
Abstract
Copper-containing enzymes perform fundamental functions by activating dioxygen (O2) and therefore allowing chemical energy-transfer for aerobic metabolism. The copper-dependence of O2 transport, metabolism and production of signalling molecules are supported by molecular systems that regulate and preserve tightly-bound static and weakly-bound dynamic cellular copper pools. Disruption of the reducing intracellular environment, characterized by glutathione shortage and ambient Cu(II) abundance drives oxidative stress and interferes with the bidirectional, copper-dependent communication between neurons and astrocytes, eventually leading to various brain disease forms. A deeper understanding of of the regulatory effects of copper on neuro-glia coupling via polyamine metabolism may reveal novel copper signalling functions and new directions for therapeutic intervention in brain disorders associated with aberrant copper metabolism.
Similar content being viewed by others


Background
Copper is a generally utilized heavy metal [1] with a toxic limit beyond 10 μM [2, 3]. At low concentrations, copper ion is an essential micronutrient that plays a variety of functions in biological systems. Copper containing enzymes and transcription factors are essential for cellular integrity, energy production, signalling, proliferation, oxidation and radiation defence. Research concerning acute or chronic toxicity of copper due to its deficiency or excess is growing rapidly and interest in the subject is pervasive [4,5,6,7,8,9,10,11,12]. Nevertheless, the pertinent redox status-dependent chelation [13,14,15,16,17,18,19] and regulatory mechanisms [20,21,22,23,24,25,26,27,28,29,30,31,32] are still being elucidated.
Recently, copper-related mechanisms have been suggested as therapeutic targets for important indications such as cancer [33], microbial defence [34,35,36,37], chronic lung inflammation [38], influenza A [39], neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD) and prion disease along with disorders linked to copper homeostasis such as Menkes disease (MD) or Wilson’s disease (WD) [40,41,42,43]. Elevated copper levels in the serum and tissue of cancer patients also suggest the involvement of copper in tumour growth [44, 45].
Our review will focus on biologically-relevant and emerging features of copper-dependent processes such as redox disproportionation, the properties of the chemical species generated (acid-base character, ligands, geometry etc. [46, 47]), the interaction between copper and sulfur redoxomes, the underlying redox signalling, along with the “dark side” where copper metabolism has been linked to compromised or fatal conditions [48,49,50].
The redox capability of copper
Evidence for the incorporation of oxygen atoms from dioxygen (O2) into oxidation products of cuproenzyme-catalyzed reactions in nature was first published in 1955 [51]. Since the pioneering work of Osamu Hayaishi, and independently Howard S. Mason a consensus has been achieved as to the involvement of Cu(I) disproportionation (redox) equilibria 2Cu(I)(aq)ᅟCu(0)(s) + Cu(II)(aq) (Eq. 1.) in the aqueous reduction of O2 to water (see [52,53,54] and citations included). The value of + 0.37 V relating K=Cu(II)]/[Cu(I)]2 = 106 M− 1 indicates that aerobic organisms can effectively utilize O2 when excess Cu(I) is sufficient [46]. This condition can be achieved within a copper concentration range of 10− 7 M to 10− 6 M (Fig. 1.) Within the 10− 4-10− 3 M range, however, the reduced Cu(I) form is minimally present, which would impair oxidative energy-transfer. The pertinent copper-containing enzymes such as cytochrome c oxidase (COX) [55] or copper, zinc superoxide dismutase (Cu, Zn-SOD1) [56,57,58] are involved in the mitochondrial electron transport chain [59] or in the dismutation of superoxide radical anion (O2• -) to hydrogen peroxide (H2O2), respectively. It is worth noting that the higher oxidation state of copper, Cu(III) may also shape the redox activation of the cytosolic copper pool and contributes to hydroxylation of phenolate substrates [60,61,62].

Disproportionation equilibria predicts Cu(I) in excess in the submicromolar to low micromolar range of ambient copper concentration. Due to the narrow non-toxic window for copper concentration, even small conditional changes may turn control into deregulation of copper signaling
Cuprous Cu(I) ion possesses both electron “donor” and “acceptor” attributes, and redox capability via the one-electron transfer charge-disproportionation between the “donor” and “acceptor” Cu(I) yielding Cu(II) and Cu(0). This ability of Cu(I) to disproportionate is fundamental, not only to vital functions related to O2 transport, regulation of respiration, neuronal differentiation and signal transmission [63, 64], but also to the instability of the copper ionome [26]. We know that uncontrolled redox reactions of copper that can be deleterious to life [12, 65,66,67,68,69,70,71,72,73,74,75], however, here we focus on and re-consider the controlled, redox capability-related signalling of copper that may be important for neurobiology.
Copper homeostasis
An evaluation of the effects of copper under normal and pathological conditions depends on an accurate knowledge of copper concentrations present in vivo. In spite of this, a bewildering feature of efforts to examine the role of copper in biological processes is the limited data available on the relative distribution of copper between organs, tissues, cell types and sub-cellular compartments in mammals [2, 44, 76,77,78,79,80,81,82,83]. From a practical viewpoint, the lack of the information makes it unrealistic to determine the recommended concentration of copper in drinking water. In addition to its biological variance, the significant differences in copper levels that exist in habitats and diets may also explain difficulties in determining the impact of copper on biological systems [2]. Moreover, multiple comparisons of existing data are compromised by the use of varying techniques, characteristically atomic absorption spectroscopy (AAS), flameless atomic absorption spectroscopic technique (FAAS), inductively coupled plasma-atomic emission spectrometry (ICP-AES) (Tables 1 and 2) and radiotracer detection or diverse sample preparing protocols. Data obtained by FAAS on brain tissue samples taken from 38 brain regions of 7 males within 2–4 h after death showing no macroscopic signs of disease [77, 78] disclosed significant copper concentration differences between brain areas, grey versus white matter cells, and between individuals. Brain copper concentrations were inversely correlated with age. It is worth noting that measurements of total copper levels may not necessarily reflect the biologically active metal pools [84].
Transition metals in biological tissues have been evaluated by atom absorption spectroscopy or radiotracer detection techniques, and more recently by the laser ablation inductively-coupled plasma mass spectrometry (LA-ICP-MS), secondary ion mass spectrometry, X-ray fluorescence microscopy (XFM), X-ray absorbance spectroscopy (XAS), micro particle-induced X-ray emission, and electron microscopy. Innovative imaging technologies of transitional metals were reviewed recently [85,86,87,88,89,90]. The recent development of recognition-based copper sensors and reaction-based copper indicators has allowed fluorescence imaging of labile copper pools [91,92,93,94,95]. Recent advances in non-destructive analytical methods will likely enable the assessment of copper dynamics over short, medium or long time scales that are relevant to signalling, metabolism and nutrition or aging.
These technologies have made possible a deeper understanding of copper dynamics and distribution. Significant relationships regarding the levels of Ctr1, Atox1, ATP7A/ATP7B and copper concentrations in the human brain have been identified by the combined application of ICP-MS spectrometry, Western blot and immunohistochemistry. Copper and ATP7A levels in the substantia nigra and in the cerebellum, respectively, have been found to be significantly greater compared to other brain regions [96]. New insights into the relative distribution of copper among elements including P, S, Cl, K, Ca, Fe, Zn within the choroid plexus (CP), ventricle system, and surrounding brain tissue have been provided by XFI techniques. In agreement with the known abundance of specific metal transporters, the elemental maps indicate that Zn, Fe and Cu are present within the CP, where the blood-cerebrospinal fluid barrier is primarily located [96,97,98,99]. Investigating the relationships between age, copper levels, and regulatory genes in the neurogenesis active sub-ventricular zone (SVZ) and CP has revealed i) age-related increases in Cu levels in both areas; ii) an age-related increase in MTs in SVZ, and iii) an age-related decrease and increase in Ctr1 in SVZ and CP, respectively [98]. These and past [100] findings suggest a specific role for copper in the development of brain tissue. The development of new imaging methods should provide a basis for further examination of the genuine labile copper pools, and related redox signalling within the brain.
From atomic structure to Speciations shaping dynamic copper Pool and Signalling
Among transition metal elements in brain, copper ranks third only to iron and zinc in pervasiveness. Yet, its disproportionation chemistry is unique due to its electronic structure (3s23p63d104s1) characterized by small energy differences between 3d and 4 s orbitals that allows for strong hybridization effects and electron tunneling [101, 102]. The easily convertible redox states Cu(I) and Cu(II) generate distinguishable bioligand variations (speciation). Indeed, axial symmetry distortion of Cu(II) aquo-complexes leads to extremely fast exchange of water (near to 1010s− 1) [103, 104]. This copper electron transfer-coupled structural alteration of coordination at copper sites in proteins [105, 106] can be envisaged as a molecular machine [107,108,109] switched on and driven by the redox disproportionation of copper. These molecular motions permit straight energy transfer from O2 to intrinsic cellular processes, potentially supporting fast neuronal signalling and remodelling of neuro-glia coupling [110] within the brain.
The extremely diverse copper speciation may be represented by a collection of copper bioligands including small ions and molecules such as sulfide ion, amino acids like His, Cys, Met, Asp, Tyr, Thr, Gly; neurotransmitters such as ATP, norepinephrine [111]; γ-aminobutyric acid (GABA) [112], and constituents of dense core vesicle cargo neurotrophins ([113] and references cited) inositol phosphates (IPs) [114], low-density lipoproteins (LDL) [115]. Redox propensity of chelates between copper and pertinent peptides (tripeptide glutathione (γ-L-glutamyl-L-cysteinylglycine: GSH) [116, 117]; peptide fragments of matricellular calcium-binding glycoprotein (secreted protein, acidic and rich in cysteine: SPARC) Gly-His-Lys (GHK) (for a recent review see [118] and proteins (metallothionein, ceruloplasmin, albumin, macroglobulin, transcuprein [3, 19, 119,120,121,122]), prion protein PrPC [65], amylin [123]) may present specific feature of transport and storage of copper. Likewise, many cuproproteins with redox, or redox-with-transport functions (mono-, di-, tetranuclear cupredoxins nitrite reductase, laccase, Cu, Zn-SOD1, amine oxidase CuAO, galactose oxidase, hemocyanin, tyrosinase, catechol oxidase, COX, N2O reductase, menaquinol NO reductase et cetera) [47], copper-transporting ATPases (Cu-ATPases, ATP7A and ATP7B) [124,125,126], divalent metal transporter DMT1 [127], copper transporters and chaperons Ctr1, Ctr2, Atox1 and CCS [128, 129], diverse group of bacterial periplasmic copper binding proteins (CopC) [130] are known. It is to note, that major molecular players of growth or metabolism DNA [131] or biogenic polyamines (pAs) [132] also bind copper. It is to note, that the four metal binding sites of albumin are partially selective, transporting not only Cu(II) but also Zn(II), Ni(II), Cd(II), Pt(II), V(IV)O and Au(I) [133]. Besides, the rather unique redox stability of Cu(II) bound to the the N-terminal albumin sequence could also be explained by the presence of the axially coordinated water [133], presenting less-distorted pyramidal symmetry [103].
Through the application of multiple complementary approaches, two subsets of total copper can be distinguished: the static, tightly bound and the dynamic, relatively weakly bound (labile or exchangeable) pools [134]. Most of the copper uptake in cells takes place through the Ctr1, whereas ATP7A and ATP7B prevent excess copper accumulation within cells [125, 126]. The membrane protein Ctr1 is considered as the major entry pathway for copper into eukaryotic cells. Although it is currently the sole identified transporter for copper uptake, the existence of Ctr1-independent copper entry by as yet unknown transporters has been suggested [135, 136]. Copper entrance requires its prior reduction by cell surface metalloreductases, as Ctr1 mediates transport of Cu(I) only, whereas ceruloplasmin, which carries half of the copper in blood plasma, delivers it as Cu(II) to the cell membrane [137]. Copper uptake is regulated mainly by Ctr1 translocation between the membrane surface and intracellular vesicles on demand, however, the Ctr1 protein has been shown to be degraded more rapidly under conditions of high copper excess [136].
Binding events in the His- and Met-rich extracellular amino terminal domain of vertebrate Ctr1 may support both reduction and transfer of copper from the carriers to the transporter [138]. Questions arise how Ctr1- bound copper moves outside-in down the peptide chain and dissociates? The human transporter is a symmetric Ctr1 trimer shaping a cone-like pore, which becomes wider in the outside-in direction from approximately 8 Å to 22 Å [129]. Cu(I) may traverse from the extracellular binding site through the cone to the HisCysHis motif near the intracellular carboxyl terminus of the protein by exchanging neighbouring Met of the conserved Met-XXX-Met Cu-binding motifs positioned along the pore interface. Higher stability of Cu(I)-Cys versus Cu(I)-Met could be the driving force for Cu(I) passage [136]. As far as the intracellular Cu(I) discharge pathways are concerned, Cys containing small peptides, such as GSH, or the “antioxidant peptide” Atox1 may contribute to Cu(I) release from the carrier. The typically high intracellular concentration of GSH (cca. 10 mM [139]) may produce shifting of the binding equilibrium towards GSH bound Cu(I) suggesting that GSH can efficiently collect copper [140, 141] bound to the intracellular HisCysHis binding crevice of Ctr1. Alternatively, Atox1 can also pick up HisCysHis bound Cu(I) and shuttles it to cytoplasmic metal-binding domains in ATP7A and ATP7B (also called MD and WD proteins, respectively) [16, 63, 142,143,144]. As suggested previously, the fast exchange of amino acid residues surrounding Cu(I) can readily explain entropy-compensation phenomena in course of dynamic interconversion of Cu-Cys coordinations during chaperon-target hopping [144].
The astonishing fact that free copper is undetectable within cells is due to the existence of copper chaperones, such as CCS which binds and transfers copper directly to its final target Cu, Zn-SOD1 [145, 146]. One can assume a novel type of protein-protein interaction delivering copper to its protein target destinations intracellularly [147]. It has been suggested that the exchange of copper between a variety of target-specific cytosolic chaperones and their targets in distinct compartments is driven by an increase in the copper binding affinity [148]. The speciation of copper sites in the CCS chaperone for target Cu, Zn-SOD1 and in the HAH1 chaperone for the soluble cytosolic domains (Menkes protein, MNK1) of the target ATP7A (Fig. 2.) [148,149,150,151] indicates, that the first domain of CCS, the sixth domain of the MNK1 and the HAH1 binds Cu(I) through two cysteines in a Cys-XX-Cys motif. The Cu, Zn-SOD1 protein binds copper via four His residues. Reportedly, the values of the apparent dissociation constants of Cu(I) towards chaperones and their intracellular targets may vary mostly in the range of 0.01pM to 0.1fM [148]. These estimates, however, turned to be erroneous as demonstrated by Shoshan and co-workers [152]. By taking into consideration oligomeric species of Cu(I)-dithiothreitol, the modified calculations conclude affinity values several orders of magnitude higher, an observation that deserves further comments. The affinities of copper sensors and indicators associated with novel imaging technologies may not allow fluorescence imaging of strongly bound copper in chaperones or targets, but possibly will permit detection of chaperone-Cu(I)-target complex formation (Fig. 2. lower panel), as characterized by several orders of magnitude lower affinity for the complex formation equilibria MNK1 + HAH1 MNK1-Cu(I)-HAH1 (Eq. 2.) [151]. In this case, variations of AAS (total pool) and fluorescence imaging (labile pool) data could give rise to proper assess of the strongly bound copper pool.
MNK1-Cu(I)-HAH1 (Eq. 2.) [151]. In this case, variations of AAS (total pool) and fluorescence imaging (labile pool) data could give rise to proper assess of the strongly bound copper pool.

