
Abstract
Background
Lung adenocarcinoma (LUAD) accounts for 50% of lung cancers, with high mortality and poor prognosis. Long non-coding RNA (lncRNA) plays a vital role in the progression of tumors. Cuproptosis is a newly discovered form of cell death that is highly investigated. Therefore, in the present study, we aimed to investigate the role of cuproptosis-related lncRNA signature in clinical prognosis prediction and immunotherapy and the relationship with drug sensitivity.
Material and methods
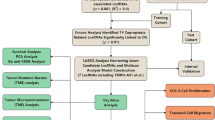
Genomic and clinical data were obtained from The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases, and cuproptosis-related genes were obtained from cuproptosis-related studies. The prognostic signature was constructed by co-expression analysis and Cox regression analysis. Patients were divided into high and low risk groups, and then, a further series of model validations were carried out to assess the prognostic value of the signature. Subsequently, lncRNAs were analyzed for gene ontology (GO), Kyoto Encyclopedia of Genes and Genomes Enrichment (KEGG), immune-related functions, and tumor mutation burden (TMB). Finally, we used tumor immune dysfunction and exclusion (TIDE) algorithms on immune escape and immunotherapy of cuproptosis-related lncRNAs, thereby identifying its sensitivity toward potential drugs for LUAD.
Results
A total of 16 cuproptosis-related lncRNAs were obtained, and a prognostic signature was developed. We found that high-risk patients had worse overall survival (OS) and progression-free survival (PFS) and higher mortality. Independent prognostic analyses, ROC, C-index, and nomogram showed that the cuproptosis-related lncRNAs can accurately predict the prognosis of patients. The nomogram and heatmap showed a distinct distribution of the high- and low-risk cuproptosis-related lncRNAs. Enrichment analysis showed that the biological functions of lncRNAs are associated with tumor development. We also found that immune-related functions, such as antiviral activity, were suppressed in high-risk patients who had mutations in oncogenes. OS was poorer in patients with high TMB. TIDE algorithms showed that high-risk patients have a greater potential for immune escape and less effective immunotherapy.
Conclusion
To conclude, the 16 cuproptosis-related lncRNAs can accurately predict the prognosis of patients with LUAD and may provide new insights into clinical applications and immunotherapy.
Similar content being viewed by others

Background
Lung adenocarcinoma (LUAD) is a common type of lung cancer. It has one of the highest rates of incidence and mortality globally, posing a serious risk to human health [1]. Treatment options for lung cancer include surgery, radiotherapy, and chemotherapy [2]. Presently, LUAD is not diagnosed easily, and most patients are already at an advanced stage during diagnosis. This reduces the scope of surgery, and the distant spread of cancer cells causes serious health damage. Therefore, the treatment of LUAD should be improved through a rational treatment plan. Nowadays, tumor risk score prediction signatures are a non-invasive technique to identify patient survival. They can help to accurately predict prognosis and are gradually being used in clinical applications [3]. Therefore, prognostic signatures should be developed urgently to predict the long-term survival of patients with LUAD.
Copper is essential to all living organisms and plays a dual, paradoxical role in the cell, which helps to maintain intracellular copper concentrations at very low levels via a homeostatic mechanism [4]. Copper-induced cell death is mediated by iron-sulfur protein, when copper binds to acylated lipid products in the TCA cycle, leading to lipid acylated protein aggregation and loss of iron-sulfur cluster proteins. This results in proteotoxic stress and ultimately cell death, and this unique form of cell death is called cuproptosis [5]. Previous animal models and clinical trials have shown that copper deficiency inhibits tumor angiogenesis and growth [6]. The imbalance in copper levels can lead to uncontrolled tumor growth [7]. However, the exact mechanism underlying cuproptosis is still not completely elucidated. In hematopoietic cancers, a copper-dependent antitumor agent (IC25 (10)) exerts potent antitumor effects in vivo and in vitro [8]. In breast cancer, the endoplasmic reticulum-targeting copper (II) complex promotes phagocytosis of cancer cells by macrophages [9]. In pancreatic cancer, copper transporter 1 (SLC31A1) and copper chelator tetrathiomolybdate (TM) can promote autophagy and inhibit growth of cancer cells by reducing copper uptake [6]. Therefore, investigating the role of cuproptosis in cancer has great potential for clinical application.
Long non-coding RNAs (lncRNAs) are a class of RNA molecules with transcripts longer than 200 nt. They do not encode proteins but regulate gene expression at multiple epigenetic, transcriptional, and post-transcriptional levels in the form of RNA [10]. Previous in-depth studies on lncRNAs have reported that lncRNAs play a role in important pathologies and physiologies such as autophagy, development, differentiation, apoptosis, and cell cycle [11]. In tumor research, lncRNAs are differentially expressed in the lung, stomach, liver, colon, breast, and pancreatic cancers and play a role in tumor cell proliferation, migration, and invasion [11,12,13,14,15]. Many studies have reported that in colorectal cancer, lncRNA SH3PXD2A-AS1 expression is upregulated and promotes the growth of cancer cells, potentially serving as a diagnostic and therapeutic target [16]. DDX11-AS1 is a prognostic lncRNA with therapeutic potential in hepatocellular carcinoma by analyzing RNA-seq data [17]. In pancreatic cancer, lncRNA reprogramming (lncRNA-ROR) acts as a typical lncRNA with antitumor effects in vivo and in vitro. lncRNA-ROR can also inhibit cancer cell growth by activating miR-145 [15]. Notably, in endometrial cancer, autophagy-related lncRNAs can predict a patient’s prognosis. However, the role of lncRNAs in LUAD still needs to be further studied.
