The percentages of positively stained cells were calculated based on three independent experiments. Chondrocyte senescence was induced using IL-1β at 10 ng/mL for 48 h.
Animal model of IVD degeneration and histological staining
A total of 18 ICR mouse, aged 6 weeks, were used for caudal vertebra degeneration. Mice were raised in two groups randomly. To develop degeneration, mice received a surgical procedure. Briefly, the mice were anesthetized with 0.3 mL of 0.6% pentobarbital sodium intraperitoneally. The levels between the sixth and seventh, seventh and eighth, and eighth and ninth coccygeal vertebrae were identified under surgical microscope. Induction of degeneration was performed by percutaneous puncture with a 1-mL syringe needle. The needle was introduced until it reached the nucleus pulposus, when it was turned 360° and maintained in the same position for 2 min. The mice were raised in double cages where animals can run through two cages.
The samples were collected at 0, 1, and 3 months after the puncture, and then the animals were executed by neck dislocation. The samples were removed and decalcified in 15% EDTA for a week, then the samples were stored in 10% formaldehyde for 48–72 h, before water flushing for 4 h followed by paraffin embedding. Finally, paraffin sections (3 µm in thickness) were stained with hematoxylin and eosin (H&E) by a standard procedure and imaged at 40–200× magnification (Nikon).
Apoptosis assays
Primary chondrocyte cells were plated at 6 × 105 cells/dish in 6-well plates and cultured for either 1 week after Lentiviral infection or 48 h of drug treatment. Cells were treated and subjected to apoptosis kit reagents as previously described [17, 19]. Cisplatin (5 μM) was used in this assay as a positive control for methodology. The data were analyzed using FlowJo software (Ashland, OR, USA).
Western blot assays
The standard procedures used in this study are described previously [16, 19, 20]. Each experiment was independently performed at least three times. Briefly, cartilage cells were lysed in Mammalian Protein Extraction Reagent (MPER™, Thermo Fisher Scientific) and lysis buffer was supplemented with Halt™ Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and Complete™ Mini Protease Inhibitor Cocktail (Roche Diagnostics). BCA Protein Assay Kits were used to determine protein concentrations. Proteins were resolved on 4–12% gradient SDS-PAGE gels (Bio-Rad, Hercules, CA) and transferred to PVDF membrane (EMD Millipore, Temecula, CA). Membranes were incubated with primary antibody, followed by HRP-conjugated secondary antibody (Cell Signaling Technology) and signal detected with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). Immunoblots were quantified using Image J as described. Each experiment was independently performed at least for three times.
Cytokine assay
Cytokines were measured using the Bio-Plex Pro mouse cytokine 23-plex assay (Bio-Rad) according to the manufacturer’s instructions for the Luminex 200 instrument. Where a value was above or below the reference range, it was assigned the value of the highest or lowest standard, respectively. Lysates were made by homogenizing organs in ice-cold protein DISC lysis buffer (Roche, 30 mM Tris–HCl (pH 7.0), 120 mM NaCl, 10% Glycerol, 1% Triton X-100, complete protease inhibitors) followed by protein level normalization using a BCA assay (Thermo Scientific).
Statistical analysis
Data are reported as mean ± SD, unless otherwise noted. Significance was analyzed by one-way ANOVA using GraphPad Prism version 5.00 (GraphPad, San Diego, CA, USA), unless otherwise specified [19]. *P < 0.05; **P < 0.01; ***P < 0.001.
Results
Signaling network of potential pathology-associated proteins in intervertebral disc diseases
Building a signaling network is one way to investigate pathology-associated proteins potentially involved in IVD diseases and to identify novel candidates. Changes in load, nutrition, cell metabolism, matrix composition, and matrix turnover can initiate imbalances in IVD homeostasis that alter downstream mediators [1], and some of these might act as signaling nodes in IVD pathologies [1, 6,7,8]. Potential pathology-associated proteins include growth factors, chemokines, inflammatory proteins, immunomodulatory proteins, cytokines, and proteolytic enzymes, and the gene names of such proteins were submitted for a meta-search of the protein–protein interaction database String. Only direct protein–protein interactions with reasonable confidence levels were included due to the limited amounts of mRNA extracted from patient specimens for qRT-PCR screening. The final built network therefore comprised 70 high-confidence genes with the strongest potential for influencing IVD disease processes (Fig. 1).