Diverse speciation of copper in chaperons and targets. Upper row left: The two Cys residues Cys22 and Cys25 of the first domain of CCS chaperone (PDB code: 2rsq) [149] bind copper (yellow) with an average distance of 2.2 Å. Upper panel right: Copper (yellow) delivered to the target enzyme Cu, Zn-SOD1 (PDB code: 2C9V) [150] is bound by four His residues His46, His48, His63 and His120, and characterized by a range of Cu-His distances from 2.1 Å to 2.5 Å. Lower panel: The position of copper in the chaperon-Cu-target complex between chaperon HAH1 (magenta) and the first domain of the target ATP7A (Menkes protein, MNK1) (green) (PDB code: 2k1r) [152]). Three Cys residues fitting in both HAH1 (Cys12, Cys15) and MNK1 (Cys15, Cys18) CXXC motifs participate in the transition of copper from HAH1 to MNK1 [152]. Specifically, Cys12 of HAH1 and Cys15 of MNK1 are required for the formation of the HAH1-Cu-MNK1 complex, while the third Cys may be either of the Cys15 of HAH1 or the Cys18 of MNK1. Three coordinating Cys side chains are shown around the copper ion, all with a distance of 2.1 Å, the fourth Cys, which does not bind the metal thus far, is shown in green
Intracellular cu(I), GSH and the concept of coupled “Redoxomes”
As outlined above, intracellular copper exists in an immense variety of static forms that involve a multiple oxidation states with favoured ligand speciations (see below) or mixed-valent copper complexes. However, it also can change by reacting with “self” (see Eq. 1.) within sub-nanometer distances of multiple copper sites of vital peptides, proteins and enzymes [101, 102, 105, 106, 153,154,155,156]. A somewhat similar distinction can be made for a sulfur “redoxome”, a redox reaction-coupled proteomic network comprising numerous sulfur oxidation states and species and reactions with sulfur-containing peptides, proteins and enzymes, as well as the reaction of GSH with “self” yielding glutathione disulfide (GSSG) (2GSH → GSSG) [157,158,159]. Importantly, these “self”-reacting copper and sulfur “redoxomes” also interact with each other through the prominent chelation of Cu(I) by thiols of either the antioxidant copper chaperone protein Atox1 or GSH [63, 142, 160,161,162]. Supporting this concept, the GSH/GSSG ratio was found to be the most sensitive indicator of copper intoxication (and subsequent oxidative stress) [11]. Moreover, sulfur-doped copper clusters are relatively stable and abundant [163]. In the Cu-S-Cu unit found within active sites of copper-sulfur proteins like COX, the S− bridging a Cu2+ component displays a short Cu-Cu distance and a small Cu-S-Cu bond angle, which are essential for the electron transport performed by COX [163,164,165]. With this in mind, toxicity of copper excess in mammalian cells is explained by obstructing the control of the “interactome” of copper-sulfur containing “redoxomes” [166,167,168,169].
Because of its charge density and polarizability, the oxidized cupric Cu(II) ion would tend to be found in complex with “hard” bases such as H2O, OH−, RNH2 etc. (N- or O-ligands), while the “soft” acid Cu(I) does favour “soft” bases such as RS− and CN− ligands [46]. These trends in the stability of coordination complexes [170, 171] predict that the reduced cuprous Cu(I) ion would prefer the formation of complexes with “S-ligands” such as GSH [172]. Importantly, GSH can also represent “N- or O-ligands” for Cu(II), assisting disproportionation and O2• − dismutase activity of Cu(I). Indeed, the complex equilibrium system GSH-Cu(I) can switch to the oxidized GSSG-Cu(II) one [173]. Taken together, these observations have been used to classify the speciation of Cu(I) with GSH as a key feature accompanying redox homeostasis [11].
Although the dissociation constant for the GSH-Cu(I) equilibrium has been predicted to be about 9pM [148] (GSH-Cu(I)), this value has been called into question [174, 175]. Specifically, in an experimental model of GSH-Cu(I), the formation of the tetranuclear [Cu4(GS)6] cluster was observed as the major species within the range of pH from 5.5 to 7.5 [175]. The cluster formation equilibrium predicts that [Cu4(GS)6] limits free Cu(I)(aq) to the sub-femtomolar concentration range in eukaryotes. These findings suggest that the affinity of GSH towards Cu(I) may be orders of magnitude higher than previously thought [148]. If valid in vivo, not only the high intracellular GSH concentration but the high-affinity formation of the [Cu4(GS)6] cluster would also force the membrane-cytosol transfer of Cu(I) from Ctr1 to GSH. It is noteworthy, that bacteria capture copper surplus through the cytosolic protein Csp3s, which forms tetranuclear Cu(I) thiolate clusters [176] [Cu4(S-Cys)5]−, [Cu4(S-Cys)6]2−, and [Cu4(S-Cys)5(O-Asn)]−. In order to avoid toxicity of cytosolic copper overload, eukaryotes gain control over excess by MTs [177], including the brain-specific MT3 (growth inhibitory factor) binding. In fact, using XRF microscopy with sub-micron resolution, Sullivan et al. [178,179,, 180] demonstrated the presence of Cu-rich aggregates in astrocytes of the dentate gyrus and rostral migratory stream in the rat brain. These aggregates contain CuxSy clusters with a sulfur/Cu(I) ratio consistent with that of the Cu-MT complex. Apparently, both age-dependent [98] and overload-evoked changes [177,178,179,180] can be related to the copper-binding capacity of MTs.
Direct and indirect effects leading to sudden and catastrophic hemolytic anemia due to the direct toxic effects of copper on red blood cells has been described in the past [181,182,183,184,185,186,187,188,189,190]. Nevertheless, the observation that during chronic copper poisoning in sheep there is decreased antioxidant capacity directly correlating with the level of serum copper [191] putting GSH at the centre of anti-ROS protection [192]. Underscoring the importance of this role, the level of GSH in erythrocytes is an inheritable trait [193]. Unfortunately, it is hard to obtain valid GSH and GSH/GSSG data from biological samples [194].
Central regulation and storage of copper: Copper deficiency and toxicity disorders
ATP7A and ATP7B are highly abundant in the liver, yet disruptions in their transport functions affect the central nervous system (CNS). This is reflected in the sex-linked recessive CNS disorder observed in males with symptoms of copper deficiency (MD) arising from a mutant ATP7A pump. In contrast, a mutant ATP7B pump leads to copper toxicity in the autosomal recessive WD. These “brain” diseases suggest that the homeostasis of copper in the liver is essential for normal brain function [195,196,197]. It has been known for a long time, that WD is characterized by the accumulation of copper in tissues, particularly in the liver and brain ([198,199,200,201] and references cited). The biosynthesis, folding, localization, turnover and protein interaction network, of the most frequent copper transporter ATP7B mutant causing toxic accumulation of copper in WD has recently been described [202]. By targeting this network with specific siRNAs, correction of the localization of ATP7B-mutant restored copper levels to an acceptable range. Decreased stability associated with increased structural dynamics has been ascribed to disease-causing point-mutations in the metal-binding domains of WD protein [203]. Another fatal liver injury, the Indian childhood cirrhosis (ICC), was also found to be associated with heavy deposits of copper, though in all other respects it was different from WD [204].
Besides their significance in the overall copper efflux and balance, ATP7A and ATP7B play a critical role in copper transport between intracellular compartments. In hepatocytes, ATP7A and ATP7B are located mainly in the trans-Golgi network and supply copper for incorporation into copper-dependent enzymes such as tyrosinase, peptidylglycine amidating monooxygenase, dopamine monooxygenase, lysyl oxidase, and ceruloplasmin [205]. At high intracellular copper concentration, the carriers are translocated reversibly to the plasma membrane (ATP7A typically to the basolateral, ATP7B to the apical surface) where they efflux excess copper from the cell [206].
In food and water, the average daily intake of copper in the US is about 1 mg [207], which is relatively low. Most humans and animals are able to control excess amounts of copper by either decreased absorption or increased excretion. Ingestion of toxic amount of copper (> 10 mg/day) or acute or chronic environmental exposure, such as occupational hazard, accidents, release from copper pipes, initially affects the liver, the first organ of copper deposit. Many factors that alter copper metabolism influence the progress of chronic copper poisoning. The toxicity remains subclinical until the copper that is stored in the liver is released in massive amounts. The lethal dose of copper is about 10-20 g [207]. Initial symptoms of acute overdoses may be metallic taste, gastrointestinal distress that can progress to cardiovascular collapse, coma and death within hours. Hepatic symptoms arise after 24 h to 72 h of exposure, and are characterized by marked elevations in serum aminotransferase levels, hepatic failure, elevation in prothrombin time and jaundice. Erosion of epithelial lining of the gastrointestinal tract, acute tubular necrosis in the kidney was also reported. Blood copper concentrations can increase suddenly, causing lipid peroxidation and intravascular hemolysis [207, 208].
The liver takes up dietary copper from the portal blood, synthesizes cuproproteins in hepatocytes, and secretes excess copper into the bile. Overall balance of copper in the body is achieved by regulation of the rate of uptake in the small intestine and of biliary excretion. The key regulators of these processes are the ATP7A and ATP7B pumps. However, many other components of the machinery for copper homeostasis have been described including ceruloplasmin, small carriers, chaperones, MTs [24, 197, 205, 209, 210]. Precise regulation of intracellular copper homeostasis is essential, which is supported by the large number of clinical syndromes linked to either copper excess or shortage [197, 210, 211]. Several reviews have summarized results of genetic, biochemical and structural approaches concerning cellular copper homeostasis and related disorders [116, 212, 213], yet the entire network of events that regulate copper transport and intracellular disposition has not been fully explored.
As Ctr1 cannot transport bivalent copper, some ingested Cu(II) avoids the liver and passes rapidly into the systemic circulation where can target albumin [135]. Following entry into hepatocytes, Cu(I) binds the initial acceptor GSH, which delivers it to the different copper chaperones, such as Atox1, CCS and COX17 that partition copper into distinct intracellular compartments [116]. Nevertheless, the landscape of Cu(I) trafficking to chaperons via GSH may change. Recent data on femtomolar [175] versus picomolar [148] affinities of GSH towards Cu(I) raise the role for [Cu4(GS)6] preserving Cu(I). In fact, the Cu(I) availability is highly associated with GSH level of the cell. Ogra et al. [214] observed that depletion of GSH led to decreased copper in the bile and blood but increased copper in the liver. The decreased GSH level resulted in an oxidative environment in the liver that made Cu(I) less bioavailable. In addition, the redox state of the cells influences the activity of copper pumps. GSH deficiency inhibits ATP7A and ATP7B resulting in the intracellular accumulation of copper [136].
Synaptic release of copper
The concentration of copper in the cerebrospinal fluid (~ 70-80 μM) is rather high in comparison to serum (12-24 μM) [215, 216], raising the possibility of specific copper signalling in the brain. As outlined in previous sections, most cellular copper is strongly bound to proteins, yet the disposition of loosely bound copper can be detected by novel imaging technologies. This labile copper pool is believed to be associated with redox signalling. Labile copper has been found in the soma of cerebellar granule and cortical pyramidal neurons, in addition to the neuropil in the cerebellar and cerebral cortices, hippocampus and spinal cord [217].
The observation on the release of zinc from brain tissue during activity published in Nature in 1984 [218] provided initial evidence that transition metals could be directly involved in signalling [112, 134, 219,220,221,222,223,224,225,226,227,228,229]. Initial evidence suggested the potential of copper to modulate brain activity by affecting central inhibition. These include findings such as the pro-convulsant effects of a hitherto unidentified endogenous substance containing copper [112, 230], or depolarization-induced co-release of endogenous copper with the major inhibitory neurotransmitter γ-aminobutyric acid (GABA) in different experimental models of nerve terminals in vitro (synaptosomal fraction) and ex vivo (median eminence) [225]. Conclusions from 67Cu uptake and release measurements performed in hypothalamic slices by the presence of action potential blocker tetrodotoxin [231, 232] or determination of depolarization-induced copper release from nerve endings by AAS [225] raised the concept of copper signalling in the brain. Findings, such as the N-methyl-D-aspartate (NMDA) receptor activation-induced ATP7A trafficking to the plasmamembrane in the hippocampus [233, 234] have have provided new support for a role for copper efflux in mechanisms of excitotoxicity.
During the past 30 years, there has been a renaissance of interest and an expanded view of the contributions of copper to brain function and pathophysiology, as reflected in follow-up statistics, and throughout the literature [18, 42, 63, 69, 235,236,237,238,239,240,241,242,243,244,245,246,247,248] (Fig. 3.). One may speculate about copper speciation and/or mechanisms of copper uptake and release. Apart from trafficking in complex with various neurotransmitters, carrier peptides and proteins, or as part of the protein cargo of extracellular vesicles [39] there is also the potential for copper uptake as a result of autophagy [29, 249].

Emerging themes of copper signalling and functions. Number (Left) and percentage (Right) of papers citing the first description of depolarization-induced synaptic copper release [225] in each subject category by 5-year intervals. From the time, copper signalling in brain have considerably been developed, including inhibitory and excitatory signalling, neuromodulation, neurotoxicity, Alzheimer’s and other brain disorders
Zinc released from brain tissue during activity has been shown to reach concentrations in the hundred micromolar range, e.g. 300 μM [218]. In contrast, the concentration of copper in the synaptic cleft has been claimed to range from 1 to 10 μM, as determined by using the fluorescent indicator tetrakis-(4-sulfophenyl)-porphine (TSPP) in bovine chromaffin cells [250]. Notwithstanding the importance of imaging heavy metals in vivo (see also section 2.1. “Imaging technologies”), the quantitative relevance of fluorescent indicators strongly depends on the affinity standards applied (see for example [148]). Furthermore, TSPP has several drawbacks that can limit the validity of data obtained: i) the sub-micromolar Kd value of the TSPP-copper complex may not provide accurate data at the point of/or above saturation; ii) TSPP-copper binding is influenced by dissociation of copper from protein binding sites which necessary to validate the data an approach that is independent of protein binding, as in the case of ICP-MS technique; and finally, iii) the weak fluorescence intensitiy of the TSPP ligand itself. Conversely, based on atomic absorption spectroscopy data on depolarization-induced release of copper from nerve endings, one can estimate an activity-dependent enhancement of copper in the synaptic cleft, in the range of 100-250 μM [225], depending on the cleft size and volume taken. Furthermore, based on the Kd (100 μM) and saturating GABA concentration (1 mM) for GABAa receptor binding and desensitization [251,252,253,254] and assuming a stoichiometry of 2 for GABA co-released with copper [112] one could also conlude that a copper concentration of 100 μM can exist transiently within the synaptic cleft.
Copper can diffuse out of the synapse driven by the lower extrasynaptic concentration (1 μM) [216]. Moreover, the extrasynaptic copper concentration has been estimated to be in the nanomolar range based on the cellular and network excitability produced by bath-applied copper in the CA1 area of the rat hippocampal slice [244]. (This effect was primarily explained by the ability of copper to interfere with Hodgkin–Huxley conductances rather than the synaptic effects of copper [255].) Using a second-generation fluorescent copper sensor in combination with XFM, Dodani et al. [256] have observed that neural activity triggers copper trafficking from the cell body toward dendrites and revealed that these copper fluxes are calcium-dependent. This work provided direct imaging evidence that complemented prior studies on bulk copper release [225, 232]. Applying fluorescent copper indicators with improved hydrophilicity Dodani et al. [256] identified labile copper sources in the developing retina, and demonstrated that they modulate spontaneous activity of neural circuits via the copper transporter Ctr1, referred to as a ‘copper ion channel’ (see also section “The source-target-physiology scheme for therapeutic intervention”).
