
Abstract
Background
Recurrent pregnancy loss (RPL) refers to two or more spontaneous abortions that occur consecutively with the same spouse. RPL severely affects human reproduction health, and causes extreme physical and mental suffering to patients and their families.
Methods
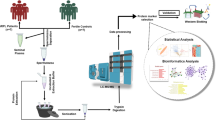
We used isobaric tags for relative and absolute quantitation (iTRAQ), which was coupled with liquid chromatography mass spectrometry (LC-MS) proteomic analysis, in order to identify differentially expressed proteins. Moreover, we used western blot to analyze differentially expressed proteins.
Results
Of the 2350 non-redundant proteins identified, 38 proteins were significantly altered and were identified as potential biomarkers for RPL. The protein-protein interaction network constructed using GeneMANIA revealed that 35.55% displayed similar co-expression, 30.87% were predicted, and 20.95% had physical interaction characteristics. Based on Gene ontology classification and KEGG pathway enrichment analyses, the majority of these differentially expressed proteins were found to be related to biological regulation, metabolic and cellular processes, protein binding and different enzymes activities, as well as disorder of fat and glucose metabolic pathways. It is noteworthy that three metabolism related biomarkers (HK1, ACLY, and FASN) were further confirmed through western blot analysis.
Conclusions
These results suggest that these differentially expressed proteins may be used as biomarkers for RPL, and related signaling pathways may play crucial roles in male induced RPL.
Similar content being viewed by others
Background
Recurrent pregnancy loss (RPL) refers to the consecutive occurrence of two or more spontaneous abortions with the same spouse in early pregnancy [1]. The incidence of RPL accounts for about 1% of all pregnancies and causes extreme physical and mental suffering to patients and their families [1]. Although many factors have been investigated, including chromosomal, endocrine and anatomical aberrations, prethrombotic state, immune disorders and infections, the causes in the half of RPL cases are still unknown which are labeled as unexplained [2]. Because the male contributes 50% of the DNA to the embryo, it is not surprising that paternal factors could also contribute to pregnancy loss [3]. The association is first studied between diminished DNA content in sperm and spontaneous abortion with special reference to male factor, which was published in 1966 [4]. Furthermore, another study also examines the relationship between recurrent pregnancy loss and sperm characteristics [5]. However, researches on RPL mainly focus on maternal factors, the male contributions have been largely unexamined and remain poorly understood [6, 7].
Advances in technological developments are expected to find new biomarkers for RPL. As an emerging research tool in the post-genome era, proteomics can be utilized for studying the protein expression, function and protein-protein interactions [8]. It has become one of the most important tools for studying sperm protein profiles. An in-depth understanding of the sperm proteomics could be conductive to explain the roles of sperm proteins in how to cause RPL. Significant advances in methods and strategies related to proteomics, including mass spectrometry and liquid chromatography, have enabled the analysis of thousands of complex cellular proteins present in sperm [9, 10]. A recent study compared the differences in the sperm proteome by liquid chromatography-tandem mass spectrometry and found that the proteins associated with sperm function and fertilization process were compromised in testicular cancer patients [11]. Up to now, there is no report of investigation the difference in the sperm proteome of males for RPL at a proteomic level.
In this study, the sperm proteome is analyzed to elucidate the proteomic expression profiles of sperm, and to further ascertain the abnormal protein biomarkers of males for RPL. We perform isobaric tags for relative and absolute quantitation (iTRAQ) to analyze sperm proteomic changes in RPL patients. We aim to uncover significantly altered sperm proteins and pathways in RPL.
Materials and methods
Ethics statement
All procedures were carried out with the approval of the Ethics Committee of the Affiliated Hospital of Weifang Medical University. All scientific experiments were conducted conforming to World Health Organization guidelines (WHO Laboratory Manual for the Examination and Processing of Human Semen, the 5th edition). Prior written consent was obtained from all males who participated in this study.