Cuproptosis-related lncRNAs can have clinical diagnostic and therapeutic implications for LUAD. In the present study, we used bioinformatics analysis to obtain cuproptosis-related lncRNAs and to analyze their biological functions and role in predicting the prognosis of patients with LUAD.
Material and methods
Data processing and identification of cuproptosis-related lncRNAs
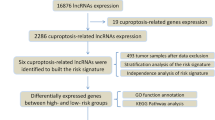
RNA-Seq data of gene expression and the corresponding clinical and mutation data of LUAD were downloaded from the cancer genome atlas (TCGA) database (https://portal.gdc.cancer.gov/), which contained information on a total of 54 normal individuals and 501 patients with LUAD. Three Gene Expression Omnibus (GEO) datasets GSE30219 (n=307), GSE31210 (n=246), and GSE37745 (n=196) were downloaded from the GEO website (https://www.ncbi.nlm.nih.gov/geo/). RNA-Seq data were obtained according to the gene annotations from TCGA to distinguish lncRNAs. To identify potential cuproptosis-related lncRNAs, we performed a co-expression correlation analysis of lncRNA and cuproptosis-related gene expression profiles using the limma package at |R| >0.4 and P < 0.001.
Construction of the prognostic cuproptosis-related lncRNA signature
A list of the cuproptosis-related genes was obtained from cuproptosis-related studies [18,19,20,21,22]. TCGA-LUAD data were randomly divided into training and testing groups in a 1:1 ratio. Univariate Cox analysis was performed in the training group, and Lasso Cox regression was performed on significantly expressed lncRNAs based on a 1000 ten-fold cross-validation to identify cuproptosis-related lncRNAs. Optimal prognostic lncRNAs were identified based on multivariate Cox regression analysis (P < 0.05), and the best model parameters were used for signature construction, followed by calculation of risk scores. Risk score = Exp lncRNA1 × β lncRNA1 + Exp lncRNA2× β lncRNA2 + Exp lncRNA3× β lncRNA3 + ••• + Exp lncRNAn × β lncRNAn.
Analysis of risk signature
Patients were divided into high- and low-risk groups based on median values of risk score to determine the prognosis of signature. We used the survival package to calculate overall survival (OS) and progression-free survival (PFS) for patients with LUAD in various groups and performed univariate and multivariate independent prognostic analyses to evaluate the independent prognostic value of the risk prediction signature. The pheatmap package was used to plot patient survival status and lncRNAs expression heatmap based on the risk scores. The survivalROC package was used to calculate the 1-, 3-, and 5-year area under the ROC curve (AUC) of signature in the training, testing, and all groups.
Construction of nomogram and validation of clinical subgroups
Nomograms were constructed for age, gender, T stage, N stage, TNM stage, and risk score using the survival and rms packages. Calibration curves were plotted to show the difference between the predicted and actual outcomes of the nomogram. C-index curves are used to verify the accuracy of the signature in predicting the survival of patients with LUAD. Finally, patients were divided into stages I–II and stages III–IV to determine if the signature can be used for patients with LUAD at different stages.
Principal Component Analysis (PCA) and functional enrichment analysis
PCA analysis was performed using the limma and scatterplot3d packages to explore the distribution of patients with different risk scores. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of cuproptosis-related lncRNAs was performed using the clusterProfiler package, and P value < 0.05 and false discovery rate (FDR) < 0.05 were considered statistically significant.
Immune-related functional analysis and tumor mutation burden (TMB) analysis
The limma GSVA package was used to analyze differences in immune-related functions in patients with LUAD, and P < 0.05 was considered statistically significant. The pheatmap package was used to visualize the results, and the maftools package was used to compare the relationship between risks core and TMB. We used the survival package to determine the difference between TMB and patient survival. A P value of <0.05 was considered statistically significant.
Immunotherapy analysis and pharmaceutical screening
We downloaded tumor immune dysfunction and exclusion (TIDE) data for non-small cell lung cancer (NSCLC) from http://tide.dfci.harvard.edu/. The TIDE algorithm is a new development that uses TIDE-scores to accurately predict the efficacy of immunotherapy drugs received by patients [23]. Jiang et al. found that TIDE scores have been shown to represent the efficacy of immune drugs (anti-PD1, anti-CTLA4) in melanoma patients, with higher TIDE scores being associated with better outcomes [23]. Using the ggpubr and limma packages, we analyzed the relationship between risk score and TMB, with P < 0.05 considered statistically significant. Screening of therapeutic agents and observation of drug sensitivity using the pRRophetic, ggplot2, and ggpubr packages with pFilter = 0.001 and corPvalue = 0.001.