Copper Dyshomeostasis and brain disorders
Chronic copper intoxication causes region-specific copper accumulation in the CNS of male Wistar rats, following intra-peritoneal injections of copper lactate (0.15 mg Cu/100 g body weight) daily for 90 days. In these animals, copper content, but not that of zinc or iron was found to be significantly elevated in the cortex, cerebellum and striatum as determined by atomic absorption spectrophotometry [257]. Remarkably, metal dis-homeostasis has been widely accepted as a hallmark of several neurodegenerative diseases, such as prion, AD, PD, amyotrophic lateral sclerosis (ALS), Huntington’s chorea (HC) ([257, 258] and references cited). The antioxidant responses to copper overloads (0–30 mg/kg) in rat brains showed markedly decreased brain GSH and GSH/GSSG ratio after chronic copper exposure. Copper overloads are characterized by a t1/2 of 9-10 h for the decrease in GSH and of 4 h for decreases in the GSH/GSSG ratio, the latter being the most sensitive indicator of copper excess [11].
Prion diseases
The mainly α-helical folded prion protein PrPC is expressed in the enteric nervous system, e.g. in enteric nerve fibers/terminals and glia within the myenteric submucosal plexuses (inguinea pigs, mice), suggesting a role in the regulation of ileal contractility [259]. Additional beneficial roles for PrPC may arise from the discovery, that prion is an agonist at the G-protein coupled Adgrg6 receptor, known to regulate demyelinization-linked neuropathy [260, 261]. Copper has long been associated with the formation of protease-resistant, β-sheet enriched “scrapie” conformation of prion protein PrPSc, which has been considered the critical step in the neurodegenerative prion diseases known as transmissible spongiform encephalopathies [43, 262]. Recently, Giachin et al. [263] proposed that there is a non-octarepeat copper binding region [264] of PrPC which switch to the infectious PrPSc under acidic conditions. The only known prion disease observed in wildlife is the chronic wasting disease (CWD). Dietary magnesium and copper have been linked to inflammatory events in CWD pathogenesis [265]. Importantly, geographical regions where CWD is absent have significantly higher concentration of magnesium, and region where CDW is endemic show a higher magnesium/copper ratio in the water. Prion diseases share characteristics of “prion-like” neurodegenerative diseases in terms of the involvement of proteins (α-synuclein, amyloid β, and tau) forming amyloid deposits [266].
Alzheimer’s disease
The metal theory of AD [43, 267,268,269,270,271,272,273] (but see the advice of Schrag et al. [274]) predicts that the disregulation of copper/zinc levels by proteins known to be involved in AD-related neurodegeneration may lead to the accumulation of amyloid fibers and oxidative stress. Indeed, by using XFM high areal concentration of copper has been detected in amyloid beta (Aβ) plaques of the hippocampal gyrus dentatus sub-region in a mouse model of AD [275]. These data corroborate previous findings on the high-affinity interaction between Cu(II) and the histidine binding motif of Aβ [276], along with the role for Aβ as a synaptic Cu(II) scavenger [277]. In addition, the experimental ‘halo’ effect in copper maps may indicate co-localization of copper with a ‘ring’ rich in lipids, observed around the Aβ plaque in AD models [278] and human AD sections [279]. This suggests a potential association between Cu-catalyzed oxidative stress and plaque formation [280]. However, the question remains as to whether changes in metal distribution are the cause or the consequence of the plaque formation and progression of AD [275] or other progressive neurodegenerative diseases. For example, the neuropathology seen in AD may also characterize individuals with Down syndrome [281, 282], ALS or HC. By supporting a common pathway for familial and sporadic ALS, the pathological inclusions containing SOD1 fibrils may hold amyloid-like properties [283]. Abnormal copper accumulation in the striatum of HC patients has been linked to the copper binding facilitated formation of amyloid- copper transporter Ctr1, rlike bodies of the huntingtin (Htt) protein [284, 285]. Differential effects of ATP7A and ATP7B regulating copper metabolism MURR1 domain protein 1 (COMMD1) on the formation of mutant Cu, Zn-SOD1 fibrils (increase) or parkin inclusions (decrease) as well as the Htt aggregates (unaltered), however, suggest mechanistic diversity [286].
Parkinson’s disease
There is evidence that alterations in copper homeostasis play a role in PD with excess copper leading to neuronal cell death and α-synuclein aggregation [121, 287]. It is noteworthy in this context, that the depletion of GSH [70] is a very early symptom in the course of PD [288]. Amyloid fibre formation in type-2 diabetes [289] may also facilitate PD, due to the acceleration of α-synuclein amyloid formation by islet amyloid polypeptide amylin [290]. Disruption of retromer, a conserved heterotrimeric protein complex consisting of VPS35, VPS29 and VPS26, has been observed in a number of diseases including PD [291], resulting in disregulation of the retrieval and recycling of vital proteins [292]. Furthermore, the mutation of VPS35 increases copper toxicity in yeast, a likely outcome of the copper transporter miss-trafficking [293]. In fact, the endosomal retrieval and recycling of the copper transporter ATP7A is retromer-dependent in human cells [294]. Protecting the cargo of regulatory membrane proteins such as copper transporters and pumps via the retromer shipment may be critical in age-related health. It will be important to consider further the link between retromer complexes and copper homeostasis.
Multiple sclerosis
Disease-specific autoantibodies against inwardly rectifying K+ ion channel 4.1 (Kir4.1) [295], have been identified in the sera of patients suffering from the chronic inflammatory CNS disorder multiple sclerosis (MS). Feeding the copper chelator bis-cyclohexanone-oxalyldihydrazone (cuprizone, CTZ) reduces the myelin sheath and activates microglial and astroglial cells in the CNS, providing a reproducible and reversible model of pathologic processes underlying white and gray matter demyelination [296,297,298,299,300,301]. Expression of Kir4.1 autoantigen has been studied in the brain of CTZ-fed mice and revealed the induction of Kir4.1 protein in microvessels of the cerebral cortex [302]. The antioxidant functions of MTs [303] may have a role in MS, as suggested by the elevated level of MTs induced by CTZ in astroglia, while the oligodendroglia express low levels of MTs, which may contribute to their oxidative stress vulnerability [304, 305]. Apart from MS modelling, several lines of evidence suggest that CTZ intoxication is an excellent paradigm to study pathology and/or therapy of epilepsy or schizophrenia as well. However, mechanistic clues claiming either copper deficiency or copper build-up (associated with hydrazide formation-dependent enzyme inhibition) remain contradictory [306].
Chelate therapy
The restoration of copper homeostasis is mostly relevant to WD [119, 307], although neurodegenerative ([308,309,310,311,312], but see [313] versus [314]) or inflammatory [38, 208] diseases can also be related. Before the disease progresses to liver and brain (WD) or lung (inflammation), the excess copper can be limited by Cu(II) reduction, Zn(II) addition and administration of Cu(II) chelating ligands such as tetrathiomolybdate (TM), triethylene tetramine (Trientine, TETA, Trien) or D-penicillamine (D-pen) [119, 207] for limiting excess copper.
Due to its high level in proliferating tissues, copper can also promote angiogenesis and cancer development [315]. Hence, the Cu(II) lowering therapy has also potential in treating cancer (breast, colorectal, leukemia, lung, prostate) by copper chelating compounds (Table 3). A range of targets and/or mechanisms of action have been suggested for the antiproliferative activity of the Cu(II) chelate forming compounds. These include proteasome inhibitors and apoptosis inducers [316], DNA and protein interactions [317, 318], ROS generation [319], oxidative stress [320], integrin β4 up-regulation [321], Schiff base copper complex formation [318, 322]. (For a comprehensive knowledge of copper ion complexes as anticancer agents we refer to reviews [323, 324]).
Considering the redox activity of potential anticancer Cu(II) chelates (Table 3 and references cited) [323,324,325,326,327] one possible consequence is that the high level of copper in proliferating tissue could also reduce oxidative redox potential which may in turn increase cancer cell proliferation [45, 50, 328, 329]. The redox imbalance could be targeted by chelate formation coupled Cu(II) reduction in the proliferating tissue. Indeed, the reversible one-electron reduction of Cu(II) does occur, as exemplified by the thiosemicarbazone complex of Cu(II) in the elesclomol [330, 331] (Table 3).
It is interesting to note the effect of metformin [50, 332,333,334], which is a first line diabetes II drug and has been shown to increase healthy life span irrespective of its anti-diabetes effect. Its copper chelating ability [335] may suggest an anti-aging role for copper.
The source-target-physiology scheme for therapeutic intervention
The advent of imaging techniques that gained insight into the dynamics of labile copper pool made it possible to look beyond the molecular-level interactions in copper homeostasis and examine network-level dynamic interplays shaping copper signalling. The source-target-nphysiology (STP) scheme suggested by Chang [336] includes labile, neuronal copper pools in the Golgi compartment as a source, signal propagation via postsynaptic membrane receptor/ion channel target (the Cu(I) transporter Ctr1), and copper-dependent spontaneous activity of the neural network (physiology). Vesicular storage of canonical neurotransmitters with copper suggesting their co-release has also been described. Furthering the interactions between compartments within neurons, we conceive that cellular-level copper signalling between neurons and astrocytes, an emerging cell type of the brain, also exists and may play a fundamental part in the brain’s information processing.
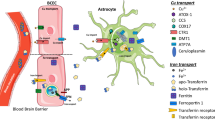
Several lines of evidence demonstrate memory deficits concurrent with copper deposition in the choroid plexus, astrocyte swelling (Alzheimer type II cells), astrogliosis and neuronal degeneration in the cerebral cortex, and augmented levels of copper and zinc in the hippocampus of chronically copper-intoxicated rats [337]. Furthermore, these and the other findings concerning the role for astrocytes in brain activity, dis-homeostasis and asscociated diseases [110, 338,339,340,341] and brain copper and pA homeostasis in particular [179, 180, 342, 343] may provide support for new astrocyte-centric directions for therapeutic intervention. It can also be depicted by the “gliocentric” alternative of the “neurocentric” STP workflow suggested by Chang [336] possibly associated with major astroglial processes and players of Glu and ammonia homeostasis underlying excitation-inhibition balance in brain [110]. Prevalent traumatic and ischaemic brain injuries are explored to validate the potential of the “gliocentric” concept of early therapeutic intervention. Now, we may add copper-dependent astroglial pA production to the list (Fig. 4.) [6, 22, 80, 179, 180, 233, 234, 236,237,238,239,240,241,242,243, 245, 254, 255, 336, 339, 340, 342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362]. GABA can be synthesized from the pA putrescine by copper-containing CuAO in astrocytes. CuAOs perform the oxidation of primary amines such as spermine, spermidine and putrescine to aldehydes and ammonia, producing H2O2 as a by-product. Putrescine-derived GABA is released by the inside-out (reverse) action of glial GABA transporter subtypes. The increased GABA release and the generated tonic inhibition thereby modulate the power of gamma range oscillation in the CA1 region in vivo. The concentration of cytosolic and extracellular putrescine has been determined to be 22 nmol/g and 12 nmol/g, respectively [339]. In contrast, copper may decrease tonic inhibition via acting on delta subunit-containing extrasynaptic GABAA receptors [235, 237, 246], thus adding a new layer to disinhibitory mechanisms in copper-rich brain areas.

Copper signaling via neuro-glia coupling. Astroglia, a previously neglected cell type of the brain [340], operate a variety of copper-dependent metabolic functions [6, 80, 240, 341, 342]. For this reason, in addition to synaptic and extrasynaptic copper signalling by way of excitatory/inhibitory receptors and ionic channels [22, 234, 235, 237,238,239,240,241,242,243,244, 246, 255, 336, 345,346,347,348,349,350,351,352,353,354,355,355], we place copper-dependent production of pAs in astrocytes [338] and correlated gap-junction modulation in the centre of this option. The proposed scheme conjectures activity-dependent changes of copper pools [179, 180] and polyamines (pAs), produced by CuAOs in astrocytes. First, an enhanced gap junction communication can be achieved by pAs [356,357,358], possibly promoting activity-dependent synchronization [339, 359]. Second, major inhibitory neurotransmitter gamma-aminobutyric acid (GABA) formed from pAs is released by astrocyte-specific GABA transporter [360]. Acting on its extrasynaptic receptor, GABA elevates tonic inhibition and enhances the fast (gamma band) neural oscillations [360]. These ways, the steady-state pA level in astrocytes determined by copper-dependent forming and consuming can be associated with neural circuit activity [244, 255, 362]
Conclusions
Despite multifaceted roles for copper observed in various brain diseases and tumours, the copper signalling theme is still in its initial stages. However, our increasing understanding of dynamic copper pools supports the idea of neuronal activity-dependent Cu(I) transmission affecting astroglia network signaling and astroglia-neuron metabolic cooperation. Rather than simply reflecting copper excess, copper-rich aggregates - likely in astrocytes and not in neurons - are a sign of a disturbed network. Brain diseases linked to oxidative stress [363] change the GSH/GSSG ratio and thereby automatically affect the copper homeostasis, as GSH is the immediate partner, along with various chaperones, that takes Cu(I) from the transporter. Therefore, Cu(I) distribution is disturbed and might in turn enhance oxidative stress at copper-containing deposits or limit Cu, Zn-SOD1 activity in regions with decreased copper level. Closer understanding of copper signalling and its vulnerability opens up new perspectives improving chelate therapy approaches against brain diseases and tumour.
Change history
12 November 2018
Following publication of the original article [1], the authors reported an error in Table 3. The correct version of Table 3 is shown below:
The publishers apologise for this error. The original article [1] has been corrected.
Abbreviations
- AAS:
-
Atomic absorption spectroscopy
- AD:
-
Alzheimer’s disease
- ALS:
-
Amyotrophic lateral sclerosis
- Atox1, CCS, HAH1, COX17:
-
Copper chaperones
- ATP7A, ATP7B, Cu-ATPases:
-
Copper-transporting P-type ATPases
- Aβ:
-
Amyloid beta
- CNS:
-
Central nervous system
- CopC:
-
Bacterial peripasmic copper binding proteins
- COX, mitochondrial complex IV:
-
Cytochrome c oxidase (EC 1.9.3.1)
- CP:
-
Choroid plexus
- Ctr1, Ctr2:
-
Copper(I) transporters
- CTZ:
-
cuprizone (bis-cyclohexanoneoxalyl-dihydrazone)
- Cu, Zn-SOD1:
-
Copper, zinc superoxide dismutase
- CuAO:
-
Copper aminooxidase
- CWD:
-
Chronic wasting disease
- DMT1:
-
Divalent metal transporter
- D-pen:
-
D-penicillamine
- EM:
-
Electron microscopy
- FAAS:
-
Flameless atomic absorption spectroscopy
- GABA:
-
Gamma-aminobutyric acid
- GHK:
-
SPARC fragment Gly-His-Lys
- GSH:
-
Gluthation (gamma-L-glutamyl-L-cysteinylglycine)
- GSSG::
-
Gluthation disulfide
- HC:
-
Huntington’s chorea
- Htt:
-
Huntingtin protein
- ICC:
-
Indian childhood cirrhosis
- ICP-AES:
-
Inductively coupled plasma - atomic emission spectrometry
- IPs:
-
Inositol phosphates
- Kir4.1:
-
Inwardly rectifying K+ ion channel 4.1;
- LA-ICP-MS:
-
Laser ablation - inductively coupled plasma - mass spectrometry
- LDL:
-
Low-density lipoproteins
- MD:
-
Menkes disease
- MNK1:
-
Menkes protein (soluble cytosolic ATP7A domain)
- MT:
-
Metallothionein
- μ-PIXE:
-
Micro - particle induced X ray emission
- MS:
-
Multiple sclerosis
- COMMD1:
-
MURR1 domain protein 1
- NMDA:
-
N-methyl-D-aspartate
- PD:
-
Parkinson’s disease
- pAs:
-
Polyamines
- PrPC :
-
Prion protein, α helical (Adgrg6 receptor agonist)
- PrPSc :
-
Prion protein, β sheet enriched (“scrapie”)
- SPARC:
-
Secreted protein, acidic and rich in cysteine
- STP:
-
Source-target-physiology
- SVZ:
-
Sub-ventricular zone
- TSPP:
-
Tetrakis-(4-sulfophenyl)-porphine
- TM:
-
Tetrathiomolybdate
- Trientine:
-
TETA, Trien (Triethylene tetramine)
- WD:
-
Wilson’s disease
- XFM:
-
X-ray fluorescence microscopy
References
Crichton RR, Pierre J-L. Old iron, young copper: from Mars to Venus. Biometals. 2001;14:99–112.