Study population and sample statement
Proteomic analyses were performed on semen obtained from two categories of donors: 7 fertile males who had fathered a child within the last 2 years (control), and 10 males whose spouse had suffered at least two miscarriages (RPL). All males recruited to this study were normozoospermic, in accordance with World Health Organization guidelines. Factors used to exclude patients were abnormal chromosomes, endocrine dysfunction, reproductive duct anomaly, antiphospholipid antibodies, immune disorders or other systemic diseases. Semen samples were ejaculated into sterile containers through masturbation. The samples were processed for 30 min to allow them to liquefy before being centrifuged in a 50% percoll gradient at 1000 g to remove seminal plasma, immature germ cells and nonsperm cells. Afterwards, the samples were frozen at − 80 °C until used.
Protein preparation and iTRAQ labeling
Proteins were extracted in a 50 mM ammonium hydrogen bicarbonate buffer containing 0.5% sodium deoxycholate, 50 mM dithiothreitol (DTT), and a protease inhibitor. We then sonicated the samples for 1 s (20 times), and centrifuged the samples at 1000 g for 15 min. The supernatant was precipitated overnight with 5vol of acetone and re-suspended in 0.5 M triethylammonium bicarbonate containing 0.5% sodium deoxycholate. Protein concentrations were tested with using the Bradford method [12, 13].
One hundred microgram of protein from each sample was reduced and alkylated, digested with trypsin, dried and reconstituted in 50 μL of 0.5 M triethyl ammonium bicarbonate. In order to avoid inter-individual variations, proteins from all males in each group were pooled in equal quantities. The iTRAQ results were compared between two pool samples. Next, we labeled the dried peptides using an iTRAQ 4-plex kit, in accordance with the AB SCIEX protocol. The protein samples obtained from the control group were labeled with iTRAQ 116, while samples from males of RPL were labeled with 121 iTRAQ reagents. Finally, the labeled samples were mixed in a single vial and dried using a rotary vacuum concentrator [14].
Liquid chromatography mass spectrometry (LC-MS) analysis
LC-MS analysis was performed on a Triple-TOF 5600 system (AB SCIEX). Mass spectra were collected (400–1250 m/z) at high resolution (> 30,000) for 250 ms per spectrum. A maximum of 50 precursors in a cycle were selected for fragmentation from each mass spectrum. Tandem mass spectra were harvested in high sensitivity mode (resolution> 15,000). 2-plex iTRAQ Labeling, strong cation exchange (SCX) and RP HPLC-MS/MS were performed by Fitgene Biological Technology Co. Ltd. (Guangzhou, China).
Protein identification and quantitation
Protein identification and quantification were conducted using Protein Pilot Software (AB SCIEX), using the algorithm of Paragon to identify the peptides, which were further analyzed using the Pro GroupTM algorithm in which isoform-specific quantification was employed to track the differences between expressions of different isoforms.
False discovery rate (FDR) was set to less than 0.01 for the identification of both peptides and proteins, and only proteins identified through peptides were used for quantitation. The cutoffod qualification was peptides with an unused confidence score larger than 1.3 and confidence level of 95%. In order to identify differentially expressed proteins, t test was employed to calculate the significant differences in protein expression differences between the RPL and the control group. Proteins with the fold changes of > 1.5 and < 0.67, as well as a p value of < 0.05 were identified as the differentially expressed proteins.
Protein-protein interaction analysis conducted on GeneMANIA
GeneMANIA [15], a user-friendly and flexible web interface for analyzing gene or protein lists, generating hypotheses about gene function, and prioritizing genes for functional assays, was utilized for protein-protein interaction analysis.
Data sets in GeneMANIA were gathered from available and public databases, which include predicted protein interaction data based on orthology from I2D [16]; genetic and physical interaction data from BioGRID [17]; co-expression data from Gene Expression Omnibus (GEO) [18]; and pathway and molecular interaction data from Pathway Commons, which contains data from MINT [19], BioGRID, Reactome [20], HumanCyc [21], IntAct [22], Memorial Sloan-Kettering Cancer Center, Systems Biology Center New York, Human Protein Reference Database [21], and NCI-Nature Pathway Interaction Database [23].
