
Abstract
Background
Traumatic brain injury (TBI) causes neuroinflammation and can lead to long-term neurological dysfunction, even in cases of mild TBI (mTBI). Despite the substantial burden of this disease, the management of TBI is precluded by an incomplete understanding of its cellular mechanisms. Sphingolipids (SPL) and their metabolites have emerged as key orchestrators of biological processes related to tissue injury, neuroinflammation, and inflammation resolution. No study so far has investigated comprehensive sphingolipid profile changes immediately following TBI in animal models or human cases. In this study, sphingolipid metabolite composition was examined during the acute phases in brain tissue and plasma of mice following mTBI.
Methods
Wildtype mice were exposed to air-blast-mediated mTBI, with blast exposure set at 50-psi on the left cranium and 0-psi designated as Sham. Sphingolipid profile was analyzed in brain tissue and plasma during the acute phases of 1, 3, and 7 days post-TBI via liquid-chromatography-mass spectrometry. Simultaneously, gene expression of sphingolipid metabolic markers within brain tissue was analyzed using quantitative reverse transcription-polymerase chain reaction. Significance (P-values) was determined by non-parametric t-test (Mann–Whitney test) and by Tukey’s correction for multiple comparisons.
Results
In post-TBI brain tissue, there was a significant elevation of 1) acid sphingomyelinase (aSMase) at 1- and 3-days, 2) neutral sphingomyelinase (nSMase) at 7-days, 3) ceramide-1-phosphate levels at 1 day, and 4) monohexosylceramide (MHC) and sphingosine at 7-days. Among individual species, the study found an increase in C18:0 and a decrease in C24:1 ceramides (Cer) at 1 day; an increase in C20:0 MHC at 3 days; decrease in MHC C18:0 and increase in MHC C24:1, sphingomyelins (SM) C18:0, and C24:0 at 7 days. Moreover, many sphingolipid metabolic genes were elevated at 1 day, followed by a reduction at 3 days and an absence at 7-days post-TBI. In post-TBI plasma, there was 1) a significant reduction in Cer and MHC C22:0, and an increase in MHC C16:0 at 1 day; 2) a very significant increase in long-chain Cer C24:1 accompanied by significant decreases in Cer C24:0 and C22:0 in MHC and SM at 3 days; and 3) a significant increase of C22:0 in all classes of SPL (Cer, MHC and SM) as well as a decrease in Cer C24:1, MHC C24:1 and MHC C24:0 at 7 days.
Conclusions
Alterations in sphingolipid metabolite composition, particularly sphingomyelinases and short-chain ceramides, may contribute to the induction and regulation of neuroinflammatory events in the early stages of TBI, suggesting potential targets for novel diagnostic, prognostic, and therapeutic strategies in the future.
Similar content being viewed by others
Introduction
Traumatic brain injury (TBI) is a leading cause of death and disability worldwide, impacting approximately 69 million individuals annually [1]. Among those who survive a TBI, consequences span a spectrum of several short and long-term impacts, including cognitive impairment, sensory abnormalities in hearing and sight, onset of psychiatric disorders, and increased susceptibility to neurodegenerative disorders such as Parkinson’s disease and Alzheimer’s disease [2,3,4,5,6]. Despite the substantial burden of this disease, diagnosis and treatment of TBI are limited by an incomplete understanding of its pathophysiology [7,8,9]. TBI involves structural damage to the brain parenchyma and vasculature occurring secondary to mechanical forces such as blunt impacts, rapid accelerations, or explosive blasts, resulting in acute neurological dysfunction, which may lead to permanent disability or death [10,11,12,13]. Transmission of these forces through solid tissue-liquid interfaces and areas of high tissue density variation between gray and white matter can cause microvascular compromise and diffuse axonal injury, contributing to acute neurological dysfunction in the acute injury phase of TBI. Secondary brain injury may occur due to subsequent disruption of brain homeostatic mechanisms, activation of inflammatory cascades, disruption of cerebral blood flow, excitotoxicity, and cerebral metabolic dysfunction [8, 9, 13, 14]. The persistent activation of secondary neuroinflammatory pathways may cause chronic, progressive neurodegeneration and neurological dysfunction in the long term, even in cases of mild TBI [15,16,17,18,19].
In 1974, the Glasgow Coma Scale (GCS) was designed as an injury severity score to predict outcomes following TBI [20]. Evaluating motor, eye, and verbal responses, the GCS is one of the most widely used clinical tools to assess the extent of impaired consciousness [21]. However, the GCS demonstrates limited predictive value of outcomes in patients with mild-to-moderately severe TBI [22, 23]. Moreover, while imaging modalities such as magnetic resonance imaging (MRI) and computer tomography (CT) are frequently employed to provide clinicians with more objective information, these also often lack sensitivity in detecting mild-to-moderate brain injury [24]. Thus, there is a strong clinical need to develop biomarkers with high specificity and sensitivity to enhance the precision of diagnosing and predicting outcomes in TBI. Over the last decade, this need has prompted research to identify a diverse range of biomarkers [25,26,27,28]. Among these biomarkers, there is a relatively limited representation of lipids, although lipids are highly abundant in the brain, constituting more than 50% of its dry weight [29].