Signaling network of pathology-associated proteins in intervertebral disc degeneration diseases. Reported pathology-associated proteins in intervertebral disc diseases were used as signaling nodes and input into the meta-search engine of protein–protein interaction database String. Interactions from experiment, database, neighborhood, gene fusion, co-expression, and co-occurrence were included in the search. The confidence score of 0.15 was used and only direct protein–protein interactions were counted. We included 70 genes with highest confidence and interaction scores in the signaling network for later experiments. Cyan, pink, green, blue, and black lines indicate known interactions from curated databases, experiments determined, gene neighborhood, gene co-occurrence, and co-expression, respectively. Gene names of these proteins were used in the figure
Potential intervertebral disc degeneration-associated proteins
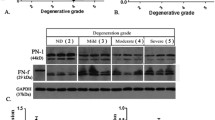
Normal and degenerative endplates were removed from patients’ spines during surgery to form bone transplantation beds to ease fusion and release stress on the spine (Fig. 2a, b). Pairwise degenerated and normal endplates were scored and diagnosed according to the Modic scoring system and the degree of disc degeneration was graded according to the MRI Pfriimann grading system (Table 1, Fig. 2c). Total RNAs were extracted from these endplates and qRT-PCR experiments were performed to evaluate the mRNA expression levels of the signaling network proteins. Fold changes were calculated based on mRNA expression levels of the degenerated versus normal endplates (Fig. 2d). We identified 26 potential IVD degeneration-associated proteins with mRNA changes of more than 20% in degenerated endplates of at least 5 patient specimens (Table 2). As expected, mRNA expression of several (matrix metallopeptidases) MMPs were significantly upregulated, possibly by the production of inflammatory mediators induced via an imbalance in anabolic and catabolic events when degeneration progressed and led to further matrix breakdown and degeneration. Polymorphisms in ESR1, estrogen receptor alpha, have been correlated with bone mass in humans since estrogens are critical for maintaining bone mineral density via diverse mechanisms in osteocytes, osteoclasts, osteoblasts, immune cells, and other cells [21]. The mRNA expression levels of several other genes were significantly changed in the degenerated endplates, including RIPK1, protein phosphatase 5 catalytic subunit (PPP5C), and 3-phosphoinositide dependent protein kinase 1 (PDPK1), none of which have been well studied in bone diseases.

Potential intervertebral disc degeneration-associated proteins identified using patient specimens. a Representative NMR images of patients’ spines with Modic grades before the surgery. b Representative NMR images of patients’ spines after the surgery. c Representative specimen images of normal and degenerative endplates from the patient surgery. d Relative mRNA expression of patients’ degenerative versus normal endplates (Patient 1–Patient 7) detected through qRT-PCR experiments with 70 genes of IVD degeneration-associated proteins in the signaling network. Color scale is shown in the corresponding figure
Construction of short hairpin RNAs for RIPK1 and short-term RIPK1 knockdown leading to inflammation in primary chondrocyte cells
Abnormal activities of RIPK1 have been indicated in several diseases, including ischemic injuries, chronic and acute inflammatory diseases, and axonal degeneration [14], and it was previously reported that RIPK1 regulates necroptosis and apoptosis. Thus, RIPK1 was chosen for further investigation with respect to IVD degeneration.
Vectors of four short-hairpin (sh) RNAs for RIPK1 were cloned into pTripz as illustrated in Fig. 3a. Primary chondrocyte cells were obtained from 6- to 10-day-old ICR mice and RIPK1 knockdown was achieved through viral transduction in primary chondrocyte cells using shRNA lentiviral transduction particles. Knockdown efficiency of these shRNAs for RIPK1 was tested via qRT-PCR (Fig. 3b) and western blot (Fig. 3c). mRNA expression of RIPK1 was significantly reduced to 0.37 and 0.29 relative to shRNA controls using shRIPK1-3 and shRIPK1-4, respectively, while protein expression of RIPK1 was significantly reduced to 0.34 and 0.27 relative to shRNA controls using shRIPK1-3 and shRIPK1-4, respectively. These experiments demonstrated that RIPK1 was successfully and efficiently knocked down with shRIPK1-4, which was therefore chosen for later experiments. RIPK1 has been previously shown to regulate RIPK3-MLKL-driven systemic inflammation [11], thus it was of interest to determine how inflammatory cytokines are regulated in primary chondrocyte cells with RIPK1 knockdown. Results showed significantly elevated levels of several inflammatory cytokines in primary chondrocyte cells after 4 days of RIPK1 knockdown by shRIPK1 (Fig. 3d), including Eotaxin, G-CSF, IL5, and MCP-1. These results indicate that inflammation was induced with short-term RIPK1 knockdown in primary chondrocyte cells.

Short-term RIPK1 knockdown led to inflammation in primary chondrocyte cells. a Construction of shRIPK1 vectors. b mRNA levels of RIPK1 5 days after shRIPK1 knockdown detected by qRT-PCR. c Representative Western blot results of shRIPK1 knockdown after 5 days. d Inflammatory cytokines levels 5 days after shRIPK1 knockdown. Data indicate the mean values calculated from triplicate samples from multiple independent experiments (n ≥ 3) (± SD). Differences were between shRIPK1 groups and the shControl group. *P < 0.05; **P < 0.01; ***P < 0.001
Long-term RIPK1 knockdown leading to apoptosis in primary chondrocyte cells
After 15 days of RIPK1 knockdown by shRIPK1 in primary chondrocyte cells, there are significantly more senescence phenotypes shown by SA-β-gal assays (Fig. 4a, b; 67.91% increase). IL-1β induced the senescence phenotypes by 51.31%, and this was significantly reversed to 26.97% by RIPK1 overexpression (Fig. 4b). After 15 days of RIPK1 knockdown, cells were analyzed using flow cytometry (Fig. 4c), indicating that both early and late apoptosis cells were significantly increased after RIPK1 knockdown (Fig. 4d).