Validation the prognostic of cuproptosis-related lncRNAs in GEO datasets
Patients were divided into high and low groups based on the expression of cuproptosis-related lncRNAs. We used the survival package for survival analysis of cuproptosis-related lncRNAs in GSE30219, GSE31210, and GSE37745 datasets, with P < 0.05 considered statistically significant.
Results
Identification of cuproptosis-related lncRNAs and construction of a prognostic signature
With the |R| >0.4 and P < 0.001 as the analysis criteria, 2244 cuproptosis-related lncRNAs were identified from 16,876 lncRNAs and 19 cuproptosis-related genes. The co-expression relationships between the cuproptosis-related genes and cuproptosis-related lncRNAs were visualized using a Sankey diagram (Fig. 1). In the training group, Lasso Cox regression analysis identified cuproptosis-related lncRNAs, univariate Cox regression analysis identified 37 lncRNAs, and multivariate COX analysis identified 16 lncRNAs as independent prognostic factors. The risk score of each sample was then calculated based on the expression of 16 lncRNAs (Fig. 2a–c). Risk score =(−0.79670054952258*AC016747.2)+(0.454977942296554*LINC00205)+(0.971038537693743*AC006947.1)+(0.466282143262505*LINC00592)+(0.580816870667386*AC020634.2)+(−0.364069056069612*AC026355.2)+(−2.94320726773469*LINC02848)+(−0.760200155891508*ZNF571-AS1)+(0.429955081610087*CRIM1-DT)+(−1.0655839243752*SEPSECS-AS1)+(0.272457956361295*HIF1A-AS3)+(1.56215847973464*AC013267.1)+(−0.364308918322871*LINC02635)+(1.84725474576194*AL162632.3)+(0.646354490290833*AC004832.5)+(1.01912906171161*AC032011.1). The correlation heatmap also showed the relationship between the cuproptosis-related genes and lncRNAs (Fig. 2d).

Sankey diagram showed the results of cuproptosis-related genes and cuproptosis-related lncRNA co-expression

Identification of the cuproptosis-related lncRNAs. a LASSO regression screened of cuproptosis-related lncRNAs at the minimum point of cross-validation. b Trajectory of each independent variable. c Forest plot showed different lncRNAs for high and low risk, with red representing high-risk lncRNAs and green representing low-risk lncRNAs. d Correlation heatmap showed the relationship between cuproptosis-related lncRNAs and cuproptosis-related genes for the signature. Red represents positive correlations and blue represents negative correlations
Survival analysis of the signature
For a better assessment of the prognostic value of the risk signature based on the median value of the risk score as the cutoff, the patients were divided into high- and low-risk groups. We found that OS and PFS were significantly shorter in the high-risk group than in the low-risk group in the training, testing, and all groups (Fig. 3). The risk curves reflect the relationship between the risk score and survival status of patients with LUAD, and we found that mortality was higher in the high-risk patients than the low-risk patients. The heatmap showed high- and low-risk levels for 16 lncRNAs. For example, LINC00205, LINC00592, and AL162632.3S were high-risk lncRNAs, whereas AC026355.2, LINC02848, and ZNF571-AS1 were low-risk lncRNAs (Fig. 4).

Kaplan–Meier survival analyses of patients. Patients were divided into high-risk and low-risk group based on the median risk score to predict overall survival (OS) and progression-free survival (PFS) in each subgroup. a OS in the training group. b OS in the testing group. c OS in all groups. d PFS in all groups

Predicting the performance of characteristics. Risk curves represented the distributed survival status of LUAD patients with different risk scores; heatmap represented the characteristics of lncRNAs in the a training group, b testing group, and c all groups
Independent analysis of prognostic factors
Univariate and multivariate Cox regression analyses were performed to determine whether the risk signature had the potential to be a prognostic factor independent of other clinical characteristics. Multivariate Cox regression results showed that stage (hazard ratio [HR] = 1.553, 1.342–1.798; P < 0.05) and risk score (HR = 1.028, 1.016–1.040; P < 0.05) were independently associated with OS, indicating that the risk signature is an independent prognostic factor for patients with LUAD (Fig. 5a, b). Further, we used ROC curves to assess the predictive accuracy of the risk score. The AUC for the risk score was 0.756, which was better than those for age (0.536), gender (0.596), and stage (0.712) (Fig. 5f). Similarly, in the all group, the AUCs for 1-, 3-, and 5-year OS were 0.756, 0.739, and 0.759, respectively (Fig. 5e), suggesting that the signature has reliable diagnostic significance. The ROC curves also showed that the signature could predict OS in the training and testing groups (Fig. 5c, d).