Georgopoulos PG, Roy A, Yonone-Lioy MJ, Opiekun RE, Lioy PJ. Environmental copper: its dynamics and human exposure issues. J Toxicol Environ Health B Crit Rev. 2001;4:341–94.
Tapia L, González-Agüero M, Cisternas MF, Suazo M, Cambiazo V, Uauy R, González M. Metallothionein is crucial for safe intracellular copper storage and cell survival at normal and supra-physiological exposure levels. Biochem J. 2004;378:617–24.
Alghobashy AA, Alkholy UM, Talat MA, Abdalmonem N, Zaki A, Ahmed IA, Mohamed RH. Trace elements and oxidative stress in children with type 1 diabetes mellitus. Diab Metab Synd Obesity Targets Ther. 2018;11:85–92.
Angelé-Martínez C, Nguyen KV, Ameer FS, Anker JN, Brumaghim JL. Reactive oxygen species generation by copper(II) oxide nanoparticles determined by DNA damage assays and EPR spectroscopy. Nanotoxicology. 2017;11:278–88.
Bulcke F, Dringen R, Scheiber IF. Neurotoxicity of copper. Adv Neurobiol. 2017;18:313–43.
Fu Y, Chang F-MJ, Giedroc DP. Copper transport and trafficking at the host−bacterial pathogen interface. Acc Chem Res. 2014;47:3605–13.
Gaetke LM, Chow-Johnson HS, Chow CK. Copper: toxicological relevance and mechanisms. Arch Toxicol. 2014;88:1929–38.
Ladomersky E, Petris MJ. Copper tolerance and virulence in bacteria. Metallomics. 2015;7:957.
Sadiq S, Ghazala Z, Chowdhury A, Büsselberg D. Metal toxicity at the synapse: presynaptic, postsynaptic, and long-term effects. J Toxicol. 2012:132671.
Semprine J, Ferrarotti N, Musacco-Sebio R, Saporito-Magriñá C, Fuda J, Torti H, Castro-Parodi M, Damiano A, Boveris A, Repetto MG. Brain antioxidant responses to acute iron and copper intoxications in rats. Metallomics. 2014;6:2083–9.
Shimberg GD, Ok K, Neu HM, Splan KE, Michel SLJ. Cu(I) disrupts the structure and function of the nonclassical zinc finger protein tristetraprolin (TTP). Inorg Chem. 2017;56:6838–48.
Bagchi P, Morgana MT, Bacsa J, Fahrni CJ. Robust affinity standards for cu(I) biochemistry. J Am Chem Soc. 2013;135:18549–59.
Ceko MJ, Aitken JB, Harris HH. Speciation of copper in a range of food types by X-ray absorption spectroscopy. Food Chem. 2014;164:50–4.
Guo M, Dong P, Feng Y, Xi X, Shao R, Tian X, Zhang B, Zhu M, Meng X. A two-photon fluorescent probe for biological cu (II) and PPi detection in aqueous solution and in vivo. Biosens Bioelectron. 2017;90:276–82.
Hatori Y, Yan Y, Schmidt K, Furukawa E, Hasan NM, Yang N, Liu C-N, Sockanathan S, Lutsenko S. Neuronal differentiation is associated with a redox-regulated increase of copper flow to the secretory pathway. Nat Commun. 2016;7:10640.
Jiang X, Chen J, Bajić A, Zhang C, Song X, Carroll SL, Cai Z-L, Tang M, Xue M, Cheng N, Schaaf CP, Li F, MacKenzie KR, Ferreon ACM, Xia F, Wang MC, Maletić-Savatić M, Wang J. Quantitative real-time imaging of glutathione. Nat Commun. 2017;8:16087.
Krishnamoorthy L, Cotruvo JA Jr, Chan J, Kaluarachchi H, Muchenditsi A, Pendyala VS, Jia S, Aron AT, Ackerman CM, Wal MNV, Guan T, Smaga L, Farhi SL, New EJ, Lutsenko S, Chang CJ. Copper regulates cyclic AMP-dependent lipolysis. Nature Chem Biol. 2016;12:586–92.
Sendzik M, Pushie MJ, Stefaniak E, Haas KL. Structure and affinity of cu(I) bound to human serum albumin. Inorg Chem. 2017;56:15057–65.
Argüello JM, Raimunda D, González-Guerrero M. Metal transport across biomembranes: emerging models for a distinct chemistry. J Biol Chem. 2012;287:13510–7.
Brown DR, Schmidt B, Kretzschmar HA. Effects of copper on survival of prion protein knockout neurons and glia. J Neurochem. 1998;70:1686–93.
Gaier ED, Eipper BA, Mains RE. Copper signaling in the mammalian nervous system: synaptic effects. J Neurosci Res. 2013;91:2–19.
Jiang WD, Liu Y, Hu K, Jiang J, Li SH, Feng L, Zhou XQ. Copper exposure induces oxidative injury, disturbs the antioxidant system and changes the Nrf2/ARE (CuZnSOD) signaling in the fish brain: protective effects of myo-inositol. Aquat Toxicol. 2014;155:301–13.
Jack H, Kaplan JH, Maryon EB. How mammalian cells acquire copper: An essential but potentially toxic metal. Biophys J. 2016;110:7–13.
Kumar D, Mains RE, Eipper BA. 60 years of POMC: from POMC and αMSH to PAM, molecular oxygen, copper and vitamin C. J Mol Endocrinol. 2016;56:T63–76.
Malinouski M, Hasan NM, Zhang Y, Seravalli J, Lin J, Avanesov A, Lutsenko S, Gladyshev VN. Genome-wide RNAi ionomics screen reveals new genes and regulation of human trace element metabolism. Nat Commun. 2014;5:3301.
Merker K, Hapke D, Reckzeh K, Schmidt H, Lochs H, Grune T. Copper related toxic effects on cellular protein metabolism in human astrocytes. Biofactors. 2005;24:255–61.
Ogra Y, Tejima A, Hatakeyama N, Shiraiwa M, Wu S, Ishikawa T, Yawata A, Anan Y, Suzuki N. Changes in intracellular copper concentration and copper-regulating gene expression after PC12 differentiation into neurons. Sci Rep. 2016;6:33007.
Seo Y, Cho YS, Huh YD, Park H. Copper ion from Cu2O crystal induces AMPK-mediated autophagy via superoxide in endothelial cells. Mol Cells. 2016;39:195–203.
Wittung-Stafshede P. Unresolved questions in human copper pump mechanisms. Q Rev Biophys. 2015;48:471–8.
Xiao Z, Wedd AG. The challenges of determining metal-protein affinities. Natural Prod Rep. 2010;27:768–89.
Young TR, Wedd AG, Xiao Z. Evaluation of cu (I) binding to the E2 domain of the amyloid precursor protein–a lesson in quantification of metal binding to proteins via ligand competition. Metallomics. 2018;10:108–19.
Blockhuys S, Wittung-Stafshede P. Roles of copper-binding proteins in breast cancer. Int J Mol Sci. 2017;18:E871.
Neumann W, Gulati A, Nolan EM. Metal homeostasis in infectious disease: recent advances in bacterial metallophores and the human metal-withholding response. Curr Op Chem Biol. 2017;37:10–8.
Srivastava S, Panda S, Li Z, Fuhs SR, Hunter T, Thiele D, Hubbard SR, Skolnik EY. Histidine phosphorylation relieves copper inhibition in the mammalian potassium channel KCa3.1. eLife. 2016;5:e16093.
Sun TS, Ju X, Gao HL, Wang T, Thiele DJ, Li JY, Wang ZY, Ding C. Reciprocal functions of Cryptococcus neoformans copper homeostasis machinery during pulmonary infection and meningoencephalitis. Nat Commun. 2014;5:5550.
Wiemann P, Perevitsky A, Lim FY, Shadkchan Y, Knox BP, Landero Figueora JA, Choera T, Niu M, Steinberger AJ, Wüthrich M, Idol RA, Klein BS, Dinauer MC, Huttenlocher A, Osherov N, Keller NP. Aspergillus fumigatus copper export machinery and reactive oxygen intermediate defense counter host copper-mediated oxidative antimicrobial offense. Cell Rep. 2017;19:1008–21.
Liu L, Geng X, McDermott J, Shen J, Corbin C, Xuan S, Kim J, Zuo L, Liu Z. Copper deficiency in the lungs of TNF-a transgenic mice. Front Physiol. 2016;7:234.
Cypryk W, Lorey M, Puustinen A, Tuula A, Nyman TA, Matikainen S. Proteomic and bioinformatic characterization of extracellular vesicles released from human macrophages upon influenza a virus infection. J Proteome Res. 2017;16:217–27 (“The Immune System and the Proteome 2016.”).
Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14:103–13.
Bánsági B, Lewis-Smith D, Pal E, Duff J, Griffin H, Pyle A, Müller JS, Rudas G, Aranyi Z, Lochmüller H, Chinnery P, Horvath R. Phenotypic convergence of Menkes and Wilson disease. Neurol Genet. 2016;2:e119.
Manto M. Abnormal copper homeostasis: mechanisms and roles in neurodegeneration. Toxics. 2014;2:327–45.
Viles JH. Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord Chem Rev. 2012;256:2271–84.
Margalioth EJ, Schenker JG, Chevion M. Copper and zinc levels in normal and malignant tissues. Cancer. 1983;52:868–72.
Gupte A, Mumper RJ. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat Rev. 2009;35:32–46.
Delhaize E, Loneragan JF, Webb J. Copper in plants. Its relation to soils and availability to animals. Chapter 1. In: McC Howell J, Gawthorne JM, editors. Copper in Animals and Man, vol. volume I and II. Boca Raton, Florida: CRC Press, Inc.; 1987. p. 1–19.
Lu Y. Cupredoxins. In: McCleverty J, Meyer TJ, editors. Comprehensive Coordination Chemistry II: From Biology to Nanotechnology, vol. 8 (Biocoordination Chemistry, Lawrence Que, Jr. and William B. Tolman, Eds.); 2003. p. 91–122.
Brewer GJ. Copper-2 ingestion, plus increased meat eating leading to increased copper absorption, are major factors behind the current epidemic of Alzheimer’s disease. Nutrients. 2015;7:10053–64.
Bulcke F, Dringen R. Handling of copper and copper oxide nanoparticles by astrocytes. Neurochem Res. 2016;41:33–43.
Watson JD. Type 2 diabetes as a redox disease. Lancet. 2014;383:841–3.
Que L. 60 years of dioxygen activation. J Biol Inorg Chem. 2017;22:171–3.
Koppenol WH, Stanbury DM, Bounds PL. Electrode potentials of partially reduced oxygen species, from dioxygen to water. Free Radic Biol Med. 2010;49:317–22.
Armstrong DA, Huie RE, Koppenol WH, Lymar SV, Merenyi G, Neta P, Ruscic B, Stanbury DM, Steenken S, Wardman P. Standard electrode potentials involving radicals in aqueous solution: inorganic radicals. Pure Appl Chem. 2015;87:1139–50.
Shepard EM, Dooley DM. Inhibition and oxygen activation in copper amine oxidases. Acc Chem Res. 2015;48:1218–26.
Lyons JA, Aragão D, Slattery O, Pisliakov AV, Soulimane T, Caffrey M. Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature. 2012;487:514–8.
Furukawa Y, O’Halloran TV. Posttranslational modifications in cu, Zn-superoxide dismutase and mutations associated with amyotrophic lateral sclerosis. Antioxid Redox Signal. 2006;8:847–67.
Lamb AL, Torres AS, O’Halloran TV, Rosenzweig AC. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone, nature Struct. Biol. 2001;8:751–5.
Zou Y, Sun Y, Zhu Y, Ma B, Nussinov R, Zhang Q. Critical nucleus structure and aggregation mechanism of the C-terminal fragment of copper−zinc superoxide dismutase protein. ACS Chem Neurosci. 2016;7:286–96.
Shimada A, Kubo M, Baba S, Yamashita K, Hirata K, Ueno G, Nomura T, Kimura T, Shinzawa-Itoh K, Baba J, Hatano YE, Miyamoto A, Murakami H, Kumasaka T, Owada S, Tono K, Yabashi M, Yamaguchi Y, Yanagisawa S, Sakaguchi M, Ogura T, Komiya R, Yan J, Yamashita E, Yamamoto M, Ago H, Yoshikawa S, Tsukihara T. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci Adv. 2017;3:e1603042.
Itoh A. Developing mononuclear copper−active-oxygen complexes relevant to reactive intermediates of biological oxidation reactions. Acc Chem Res. 2015;48:2066–74.
Keown W, Gary JB, Stack TDP. High-valent copper in biomimetic and biological oxidations. J Biol Inorg Chem. 2017;22:289–305.
Pham AN, Xing G, Miller CJ, Waite TD. Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J Catal. 2013;301:54–67.
Hatori Y, Lutsenko S. The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants. 2016;5:25.
Quist DA, Diaz DE, Liu JJ, Karlin KD. Activation of dioxygen by copper metalloproteins and insights from model complexes. J Biol Inorg Chem. 2017;22:253–88.
Haigh CL, Brown DR. Prion protein reduces both oxidative and non-oxidative copper toxicity. J Neurochem. 2006;98:677–89.
Kovács R, Schuchmann S, Gabriel S, Kardos J, Heinemann U. Ca2+ signalling and changes of mitochondrial function during low-Mg2+-induced epileptiform activity in organotypic hippocampal slice cultures. Eur J Neurosci. 2001;13:1311–9.
Kovács R, Schuchmann S, Gabriel S, Kann O, Kardos J, Heinemann U. Free radical-mediated cell damage after experimental status epilepticus in hippocampal slice cultures. J Neurophysiol. 2002;88:2909–18.
Kovács R, Kardos J, Heinemann U, Kann O. Mitochondrial calcium ion and membrane potential transients follow the pattern of epileptiform discharges in hippocampal slice cultures. J Neurosci. 2005;25:4260–9.
Li Y, Mayer FP, Hasenhuet PS, Burtscher V, Schicker K, Sitte HH, Freissmuth M, Sandtner W. Occupancy of the zinc-binding site by transition metals decreases the substrate affinity of the human dopamine transporter by an allosteric mechanism. J Biol Chem. 2017b;292:4235–43.
Liddell JR, White AR. Nexus between mitochondrial function, iron, copper and glutathione in Parkinson’s disease. Neurochem Int. 2017; pii: S0197–0186(17)30250–4.