Long-term RIPK1 knockdown led to apoptosis in primary chondrocyte cells. a Representative pictures of SA-β-gal staining at 15 days after shRIPK1 knockdown. b Statistical analysis of A and of SA-β-gal positive cells in IL-1β induced senescence with or without overexpression of RIPK1. c Representative flow cytometry plots for primary chondrocyte cells with RIPK1 knockdown for 15 days. Cells were stained with Annexin V-FITC/propidium iodide and analyzed for cell apoptosis distribution. d Statistical analysis of C for the presence of Annexin V (+)/PI (−) (early apoptosis) and Annexin V (+)/PI (+) (late apoptosis)
Additionally, qRT-PCR experiments demonstrated that TNF mRNA expression significantly increased (8.78-fold) after RIPK1 knockdown (Fig. 5a), prompting us to identify potential apoptosis mechanisms caused by RIPK1 knockdown and sequential TNF up-regulation. To do this, mRNA levels of NF-κB and MAPK cascades, TNFAIP3, CCL2, and IκBα were assessed following TNF stimulation. Notably, RIPK1 knockdown significantly decreased mRNA levels of TNFAIP3 (11.89 vs. 7.45 at 25 min and 13.67 vs. 5.41 at 60 min), CCL2 (22.16 vs. 12.23 at 25 min and 23.66 vs. 13.64 at 60 min), and IκBα (35.64 vs. 18.34 at 25 min and 40.31 vs. 21.37 at 60 min) after both 25- and 60-min TNF stimulations. Western blotting further proved that protein expression of NF-κB and MAPKs cascades, IKKα/β, JNK, and p38, significantly decreased with RIPK1 knockdown and TNF stimulation, while protein expression of cleaved caspase-3 increased (Fig. 5c). These results suggest that longer term RIPK1 knockdown can trigger apoptosis through the cleaved caspase 3 pathway while downregulating the NF-κB and MAPKs cascades and decreasing cell survival and inflammation.

Long-term RIPK1 knockdown triggered apoptosis through the cleaved caspase 3 pathway, while downregulating NF-κB and MAPKs cascades in primary chondrocyte cells. a mRNA levels of TNF 15 days after shRIPK1 knockdown detected by qRT-PCR. b mRNA levels of TNFAIP3, CCL2, and IκBα 15 days after shRIPK1 knockdown and at the indicated time after TNF stimulation, detected by qRT-PCR. c Detection of p-IKKα/β, p-JNK, p-p38, and cleaved caspase 3 in the protein lysates using western blotting. Data indicate the mean values calculated from triplicate samples from multiple independent experiments (n ≥ 3) (± SD). *P < 0.05; **P < 0.01
Mechanisms of intervertebral disc degeneration in animal models
Animal models of caudal vertebra IVD degeneration were used to further assess mechanisms and functions of the RIPK1 gene. Mice received surgical procedures with needle puncture to induce artificial damage and were raised in double cages to speed up degeneration. Primary chondrocyte cells were collected from the endplates of these animals at the indicated time points and cultured (Fig. 6a). The mRNA expression of TNF, RIPK1, JNK, and IKKα were then assessed in primary chondrocyte cells (Fig. 6b). mRNA levels of TNF were significantly increased by 5.16-fold at 3 months after surgery compared to controls at time 0 and by 5.67-fold compared to controls at 3 months. On the contrary, levels of RIPK1, JNK, and IKKα significantly decreased (by 47.62%, 30.56%, and 35.58%, respectively) at 3 months after surgery compared to controls.

Apoptosis induced in animal models of caudal vertebra intervertebral disc degeneration through TNF-induced caspase-dependent RIPK1 pathway. a H&E staining of endplates of animal models of caudal vertebra intervertebral disc degeneration and control group at the indicated time. b mRNA levels of TNF, RIPK1, JNK, and IKKα in primary chondrocyte cells from endplates of animal models as indicated. c Apoptotic cells of primary chondrocyte cells from endplates of animal models treated as indicated and detected by flow cytometry. Data indicate the mean values calculated from triplicate samples from multiple independent experiments (n ≥ 3) (± SD). *P < 0.05; **P < 0.01
To further investigate the apoptosis occurring in animal models of caudal vertebra IVD and how that might correlate with the mechanism of RIPK1 downregulation, we utilized some tool molecules (Fig. 6c), including cycloheximide (CHX, apoptosis reagent), zVAD (N-benzyloxycarbonyl-Val-Ala-Asp (O-Me) fluoromethyl ketone, caspase inhibitor), and necrostatin-1 (Nec1, RIPK1 inhibitor). Results demonstrated that apoptosis was induced by TNF and CHX in RIPK1-downregulated primary chondrocytes of caudal vertebra IVD animal models, but blocked by zVAD. These findings suggested that the apoptosis is dependent on caspase. TNF plus CHX triggered cell death in controls, but more so in caudal vertebra IVD when RIPK1 was downregulated, indicating that RIPK1 suppressed TNF-induced apoptosis. These results are consistent with the earlier findings that RIPK1−/− primary chondrocyte cells are sensitized for cell death induced by TNF plus CHX [22] and that RIPK1 functions to inhibit caspase-mediated apoptosis [9].