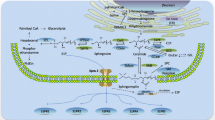
Lipids are critical contributors to both physiology and pathology in the central nervous system (CNS) [30,31,32,33]. They serve as structural components of cell membranes, function as energy storehouses, and act as important signaling molecules [34]. Given the relatively high concentrations of bioactive lipids, lipid precursors, and metabolites within the CNS, tight regulation of lipid metabolism is crucial for maintaining homeostasis in the brain and reacting to injury or other insults [35]. Sphingolipids (SPL), a significant component of CNS lipids, have emerged as integral mediators of biological processes relating to tissue injury, neuroinflammation, and resolution of inflammation [36]. Though a relatively minor component of total cellular lipids, sphingolipids have emerged as one of the most pathogenically associated lipids, and abnormal sphingolipid metabolism has been linked with numerous neurodegenerative and psychiatric diseases in the literature [37,38,39,40,41,42,43,44]. Recent advances in lipidomic technology have enabled the measurement of individual sphingolipid species within the brain, blood, and cerebrospinal fluid with high accuracy [45,46,47].
Plasma SPL has been investigated as a biomarker for neurodegeneration, neuroinflammation, and psychiatric diseases; cardiovascular and coronary diseases; in various types of cancers; and for type 1 and type 2 diabetes [37,38,39,40,41,42,43,44]. In addition, SPL markers are being developed as a next-generation “cholesterol” for human cardiac diseases, and testing of plasma ceramides has begun being utilized as a diagnostic marker for cardiac myopathy (MI-HEART ceramides) by the Mayo Clinic. Alterations in plasma SPL profiles have been documented in models of TBI and cerebral ischemia [48,49,50]. Over the past decade, our research team has developed a mouse model of TBI, thoroughly characterizing its pathology to closely mirror mild TBI in humans [51]. Recent studies revealed significant elevations in ceramide levels in the brains of mice one month following exposure to blast-induced TBI [52]. Findings such as these lend support to the theory that regulation of SPL metabolism and signaling may be critical pathways of TBI pathophysiology. However, their extreme diversity in structure, metabolism, and function necessitates a deeper understanding of the association between specific sphingolipid profiles with specific disease pathology. Currently, there is little information regarding changes in SPL profiles in the timeframe immediately following exposure to TBI. The present study adds novel information to the literature by analyzing the SPL profiles in both the brains and plasma of mice exposed to mild TBI, activity of major SPL metabolic enzymes, and gene expression of SPL enzymes at 1, 3, and 7 days post-TBI.
Materials and methods
Animals and TBI methods
All animal handling protocols and experiments were performed according to the rules and regulations of the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, the National Institutes of Health (NIH), the Society for Neuroscience, and the University of Tennessee Health Science Center Guidelines for Animals in Research. Euthanasia, tissue harvesting, and other protocols were approved by the UTHSC Institutional Animal Care and Use Committee (UTHSC IACUC). C57BL/6 J mice, used as wild type (WT) control mice were born and raised in the UTHSC LACU (Laboratory Animal Care Unit) vivarium and maintained from birth under cyclic light (50–100 lx, 12 h. on/off, 7 a.m. to 7 p.m. CST). The parents and the offspring were on a regular mouse chow diet.
All the experimental mice were exposed to an air blast to generate the mild TBI as per earlier published protocol [51, 53]. A modified paintball gun was used by a small horizontally mounted air canon system to generate the mild TBI (Invert Mini, Empire Paintball, Sewell, New Jersey, USA). A 0-psi blast was referred to as “sham blast,” and all experimental outcomes were compared between sham and 50-psi blasts. Mice were anesthetized with Avertin (400 mg/kg body weight) and exposed to the blast on the left side of the cranium between ear and eye. Before the blast, the targeted head region was shaved, and a white dot was painted in the middle of the region. A foam rubber sleeve was used to place the animal inside a Plexiglass tube in such a way the targeted head region was positioned in the center of a 7.5 mm diameter hole of the tube, facing the blast cannon barrel tip. The system is arranged to expose the parietal region of the mouse head between the ear and eye to be blasted with specific air pressure, which is set by the transducer and analyzed by a LabVIEW software (National Instruments, Austin, Texas, USA).