The prognostic value of the signature for LUAD. a Univariate and b multivariate independent prognostic analysis to analyze that the risk score was independently associated with OS. 1-, 3-, and 5-year area under the ROC curve (AUC) of signature in the c training, d testing, and e all groups. f ROC curves for the riskscore (AUC=0.756) and other clinical features
Construction of a predictive nomogram and PCA
We constructed a nomogram using factors such as age, gender, TNM stage, T stage, risk score, and N stage from the signature (the M stage showed several uncertain values, which were not included in this study). The nomogram could reliably predict the 1-, 3-, and 5-year OS of patients with LUAD (Fig. 6a, b). Furthermore, we found that the C-index values of the risk score were higher than those of other clinical characteristics, such as age, gender, and stage (Fig. 6c). We further analyzed the significant differences (P < 0.05) in OS between the high-risk and low-risk groups of patients at different stages (stages I–II and III–IV) (Fig. 6d, e), which suggested that the signature has high predictive accuracy and can be used to compare the survival of patients at different stages. Finally, we performed PCA to observe the distribution of patients for all genes, cuproptosis-related genes, cuproptosis-related lncRNAs, and risk lncRNAs, and the results showed a clear distribution, indicating that these lncRNAs can be reliably used to construct the signature (Fig. 7).

Nomogram and clinical subgroups for predicting LUAD outcomes. a Prognostic nomogram to predict the OS in LUAD. b Calibration curves for 1, 3, and 5 years. c C-Index curve analyzed the concordance index of the risk score. Patients were grouped to see if the model was applicable to LUAD patients at d stages I–II and e stages III–IV

PCA analysis. PCA analysis observed the distribution of patients according to a all genes. b Genes associated with cupulocytosis. c lncRNAs associated with cupulosis. d Risk lncRNAs
Functional enrichment analysis and immune-related functional analysis
GO analysis results showed that the cuproptosis-related lncRNAs enriched in the negative regulation of proteolysis, regulation of peptidase activity, and negative regulation of hydrolase activity (Fig. 8a). KEGG analysis showed that these cuproptosis-related lncRNAs may be related to the cytokine−cytokine receptor interaction, neutrophil extracellular trap formation, and mitogen-activated protein kinase signaling pathways, suggesting that these lncRNAs are involved in tumor development (Fig. 8b). Furthermore, we analyzed immune-related functions to evaluate the immune status of the low-risk and high-risk groups, and the results showed that the type III interferon (IFN) response was significantly more active in the low-risk group than in the high-risk group, but no significant differences were noted for other immune functions (P > 0.05) (Fig. 8c).

Functional enrichment analysis and immune-related functional analysis. a GO enrichment analyses of the 16 cuproptosis-related lncRNAs. b KEGG enrichment analyses of the 16 cuproptosis-related lncRNAs. c Immune-related functions of the 16 cuproptosis-related lncRNAs
TMB analysis and drug sensitivity analysis
We used the maftools algorithm to observe mutations in the high- and low-risk groups and showed that for most genes, the frequency of mutations was higher in the high-risk group than in the low-risk group (TP53: low risk, 43%; high risk 52%. TTN: low risk, 44%; high risk, 47%; MUC16: low risk, 38%; high risk, 42%) (Fig. 9a). Furthermore, the difference in TMB between the high-risk and low-risk groups was not significant (P = 0.84) (Fig. 9b). The reason behind this result needs to be further explored. We further investigated a probable difference in survival between patients with high and low TMB. As shown in Fig. 9c, OS was significantly better in the high TMB group than in the low TMB group (P < 0.05). In addition, the difference in sensitivity to immunotherapy between patients in the high- and low-risk groups was further investigated using the TIDE algorithm (http://tide.dfci.harvard.edu/). The TIDE algorithm is a recently developed tool for determining the efficacy of tumor immune checkpoint therapy [23]. In the present study, we found a higher TIDE score in the low-risk group than in the high-risk group (Fig. 10a). However, whether immunotherapy is better for low-risk patients with LUAD than high-risk patients with LUAD needs to be further explored. We used the pRRophetic packages to screen the potentially effective antitumor drugs, including masitinib, tipifarnib, bexarotene, 5-fluorouracil, midostaurin, vinorelbine, etoposide, and doxorubicin (Table 1). We further analyzed the sensitivity of these drugs and found that patients in the high-risk group had lower IC50 values (concentration that inhibits cell growth by 50%), representing a higher sensitivity of the drugs in patients at high risk (Fig. 10b–i).