Discussion
IVD degeneration is a complicated issue involving a myriad of factors. Pathological changes have been recognized and classified as ‘degeneration’ as early as in the second decade of life. Recent studies have focused on understanding the molecular and genetic aspects of disc degeneration to diagnosis the degeneration early, identify the optimal time to start therapeutic intervention, and halt or slow down the degenerative process. Nonetheless, apoptosis or programmed cell death appears to correlate with age-related degeneration, with a higher percentage of apoptosis present in older people [23]. Identifying the molecular causes or capturing early events in apoptosis during disc degeneration could revolutionize current treatment of back pain. To this end, the Fas receptor is expressed shortly after the onset of disc degeneration [24], while a high mechanical load, decreased production of important matrix proteins (such as type II collagen and aggrecan), and increased production of degradative, inflammatory, and catabolic molecules (such as TNF, ILs, MMPs, cathepsin, aggrecanase, lysozyme, nitric oxide, and free radicals) are also implicated causally [25].
Since IVD degeneration is a complex disease and there are no prominent therapeutic agents identified to target pathological molecules, the signaling network based on systems biology and protein–protein interaction database analysis might offer new opportunities to implicate targets and biomarkers for IVD degeneration. The signaling network built by us was based on identified pathological molecules as well as proteins not yet implicated in IVD degeneration, but that show interactions with the known pathological molecules in IVD degeneration disease. Since the cells on patient endplates are very limited, we only had enough for qRT-PCR screening to look for differentially expressed mRNAs of the network proteins in degenerated versus normal specimens. Our screening identified ESR1, a known regulator in multiple degenerative and aging diseases such as Alzheimer’s [26, 27], radiographic hip osteoarthritis [28], aging macula disorder [29], and cancers [30]. More importantly, ESR1 has been implicated to play a role in the bone metabolism of osteoporosis, osteocytes, osteoclasts, osteoblasts, immune cells, and other cells to maintain bone mineral density with varieties of mechanisms [21]. Therefore, it was not surprising to detect abnormal expression of ESR1 in patients with IVD disease and it proved that our signaling network and qRT-PCR screening was effective, informative, and identified potential molecules. Another strong hit, PPP5C, inhibits cell growth when it is knocked down in several cell types. We also investigated the strong hit of RIPK1, a mediator of necroptosis, apoptosis, and inflammation [14]. Multiple RIPK1 deficiency studies have shown persuasive evidence that RIPK1 suppresses FADD/caspase-8-dependent apoptosis in some cell types, and RIPK3/MLKL-dependent necroptosis in others [9,10,11,12, 31,32,33]. We showed herein that short-term RIPK1 knockdown can increase inflammatory cytokines, while long-term RIPK1 knockdown led to apoptosis in primary chondrocyte cells. We further demonstrated that long-term RIPK1 knockdown triggered apoptosis through the cleaved caspase 3 pathway while downregulating NF-κB and MAPKs cascades and decreasing inflammation and cell survival. Our capture of both short- and long-term results of RIPK1 knockdown relatively represent the complex and lengthy degeneration process, which in turn seems to be associated with inflammation and infection. Possibly, the impact of apoptosis is more predominant after inflammation and infection to worsen the IVD degeneration, and in animal disease models, the mRNA expression of RIPK1 was lower in degeneration models after 3 months, while that of TNF was significantly higher.
Several caspase proteins have been implicated in the apoptosis of disc degeneration and thus proposed as therapeutic targets [34,35,36]. The apoptosis we observed is also dependent on caspase as shown by zVAD blockage, and zVAD significantly inhibited apoptosis induced by THF and CHX in primary chondrocyte cells cultured from the animal models. These findings further confirmed that the apoptosis is caspase-mediated and RIPK1 dependent. Our studies suggest that inhibitors of caspase and overexpression of RIPK1 could have therapeutic potential in halting or delaying degeneration in IVD diseases or reversing the IL-1β induced senescence, and thus for developing RIPK1 agitators with low toxicity for the treatment and/or delay of IVD degeneration.
A great number of biomolecular therapies, gene and interfering RNA therapies have been broadly investigated in IVD degeneration. Injection of various growth factors, such as BMPs, EGF, TGF-βs, has shown promising results in delaying degeneration [37]. Viral and non-viral gene delivery of genes, such as Sox-9, OP-1, TIMP-1, and BMP-2, dramatically increased disc height, gene expression, and matrix molecules [37, 38]. Despite harsh environment where these biological molecules are injected and further improvement are required for clinical application, these studies have provided encouraging results about delaying apoptosis and long-term promotion of regeneration. Both natural and synthetic materials can provide favorable scaffolds for tissue engineering and bioactive agent delivery. Natural materials have the advantages such as low toxicity, similarity to native tissue, and easy large-scale production, and synthetic materials are highly reproducible and their mechanical and physicochemical properties can be finely adjusted. All such development will surely contribute to combating IVD degeneration with gene therapy strategies, e.g. gene therapy with RIPK1, in the near future.
Conclusion
These results demonstrated that RIPK1 is a promising IVD degeneration-associated protein and implicated RIPK1-regulated apoptosis in such degeneration. They thus provide a proof-of-concept method for developing novel therapies to combat IVD degeneration through interfering with RIPK1-mediated apoptosis signaling pathways especially in patients with RIPK1 abnormality. Realistically, the goal of our study is not to reverse the disc degeneration completely, but to offer a new insight into the molecular pathology of endplate degeneration and explore the potential to delay the inevitable degeneration.