At the same h post-TBI (day 1, day 3, or day 7), the mice were sacrificed, and the brain tissue was immediately harvested and snap-frozen in liquid nitrogen and stored at -80 °C. The frozen brains were thawed in ice and homogenized with ice-cold RNAse-free phosphate buffer, fractionated in five parts, snap-frozen in liquid N2, and stored back to -80 °C until used for analysis. Whole blood was immediately collected in EDTA-containing (anti-coagulant) microcentrifuge tubes and placed on ice. The blood sample was then centrifuged at 4 °C within 1 h of collection, and the plasma was isolated from the particulate matter and snap-frozen in liquid N2 and stored at -80 °C until analysis.
Sphingomyelinase assay
The entire brain of a mouse was homogenized in RNAse-free phosphate buffer and divided into 5 fractions. One of the fractions was then used to isolate proteins and quantify them using a bicinchoninic acid assay. Neutral and Acidic sphingomyelinase (nSMase, and aSMase, respectively) activity was measured from isolated protein samples from mouse brain tissue of Sham and Blast-exposed mice harvested a 1, 3 and 7 days post-TBI by using Amplex Red Sphingomyelinase Assay kit (Thermo Fisher, Waltham, Massachusetts, USA) as previously described [54].
Sphingolipids analysis: sphingolipid preparation
Plasma (50 uL) was extracted using a modified Bligh and Dyer Extraction as previously described [55,56,57,58,59,60,61]. Samples were spiked with 250 pmol of C1P, sphingomyelin, ceramide, and monohexosyl ceramide (d18:1/12:0 species), and sphingansine (So), sphinganine (Sa), sphingosine-1-phosphate (S1P), sphinganine-1-phosphate (Sa1P) (d17:0 sphinganine/d17:1 sphingosine) as internal standard (Avanti Polar Lipids, Alabaster, Alabama, USA). Following addition of IS, 3 mL of MeOH:CHCl3 (2:1) was added to the plasma and the mixture was sonicated to disperse plasma clumps. Samples were then incubated for 6 h at 48 °C. Extracts were then centrifuged at 5000 rpm for 20 min, transferred to a new glass tube, dried down and reconstituted in methanol (500 uL) by sonicating. Extracts were again centrifuged at 5000 rpm for 20 min and transferred to injection vials for mass spec analysis. Tissue samples (ranging from 4.6–48.5 mg in weight) were extracted by transferring sample tissue to tubes with ceramic beads and a 1 mL solution of 50:50 Methanol:Water with 250 pmol of the above described sphingolipid standard. Samples were then homogenized via Bead Rupter Elite (Omni International, Miami, Florida, USA) for 5 min per samples. The entire homogenate was transferred to glass borosilicate tubes and 3 mL of MeOH:CHCl3 (2:1) was added to the tissue. The rest of the extraction was carried identically to the plasma as described previously and final tissue concentrations were normalized by their weights.
Liquid chromatography/mass spectrometry
Sphingolipids were separated using a Shimadzu Nexera X2 LC-30AD coupled to a SIL-30AC auto injector, coupled to a DGU-20A5R degassing unit in the following way. An 8 min, reversed phase LC method utilizing an Acentis Express C18 column (5 cm × 2.1 mm, 2.7 µm) was used to separate the eicosanoids at a 0.5 mL/min flow rate at 60 °C. The column was equilibrated with 100% Solvent A [methanol:water:formic acid (58:44:1, v/v/v) with 5 mM ammonium formate] for 5 min and then 10 µl of sample was injected. 100% Solvent A was used for the first 0.5 min of elution. Solvent B [methanol:formic acid (99:1, v/v) with 5 mM ammonium formate] was increased in a linear gradient to 100% Solvent B from 0.5 min to 3.5 min. Solvent B was held constant at 100% from 3.5 min to 6 min. From 6 min to 6.1 min solvent B was reduced to 0%, and solvent A returned to 100%. Solvent A was held constant at 100% from 6.1 min to 8 min. Sphingolipids were analyzed via mass spec using an AB Sciex Triple Quad 5500 Mass Spectrometer. Q1 and Q3 were set to detect distinctive precursor and product ion pairs. Ions were fragmented in Q2 using N2 gas for collisionally induced dissociation. Analysis used multiple-reaction monitoring in positive-ion mode. Sphingolipids were monitored using precursor → product MRM pairs. The mass spectrometer parameters used were: Curtain Gas: 30 psi; CAD: Medium; Ion Spray Voltage: 5500 V; Temperature: 500 °C; Gas 1: 60 psi; Gas 2: 40 psi; Declustering Potential, Collision Energy, and Cell Exit Potential vary per transition. The species of sphingomyelin (SM); ceramide (Cer), hexosyl-ceramide (Hex-Cer), sphingoid lipids such as sphingosine (Sph); DHS1P dihydro-sphingosine (DH-Sph), and S1P were identified based on their retention time and mass-to-charge (m/z) ratio, and quantified as described in previous publications [62]. In this targeted analysis, species of sphingolipids are measured semi-quantitatively in pmols by comparing their peak area values with the internal standards (which are used at 250 pmol/ sample). Mole % comparison is the most common way of comparing two or many groups of samples in which picomol values of each species are converted to the % of total picomol of that particular group. For example, the Mole % of Cer C18:0 = (pmol of Cer C18:0)/ (pmol total Cer) × 100 [63, 64].