Given a query list, GeneMANIA provides a list of genes or proteins that are functionally similar, or have shared properties with the initial query genes, and displays a functional association network, illustrating the relationships among the list and the curated genomics and proteomics data. The previously identified differentially expressed proteins were added to the search bar, after selecting Homo sapiens as the optional organism.
Protein function and pathway enrichment analysis
In order to better study the biological functions and pathways of differentially expressed proteins, Web-based Gene set analysis toolkit (WebGestalt) was utilized to fully and deeply understand the functional and pathway enrichment information of the interesting proteins [24]. Differentially expressed proteins were uploaded to the WebGestalt server using the overrepresentation enrichment analysis (ORA) method with the Gene Oncology and KEGG databases.
Gene ontology (GO) analysis is a widely used method for analyzing genes and gene products relating to functions including biological processes, molecular functions, and cellular components [25]. Kyoto Encyclopedia of Genes and Genomes pathway (KEGG) is a useful resource for the systematic annotation of gene functions and related high-level genomic functional information [26, 27].
Protein selection and validation using western blot
Sperm proteins involved in fat and glucose metabolic pathways were selected for validation using western blot. Three key proteins hexokinase-1 (HK1), ATP citrate lyase (ACLY) and fatty acid synthase (FASN) were applied on individual samples obtained from RPL patients (n = 10) and the control group (n = 7). Proteins from each individual sample of the control and RPL sperm were used for western blot analysis. Primary antibodies: anti-β-actin mouse antibody (Sigma, A5441), anti-HK1 rabbit polyclonal antibody (Abcam, ab150423), anti-ACLY rabbit polyclonal antibody (Abcam, ab40793) and anti-FASN rabbit monoclonal antibody (Abcam, ab128856). Bands were revealed using a chemiluminescence reagent (ECL kit, PerkinElmer, Boston, MA, USA).
Results
Protein expression profiling and identification of differentially expressed proteins
Sperm concentration and motility were assessed in the RPL group and the healthy group (Table 1). Statistical analysis showed no difference in sperm concentration and motility between the two groups. Using an iTRAQ based quantitative proteomic method, we identified 2350 proteins in total (Additional file 1: Table S1), and each had peptides with 95% confidence of a minimum unused score of 1.3. The basic statistics of these proteins, such as the isoelectric point, protein mass and peptide number were shown in Additional files 2, 3, 4, 5, 6, 7: Figure S1-S6. Among the 2350 identified proteins, 38 proteins were differentially expressed between the RPL group and the healthy group, with the cutoff fold change of > 1.5 and < 0.67, and a p value of < 0.05. To be specific, 25 of these proteins were upregulated, while the other 13 proteins were downregulated in the RPL patients (Fig. 1 and Table 1).

Protein expression profiling and identification of differentially expressed proteins. Volcano plot of differentially expressed proteins. The x axis is the log2 level of protein expression, while the y axis is the log10 level of the p value between the RPL and the control group. Each point represents a protein and the different colors denote the differential expression pattern: red for up regulation, grey for flat regulation, and blue for down regulation
Protein-protein interaction (PPI) of differentially expressed proteins
To gain a comprehensive view thorough, we performed a PPI analysis using GeneMANIA. Among the 38 differentially expressed proteins and their interacting proteins, we found that 35.55% displayed similar co-expression, 30.87% were predicted, and 20.95% had physical interaction characteristics. Other results including shared protein domains, co-localization and genetic interaction are shown in Fig. 2.