Abbreviations
- RIPK1:
-
receptor interacting serine/threonine kinase 1
- IVD:
-
intervertebral disc
- AF:
-
annulus fibrosus
- NP:
-
nucleus pulposus
- RIPK:
-
receptor-interacting protein kinase
- MLKL:
-
mixed lineage kinase domain like pseudokinase
- TLR:
-
Toll-like receptor
- TNF:
-
tumor necrosis factor
- FADD:
-
Fas-associating protein with a novel death domain
- NF-κb:
-
nuclear factor kappa beta
- IKKα/β:
-
inhibitor of nuclear factor kappa-B kinase subunit beta
- JNK:
-
C-Jun N-terminal kinase
- p38:
-
P38 mitogen-activated protein kinases
- X-Gal:
-
5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside
- zVAD:
-
N-benzyloxycarbonyl-Val-Ala-Asp (O-Me) fluoromethyl ketone
- Nec1:
-
necrostatin-1
- qRT-PCR:
-
quantitative real-time PCR
- ICR:
-
Institute of Cancer Research
- Xbal:
-
restriction enzyme isolated from the bacterium Xanthomonas badrii
- BamHI:
-
(from Bacillus amyloliquefaciens) is a type II restriction endonuclease
- β-gal:
-
β-galactosidase
- IL-1β:
-
interleukin 1 beta
- H&E:
-
hematoxylin and eosin
- MMP:
-
matrix metalloprotein
- ESR1:
-
estrogen receptor alpha
- PPP5C:
-
serine/threonine-protein phosphatase 5
- PDPK1:
-
phosphoinositide-dependent kinase-1
- G-CSF:
-
granulocyte-colony stimulating factor
- IL5:
-
interleukin 5
- MCP-1:
-
monocyte chemoattractant protein 1
- MAPK:
-
mitogen-activated protein kinase
- CHX:
-
cycloheximide
References
Sakai D, Grad S. Advancing the cellular and molecular therapy for intervertebral disc disease. Adv Drug Deliv Rev. 2015;84:159–71.
Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine. 2006;31(18):2151–61.
Przybyla A, et al. Outer annulus tears have less effect than endplate fracture on stress distributions inside intervertebral discs: relevance to disc degeneration. Clin Biomech. 2006;21(10):1013–9.
Moore RJ. The vertebral endplate: disc degeneration, disc regeneration. Eur Spine J. 2006;15(Suppl 3):S333–7.
Ariga K, et al. The relationship between apoptosis of endplate chondrocytes and aging and degeneration of the intervertebral disc. Spine. 2001;26(22):2414–20.
Zhang F, et al. Molecular mechanisms of cell death in intervertebral disc degeneration (review). Int J Mol Med. 2016;37(6):1439–48.
Ito K, Creemers L. Mechanisms of intervertebral disk degeneration/injury and pain: a review. Glob Spine J. 2013;3(3):145–52.
Hughes SP, et al. The pathogenesis of degeneration of the intervertebral disc and emerging therapies in the management of back pain. J Bone Joint Surg Br. 2012;94(10):1298–304.
Dillon CP, et al. RIPK1 blocks early postnatal lethality mediated by caspase-8 and RIPK3. Cell. 2014;157(5):1189–202.
Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513(7516):90–4.
Rickard JA, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175–88.
Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–9.
Polykratis A, et al. Cutting edge: RIPK1 kinase inactive mice are viable and protected from TNF-induced necroptosis in vivo. J Immunol. 2014;193(4):1539–43.
Wegner KW, Saleh D, Degterev A. Complex pathologic roles of RIPK1 and RIPK3: moving beyond necroptosis. Trends Pharmacol Sci. 2017;38(3):202–25.
Szklarczyk D, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–8.
Liu H, et al. Network analysis identifies an HSP90-central hub susceptible in ovarian cancer. Clin Cancer Res. 2013;19(18):5053–67.
Shang D, et al. Identification of a pyridine derivative inducing senescence in ovarian cancer cell lines via P21 activation. Clin Exp Pharmacol Physiol. 2018;45(5):452–60.
Tu Z, et al. BRG1 is required for formation of senescence-associated heterochromatin foci induced by oncogenic RAS or BRCA1 loss. Mol Cell Biol. 2013;33(9):1819–29.
Liu H, et al. Targeting heat-shock protein 90 with ganetespib for molecularly targeted therapy of gastric cancer. Cell Death Dis. 2015;6:e1595.
Tu Z, et al. Oncogenic RAS regulates BRIP1 expression to induce dissociation of BRCA1 from chromatin, inhibit DNA repair, and promote senescence. Dev Cell. 2011;21(6):1077–91.
Khalid AB, Krum SA. Estrogen receptors alpha and beta in bone. Bone. 2016;87:130–5.
Kelliher MA, et al. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303.
Gruber HE, Hanley EN Jr. Analysis of aging and degeneration of the human intervertebral disc. Comparison of surgical specimens with normal controls. Spine. 1998;23(7):751–7.