Lőrincz T, Jemnitz K, Kardon T, Mandl J, Szarka A. Ferroptosis is involved in acetaminophen induced cell death. Pathol Oncol Res. 2015;21:1115–21.
Macomber L, Imlay JA. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc Natl Acad Sci U S A. 2009;106:8344–9.
Szárics É, Kovács R, Hajós F, Kardos J. Ca2+ ion accumulation precedes formation of O2 −· in isolated brain mitochondria. Neuroreport. 2006;17:1767–71.
Tan G, Yang J, Li T, Zhao J, Sun S, Li X, Lin C, Li J, Zhou H, Lyu J, Ding H. Anaerobic copper toxicity and iron-sulfur cluster biogenesis in Escherichia coli. Appl Environ Microbiol. 2017. https://doi.org/10.1128/AEM.00867-17.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Diff. 2016;23:369–79.
Bárány E, Bergdahl IA, Bratteby LE, Lundh T, Samuelson G, Schütz A, Skerfving S, Oskarsson A. Trace elements in blood and serum of Swedish adolescents: relation to gender, age, residential area, and socioeconomic status. Environ Res. 2002;89:72–84.
Bonilla E, Salazar E, Villasmil JJ, Villalobos R, Gonzalez M, Davila JO. Copper distribution in the normal human brain. Neurochem. Res. 1984;9:1544–8.
Harrison WW, Netsky MG, Brown MED. Trace elements in human brain: copper, zinc, iron, and magnesium. Clin Chem Acta. 1968;21(1968):55–60.
Haswell SJ. Atomic absorption spectrometry. Theory, design and applications. Amsterdam: Elsevier; 1991. p. 368–70.
Pal A, Prasad R. Regional distribution of copper, zinc and iron in brain of Wistar rat model for non-Wilsonian brain copper toxicosis. Ind J Clin Biochem. 2016;31:93–8.
Ramos P, Santos A, Pinto NR, Mendes R, Magalhães T, Almeida A. Anatomical region differences and age-related changes in copper, zinc, and manganese levels in the human brain. Biol Trace Elem Res. 2014;161:190–201.
Sumino K, Hayakawa K, Shibata T, Kitamura S. Heavy metals in normal Japanese tissues. Amounts of 15 heavy metals in 30 subjects. Arch Environ Health. 1975;30:487–94.
Yoo Y, Lee S, Yang J, In S, Kim K, Kim S, Kwon T, Ko Y, Chung K. Distribution of heavy metals in normal Korean tissues. Probl Forensic Sci. 2000;43:283–9.
Thomason RT, Pettiglio MA, Herrera C, Kao C, Gitlin JD, Bartnikas TB. Characterization of trace metal content in the developing zebrafish embryo. PLoS One. 2017;12:e0179318.
Ackerman CM, Le S, Chang CJ. Analytical methods for imaging metals in biology: from transition metal metabolism to transition metal signalling. Anal Chem. 2017;89:22–41.
Davies KM, Hare DJ, Bohic S, James SA, Billings JL, Finkelstein DI, Doble PA, Double KL. Comparative study of metal quantification in neurological tissue using laser ablation-inductively coupled plasma-mass spectrometry imaging and X-ray fluorescence microscopy. Anal Chem. 2015;87:6639–45.
Hare DJ, Kysenius K, Paul B, Knauer B, Hutchinson RW, O’Connor C, Fryer F, Hennessey TP, Bush AI, Crouch PJ, Doble PA. Imaging metals in brain tissue by laser ablation - inductively coupled plasma - mass spectrometry (LA-ICP-MS). J Vis Exp. 2017;119:e55042.
Jin Q, Paunesku T, Lai B, Gleber SC, Chen SI, Finney L, Vine D, Vogt S, Woloschak G, Jacobsen C. Preserving elemental content in adherent mammalian cells for analysis by synchrotron-based x-ray fluorescence microscopy. J Microsc. 2017;265:81–93.
Smith SV. Molecular imaging with copper-64 in the drug discovery and development arena. Exp Op Drug Disc. 2007;2:659–72.
Vogt S, Ralle M. Opportunities in multi dimensional trace metal imaging: taking copper associated disease research to the next level. Anal Bioanal Chem. 2013;405:1809–20.
Aron AT, Ramos-Torres KM, Cotruvo JA Jr, Chang CJ. Recognition- and reactivity-based fluorescent probes for studying transition metal signaling in living systems. Acc Chem Res. 2015;48:2434–42.
Carter KP, Young AM, Palmer AE. Fluorescent sensors for measuring metal ions in living systems. Chem Rev. 2014;114:4564–601.
Chang PV, Bertozzi CR. Imaging beyond the proteome. Chem Commun. 2012;48:8864–79.
Cotruvo JA Jr, Arona AT, Ramos-Torresa KM, Chang CJ. Synthetic fluorescent probes for studying copper in biological systems. Chem Soc Rev. 2015;44:4400–14.
Hirayama T, Van de Bittner GC, Gray LW, Lutsenko S, Chang CJ. Near-infrared fluorescent sensor for in vivo copper imaging in a murine Wilson disease model. Proc Natl Acad Sci U S A. 2012;109:2228–33.
Davies KM, Hare DJ, Cottam V, Chen N, Hilgers L, Halliday G, Mercer JF, Double KL. Localization of copper and copper transporters in the human brain. Metallomics. 2013;5:43–51.
Choi B-S, Zheng W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009;1248:14–21.
Fu S, Jiang W, Zheng W. Age-dependent increase of brain copper levels and expressions of copper regulatory proteins in the subventricular zone and choroid plexus. Front Mol Neurosci. 2015;8:22.
Lins BR, Pushie JM, Jones M, Howard DL, Howland JG, Hackett MJ. Mapping alterations to the endogenous elemental distribution within the lateral ventricles and choroid plexus in brain disorders using x-ray fluorescence imaging. PLoS One. 2016;11:e0158152.
Kozma M, Ferke A. Trace element localization and changes in zinc and copper concentrations during postnatal development of the rat CNS. Acta Histochem. 1979;65:219–27.
Ghosh S, Xiea X, Deya A, Sun Y, Scholes CP, Solomon EI. Thermodynamic equilibrium between blue and green copper sites and the role of the protein in controlling function. Proc Natl Acad Sci U S A. 2009;106:4969–74.
Moser CC, Page CC, Dutton PL. Darwin at the molecular scale: selection and variance in electron tunnelling proteins including cytochrome c oxidase. Phil Trans R Soc B. 2006;361:1295–305.
Halcrow MA. Jahn–teller distortions in transition metal compounds, and their importance in functional molecular and inorganic materials. Chem Soc Rev. 2013;42:1784–95.
Pasquarello A, Petri I, Salmon PS, Parisel O, Car R, Toth E, Powell DH, Fischer HE, Helm L, Merbach A. First solvation shell of the cu(II) aqua ion: Evidence for fivefold coordination. Science. 2001;291:856–9.
Solomon EI, LaCroix LB, Randall DW. Electronic structure contributions to function in bioinorganic chemistry: the blue copper active site. Pure Appl Chem. 1998;70:799–808.
Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper active sites in biology. Chem Rev. 2014;114:3659–853 (Special issue on Bioinorganic Enzymology).
Bissell RA, Córdova E, Kaifer AE, Stoddart JFA. Chemically and electrochemically switchable molecular shuttle. Nature. 1994;369:133–7.
Collin J-P, Dietrich-Buchecker C, Gaviña P, Jimenez-Molero MC, Sauvage J-P. Shuttles and muscles: linear molecular machines based on transition metals. Acc Chem Res. 2001;34:477–87.
Durola F, Lux J, Sauvage J-P. A fast-moving copper-based molecular shuttle: synthesis and dynamic properties. Chem Eur J. 2009;15:4124–34.
Kardos J, Héja L, Jemnitz K, Kovács R, Palkovits M. The nature of early astroglial protection - fast activation and signalling. Prog Neurobiol. 2017;153:86–99.
Colburn RW, Maas JW. Adenosine triphosphate-metal-norepinephrine ternary complexes and catecholamine binding. Nature. 1965;208:37–41.
Kardos J, Samu J, Ujszászi K, Nagy J, Kovács I, Visy J, Maksay G, Simonyi M. Cu2+ is the active principle of an endogenous substance from porcine cerebral cortex which antagonizes the anticonvulsant effect of diazepam. Neurosci Lett. 1984;52:67–72.
van de Bospoort R, Farina M, Schmitz SK, de Jong A, de Wit H, Verhage M, Toonen RF. Munc13 controls the location and efficiency of dense-core vesicle release in neurons. J Cell Biol. 2012;199:883.
Persson H, Türk M, Nyman M, Sandberg A-S. Binding of Cu2+, Zn2+, and Cd2+ to inositol tri-, tetra-, penta-, and hexaphosphates. J Agric Food Chem. 1998;46:3194–200.
Kuzuya M, Yamada K, Hayashi T, Funaki C, Naito M, Asai K, Kuzuya F. Role of lipoprotein-copper complex in copper catalyzed-peroxidation of low-density lipoprotein. Biochim Biophys Acta, Lipids Lipid Metab. 1992;1123:334–41.
Bhattacharjee A, Chakraborty K, Shukla A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics. 2017;9:1376–88.
Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012; art. 736837.
Pickart L, Vasquez-Soltero JM, Margolina A. The effect of the human peptide GHK on gene expression relevant to nervous system function and cognitive decline. Brain Sci. 2017;7:20.
Langley A, Dameron CT. Copper and anesthesia: clinical relevance and management of copper related disorders. Anesthesiol Res Pract. 2013; art. 750901.
Linder MC. Ceruloplasmin and other copper binding components of blood plasma and their functions: an update. Metallomics. 2016;8:887–905 (“Mammalian Copper Transport and Related Diseases.”).
Okita Y, Rcom-H’cheo-Gauthier AN, Goulding M, Chung RS, Faller P, Pountney DL. Metallothionein, copper and alpha-synuclein in alpha-synucleinopathies. Front Neurosci. 2017;11:114.
Liu N, Lo LS, Askary SH, Jones L, Kidane TZ, Trang T, Nguyen M, Goforth J, Chu YH, Vivas E, Tsai M, Westbrook T, Linder MC. Transcuprein is a macroglobulin regulated by copper and iron availability. J Nutr Biochem. 2007;18:597–608.
Caruso G, Distefano DA, Parlascino P, Fresta CG, Lazzarino G, Lunte SM, Nicoletti VG. Receptor-mediated toxicity of human amylin fragment aggregated by short- and long-term incubations with copper ions. Mol Cell Biochem. 2017;425:85–93.
Gourdon P, Liu X-Y, Skjørringe T, Morth JP, Møller LB, Pedersen BP, Nissen P. Crystal structure of a copper-transporting PIB-type ATPase. Nature. 2011;475:59–64.
Gupta A, Lutsenko S. Human copper transporters: mechanism, role in human diseases and therapeutic potential. Future Med Chem. 2009;1:1125–42.
Wang Y, Hodgkinson V, Zhu S, Weisman GA, Petris MJ. Advances in the understanding of mammalian copper transporters. Adv Nutr. 2011;2:129–37.
Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF, Nussberger S, Gollan JL, Hediger MA. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482–8.
Bompiani KM, Tsai C-Y, Achatz FP, Liebig JK, Howell SB. Copper transporters and chaperones CTR1, CTR2, ATOX1, and CCS as determinants of cisplatin sensitivity. Metallomics. 2016;8:951–62.
De Feo CJ, Aller SG, Siluvai GS, Blackburn NJ, Unger VM. Three-dimensional structure of the human copper transporter hCTR1. Proc Natl Acad Sci U S A. 2009;106:4237–42.
Lawton TJ, Kenney GE, Hurley JD, Rosenzweig AC. The CopC family: structural and bioinformatic insights into a diverse group of periplasmic copper binding proteins. Biochemistry. 2016;55:2278–90.
Zhou W, Saran R, Liu J. Metal sensing by DNA. Chem Rev. 2017;117:8272–325.
Løvaas E. Antioxidative and metal-chelating effects of polyamines. Adv Pharmacol. 1997;38:119–49.
Bal W, Sokołowska M, Kurowska E, Faller P. Binding of transition metal ions to albumin: sites, affinities and rates. Biochim Biophys Acta. 2013;1830:5444–55.
Ackerman CM, Chang CJ. Copper signaling in the brain and beyond. J Biol Chem. 2018;293:4628–35.
Lee J, Petris M, Thiele DJ. Characterization of mouse embryonic cells deficient in the Ctr1 high affinity transporter. Identification of a Ctr1-independent copper transport system. J Biol Chem. 2002;277:4380–7.
Nevitt T, Öhrvik H, Thiele DJ. Charting the travels of copper in eukaryotes from yeast to mammals. Biochim Biophys Acta. 2012;1823:1580–93.
Ramos D, Mar D, Ishida M, Vargas R, Gaite M, Montgomery A, Linder MC. Mechanism of copper uptake from blood plasma ceruloplasmin by mammalian cells. PLoS One. 2016;11:e0149516.
Bossak K, Drew SC, Stefaniak E, Płonka D, Bonna A, Bal W. The cu(II) affinity of the N-terminus of human copper transporter CTR1: comparison of human and mouse sequences. J Inorg Biochem. 2018; pii: S0162–0134(17)30808–5.
Ostergaard H, Tachibana C, Winther JR. Monitoring disulfide bond formation in the eukaryotic cytosol. J Cell Biol. 2004;166:337–45.
Freedman JH, Ciriolo MR, Peisach J. The role of glutathione in copper metabolism and toxicity. J Biol Chem. 1989;264:5598–605.
Maryon EB, Molloy SA, Kaplan JH. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am J Physiol Cell Physiol. 2013;304:C768–79.
Hatori Y, Inouye S, Akagi R. Thiol-based copper handling by the copper chaperone Atox1. IUBMB Life. 2017;69:246–54 (“Copper Homeostasis”.).
Kahra D, Kovermann M, Wittung-Stafshede P. The C-terminus of human copper importer Ctr1 acts as a binding site and transfers copper to Atox1. Biophys J. 2016;110:95–102.
Niemiec MS, Artur PG, Dingeldein APG, Wittung-Stafshede P. Enthalpy-entropy compensation at play in human copper ion transfer. Sci Rep. 2015;5:10518.
Boal AK, Rosenzweig AC. Structural biology of copper trafficking. Chem Rev. 2009;109:4760–79.
Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O’Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–8.
Palumaa P. Copper chaperones. The concept of conformational control in the metabolism of copper. FEBS Lett. 2013;587:1902–10.
Banci L, Bertini I, Ciofi-Baffoni S, Kozyreva T, Zovo K, Paluma P. Affinity gradients drive copper to cellular destinations. Nature. 2010;465:645–8.
Banci L, Cantini F, Kozyreva T, Rubino JT. Mechanistic aspects of hSOD1 maturation from the solution structure of cu(I)-loaded hCCS domain 1 and analysis of disulfide-free hSOD1 mutants. Chembiochem. 2013;14:1839–44.
Strange RW, Antonyuk SV, Hough MA, Douctte PA, Valentine JS, Hasnian SS. Variable metallation of human superoxide dismutase: atomic resolution crystal structures of cu–Zn, Zn–Zn and as-isolated wild-type enzymes. J Mol Biol. 2005;356.
Banci L, Bertini I, Calderone V, Della-Malva N, Felli IC, Neri S, Pavelkova A, Rosato A. Copper(I)-mediated protein-protein interactions result from suboptimal interaction surfaces. Biochem J. 2009;422:37–42.