Protein-protein interaction (PPI) network of the differentially expressed proteins. PPI Network of the differentially expressed proteins. Black protein nodes represent target proteins, and the different connecting colors indicate different correlations. Functional association between differentially expressed proteins was investigated using GeneMANIA. Proteins in black circles were the query terms while those in grey circle were indicated proteins
Gene ontology enrichment analysis of differentially expressed proteins
In order to understand the functional basis of the 38 differentially expressed proteins, GO enrichment analyses were performed using WebGestalt. The proteins identified were sorted into categories based on the ontology as determined from their GO annotation terms. Interestingly, the differentially expressed proteins were primarily involved in biological regulation, metabolic and cellular processes, protein binding and different enzymes activities, reproduction, and development processes (Fig. 3). Furthermore, most of these proteins were located in vesicle, the nucleuses, membranes and cytosol.

Gene ontology enrichment analysis of differentially expressed proteins. GO enrichment of differentially expressed proteins. a: upregulated proteins. b: downregulated proteins
KEGG pathway enrichment analysis of differentially expressed proteins
To gain insights into the biological pathways of the differentially expressed proteins which were identified through iTRAQ technology, we performed KEGG pathway analyses. Interestingly, it was demonstrated that the upregulated proteins were primarily enriched in fatty acid biosynthesis, while the downregulated proteins were significantly enriched in glucose metabolism (Fig. 4a and b). HK1 was involved in glucose metabolic pathway, while FASN and ACLY were enriched in fat metabolic pathway. So we tested the expressions of the three proteins in RPL samples by western blot (Fig. 4c).

KEGG pathway enrichment analysis of differentially expressed proteins. KEGG enrichment of differentially expressed proteins. a: upregulated proteins and b: downregulated proteins
Discussion
The leading causes for RPL in males are still unknown and the use of the novel proteomic technology may hold the key to more accurately diagnosing and treating male induced RPL [28]. One method of revealing involvement of sperm proteins in the development of RPL is to compare sperm samples from RPL males with samples from healthy individuals.
In this study, we first carried out an iTRAQ proteomic analysis to identify proteins that were differentially expressed between the RPL and the control group. A total of 38 proteins were found to have different expressions between the two groups. It was of great importance to understand why these proteins exerted different expression pattern and how the abnormal expression of the identified proteins could result in male induced RPL. Gene ontology analysis showed that the functions of these proteins could be classified into several important categories, such as biological regulation, metabolic and cellular processes, protein binding and different enzymes activities. More interestingly, we found that differentially expressed proteins were significantly enriched in fatty acid biosynthesis and glucose metabolism pathways. These results demonstrate that disorders of fat and glucose metabolism may contribute to male induced RPL.
Cellular energy metabolism is correlated with cell fate and a few studies report the association between glycogen and the nuclear envelope, the endoplasmic reticulum, as well as the annulate lamellae of embryonic and transformed cells [29, 30]. HK1 protein localizes on the outer membrane of mitochondria and phosphorylates glucose to produce glucose-6-phosphate, which is the first step of glucose metabolism pathways [31]. Spermatogenic cell-specific type 1 hexokinase (HK1S), an isoform of HK1, is found abundantly in sperm, which is mainly expressed in area of the sperm flagellum [32]. HK1 is found to be associated with active spermatogenesis in mice, and might abrogate the process of spermatogenesis leading to infertility [33]. Since glucose metabolism is very important in embryonic development, the downregulation expression of the HK1 might induce miscarriage. Therefore, HK1 functions in RPL need to be further investigated. Furthermore, de novo fatty acid synthesis is activated during embryogenesis, which plays a critical role in embryonic development [34]. FASN is a limited enzyme in fatty acid synthesis, which could catalyze the synthesis of palmitate from acetyl-CoA and malonyl-CoA, in the presence of NADPH [35]. The FASN−/− mutant embryos are died in generated FASN knockout mice [34]. So FASN might play an important role in male induced abortion. Moreover, ACLY is responsible for the synthesis of cytosolic acetyl-CoA in many tissues, and is identified as a potential heat sensitive target in germ cells [36]. The relationship between ACLY and abortion has not been reported, so further research need to be studied.
Conclusions
We used iTRAQ technology to identify 38 differentially expressed proteins that may be utilized as candidate biomarkers for RPL. The identified proteins and related signaling pathways might play crucial roles in male induced RPL.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article.