Anderson DG, et al. Comparative gene expression profiling of normal and degenerative discs: analysis of a rabbit annular laceration model. Spine. 2002;27(12):1291–6.
Walker MH, Anderson DG. Molecular basis of intervertebral disc degeneration. Spine J. 2004;4(6 Suppl):158S–66S.
Boada M, et al. Estrogen receptor alpha gene variants are associated with Alzheimer’s disease. Neurobiol Aging. 2012;33(1):198/e15–24.
Goumidi L, et al. Study of estrogen receptor-alpha and receptor-beta gene polymorphisms on Alzheimer’s disease. J Alzheimers Dis. 2011;26(3):431–9.
Lian K, et al. Estrogen receptor alpha genotype is associated with a reduced prevalence of radiographic hip osteoarthritis in elderly Caucasian women. Osteoarthritis Cartilage. 2007;15(8):972–8.
Boekhoorn SS, et al. Estrogen receptor alpha gene polymorphisms associated with incident aging macula disorder. Invest Ophthalmol Vis Sci. 2007;48(3):1012–7.
Li LC, et al. Age-dependent methylation of ESR1 gene in prostate cancer. Biochem Biophys Res Commun. 2004;321(2):455–61.
Kaiser WJ, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci USA. 2014;111(21):7753–8.
Kearney CJ, et al. RIPK1 can function as an inhibitor rather than an initiator of RIPK3-dependent necroptosis. FEBS J. 2014;281(21):4921–34.
Orozco S, et al. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014;21(10):1511–21.
Chen ZH, et al. Enhanced NLRP3, caspase-1, and IL-1beta levels in degenerate human intervertebral disc and their association with the grades of disc degeneration. Anat Rec. 2015;298(4):720–6.
Ding L, et al. Lentiviral-mediated RNAi targeting caspase-3 inhibits apoptosis induced by serum deprivation in rat endplate chondrocytes in vitro. Braz J Med Biol Res. 2014;47(6):445–51.
Sudo H, Minami A. Caspase 3 as a therapeutic target for regulation of intervertebral disc degeneration in rabbits. Arthritis Rheum. 2011;63(6):1648–57.
Chu G, et al. Strategies for annulus fibrosus regeneration: from biological therapies to tissue engineering. Front Bioeng Biotechnol. 2018;6:90.
Ren S, et al. Treatment of rabbit intervertebral disc degeneration with co-transfection by adeno-associated virus-mediated SOX9 and osteogenic protein-1 double genes in vivo. Int J Mol Med. 2013;32(5):1063–8.
Zhao K, et al. Epigenetic silencing of miRNA-143 regulates apoptosis by targeting BCL2 in human intervertebral disc degeneration. Gene. 2017;628:259–66.
Sanchez-Sabate E, et al. Identification of differentially expressed genes in trabecular bone from the iliac crest of osteoarthritic patients. Osteoarthritis Cartilage. 2009;17(8):1106–14.
Chandar N, et al. Relationship of bone morphogenetic protein expression during osteoblast differentiation to wild type p53. J Orthop Res. 2005;23(6):1345–53.
Pan Z, et al. Therapeutic effects of gefitinib-encapsulated thermosensitive injectable hydrogel in intervertebral disc degeneration. Biomaterials. 2018;160:56–68.
Zhang X, et al. Reduced EGFR signaling enhances cartilage destruction in a mouse osteoarthritis model. Bone Res. 2014;2:14015.
Joiner DM, et al. Accelerated and increased joint damage in young mice with global inactivation of mitogen-inducible gene 6 after ligament and meniscus injury. Arthritis Res Ther. 2014;16(2):R81.
Pan Z, et al. Delivery of epidermal growth factor receptor inhibitor via a customized collagen scaffold promotes meniscal defect regeneration in a rabbit model. Acta Biomater. 2017;62:210–21.
Shin SY, et al. Integrin alpha1beta1 protects against signs of post-traumatic osteoarthritis in the female murine knee partially via regulation of epidermal growth factor receptor signalling. Osteoarthritis Cartilage. 2016;24(10):1795–806.
Yuan FL, et al. Epidermal growth factor receptor (EGFR) as a therapeutic target in rheumatoid arthritis. Clin Rheumatol. 2013;32(3):289–92.
Guo W, et al. Circular RNA GRB10 as a competitive endogenous RNA regulating nucleus pulposus cells death in degenerative intervertebral disk. Cell Death Dis. 2018;9(3):319.
Hallbeck AL, et al. TGF-alpha and ErbB2 production in synovial joint tissue: increased expression in arthritic joints. Scand J Rheumatol. 2005;34(3):204–11.
Kitamura T, et al. Involvement of poly(ADP-ribose) polymerase 1 in ERBB2 expression in rheumatoid synovial cells. Am J Physiol Cell Physiol. 2005;289(1):C82–8.
Richards JB, et al. Collaborative meta-analysis: associations of 150 candidate genes with osteoporosis and osteoporotic fracture. Ann Intern Med. 2009;151(8):528–37.
Peach CA, Carr AJ, Loughlin J. Recent advances in the genetic investigation of osteoarthritis. Trends Mol Med. 2005;11(4):186–91.