Shoshan M., N. Dekel, W. Goch, D. Shalev, T. Danieli, M. Lebendiker, W. Bal, E. Tshuva. Unbound position II in MXCXXC metallochaperone model peptides impacts metal binding mode and reactivity: Distinct similarities to whole proteins, J. Inorg. Biochem. 2016;159:29–36.
Cao R, Taylor Elrod L, Lehane RL, Kim E, Karlin KD. A peroxynitrite dicopper complex: formation via cu−NO and cu−O2 intermediates and reactivity via O−O cleavage chemistry. J Am Chem Soc. 2016;138:16148–58.
Johnston EM, Dell’Acqua S, Pauleta SR, Mourac I, Solomon EI. Protonation state of the Cu4S2 CuZ site in nitrous oxide reductase: redox dependence and insight into reactivity. Chem Sci. 2015;6:5670–9.
Johnston EM, Carreira C, Dell’Acqua S, Ghosh Dey S, Pauleta SR, Moura I, Solomon EI. Spectroscopic definition of the CuZ° intermediate in turnover of nitrous oxide reductase and molecular insight into the catalytic mechanism. J Am Chem Soc. 2017;139:4462–76.
Page CC, Moser CC, Dutton PL. Mechanism for electron transfer within and between proteins. Curr Op Chem Biol. 2003;7:551–6.
Flohé L. The fairytale of the GSSG/GSH redox potential. Biochim Biophys Acta. 2013;1830:3139–42.
Giles GI, Nasim MJ, Ali W, Jacob C. The reactive sulfur species concept: 15 years on. Antioxidants. 2017;6:E38.
Travasso RDM, dos Aidosa FS, Bayani A, Abranches P, Salvador A. Localized redox relays as a privileged mode of cytoplasmic hydrogen peroxide signaling. Redox Biol. 2017;12:233–45.
Brose J, La Fontaine S, Wedd AG, Xiao Z. Redox sulfur chemistry of the copper chaperone Atox1 is regulated by the enzyme glutaredoxin 1, the reduction potential of the glutathione couple GSSG/2GSH and the availability of cu(I). Metallomics. 2014;6:793–808.
Hatori Y, Clasen S, Hasan NM, Barry AN, Lutsenko S. Functional partnership of the copper export machinery and glutathione balance in human cells. J Biol Chem. 2012;287:26678–87.
Polishchuk R, Lutsenko S. Golgi in copper homeostasis: a view from the membrane trafficking field. Histochem Cell Biol. 2013;140:285–95.
Li C-G, Shen Z-G, Hu Y-F, Tang Y-N, Chen W-G, Ren B-Z. Insights into the structures and electronic properties of Cun+1 μ and CunSμ (n=1–12; μ=0, ±1) clusters. Sci Rep. 2017;7:1345.
Andrew CR, Sanders-Loehr J. Copper-sulfur proteins: using Raman spectroscopy to predict coordination geometry. J Acc Chem Res. 1996;29:365–72.
Scott RA. Functional significance of cytochrome c oxidase structure. Structure. 1995;3:981–6.
Cortese-Krott MM, Koning A, Kuhnle GGC, Nagy P, Bianco CL, Pasch A, Wink DA, Fukuto JM, Jackson AA, van Goor H, Olson KR, Feelisch M. The reactive species interactome: evolutionary emergence, biological significance, and opportunities for redox metabolomics and personalized medicine. Antioxid Redox Signal. 2017;27:684–712.
Pompella A, Visvikis A, Paolicchi A, De Tata V, Casini AF. The changing faces of glutathione, a cellular protagonist. Biochem Pharmacol. 2003;66:1499–503.
Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad Biol Med. 2001;30:1191–212.
Timme-Laragy AR, Hahn ME, Hansen JM, Rastogi A, Roy MA. Redox stress and signaling during vertebrate embryonic development: Regulation and responses. Semin Cell Dev Biol. 2017; pii: S1084–9521(17)30279–3.
Irving H, Williams RJP. Order of stability of metal complexes. Nature. 1948;162:746–7.
Pearson RG, editor. Hard and Soft Acids and Bases. Stroudsburg, Pa.: Dowden, Hutchinson & Ross; 1973.
Jiang L, Tu Y, Hu X, Bao A, Chen H, Ma X, Doyle T, Shi H, Cheng Z. Pilot study of 64Cu(I) for PET imaging of melanoma. Sci Rep. 2017;7:2574.
Aliaga ME, López-Alarcón C, Bridi R, Speisky H. Redox-implications associated with the formation of complexes between copper ions and reduced or oxidized glutathione. J Inorg Biochem. 2016;154:78–88.
Deponte M. The incomplete glutathione puzzle: Just guessing at numbers and figures? Antioxid Redox Signal. 2017;27:1130–61.
Morgan MT, Anh L, Nguyen H, Hancock HL, Fahrni CJ. Glutathione limits aquacopper(I) to sub-femtomolar concentrations through cooperative assembly of a tetranuclear cluster. J Biol Chem. 2017;292(52):21558–67.
Baslé A, Platsaki S, Dennison C. Visualizing biological copper storage: the importance of thiolate-coordinated tetranuclear clusters. Angew Chem Int Ed. 2017;56:8697–700.
Sakurai H, Fukudome A, Tawa R, Kito M, Takeshima S, Kimura M, Otaki N, Nakajima K, Hagino T, Kawano K, Hirai S, Suzuki S. Metallothionein in the liver of LEC rats. Biochem Biophys Res Commun. 1992;184:393–1397.
Sutherland DE, Stillman MJ. The “magic numbers” of metallothionein. Metallomics. 2011;3:444–63.
Sullivan B, Robison G, Osborn J, Kay M, Thompson P, Davis K, Zakharova T, Antipova O, Pushkar Y. On the nature of the cu-rich aggregates in brain astrocytes. Redox Biol. 2017;11:231–9.
Sullivan B, Robison G, Pushkar Y, Young JK, Manaye KF. Copper accumulation in rodent brain astrocytes: a species difference. J Trace Elem Med Biol. 2017;39:6–13.
Adams KF, Johnson G Jr, Hornowski KE, Lineberger TH. The effect of copper on erythrocyte deformability a possible mechanism of hemolysis in acute copper intoxication. Biochim Biophys Acta. 1979;550:279–87.
Boulard M, Blume KG, Seutler E. The effect of copper on red cell enzyme activities. J Clin Invest. 1972;51:459–61.
Fernandes A, Mira ML, Azevedo MS, Manso C. Mechanisms of hemolysis induced by copper. Free Rad Res Commun. 1988;4:291–8.
Han Y-H, Kim S-U, Kwon T-H, Lee D-S, Ha H-L, Park D-S, Woo E-J, Lee S-H, Kim J-M, Chae H-B, Lee SY, Kim BY, Yoon DY, Rhee SG, Fibach E, Yu D-Y. Peroxiredoxin II is essential for preventing hemolytic anemia from oxidative stress through maintaining hemoglobin stability. Biochem Biophys Res Commun. 2012;426:427–32.
Kaiserová K, Lakatos B, Peterajová E, Orlický J, Varecka L. Investigation of properties of the Ca2+ influx and of the Ca2+-activated K+ efflux (Gárdos effect) in vanadate-treated and ATP-depleted. Gen Physiol Biophys. 2002;21:429–42.
Lubin A, Desforges JF. Effect of Heinz bodies on red cell deformability. Blood. 1972;39:658–65.
Mentz EN, Sagone AL Jr. The effect of copper on the erythrocyte hexose monophosphate shunt pathway. J Lab Clin Med. 1972;80:405–13.
Piriou A, Tallineau C, Chahboun S, Pontcharraud R, Guillard O. Copper-induced lipid peroxidation and hemolysis in whole blood: evidence for a lack of correlation. Toxicology. 1987;47:351–61.
Sneddon J. Action of di- and tri-valent cations on calcium-activated K+-efflux in rat erythrocytes. Biochem Pharmacol. 1987;36:3723–30.
Varecka L, Peterajová E, Pogády J. Inhibition by divalent cations and sulphydryl reagents of the passive Ca2+ transport in human red blood cells observed in the presence of vanadate. Biochim Biophys Acta. 1986;856:585–94.
Sansinanea AS, Cerone SI, Quiroga M, Auza N. Antioxidant capacity of erythrocytes from sheep chronically poisoned by copper. Nutr Res. 1993;13:891–9.
Raftos JE, Whillier S, Kuchel PW. Glutathione synthesis and turnover in the human erythrocyte. Alignment of a model based on detailed enzyme kinetics with experimental data. J. Biol Chem. 2010;285:23557–67.
van’t Erve TJ, Wagner BA, Ryckman KK, Raife TJ, Buettner GR. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic Biol Med. 2013;65:742–9.
Giustarini D, Tsikas D, Colombo G, Milzani A, Dalle-Donne I, Fanti P, Rossi R. Pitfalls in the analysis of the physiological antioxidant glutathione (GSH) and its disulfide (GSSG) in biological samples: An elephant in the room. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1019:21–8.
Kaler SG. Inborn errors of copper metabolism. Handb Clin Neurol. 2013;113:1745–54.
Patil M, Sheth KA, Krishnamurthy AC, Devarbhavi H. A review and current perspective on Wilson disease. J Clin Exp Hepatol. 2013;3:321–36.
Yahata S, Yung S, Mandai M, Nagahara T, Kuzume D, Sakaeda H, Wakusawa S, Kato A, Tatsumi Y, Kato K, Hayashi H, Isaji R, Sasaki Y, Yano M, Hayashi K, Ishigami M, Goto H. Phenotypes and chronic organ damage may be different among siblings with Wilson’s disease. J Clin Transl Hepatol. 2017;28:27–30.
Cumings JN. The copper and iron content of brain and liver in the normal and in hepatolenticular degeneration. Brain. 1948;71:410–5.
Li Y, Togashi Y, Sato S, Emoto T, Kang J-H, Takeichi N, Kobayashi H, Kojima Y, Une Y, Uchino J. Spontaneous hepatic copper accumulation in long-Evans cinnamon rats with hereditary hepatitis. J Clin Invest. 1991;87:1858–61.
Solioz M. The copper rush of the nineties. Metallomics. 2016;8:824.
Telianidis J, Hung YH, Materia S, LaFontaine S. Role of the P-type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front Aging Neurosci. 2013;5:44.
Concilli M, Iacobacci S, Chesi G, Carissimo A, Polishchuk R. A systems biology approach reveals new endoplasmic reticulum-associated targets for the correction of the ATP7B mutant causing Wilson disease. Metallomics. 2016;8:920.
Kumar R, Ariöz C, Li Y, Rocha S, Bosaeus N, Wittung-Stafshede P. Disease-causing point-mutations in metal-binding domains of Wilson disease protein decrease stability and increase structural dynamics. Biometals. 2017;30:27–35.
Nayak NC, Chitale AR. Indian childhood cirrhosis (ICC) & ICC-like diseases: the changing scenario of facts versus notions, Indian. J Med Res. 2013;137:1029–42.
Scheiber IV, Bruha R, Dusek P. Pathogenesis of Wilson disease. In: Członkowska A, Schilsky ML, editors. Handbook of clinical neurology, Vol. 142, Chapter 5: Elsevier B.V; 2017.
Kaler SG. ATP7A-related copper transport diseases - emerging concepts and future trends. Nat Rev Neurol. 2011;7:15–29.
Gaetke LM, Chow CK. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology. 2003;189:147–63.
Brewer GJ. The promise of copper lowering therapy with tetrathiomolybdate in the cure of cancer and in the treatment of inflammatory disease. J Trace Elements Med Biol. 2014;28:372–8.
Calvo J, Jung H, Meloni G. Copper metallothioneins. IUBMB Life. 2017;69:236–45.
Materia S, Michael A, Cater MA, Klomp LWJ, Mercer JFB, La Fontaine S. Clusterin (Apolipoprotein J), a molecular chaperone that facilitates degradation of the copper-ATPases ATP7A and ATP7B. J Biol Chem. 2011;286:10073–83.
Morrell S, Tallino L, Lei Y, Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB Life. 2017;69:263–70.
Inesi G, Pilankatta R, Tadini-Buoninsegni F. Biochemical characterization of P-type copper ATPases. Biochem J. 2014;463:167–76.
Lutsenko S. Human copper homeostasis: a network of interconnected pathways. Curr Opinion Chem Biol. 2010;14:211–7.
Ogra Y, Miyayama T, Anan Y. Effect of glutathione depletion on removal of copper from LEC rat livers by tetrathiomolybdate. J Inorg Biochem. 2010;104:858–62.
McCranor BJ, Szmacinski H, Zeng HH, Stoddard AK, Hurst T, Fierke CA, Lakowicz JR, Thompson RB. Fluorescence lifetime imaging of physiological free cu(II) levels in live cells with a cu(II)-selective carbonic anhydrase-based biosensor. Metallomics. 2014;6:1034–42.
Stuerenburg HJ. CSF copper concentrations, blood-brain barrier function and coeruloplasmin synthesis during the treatment of Wilson’s disease. J Neural Transm. 2000;107:321–9.
Palm R, Wahlstrom G, Hallmans G. Age related changes in weight and the concentrations of zinc and copper in the brain of the adult rat. Lab Anim. 1990;24:240–5.
Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–6.
Bush AI. Metals and neuroscience. Curr Opin Chem Biol. 2000;4:184–91.
Carver CM, Chuang SH, Reddy DS. Zinc selectively blocks neurosteroid-sensitive extrasynaptic δGABAa receptors in the hippocampus. J Neurosci. 2016;36:8070–7.
Elsas S-M, Hazany S, Gregory WL, Mody I. Hippocampal zinc infusion delays the development of afterdischarges and seizures in a kindling model of epilepsy. Epilepsia. 2009;50:870–9.
Frederickson CJ, Hernandez MD, Goik SA, Morton JD, McGinty JF. Loss of zinc staining from hippocampal mossy fibers during kainic acid induced seizures: a histofluorescence study. Brain Res. 1988;446:383–6.
Frederickson CJ, Bush AI. Synaptically released zinc: physiological functions and pathological effects. Biometals. 2001;14:353–66.
Frederickson CJ, Koh J-Y, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–62.
Kardos J, Kovács I, Hajós F, Kálmán M, Simonyi M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci Lett. 1989;103:139–44.
Khana M, Goldsmith CR, Huang Z, Georgiou J, Luyben TT, Rodera JC, Lippard SJ, Okamoto K. Two-photon imaging of Zn2+ dynamics in mossy fiber boutons of adult hippocampal slices. Proc Natl Acad Sci U S A. 2014;111:6786–91.
Palló A, Simon Á, Bencsura Á, Héja L, Kardos J. Substrate-Na+ complex formation: coupling mechanism for gamma-aminobutyrate symporters. Biochem Biophys Res Commun. 2009;385:210–4.
Pan E, Zhang XA, Huang Z, Krezel A, Zhao M, Tinberg CE, Lippard SJ, McNamara JO. Vesicular zinc promotes presynaptic and inhibits postsynaptic longterm potentiation of mossy fiber-CA3 synapse. Neuron. 2011;71:1116–26.
Vogt K, Mellor J, Nicoll R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron. 2000;26:187–96.
Nagy J, Kardos J, Maksay G, Simonyi M. An endogenous substance from porcine brain antagonizes the anticonvulsant effect of diazepam. Neuropharmacology. 1981;20:529–33.
Hartter DE, Barnea A. Brain tissue accumulates 67Copper by two ligand-dependent saturable processes. A high affinity, low capacity and a low affinity, high capacity process. J Biol Chem. 1988;263:799–805.
Hartter DE, Barnea A. Evidence for release of copper in the brain: depolarisation-induced release of newly taken-up 67Copper. Synapse. 1988;2:412–5.