The relationship between TMB and the signature. a Waterfall plot revealed the top 15 mutation genes in LUAD for the low-risk (221 samples) and high-risk (240 samples) groups. b Differential TMB in high-risk and low-risk groups in LUAD. c Survival curves for the high-TMB and low-TMB groups in LUAD and a combined TMB-risk survival curve

Immunotherapy and drug sensitivity. a Tumor immune dysfunction and exclusion (TIDE) algorithm analysis for high-risk and low-risk groups. *Represents statistically significant differences. Observed the drug sensitivity of b CX-5461, c Tipifarnib, d Bexarotene, e 5-Fluorouracil, f Midostaurin, g Vinorelbine, h Etoposide, and i Doxorubicin
Prognostic analysis of the GEO datasets
Among the 16 Cuproptosis-related lncRNAs (AC016747.2, LINC00205, AC006947.1, LINC00592, AC020634.2, AC026355.2, LINC02848, ZNF571-AS1, CRIM1-DT, SEPSECS-AS1, HIF1A-AS3, AC013267.1, LINC02635, AL162632.3, AC004832.5, AC032011.1) in GSE30219, GSE31210, GSE37745, only LINC00592, ZNF571-AS1, and SEPSECS-AS1 was observed, and the remaining 13 cuproptosis-related lncRNAs had not been observed. Then, we performed the survival analysis of single cuproptosis-related lncRNAs in GSE30219, GSE31210, and GSE37745 for LINC00592, ZNF571-AS1, and SEPSECS-AS1, respectively, and the results are shown in Fig. 11.

Prognostic validation of single cuproptosis-related lncRNAs in the GEO datasets. a LINC00592, b SEPSECS-AS1, and c ZNF571-AS1 in GSE30219. d LINC00592, e SEPSECS-AS1, and f ZNF571-AS1 in GSE31210. g LINC00592, h SEPSECS-AS1, and i ZNF571-AS1 in GSE37745
Discussion
Lung cancer has the highest incidence and mortality rate among malignant tumors in China, and LUAD, a type of lung cancer, has a very poor prognosis [24]. Currently available lung cancer screening tools, such as low-dose computed tomography, are effective in reducing lung cancer mortality but have a high false-positive rate. Therefore, the construction of a reliable lung cancer risk signature to accurately determine the prognosis and survival of patients with LUAD is of great importance in the prevention and control of LUAD [25]. lncRNAs are a class of non-protein-coding RNAs that are >200 nt in length and account for more than 80% of lncRNAs [26]. lncRNAs play an important regulatory role in lung cancer. A study found that lncRNA KTN1-AS1 acts as a pro-oncogene in NSCLC and can affect the NSCLC cell cycle by regulating cyclin-dependent kinase (CDK) 1, suggesting that lncRNAs may act as a novel lung cancer biomarker and therapeutic target [27]. Cuproptosis is a unique form of cell death that has been identified recently [18,19,20,21,22]. This copper-dependent cell death occurs via the direct binding of copper to acylated lipid components of the TCA cycle in mitochondrial respiration, leading to the aggregation of acylated proteins and the subsequent decrease in iron-sulfur proteins, resulting in proteotoxicity and ultimately cell death [28]. Although clinical trials of the copper ionophore micromolecule anticancer drug elesclomol [29] have been conducted, their results have been unsatisfactory. However, the co-regulatory role of cuproptosis and lncRNAs in LUAD needs to be further investigated.
In the present study, cuproptosis-related lncRNAs were obtained by the co-expression of lncRNAs and cuproptosis-related genes. A total of 16 prognostic cuproptosis-related lncRNAs, including including AC016747.2, LINC00205, AC006947.1, LINC00592, AC020634.2, AC026355.2, LINC02848, ZNF571-AS1, CRIM1-DT, SEPSECS-AS1, HIF1A-AS3, AC013267.1, LINC02635, AL162632.3, AC004832.5, and AC032011.1 were obtained by univariate and multivariate Cox regression analyses, and the prognostic signature was constructed. The results of ROC, survival, nomogram, and pheatmap analyses showed that the prognostic features of the 16 cuproptosis-related lncRNAs were accurately distinguished between the high-risk and low-risk groups and early-stage and late-stage patients, reliably predicting outcomes in patients with LUAD, and these lncRNAs were prognostic factors independent of other common clinical characteristics. Of the 16 cuproptosis-related lncRNAs in the LUAD signature, only LINC00205 has been shown to function in cancer. LINC00205 was an overexpressed oncogene involved in tumor progression in lung, liver, gastric cancers, and retinoblastoma [30,31,32,33,34,35]. In hepatocellular carcinoma, LINC00592 was associated with patient prognosis and could target the cold shock domain-containing E1 protein, CDK6, miR-122-5p, and epoxide hydrolase 1, promoting the proliferation of hepatocellular carcinoma cells [30,31,32,33]. Li et al. found that LINC00205 targets the oncogene miR-185-5p as a potential therapeutic target in lung and gastric cancers. LINC00205 promoted the proliferation and migration of gastric cancer cells by inhibiting miR-26a [36]. Zhang et al. suggested that LINC00205 targeting the high mobility group box 1 protein promoted the proliferation of neuroblastoma, which was correlated with OS [34]. The roles of other 15 lncRNAs were first investigated in cancers. Further, the GO and KEGG pathway analyses revealed that cuproptosis-related lncRNAs may be associated with LUAD development.