Song XX, et al. Estrogen receptor expression in lumbar intervertebral disc of the elderly: gender- and degeneration degree-related variations. Joint Bone Spine. 2014;81(3):250–3.
Ishimaru T, et al. Cinchona alkaloid catalyzed enantioselective fluorination of allyl silanes, silyl enol ethers, and oxindoles. Angew Chem Int Ed Engl. 2008;47(22):4157–61.
Bocelli-Tyndall C, et al. FGF2 induces RANKL gene expression as well as IL1beta regulated MHC class II in human bone marrow-derived mesenchymal progenitor stromal cells. Ann Rheum Dis. 2015;74(1):260–6.
Wang X, et al. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthritis Cartilage. 2004;12(12):963–73.
Rodrigues-Pinto R, et al. Human notochordal cell transcriptome unveils potential regulators of cell function in the developing intervertebral disc. Sci Rep. 2018;8(1):12866.
McAlindon TE, Teale JD, Dieppe PA. Levels of insulin related growth factor 1 in osteoarthritis of the knee. Ann Rheum Dis. 1993;52(3):229–31.
Yan J, et al. Gut microbiota induce IGF-1 and promote bone formation and growth. Proc Natl Acad Sci USA. 2016;113(47):E7554–63.
Canalis E. Management of endocrine disease: novel anabolic treatments for osteoporosis. Eur J Endocrinol. 2018;178(2):R33–44.
Singh PK, Kashyap A, Silakari O. Exploration of the therapeutic aspects of Lck: a kinase target in inflammatory mediated pathological conditions. Biomed Pharmacother. 2018;108:1565–71.
Gerlag DM, et al. Real-time quantitative PCR to detect changes in synovial gene expression in rheumatoid arthritis after corticosteroid treatment. Ann Rheum Dis. 2007;66(4):545–7.
Teixeira GQ, et al. Anti-inflammatory chitosan/poly-gamma-glutamic acid nanoparticles control inflammation while remodeling extracellular matrix in degenerated intervertebral disc. Acta Biomater. 2016;42:168–79.
Zhang Q, et al. Differential Toll-like receptor-dependent collagenase expression in chondrocytes. Ann Rheum Dis. 2008;67(11):1633–41.
Sieker JT, et al. Transcriptional profiling of articular cartilage in a porcine model of early post-traumatic osteoarthritis. J Orthop Res. 2018;36(1):318–29.
van Kuijk AW, et al. A prospective, randomised, placebo-controlled study to identify biomarkers associated with active treatment in psoriatic arthritis: effects of adalimumab treatment on synovial tissue. Ann Rheum Dis. 2009;68(8):1303–9.
Nurs Stand. 1991:5(40):14.
Ahmad R, et al. Inhibition of interleukin 1-induced matrix metalloproteinase 13 expression in human chondrocytes by interferon gamma. Ann Rheum Dis. 2007;66(6):782–9.
Mosig RA, et al. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet. 2007;16(9):1113–23.
Gao W, et al. Notch signalling pathways mediate synovial angiogenesis in response to vascular endothelial growth factor and angiopoietin 2. Ann Rheum Dis. 2013;72(6):1080–8.
Yin J, et al. Exploration about changes of IL-10, NF-kappaB and MMP-3 in a rat model of cervical spondylosis. Mol Immunol. 2018;93:184–8.
Kim JH, et al. Regulation of the catabolic cascade in osteoarthritis by the zinc-ZIP8-MTF1 axis. Cell. 2014;156(4):730–43.
Flannelly J, et al. Metalloproteinase and tissue inhibitor of metalloproteinase expression in the murine STR/ort model of osteoarthritis. Osteoarthritis Cartilage. 2002;10(9):722–33.
Phillips KL, et al. Interleukin-1 receptor antagonist deficient mice provide insights into pathogenesis of human intervertebral disc degeneration. Ann Rheum Dis. 2013;72(11):1860–7.
Ra HJ, et al. Effects of salmon DNA fraction in vitro and in a monosodium iodoacetate-induced osteoarthritis rat model. Korean J Physiol Pharmacol. 2018;22(2):163–72.
Jackson CJ, Arkell J, Nguyen M. Rheumatoid synovial endothelial cells secrete decreased levels of tissue inhibitor of MMP (TIMP1). Ann Rheum Dis. 1998;57(3):158–61.
Li L, et al. Macrolactin F inhibits RANKL-mediated osteoclastogenesis by suppressing Akt, MAPK and NFATc1 pathways and promotes osteoblastogenesis through a BMP-2/smad/Akt/Runx2 signaling pathway. Eur J Pharmacol. 2017;815:202–9.
Chen YT, et al. The effects of amphiregulin induced MMP-13 production in human osteoarthritis synovial fibroblast. Mediat Inflamm. 2014;2014:759028.
Poveda L, et al. Peroxynitrite induces gene expression in intervertebral disc cells. Spine. 2009;34(11):1127–33.
Iotsova V, et al. Osteopetrosis in mice lacking NF-kappaB1 and NF-kappaB2. Nat Med. 1997;3(11):1285–9.