Schlief ML, Craig AM, Gitlin JD. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J Neurosci. 2005;25:239–46.
Schlief ML, West T, Craig AM, Holtzman DM, Gitlin JD. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc Natl Acad Sci U S A. 2006;103:14919–24.
Fisher JL, Macdonald RL. The role of an α subtype M2-M3 his in regulating inhibition of GABAA receptor current by zinc and other divalent cations. J Neurosci. 1998;18:2944–53.
Schlief ML, Gitlin JD. Copper homeostasis in the CNS - a novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol Neurobiol. 2006;33:81–90.
McGee TP, Houston CM, Brickley SG. Copper block of extrasynaptic GABAA receptors in the mature cerebellum and striatum. J Neurosci. 2013;33:13431–5.
Marchetti C. Interaction of metal ions with neurotransmitter receptors and potential role in neurodiseases. Biometals. 2014;17:1097–113.
Narahashi T, Ma JY, Arakawa O, Reuveny E, Nakahiro M. GABA receptor-channel complex as a target site of mercury, copper, zinc, cell. Mol. Neurobiol. 1994;14:599–621.
Opazo CM, Greenough MA, Bush AI. Copper: From neurotransmission to neuroproteostasis. Front Aging Neurosci. 2014;6:143.
Scheiber IF, Mercer JFB, Dringen R. Metabolism and functions of copper in brain. Prog Neurobiol. 2014;116:33–57.
Stojilkovic SS, Leiva-Salcedo E, Rokic MB, Coddou C. Regulation of ATP-gated P2X channels: from redox signaling to interactions with other proteins. Antioxid Redox Signal. 2014;21:953–70.
D’Ambrosi N, Rossi L. Copper at synapse: release, binding and modulation of neurotransmission. Neurochem Int. 2015;90:36–45.
Maureira C, Letelier JC, Alvarez O, Delgado R, Vergara C. Copper enhances cellular and network excitabilities, and improves temporal processing in the rat hippocampus. Eur J Neurosci. 2015;42:3066–80.
Neumaier F, Dibue-Adjei M, Hescheler J, Schneider T. Voltage-gated calcium channels: determinants of channel function and modulation by inorganic cations, Progr. Neurobiol. 2015;129:1–36.
Villumsen IS, Wellendorph P, Smart TG. Pharmacological characterisation of murine α4β1δ GABAA receptors expressed in Xenopus oocytes. BMC Neurosci. 2015;16:8.
Comstra HS, McArthy J, Rudin-Rush S, Hartwig C, Gokhale A, Zlatic SA, Blackburn JB, Werner E, Petris M, D’Souza P, Panuwet P, Boyd Barr D, Lupashin V, Vrailas-Mortimer A, Faundez V. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. eLife. 2017;6:e24722.
White AR, Aschner M, Costa LG, Bush AI, editors. Biometals in neurodegenerative diseases: mechanisms and therapeutics: Elsevier; 2017.
Su P, Aschner M, Chen J, Luo W. Metals and autophagy in neurotoxicity. Chapter 19. Molecular features of copper binding proteins involved in copper homeostasis. In: White AR, Aschner M, Costa LG, Bush AI, editors. Biometals in neurodegenerative diseases: Mechanisms and Therapeutics: Elsevier; 2017. p. 378–98.
Hopt A, Korte S, Fink H, Panne U, Niessner R, Jahn R, Kretzschmar H, Herms J. Methods for studying synaptosomal copper release. J Neurosci Methods. 2003;128:159–72.
Cash DJ, Subbarao K. Channel opening of gamma-aminobutyric acid receptor from rat brain: molecular mechanisms of the receptor responses. Biochemistry. 1987a;26:7562–70.
Cash DJ, Subbarao K. Desensitization of gamma-aminobutyric acid receptor from rat brain: two distinguishable receptors on the same membrane. Biochemistry. 1987b;26:7556–62.
Kardos J, Cash DJ. Transmembrane 36Cl− flux measurements and desensitization of the gamma-aminobutyric acidA receptor. J Neurochem. 1990;55:1095–9.
Kardos J. The GABAA receptor channel mediated chloride-ion translocation through the plasma-membrane - new insights from 36Cl− ion flux measurements. Synapse. 1993;13:74–93.
Delgado R, Vergara C, Wolff D. Divalent cations as modulators of neuronal excitability: emphasis on copper and zinc. Biol Res. 2006;39:173–82.
Dodani SC, Domaille DW, Nam CI, Miller EW, Finney LA, Vogt S, Chang CJ. Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc Natl Acad Sci U S A. 2011;108:5980–5.
Pal A, Prasad R. Recent discoveries on the functions of astrocytes in the copper homeostasis of the brain: a brief update. Neurotox Res. 2014;26:78–84.
Kozlowski H, Luczkowski M, Remelli M, Valensin D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases). Coord Chem Rev. 2012;256:2129–41.
Martin GR, Alvarez AL, Bashashati M, Keenan CM, Jirik FR, Sharkey KA. Endogenous cellular prion protein regulates contractility of the mouse ileum. Neurogastroenterol Motil. 2012;24:e412–24.
Küffer A, Lakkaraju AK, Mogha A, Petersen SC, Airich K, Doucerain C, Mrpakwar R, Bakirci P, Senatore A, Monnard A, Schiavi C, Nuvolone M, Grosshans B, Hornemann S, Bassilana F, Monk KR, Aguzzi A. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature. 2016;536:464–8.
Wulf M-A, Senatore A, Aguzzi A. The biological function of the cellular prion protein: an update. BMC Biol. 2017;15:34.
Yen C-F, Harischandra DS, Kanthasamy A, Sivasankar S. Copper-induced structural conversion templates prion protein oligomerization and neurotoxicity. Sci Adv. 2016;2:e1600014.
Giachin G, Mai PT, Tran TH, Salzano G, Benetti F, Migliorati V, Arcovito A, Della Longa S, Mancini G, D’Angelo P, Legname G. The non-octarepeat copper binding site of the prion protein is a key regulator of prion conversion. Sci Rep. 2015;5:15253.
Stanyon HF, Patel K, Begum N, Viles JH. Copper(II) sequentially loads onto the N-terminal amino group of the cellular prion protein before the individual octarepeats. Biochemistry. 2014;53:3934–9.
Nichols TA, Spraker TR, Gidlewski T, Cummings B, Hill D, Kong Q, Balachandran A, VerCauteren KC, Zabel MD. Dietary magnesium and copper affect survival time and neuroinflammation in chronic wasting disease. Prion. 2016;10:228–50.
Toni M, Massimino ML, DeMario A, Angiulli E, Spisni E. Metal dyshomeostasis and their pathological role in prion and prion-like diseases: the basis for a nutritional approach. Front Neurosci. 2017;11:3.
Bush AI. The metal theory of Alzheimer’s disease. J Alzheimers Dis. 2013;33:S277–81.
Greenough MA, Munoz AR, Bush AI, Opazo CM. Metallo-pathways to Alzheimer’s disease: lessons from genetic disorders of copper trafficking. Metallomics. 2016;8:831–9 („Mammalian Copper Transport and Related Diseases.”).
Gray EH, De Vos KJ, Dingwall C, Perkinton MS, Miller CCJ. Deficiency of the copper chaperone for superoxide dismutase increases amyloid-β production. J Alzheimers Dis. 2010;21:1101–5.
Hureau C. Coordination of redox active metal ions to the amyloid precursor protein and to amyloid-β peptides involved in Alzheimer disease. Part 1: an overview. Coord Chem Rev. 2012;256:2164–74.
Kepp KP. Bioinorganic chemistry of Alzheimer’s disease. Chem Rev. 2012;112:5193–239.
Tõugu V, Tiiman A, Palumaa P. Interactions of Zn(II) and cu(II) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics. 2011;3:250–61.
Wild K, August A, Pietrzik CU, Kins S. Structure and synaptic function of metal binding to the amyloid precursor protein and its proteolytic fragments. Front Mol Neurosci. 2017;17:21.
Schrag M, Mueller C, Oyoyo U, Kirsch WM. Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol. 2011;94:296–306.
James SA, Churches QI, de Jonge MD, Birchall IE, Streltsov V, McColl G, Adlard PA, Hare DJ. Iron, copper, and zinc concentration in Aβ plaques in the APP/PS1 mouse model of Alzheimer’s Disease correlates with metal levels in the surrounding neuropil. ACS Chem Neurosci. 2017;8:629–637.
Mital M, Wezynfeld NE, Frączyk T, Wiloch MZ, Wawrzyniak UE, Bonna A, Tumpach C, Barnham KJ, Haigh CL, Bal W, Drew SC. A functional role for Aβ in metal homeostasis? N-truncation and high-affinity copper binding. Angew Chem Int Ed. 2015;54:10460–4.
Wezynfeld NE, Stefaniak E, Stachucy K, Drozd A, Płonka D, Drew SC, Krężel A, Bal W. Resistance of cu(Aβ4−16) to copper capture by metallothionein-3 supports a function for the Aβ4−42 peptide as a synaptic cu(II) scavenger. Angew Chem Int Ed. 2016;55:8235–8.
Liao CR, Rak M, Lund J, Unger M, Platt E, Albensi BC, Hirschmugl CJ, Gough KM. Synchrotron FTIR reveals lipid around and within amyloid plaques in transgenic mice and Alzheimer’s disease brain. Analyst. 2013;138:3991–7.
Benseny-Cases N, Klementieva O, Cotte M, Ferrer I, Cladera J. Microspectroscopy (μFTIR) reveals co-localization of lipid oxidation and amyloid plaques in human alzheimer disease brains. Anal Chem. 2014;86:12047–54.
Furman R, Murray IVJ, Schall HE, Liu Q, Ghiwot Y, Axelsen PH. Amyloid plaque-associated oxidative degradation of uniformly radiolabeled arachidonic acid. ACS Chem Neurosci. 2016;7:367–77.
Malakooti N, Pritchard MA, Adlard PA, Finkelstein DI. Role of metal ions in the cognitive decline of Down syndrome. Front Aging Neurosci. 2014;6:136.
Mann DM, Jones D, Prinja D, Purkiss MS. The prevalance of amyloid (A4) protein deposits within the cerebral and cerebellar cortex in Down’s syndrome and Alzheimer’s disease. Acta Neuropathol. 1990;80:318–27.
Ivanova MI, Sievers SA, Guenther EL, Johnson LM, Winkler DD, Galaleldeen A, Sawaya MR, Hart PJ, Eisenberg DS. Aggregation-triggering segments of SOD1 fibril formation support a common pathway for familial and sporadic ALS. Proc Natl Acad Sci U S A. 2014;111:197–201.
Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–58.
Xiao G, Fan Q, Wang X, Zhou B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A. 2013;110:14995–5000.
Vonk WIM, Kakkar V, Bartuzi P, Jaarsma D, Berger R, Hofker MH, Klomp LWJ, Wijmenga C, Kampinga HH, van de Sluis B. The copper metabolism MURR1 domain protein 1 (COMMD1) modulates the aggregation of misfolded protein species in a client-specific manner. PLoS One. 2014;9:e92408.
Davies KM, Mercer JFB, Chen JN, Double KL. Copper dyshomoeostasis in Parkinson’s disease: implications for pathogenesis and indications for novel therapeutics. Clin Sci. 2016;130:565–74.
Mischley LK, Conley KE, Shankland EG, Kavanagh TJ, Rosenfeld ME, Duda JE, White CC, Wilbur TK, De La Torre PU, Padowski JM. Central nervous system uptake of intranasal glutathione in Parkinson’s disease. NPJ Parkinson's Dis. 2016;2:16002.
Sinopoli A, Magrı A, Milardi D, Pappalardo M, Pucci P, Flagiello A, Titman JJ, Nicoletti VG, Caruso G, Pappalardo G, Grasso G. The role of copper(II) in the aggregation of human amylin. Metallomics. 2014;6:1841.
Horvath I, Wittung-Stafshede P. Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease. Proc Natl Acad Sci U S A. 2016;113:12473–7.
Small SA, Petsko GA. Retromer in Alzh eimer disease, Parkinson disease and other neurological disorders. Nat Rev Neurosci. 2015;16:126–32.
McMillan KJ, Korswagen HC, Cullen PJ. The emerging role of retromer in neuroprotection. Curr Opin Cell Biol. 2017;47:72–82.
Sowada N, Stiller B, Kubisch C. Increased copper toxicity in Saccharomyces cerevisiae lacking VPS35, a component of the retromer and monogenic Parkinson disease gene in humans. Biochem Biophys Res Commun. 2016;476:528–33.
Steinberg F, Gallon M, Winfield M, Thomas EC, Bell AJ, Heesom KJ, Tavare JM, Cullen PJ. A global analysis of SNX27-retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15:461–71.
Srivastava R, Aslam M, Kalluri SR, Schirmer L, Buck D, Tackenberg B, Rothhammer V, Chan A, Gold R, Berthele A, Bennett JL, Korn T, Hemmer B. Potassium channel Kir4.1 as an immune target in multiple sclerosis. N Engl J Med. 2012;367:115–23.
Ács P, Kálmán B. Pathogenesis of multiple sclerosis: what can we learn from the cuprizone model. Methods Mol Biol. 2012;900:403–31.
Ács P, Selak MA, Komoly S, Kálmán B. Distribution of oligodendrocyte loss and mitochondrial toxicity in the cuprizone-induced experimental demyelination model. J Neuroimmunol. 2013;262:128–31.
Gudi V, Moharregh-Khiabani D, Skripuletz T, Koutsoudaki PN, Kotsiari A, Skuljec J, Trebst C, Stangel M. Regional differences between grey and white matter in cuprizone induced demyelination. Brain Res. 2009;1283:127–38.
Khodanovich MY, Sorokina IV, Glazacheva VY, Akulov AE, Nemirovich-Danchenko NM, Romashchenko AV, Tolstikova TG, Mustafina LR, Yarnykh VL. Histological validation of fast macromolecular proton fraction mapping as a quantitative myelin imaging method in the cuprizone demyelination model. Sci Rep. 2017;7:46686.
Kipp M, Clarner T, Dang J, Copray S, Beyer C. The cuprizone animal model: new insights into an old story. Acta Neuropathol. 2009;118:723–36.
Skripuletz T, Gudi V, Hackstette D, Stangel M. De- and remyelination in the CNS white and grey matter induced by cuprizone: the old, the new, and the unexpected. Histol Histopathol. 2011;26:1585–97.
Nakajima M, Kawamura T, Tokui R, Furuta K, Suginoa M, Nakanishi M, Okuyama S, Furukawa Y. Enhanced accumulation of Kir4.1 protein, but not mRNA, in a murine model of cuprizone-induced demyelination. Brain Res. 2013;1537:340–9.
Kang YJ. Metallothionein redox cycle and function. Exp Biol Med. 2006;231:1459–67.
Biancotti JC, Kumar S, de Vellis J. Activation of inflammatory response by a combination of growth factors in cuprizone-induced demyelinated brain leads to myelin repair. Neurochem Res. 2008;33:2615–28.
Witherick J, Wilkins A, Scolding N, Kemp K. Mechanisms of oxidative damage in multiple sclerosis and a cell therapy approach to treatment. Autoimmune Dis. 2011;2011:164608.
Praet J, Guglielmetti C, Berneman Z, Van der Linden A, Ponsaerts P. Cellular and molecular neuropathology of the cuprizone mouse model: clinical relevance for multiple sclerosis. Neurosci Biobehav Rev. 2014;47:485–505.