We then analyzed the relationship between immune-related function, TMB, and risk scores in patients with LUAD and found that type III-IFN response was inhibited in high-risk patients. IFNs interfere with virus replication in vitro, and type III-IFNs are an essential component of antiviral immunity [37]. Type III-IFN-response inhibition can be one of the main causes of immune escape, and its activation is essential to maintain immune potency. TMB is often used as a predictive biomarker for an immune checkpoint blockade in melanoma, lung, and bladder cancers [38,39,40]. We found a significant decrease in the survival of patients with high TMB (P < 0.05) and an increase in TP53 and TTN expression in patients at high risk. TP53 is a frequently mutated oncogene in human cancers, affecting the development of breast, lung, bladder, esophageal, prostate, pancreatic, and colorectal cancers and is involved in the normal physiology and metabolism of diabetes, liver, and cardiovascular diseases [40,41,42,43,44]. TP53 is also associated with the survival of patients with multiple cancers [45]. TTN participates in the development of multiple cancers. Shen et al. found that TTN can target the miR-376a-3p/PUM2 axis and promote the growth of endometrial cancer cells, suggesting that TTN may be a therapeutic target for endometrial cancer [46]. Fu et al. found that TTN acts as a pro-oncogene in osteosarcoma by targeting miR-134-5p and promoting the expression of the malignant brain tumor domain-containing 1 gene, which ultimately promotes the growth of osteosarcoma cells [47]. Xiao et al. found that in bladder cancer, downregulated TTN expression inhibited the proliferative capacity of breast cancer cells [47]. Our results are consistent with those of previous studies [40,41,42,43,44,45,46,47,48], suggesting that TP53 and TTN can act as targets for cancer immunotherapy. In the present study, we found that the TIDE score in the low-risk group was higher than that in the high-risk group; however, the difference in the effect of immunotherapy on high-risk patients with LUAD and low-risk patients with LUAD still needs to be further explored. PD-L1/PD-1, CD24/Siglec-10, epithelial-mesenchymal transition signaling, hypoxia-/hypoxia-inducible factor 1α drivers, and CCAAT/enhancer-binding protein β transcription factors affect immunotherapy, a recently prevalent oncology treatment [49,50,51,52,53]. Notably, the role of ncRNAs in immunotherapy has been widely researched. Zhou et al. found that miR-1468-5p could suppress the immune system by targeting lymphatic PD-L1, causing cervical cancer cells to evade treatment [54]. In neuroblastoma, the oncogenic factor miR-186 attenuates the efficacy of immunotherapy by inhibiting natural killer cells [55]. Zhang et al. identified lncRNA GATA3-AS1 as a potential therapeutic target for breast cancer, promoting immune escape by targeting GATA3 [56]. However, the relationship between lncRNAs and immune-related functions is not fully understood, and in the future, we will evaluate the prognosis of patients with lung cancer regarding immune cell infiltration and explore the role of immune cells in targeted therapy for patients with LUAD. We then used the pRRophetic algorithm to screen for effective drugs for tumor immunotherapy and explored the sensitivity of these drugs, which have been used in lung, lymphoma, pancreatic, breast, kidney, and bile duct cancers, followed by the observation that patients at high risk are more sensitive to anticancer drugs. However, the drug mechanisms and their impact on LUAD progression need further study. Despite the prognostic value of the signature, this study still has some limitations. First, the prognostic results of cuproptosis-related lncRNAs in the GEO dataset were less than favorable. Second, our findings are based on public databases and lack available information on clinical specimens. Moreover, we will continue to focus on and improve this limitation in the future.
Conclusion
In the present study, we constructed a cuproptosis-related lncRNA signature for LUAD and analyzed the relationship between risk score-based groups and TMB, immunotherapy, and drug sensitivity. This study provides new insights into the prediction of survival of patients with LUAD and the efficacy of clinical treatment.
Availability of data and materials
Data supporting the findings of this study are available from the respective authors upon reasonable request.
References
Hamann HA, Ver Hoeve ES, Carter-Harris L, Studts JL, Ostroff JS. Multilevel opportunities to address lung cancer stigma across the cancer control continuum. J Thorac Oncol. 2018;13:1062–75.
Subramanian J, Govindan R. Molecular genetics of lung cancer in people who have never smoked. Lancet Oncol. 2008;9:676–82.
Senan S, Paul MA, Lagerwaard FJ. Treatment of early-stage lung cancer detected by screening: surgery or stereotactic ablative radiotherapy? Lancet Oncol. 2013;14:e270–4.
Williams DM. Copper deficiency in humans. Semin Hematol. 1983;20:118–28.
Chang W-H, Lin H-H, Tsai I-K, Huang S-H, Chung S-C, Tu I-P, et al. Copper centers in the Cryo-EM structure of particulate methane monooxygenase reveal the catalytic machinery of methane oxidation. J Am Chem Soc. 2021;143:9922–32.
Yu Z, Zhou R, Zhao Y, Pan Y, Liang H, Zhang J-S, et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif. 2019;52:e12568.
Jiang Y, Huo Z, Qi X, Zuo T, Wu Z. Copper-induced tumor cell death mechanisms and antitumor theragnostic applications of copper complexes. Nanomedicine (London). 2022;17:303–24.
Chen G, Niu C, Yi J, Sun L, Cao H, Fang Y, et al. Novel triapine derivative induces copper-dependent cell death in hematopoietic cancers. J Med Chem. 2019;62:3107–21.