References
Sugiura-Ogasawara M, Ozaki Y, Suzumori N. Management of recurrent miscarriage. J Obstet Gynaecol Res. 2014;40:1174–9.
Li TC, Makris M, Tomsu M, Tuckerman E, Laird S. Recurrent miscarriage: aetiology, management and prognosis. Hum Reprod Update. 2002;8:463–81.
Larsen EC, Christiansen OB, Kolte AM, Macklon N. New insights into mechanisms behind miscarriage. BMC Med. 2013;11:154.
Joel CA. New etiologic aspects of habitual abortion and infertility, with special reference to the male factor. Fertil Steril. 1966;17:374–80.
Niederberger C. Re: relationship between sperm aneuploidy, sperm DNA integrity, chromatin packaging, traditional semen parameters, and recurrent pregnancy loss. J Urol. 2016;196:1232.
Coomarasamy A, Williams H, Truchanowicz E, Seed PT, Small R, Quenby S, Gupta P, Dawood F, Koot YE, Bender Atik R, et al. A randomized trial of progesterone in women with recurrent miscarriages. N Engl J Med. 2015;373:2141–8.
Feinberg EC. The relationship between recurrent pregnancy loss and the male contribution. Fertil Steril. 2016;105:1160–1.
Pan HT, Ding HG, Fang M, Yu B, Cheng Y, Tan YJ, Fu QQ, Lu B, Cai HG, Jin X, et al. Proteomics and bioinformatics analysis of altered protein expression in the placental villous tissue from early recurrent miscarriage patients. Placenta. 2018;61:1–10.
Oliva R, de Mateo S, Estanyol JM. Sperm cell proteomics. Proteomics. 2009;9:1004–17.
Amaral A, Castillo J, Ramalho-Santos J, Oliva R. The combined human sperm proteome: cellular pathways and implications for basic and clinical science. Hum Reprod Update. 2014;20:40–62.
Panner Selvam MK, Agarwal A, Pushparaj PN. A quantitative global proteomics approach to understanding the functional pathways dysregulated in the spermatozoa of asthenozoospermic testicular cancer patients. Andrology. 2019. https://doi.org/10.1111/12620.
Yang T, Ren C, Qiao P, Han X, Wang L, Lv S, Sun Y, Liu Z, Du Y, Yu Z. PIM2-mediated phosphorylation of hexokinase 2 is critical for tumor growth and paclitaxel resistance in breast cancer. Oncogene. 2018;37:5997–6009.
Ren C, Yang T, Qiao P, Wang L, Han X, Lv S, Sun Y, Liu Z, Du Y, Yu Z. PIM2 interacts with tristetraprolin and promotes breast cancer tumorigenesis. Mol Oncol. 2018;12:690–704.
Wang L, Dai Y, Qi S, Sun B, Wen J, Zhang L, Tu Z. Comparative proteome analysis of peripheral blood mononuclear cells in systemic lupus erythematosus with iTRAQ quantitative proteomics. Rheumatol Int. 2012;32:585–93.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20.
Brown KR, Jurisica I. Online predicted human interaction database. Bioinformatics. 2005;21:2076–82.
Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, Oughtred R, Lackner DH, Bahler J, Wood V, et al. The BioGRID interaction database: 2008 update. Nucleic Acids Res. 2008;36:D637–40.
Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, et al. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–90.
Ceol A, Chatr Aryamontri A, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–9.
Vastrik I, D'Eustachio P, Schmidt E, Gopinath G, Croft D, de Bono B, Gillespie M, Jassal B, Lewis S, Matthews L, et al. Reactome: a knowledge base of biologic pathways and processes. Genome Biol. 2007;8:R39.
Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. Human protein reference database--2009 update. Nucleic Acids Res. 2009;37:D767–72.
Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–31.
Schaefer CF, Anthony K, Krupa S, Buchoff J, Day M, Hannay T, Buetow KH. PID: the pathway interaction database. Nucleic Acids Res. 2009;37:D674–9.