Katerji M, Barada K, Jomaa M, Kobeissy F, Makkawi A-K, Abou-Kheir W, Usta J. Chemosensitivity of U251 cells to the co-treatment of D-penicillamine and copper: possible implications on Wilson disease patients. Front Mol Neurosci. 2017;10:10.
Barnham KJ, Bush AI. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev. 2014;43:6727–49.
Chen D, Cui QC, Yang H, Barrea RA, Sarkar FH, Sheng S, Yan B, Reddy GP, Dou QP. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor–suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007;67:1636–44.
Duncan C, White AR. Copper complexes as therapeutic agents. Metallomics. 2012;4:127–38.
Hilton JB, Mercer SW, Lim NKH, Faux NG, Buncic G, Beckman JS, Roberts BR, Donnelly PS, White AR, Crouch PJ. CuII(atsm) improves the neurological phenotype and survival of SOD1G93A mice and selectively increases enzymatically active SOD1 in the spinal cord. Sci Rep. 2017;7:42292.
Soon CPW, Donnelly PS, Turner BJ, Hung LW, Crouch PJ, Sherratt NA, Tan J-L, Lim NK-H, Lam L, Bica L, Lim S, Hickey JL, Morizzi J, Powell A, Finkelstein DI, Culvenor JG, Masters CL, Duce J, White AR, Barnham KJ, Li Q-X. Diacetylbis (N(4) methylthiosemi-carbazonato) copper(II) (CuII(atsm)) protects against peroxynitrite-induced nitrosative damage and prolongs survival in amyotrophic lateral sclerosis mouse model. J Biol Chem. 2011;286:44035–44.
Drew SC. The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease. Front Neurosci. 2017;11:317.
Squitti R, Salustri C, Rongioletti M, Siotto M. Commentary: The case for abandoning therapeutic chelation of copper ions in Alzheimer’s disease, Front Neurol. 2017;8:503.
Wang F, Jiao P, Qi M, Frezza M, Dou QP, Yan B. Turning tumor-promoting copper into an anti-cancer weapon via high-throughput chemistry. Curr Med Chem. 2010;17:2685–98.
Daniel KG, Chen D, Orlu S, Cui QC, Miller FR, Dou QP. Clioquinol and pyrrolidine dithiocarbamate complex with copper to form proteasome inhibitors and apoptosis inducers in human breast cancer cells. Breast Cancer Res. 2005;7:R897–908.
Bisceglie F, Baldini M, Belicchi-Ferrari M, Buluggiu E, Careri M, Pelosi G, Pinelli S, Tarasconi P. Metal complexes of retinoid derivatives with antiproliferative activity: synthesis, characterization and DNA interaction studies. Eur J Med Chem. 2007;42:627–34.
Lian W-J, Wang X-T, Xie C-Z, Tian H, Song X-Q, Pan H-T, Qiao X, Xu J-Y. Mixed-ligand copper(II) Schiff base complexes: the role of the co-ligand in DNA binding, DNA cleavage, protein binding and cytotoxicity. Dalton Trans. 2016;45:9073–87.
Gupte A, Mumper RJ. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic Biol Med. 2007;43:1271–8.
Kirshner JR, He S, Balasubramanyam V, Kepros J, Yang C-Y, Zhang M, Du Z, Barsoum J, Bertin J. Elesclomol induces cancer cell apoptosis through oxidative stress. Mol Cancer Ther. 2008;7:2319–27.
Fan CD, Su H, Zhao J, Zhao BX, Zhang SL, Miao JY. A novel copper complex of salicylaldehyde pyrazole hydrazone induces apoptosis through up-regulating integrin β4 in H322 lung carcinoma cells. Eur J Med Chem. 2010;45:1438–46.
Chakraborty A, Kumar P, Ghosh K, Roy P. Evaluation of a Schiff base copper complex compound as potent anticancer molecule with multiple targets of action. Eur J Pharmacol. 2010;647:1–12.
Santini C, Pellei M, Gandin V, Porchia M, Tisato F, Marzano C. Advances in copper complexes as anticancer agents. Chem Rev. 2014;114:815–62.
Weekley CM, He C. Developing drugs targeting transition metal homeostasis. Curr Opin Chem Biol. 2017;37:26–32.
Barilli A, Atzeri C, Bassanetti I, Ingoglia F, Dall’Asta V, Bussolati O, Maffini M, Mucchino C, Marchiò L. Oxidative stress induced by copper and iron complexes with 8-hydroxyquinoline derivatives causes paraptotic death of HeLa cancer cells. Mol Pharm. 2014;11:1151–63.
Gaál A, Orgován G, Polgári Z, Réti A, Mihucz VG, Bősze S, Szoboszlai N, Streli C. Complex forming competition and in-vitro toxicity studies on the applicability of di-2-pyridylketone-4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) as a metal chelator. J Inorg Biochem. 2014;130:52–8.
Pape VFS, Türk D, Szabó P, Wiese M, Enyedy EA, Szakács G. Synthesis and characterization of the anticancer and metal binding properties of novel pyrimidinylhydrazone derivatives. J Inorg Biochem. 2015;144:18–30.
Jorgenson TC, Zhong W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013;73:6118–23.
Kim J, Kim J, Bae J-S. ROS homeostasis and metabolism: a critical liaison for cancer therapy. Exp Mol Med. 2016;48:e269.
Franz KJ. Clawing back: broadening the notion of metal chelators in medicine. Curr Opin Chem Biol. 2013;17:143–9.
Vo NH, Xia Z, Hanko J, Yun T, Bloom S, Shen J, Koya K, Sun L, Chen S. Synthesis, crystallographic characterization and electrochemical property of a copper(II) complex of the anticancer agent elesclomol. J Inorg Biochem. 2014;130:69–73.
Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s-like changes. Neuropharmacology. 2011;60:910–20.
Li J, Deng J, Sheng W, Zuo Z. Metformin attenuates Alzheimer’s disease-like neuropathology in obese, leptin-resistant mice. Pharmacol Biochem Behav. 2012;101:564–74.
Novelle MG, Ali A, Diéguez C, Bernier M, de Cabo R. Metformin: a hopeful promise in aging research. Cold Spring Harb Perspect Med. 2016;6:a025932.
Lanza V, Milardi D, Di Natale G, Pappalardo G. Repurposing of copper(II)-chelating drugs for the treatment of neurodegenerative diseases. Curr Med Chem. 2018;25:525–39.
Chang CJ. Searching for harmony in transition-metal signalling. Nature Chem Biol. 2015;11:744–7.
Pal A, Badyal RK, Vasishta RK, Attri SV, Thapa BR, Prasad R. Biochemical, histological, and memory impairment effects of chronic copper toxicity: a model for non-Wilsonian brain copper toxicosis in wistar rat. Biol Trace Elem Res. 2013;153:257–68.
Oksanen M, Petersen AJ, Naumenko N, Puttonen K, Lehtonen S, Olivé MG, Shakirzyanova A, Leskelä S, Sarajärvi T, Viitanen M, Rinne JO, Hiltunen M, Haapasalo A, Giniatullin R, Tavi P, Zhang S-C, Kanninen KM, Hämäläinen RH, Koistinaho J. PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep. 2017;9:1885–97.
Szabó* Z, Héja* L, Szalay G, Kékesi O, Füredi A, Szebényi K, Dobolyi Á, Orbán TI, Kolacsek O, Tompa T, Miskolczy Z, Biczók L, Rózsa B, Sarkadi B, Kardos J. Extensive astrocyte synchronization advances neuronal coupling in slow wave activity in vivo. Sci Rep. 2017;7:6018.
Verkhratsky A, Nedergaard M. Physiology of astroglia. Physiol Rev. 2018;98:239–389.
Wu X-M, Qian C, Zhou Y-F, Yan Y-C, Luo Q-Q, Yung W-H, Zhang F-L, Jiang L-R, Qian ZM, Ke Y. Bi-directionally protective communication between neurons and astrocytes under ischemia. Redox Biol. 2017;13:20–31.
Skatchkov SN, Antonov SM, Eaton MJ. Glia and glial polyamines. Role in brain function in health and disease. Biochemistry (Moscow) Suppl A Membrane Cell Biol. 2016;10:73–98.
Scheiber IF, Dringen R. Astrocyte functions in the copper homeostasis of the brain. Neurochem Int. 2013;62:556–65.
Dringen R, Scheiber IF, Mercer JF. Copper metabolism of astrocytes. Front Aging Neurosci. 2013;5:9.
Kim HJ, Macdonald RL. An N-terminal histidine is the primary determinant of alpha subunit dependent Cu2+ sensitivity of alpha beta3 gamma2L GABA(A). Mol Pharmacol. 2003;64:1145–52.
Kumamoto E, Murata Y. Characterization of GABA current in rat septal cholinergic neurons in culture and its modulation by metal-cations. J Neurophys. 1995;74:2012–27.
Liu X, Surprenant A, Mao HJ, Roger S, Xia R, Bradley H, Jiang LH. Identification of key residues coordinating functional inhibition of P2X(7) receptors by zinc and copper. Mol Pharmacol. 2008;73:252–9.
Lovinger DM. Inhibition of 5-HT3 receptor-mediated ion current by divalent metal- cations in NCB-20 neuroblastoma-cells. J Neurophysiol. 1991;66:1329–37.
Ma JY, Narahashi T. Differential modulation of GABA-A receptor-channel complex by polyvalent cations in rat dorsal-root ganglion neurons. Brain Res. 1993;607:222–32.
Ma ZM, Wong KY, Horrigan FT. An extracellular Cu2+ binding site in the voltage sensor of BK and shaker potassium channels. J Gen Physiol. 2008;131:483–502.
Ruthstein S, Stone KM, Cunningham TF, Ji M, Cascio M, Saxena S. Pulsed electron spin resonance resolves the coordination site of cu(2+) ions in alpha 1-glycine receptor. Biophys J. 2010;99:2497–506.
Schumann T, Grudzinska J, Kuzmin D, Betz H, Laube B. Binding-site mutations in the α1 subunit of the inhibitory glycine receptor convert the inhibitory metal ion Cu2+ into a positive modulator. Neuropharmacology. 2009;56:310–7.
Shcheglovitov A, Vitko I, Lazarenko RM, Orestes P, Todorovic SM, Perez-Reyes E. Molecular and biophysical basis of glutamate and trace metal modulation of voltage-gated ca(v)2.3 calcium channels. J Gen Physiol. 2012;139:219–34.
Wang W, Yu Y, Xu TL. Modulation of acid-sensing ion channels by Cu2+ in cultured hypothalamic neurons of the rat. Neurosci. 2007;145:631–41.
Xie XM, Hider RC, Smart TG. Modulation of GABA-mediated synaptic transmission by endogenous zinc in the immature rat hippocampus in-vitro. J Physiol Lond. 1994;478:75–86.
Benedikt J, Inyushin M, Kucheryavykh YV, Rivera Y, Kucheryavykh LY, Nichols CG, Eaton MJ, Skatchkov SN. Intracellular polyamines enhance astrocytic coupling. Neuroreport. 2012;23:1021–5.
Desforges B, Curmi PA, Bounedjaha O, Nakib S, Hamon L, De Bandt J-P, Pastré D. An intercellular polyamine transfer via gap junctions regulates proliferation and response to stress in epithelial cells. Mol Biol Cell. 2013;24:1529–43.
Shore L, McLean P, Gilmour SK, Hodgins MB, Finbow ME. Polyamines regulate gap junction communication in connexin 43-expressing cells. Biochem J. 2001;357:489–95.
Kékesi O, Ioja E, Szabó Z, Kardos J, Héja L. Recurrent seizure-like events are associated with coupled astroglial synchronization. Front Cell Neurosci. 2015;9:215.
Héja L, Nyitrai G, Kékesi O, Dobolyi A, Szabó P, Fiáth R, Ulbert I, Pál-Szenthe B, Palkovits M, Kardos J. Astrocytes convert network excitation to tonic inhibition of neurons. BMC Biol. 2012;10:26.
Dodani SC, Firl A, Chan J, Nam CI, Aron AT, Onak CS, Ramos-Torres KM, Paek J, Webster CM, Feller MB, Chang CJ. Copper is an endogenous modulator of neural circuit spontaneous activity. Proc Natl Acad Sci U S A. 2014;111:16280–5.
Mathie A, Sutton GL, Clarke CE, Veale EL. Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther. 2006;111:567–83.
Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490–503.
Hamilton EI, Sabbioni E, Van der Venne MT. Element reference values in tissues from inhabitants of the European Community. VI. Review of elements in blood, plasma and urine and a critical evaluation of reference values for the United Kingdom population. Sci Total Environ. 1994;158:165–90.
Lech T, Sadlik JK. Copper concentration in body tissues and fluids in Normal subjects of southern Poland. Biol Trace Elem Res. 2007;118:10–5.
Yoshiji H, Yoshii J, Kuriyama S, Ikenaka Y, Noguchi R, Yanase K, Namisaki T, Kitade M, Yamazaki M, Fukui H. Combination of copper-chelating agent, trientine, and methotrexate attenuates colorectal carcinoma development and angiogenesis in mice. Oncol Rep. 2005;14:213–8.
Kotsaki-Kovatsi VP, Koehler-Samouilidis G, Kovatsis A, Rozos G. Fluctuation of zinc, copper, magnesium and calcium concentrations in Guinea pig tissues after administration of captopril (SQ14225). J Trace Elem Med Biol. 1997;11:32–6.
Small W Jr, Molteni A, Kim YT, Taylor JM, Ts’ao C-H, Ward WF. Mechanism of captopril toxicity to a human mammary ductal carcinoma cell line in the presence of copper. Breast Cancer Res Treat. 1999;55:223–9.
Duan L, Shen H, Zhao G, Yang R, Cai X, Zhang L, Jin C, Huang Y. Inhibitory effect of disulfiram/copper complex on non-small cell lung cancer cells. Biochem Biophys Res Commun. 2014;446:1010–6.
Xie F, Peng F. Anti-prostate cancer activity of 8-hydroxyquinoline-2-carboxaldehyde-thiosemicarbazide copper complexes by fluorescent microscopic imaging. J Fluoresc. 2017;27:1937–41.
Paterson BM, Donnelly PS. Copper complexes of bis (thiosemicarbazones): from chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem Soc Rev. 2011;40:3005–18.
Shrestha S, Maharjan S. Synthesis and characterization of copper complex of salicylaldehyde benzoyl hydrazone. J Nepal Chem Soc. 2012;29:11–7.
Ainscough EW, Brodie AM, Denny WA, Finlay GJ, Gothe SA, Ranford JD. Cytotoxicity of salicylaldehyde benzoylhydrazone analogs and their transition metal complexes: quantitative structure-activity relationships. J Inorg Biochem. 1999;77:125–33.
Acknowledgements
We do thank Susan Amara for her valuable comments and criticism.
Funding
This work was supported by grants KMR_12-1-2012-0112 TRANSRAT, VEKOP-2.1.1-15-2016-00156 and OTKA K124558.
Author information
Authors and Affiliations
Contributions
LH participated in the design, helped to draft the manuscript, and carried out documentation materials. AS participated assessed relevant bioinformatics. IJ and KJ evaluated organic chemistry and liver toxicity studies, respectively. RK participated in the design and helped to draft the manuscript. JK participated in the design, coordinated the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not relevant.
Consent for publication
Not relevant.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional information
The original version of this article was revised: An error in the structures was noticed in Table 3. The structure for TM was incorrect and in the PDF version of the article, some of the structures were overlapping.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Kardos, J., Héja, L., Simon, Á. et al. Copper signalling: causes and consequences. Cell Commun Signal 16, 71 (2018). https://doi.org/10.1186/s12964-018-0277-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0277-3