Kaur P, Johnson A, Northcote-Smith J, Lu C, Suntharalingam K. Immunogenic cell death of breast cancer stem cells induced by an endoplasmic reticulum-targeting copper(II) complex. Chembiochem. 2020;21:3618–24.
Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet. 2009;10:155–9.
Meryet-Figuière M, Lambert B, Gauduchon P, Vigneron N, Brotin E, Poulain L, et al. An overview of long non-coding RNAs in ovarian cancers. Oncotarget. 2016;7:44719–34.
Wang L, Chen Z, An L, Wang Y, Zhang Z, Guo Y, et al. Analysis of long non-coding RNA expression profiles in non-small cell lung cancer. Cell Physiol Biochem. 2016;38:2389–400.
Yuan J, Yue H, Zhang M, Luo J, Liu L, Wu W, et al. Transcriptional profiling analysis and functional prediction of long noncoding RNAs in cancer. Oncotarget. 2016;7:8131–42.
Prensner JR, Sahu A, Iyer MK, Malik R, Chandler B, Asangani IA, et al. The lncRNAs PCGEM1 and PRNCR1 are not implicated in castration resistant prostate cancer. Oncotarget. 2014;5:1434–8.
Gao S, Wang P, Hua Y, Xi H, Meng Z, Liu T, et al. ROR functions as a ceRNA to regulate Nanog expression by sponging miR-145 and predicts poor prognosis in pancreatic cancer. Oncotarget. 2016;7:1608–18.
Ma Z, Peng P, Zhou J, Hui B, Ji H, Wang J, et al. Long non-coding RNA SH3PXD2A-AS1 promotes cell progression partly through epigenetic silencing P57 and KLF2 in colorectal cancer. Cell Physiol Biochem. 2018;46:2197–214.
Shi M, Zhang X-Y, Yu H, Xiang S-H, Xu L, Wei J, et al. DDX11-AS1 as potential therapy targets for human hepatocellular carcinoma. Oncotarget. 2017;8:44195–202.
Polishchuk EV, Merolla A, Lichtmannegger J, Romano A, Indrieri A, Ilyechova EY, et al. Activation of autophagy, observed in liver tissues from patients with Wilson disease and from ATP7B-deficient animals, protects hepatocytes from copper-induced apoptosis. Gastroenterology. 2019;156:1173–1189.e5.
Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J, et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat Commun. 2020;11:3701.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Dong J, Wang X, Xu C, Gao M, Wang S, Zhang J, et al. Inhibiting NLRP3 inflammasome activation prevents copper-induced neuropathology in a murine model of Wilson’s disease. Cell Death Dis. 2021;12:87.
Ren X, Li Y, Zhou Y, Hu W, Yang C, Jing Q, et al. Overcoming the compensatory elevation of NRF2 renders hepatocellular carcinoma cells more vulnerable to disulfiram/copper-induced ferroptosis. Redox Biol. 2021;46:102122.
Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med. 2018;24:1550–8.
Neal JW, Gainor JF, Shaw AT. Developing biomarker-specific end points in lung cancer clinical trials. Nat Rev Clin Oncol. 2015;12:135–46.
Duncan MW. Place for biochemical markers in early-stage lung cancer detection? J Clin Oncol. 2009;27:2749–50.
Deng Z, Li X, Shi Y, Lu Y, Yao W, Wang J. A novel autophagy-related lncRNAs signature for prognostic prediction and clinical value in patients with pancreatic cancer. Front Cell Dev Biol. 2020;8:606817.
Wu F, Wang L, Xu L, Song S, Liang M. lncRNA KTN1-AS1 silencing inhibits non-small-cell lung cancer cell proliferation and KTN1-AS1 expression predicts survival. Crit Rev Eukaryot Gene Expr. 2022;32:39–46.
Koh E-I, Robinson AE, Bandara N, Rogers BE, Henderson JP. Copper import in Escherichia coli by the yersiniabactin metallophore system. Nat Chem Biol. 2017;13:1016–21.
Soma S, Latimer AJ, Chun H, Vicary AC, Timbalia SA, Boulet A, et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc Natl Acad Sci U S A. 2018;115:8161–6.
Xie P, Guo Y. LINC00205 promotes malignancy in lung cancer by recruiting FUS and stabilizing CSDE1. Biosci Rep. 2020;40:BSR20190701.
Cheng T, Yao Y, Zhang S, Zhang X-N, Zhang A-H, Yang W, et al. LINC00205, a YY1-modulated lncRNA, serves as a sponge for miR-26a-5p facilitating the proliferation of hepatocellular carcinoma cells by elevating CDK6. Eur Rev Med Pharmacol Sci. 2021;25:6208–19.
Zhang L, Wang Y, Sun J, Ma H, Guo C. LINC00205 promotes proliferation, migration and invasion of HCC cells by targeting miR-122-5p. Pathol Res Pract. 2019;215:152515.