Wang J, Duncan D, Shi Z, Zhang B. WEB-based GEne SeT AnaLysis toolkit (WebGestalt): update 2013. Nucleic Acids Res. 2013;41:W77–83.
The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45:D331–8.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–61.
Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30.
van den Berg MMJ, Dancet EAF, Erlikh T, van der Veen F, Goddijn M, Hajenius PJ. Patient-centered early pregnancy care: a systematic review of quantitative and qualitative studies on the perspectives of women and their partners. Hum Reprod Update. 2018;24:106–18.
Eyal-Giladi H, Feinstein N, Friedlander M, Raveh D. Glycogen metabolism and the nuclear envelope-annulate lamella system in the early chick embryo. J Cell Sci. 1985;73:399–407.
Kessel RG. The annulate lamellae--from obscurity to spotlight. Electron Microsc Rev. 1989;2:257–348.
Kumar S, Parameswaran S, Sharma RK. Novel myristoylation of the sperm-specific hexokinase 1 isoform regulates its atypical localization. Biol Open. 2015;4:1679–87.
Nakamura N, Shibata H, O'Brien DA, Mori C, Eddy EM. Spermatogenic cell-specific type 1 hexokinase is the predominant hexokinase in sperm. Mol Reprod Dev. 2008;75:632–40.
Sarkar H, Arya S, Rai U, Majumdar SS. A study of differential expression of testicular genes in various reproductive phases of Hemidactylus flaviviridis (wall lizard) to derive their association with onset of spermatogenesis and its relevance to mammals. PLoS One. 2016;11:e0151150.
Chirala SS, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, Finegold M, Wakil SJ. Fatty acid synthesis is essential in embryonic development: fatty acid synthase null mutants and most of the heterozygotes die in utero. Proc Natl Acad Sci U S A. 2003;100:6358–63.
Buckley D, Duke G, Heuer TS, O'Farrell M, Wagman AS, McCulloch W, Kemble G. Fatty acid synthase - modern tumor cell biology insights into a classical oncology target. Pharmacol Ther. 2017;177:23–31.
Yadav SK, Pandey A, Kumar L, Devi A, Kushwaha B, Vishvkarma R, Maikhuri JP, Rajender S, Gupta G. The thermo-sensitive gene expression signatures of spermatogenesis. Reprod Biol Endocrinol. 2018;16:56.
Acknowledgments
Not applicable.
Funding
The study was supported by research grants from the Innovation fund of National Natural Science Foundation of China (Grant no. 81602440), Weifang Science and Technology Bureau (Weifang Soft Science Research Program) (Grant no. 2017RKX088).
Author information
Authors and Affiliations
Contributions
ZHY and DNX: project development, data collection, data analysis, and manuscript writing. YZ, YXW, JW, FXA and XWS: data collection. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in this study involving were in accordance with the ethical standards of the institutional research committee of Weifang medical university and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. The total proteins were identified using an iTRAQ based quantitative proteomic method. (XLSX 454 kb)
Additional file 2:
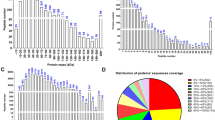
Figure S1. Isoelectic point distribution. (JPG 783 kb)
Additional file 3:
Figure S2. Protein mass distribution. (JPG 965 kb)
Additional file 4:
Figure S3. Peptide number distribution. (JPG 1077 kb)
Additional file 5:
Figure S4. Distribution of spectral quality matching error. (JPG 2643 kb)
Additional file 6:
Figure S5. Distribution of proteins’ sequences coverage. (JPG 1699 kb)
Additional file 7:
Figure S6. Peptide length distribution. (JPG 1118 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Xue, D., Zhang, Y., Wang, Y. et al. Quantitative proteomic analysis of sperm in unexplained recurrent pregnancy loss. Reprod Biol Endocrinol 17, 52 (2019). https://doi.org/10.1186/s12958-019-0496-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-019-0496-5