Ishijima M, et al. Osteopontin is associated with nuclear factor kappaB gene expression during tail-suspension-induced bone loss. Exp Cell Res. 2006;312(16):3075–83.
Aoki Y, et al. Increase of nerve growth factor levels in the human herniated intervertebral disc: can annular rupture trigger discogenic back pain? Arthritis Res Ther. 2014;16(4):R159.
Lane NE, Corr M. Osteoarthritis in 2016: anti-NGF treatments for pain—two steps forward, one step back? Nat Rev Rheumatol. 2017;13(2):76–8.
Yasui M, et al. Nerve growth factor and associated nerve sprouting contribute to local mechanical hyperalgesia in a rat model of bone injury. Eur J Pain. 2012;16(7):953–65.
Chen S, et al. Critical contribution of RIPK1 mediated mitochondrial dysfunction and oxidative stress to compression-induced rat nucleus pulposus cells necroptosis and apoptosis. Apoptosis. 2018;23(5–6):299–313.
Cuchet-Lourenco D, et al. Biallelic RIPK1 mutations in humans cause severe immunodeficiency, arthritis, and intestinal inflammation. Science. 2018;361(6404):810–3.
Wagner PN, Shi Q, Salisbury-Ruf CT, Zou J, Savona MR, Fedoriw Y, Zinkel SS. Increased Ripk1-mediated bone marrow necroptosis leads to myelodysplasia and bone marrow failure in mice. Blood. 2019;133(2):107–20.
Olsen BR. Mutations in collagen genes resulting in metaphyseal and epiphyseal dysplasias. Bone. 1995;17(2 Suppl):45S–9S.
Blasi E, et al. Selective immortalization of murine macrophages from fresh bone marrow by a raf/myc recombinant murine retrovirus. Nature. 1985;318(6047):667–70.
Tanaka S, et al. c-Cbl is downstream of c-Src in a signalling pathway necessary for bone resorption. Nature. 1996;383(6600):528–31.
Li K, et al. Tyrosine kinase Fyn promotes osteoarthritis by activating the beta-catenin pathway. Ann Rheum Dis. 2018;77(6):935–43.
Yu X, et al. Inhibition of JAK2/STAT3 signaling suppresses bone marrow stromal cells proliferation and osteogenic differentiation, and impairs bone defect healing. Biol Chem. 2018;399(11):1313–23.
Latourte A, et al. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann Rheum Dis. 2017;76(4):748–55.
Ji ML, et al. Dysregulated miR-98 contributes to extracellular matrix degradation by targeting IL-6/STAT3 signaling pathway in human intervertebral disc degeneration. J Bone Miner Res. 2016;31(4):900–9.
Ohba S, Lanigan TM, Roessler BJ. Leptin receptor JAK2/STAT3 signaling modulates expression of frizzled receptors in articular chondrocytes. Osteoarthritis Cartilage. 2010;18(12):1620–9.
Schlichting N, et al. Suitability of porcine chondrocyte micromass culture to model osteoarthritis in vitro. Mol Pharm. 2014;11(7):2092–105.
Hicks KL, et al. Testing patterns for genetically triggered aortic and arterial aneurysms and dissections at an academic center. J Vasc Surg. 2018;68(3):701–11.
Wen Y, et al. Integrating genome-wide DNA methylation and mRNA expression profiles identified different molecular features between Kashin-Beck disease and primary osteoarthritis. Arthritis Res Ther. 2018;20(1):41.
He J, et al. Identification of the potential molecular targets for human intervertebral disc degeneration based on bioinformatic methods. Int J Mol Med. 2015;36(6):1593–600.
Yu J, et al. Zoledronate induces apoptosis in cells from fibro-cellular membrane of unicameral bone cyst (UBC). J Orthop Res. 2005;23(5):1004–12.
Qian A, et al. cDNA microarray reveals the alterations of cytoskeleton-related genes in osteoblast under high magneto-gravitational environment. Acta Biochim Biophys Sin. 2009;41(7):561–77.
Authors’ contributions
XQ, ZLu, and DC designed and performed experiments, XQ and MZ analyzed the data, CZ participated in the patients’ surgery, XQ, ZLiu, and JL drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We appreciate staff members and students who offered favorable help to our research at the Third Affiliated Hospital of Soochow University and Jiangsu University.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All data and results of this study are available from the corresponding author upon reasonable request.
Consent for publication
All authors have read and approved the final manuscript. All data/images may be seen by the general public.
Ethics approval and consent to participate
Approval for the study was obtained from the Ethics Committee at Soochow University and informed consent was obtained from each patient according to the Declaration of Soochow University. All procedures involving mice were approved by Jiangsu University Institutional Animal Care and Use Committee.
Funding
This work was supported by the National Natural Science Foundation of China 81471263 (to J. Liu) and Natural Science Foundation of Jiangsu Province, Grant (BK20151177).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Primer sequences for qRT-PCR experiments.
Additional file 2: Table S2.
Sequences of shRIPK1s.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Qiu, X., Zhuang, M., Lu, Z. et al. RIPK1 suppresses apoptosis mediated by TNF and caspase-3 in intervertebral discs. J Transl Med 17, 135 (2019). https://doi.org/10.1186/s12967-019-1886-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-019-1886-3