Long X, Li Q, Zhi L-J, Li J-M, Wang Z-Y. LINC00205 modulates the expression of EPHX1 through the inhibition of miR-184 in hepatocellular carcinoma as a ceRNA. J Cell Physiol. 2020;235:3013–21.
Zhang S, Long J, Hu Y. Long noncoding RNA LINC00205 enhances the malignant characteristics of retinoblastoma by acting as a molecular sponge of microRNA-665 and consequently increasing HMGB1 expression. Biochem Biophys Res Commun. 2020;526:396–403.
Li Y, Hu Y, Wu Y, Zhang D, Huang D. LINC00205 promotes tumor malignancy of lung adenocarcinoma through sponging miR-185-5p. Lab Med. 2022;53:39–46.
Huangfu L, Fan B, Wang G, Gan X, Tian S, He Q, et al. Novel prognostic marker LINC00205 promotes tumorigenesis and metastasis by competitively suppressing miRNA-26a in gastric cancer. Cell Death Dis. 2022;8:5.
Zhang S-Y, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, et al. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40.
Jardim DL, Goodman A, de Melo GD, Kurzrock R. The challenges of tumor mutational burden as an immunotherapy biomarker. Cancer Cell. 2021;39:154–73.
Fusco MJ, West HJ, Walko CM. Tumor mutation burden and cancer treatment. JAMA Oncol. 2021;7:316.
McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32:661–72.
Mogi A, Kuwano H. TP53 mutations in nonsmall cell lung cancer. J Biomed Biotechnol. 2011;2011:583929.
Siracusano S, Rizzetto R, Porcaro AB. Bladder cancer genomics. Urologia. 2020;87:49–56.
Liu Z, Guo H, Zhu Y, Xia Y, Cui J, Shi K, et al. TP53 alterations of hormone-naïve prostate cancer in the Chinese population. Prostate Cancer Prostatic Dis. 2021;24:482–91.
Du L, Kim JJ, Shen J, Chen B, Dai N. KRAS and TP53 mutations in inflammatory bowel disease-associated colorectal cancer: a meta-analysis. Oncotarget. 2017;8:22175–86.
Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019;28:1370–84 e5.
Shen L, Wu Y, Li A, Li L, Shen L, Jiang Q, et al. lncRNA TTN-AS1 promotes endometrial cancer by sponging miR-376a-3p. Oncol Rep. 2020;44:1343–54.
Fu D, Lu C, Qu X, Li P, Chen K, Shan L, et al. lncRNA TTN-AS1 regulates osteosarcoma cell apoptosis and drug resistance via the miR-134-5p/MBTD1 axis. Aging (Albany NY). 2019;11:8374–85.
Lacroix M, Riscal R, Arena G, Linares LK, Le Cam L. Metabolic functions of the tumor suppressor p53: implications in normal physiology, metabolic disorders, and cancer. Mol Metab. 2020;33:2–22.
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18:10.
Yin S-S, Gao F-H. Molecular mechanism of tumor cell immune escape mediated by CD24/Siglec-10. Front Immunol. 2020;11:1324.
Jiang Y, Zhan H. Communication between EMT and PD-L1 signaling: new insights into tumor immune evasion. Cancer Lett. 2020;468:72–81.
Vaupel P, Multhoff G. Hypoxia-/HIF-1α-driven factors of the tumor microenvironment impeding antitumor immune responses and promoting malignant progression. Adv Exp Med Biol. 2018;1072:171–5.
Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, et al. Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32:790–802.
Zhou C, Wei W, Ma J, Yang Y, Liang L, Zhang Y, et al. Cancer-secreted exosomal miR-1468-5p promotes tumor immune escape via the immunosuppressive reprogramming of lymphatic vessels. Mol Ther. 2021;29:1512–28.
Neviani P, Wise PM, Murtadha M, Liu CW, Wu C-H, Jong AY, et al. Natural killer-derived exosomal miR-186 inhibits neuroblastoma growth and immune escape mechanisms. Cancer Res. 2019;79:1151–64.
Zhang M, Wang N, Song P, Fu Y, Ren Y, Li Z, et al. lncRNA GATA3-AS1 facilitates tumour progression and immune escape in triple-negative breast cancer through destabilization of GATA3 but stabilization of PD-L1. Cell Prolif. 2020;53:e12855.
Acknowledgements
We are grateful to the TCGA database for providing the platform and to the contributors for uploading their meaningful datasets. We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript.
Funding
This research was supported by Guangxi Natural Science Foundation under Grant NO. 2020GXNSFDA238003. This work was done at The First Affiliated Hospital of Guangxi Medical University.
Author information
Authors and Affiliations
Contributions
WF Wang and CQ Li designed the implementation of the research, drafted the preliminary papers, and participated in the investigations. WF Wang, QS Su, and LH Sheng participated in the research design and implementation, manuscript revision, manuscript submission, and fund acquisition. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, F., Lin, H., Su, Q. et al. Cuproptosis-related lncRNA predict prognosis and immune response of lung adenocarcinoma. World J Surg Onc 20, 275 (2022). https://doi.org/10.1186/s12957-022-02727-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-022-02727-7