mRNA analysis by RT-PCR
Total RNA was extracted from one of the fractions of the mouse brain using a Life Technologies RNA extraction kit (Sigma-Aldrich Inc. St. Louis, Missouri, USA), and quantitative RT-PCR was performed as previously reported protocol [65]. Then mRNA level was measured for the sphingolipid metabolic genes that include Serine palmitoyl transferase 1 (Spt1) and Spt2; Sphingosine kinase 1 (Sphk1), and Sphk2; Ceramide synthase 2 (CerS2), CerS4, CerS5, and CerS6; Sphingomyelin phosphodiesterase 1 (Smpd1), and Smpd2; Ceramide kinase (Cerk), Acyl-sphingosine amido-hydrolase-1(acid-Ceramidase) (Asah1), Glucosylceramide synthase (Gcs). Detailed information on primer sequences is listed in the Supplementary Table 1.
Statistical analysis
All results were analyzed by using GraphPad Prism 8 software. Single statistical comparisons of a treatment group versus the control group were evaluated using Non-parametric t-test (Mann–Whitney test), whereas multiple group comparisons were evaluated using a one-way ANOVA test followed by a Tukey’s multiple comparison for adjustments. The p < 0.05 values were considered significant.
Results
Acute TBI and activation of sphingomyelinases (SMases) in the brains
The activation of sphingomyelinase enzymes (SMases) plays a crucial role in inducing inflammation [66]. This activation hydrolyzes sphingomyelin, releasing a cascade of bioactive lipids including ceramides, sphingosine, ceramide-1-phosphate and sphingosine-1-phosphate [66, 67]. Therefore, the study measured the enzymatic activity of sphingomyelinases, both acidic (aSMase) and neutral (nSMase), in the brain tissue of wildtype mice exposed to either Sham (Control) or Blast (TBI) and analyzed at 1, 3, and 7-days post-TBI. At 1 day post-TBI, Blast-exposed mice showed a significant elevation in aSMase activity compared to Sham mice; however, no changes were observed with nSMase activity (Fig. 1A). At 3 days post-TBI, a similar pattern was observed for both aSMase and nSMase, where aSMase activity increased significantly but not nSMase (Fig. 1B). However, at 7 days post-TBI, nSMase activity was found to be significantly higher in Blast-exposed brains, but not aSMase (Fig. 1C).

Analysis of enzymatic activity of sphingomyelinases (acidic sphingomyelinase: aSMase and basic sphingomyelinase: nSMase) in brain tissue of wild type (WT) mice at A) 1 day after treatment with 0-psi (sham) or 50-psi (blast) TBI, B) 3 days after treatment with 0-psi (sham) or 50-psi (blast) TBI, C) 7 days after treatment with 0-psi (sham) or 50-psi (blast) TBI. (n = 6; p < 0.05; ANOVA)
Sphingolipid profile changes in the acute stages in post-TBI brains
In an earlier study, our group reported significant alterations of ceramide levels in brain tissue of C57 mice one month after blast-induced TBI [52]. To identify mechanistic connections between sphingolipid changes and the development of TBI neuropathology, the present study focused on analyzing sphingolipids at the very acute stages of TBI. Using a targeted sphingolipids (SPLs) analysis by LC–MS/MS, the total tissue levels and composition of 12 major species of Ceramides C, monohexosylceramides (MHC), and sphingomyelins (SM) were determined. Levels of sphingosine (Sph), sphingosine-1-phosphate (S1P), and dihydrosphingosine or sphinganine (Sa) and ceramide-1-phosphate (C1P) were also measured. At 1 day post-TBI, there were no significant changes in total SPL as well in the major SPL classes, including total Cer, total MHC, and total SM (Fig. 2). However, an increase in total MHC levels was noticed at 7 days post-TBI (Fig. 2C). Additionally, there was an elevation in the levels of total ceramide-1-phosphate (C1P) at 1 day post-TBI (Fig. 2D) and sphingosine (Sph) at 7 days post-TBI (Fig. 2F).

Analysis of different sphingolipids in the brain tissues of wild type (WT) mice 1 to 7 days after treatment with 0-psi (sham) or 50-psi (blast) TBI. A Ceramide (Cer), Monohexosylceramides (MHC), Sphingomyelins (SM), and total level of these sphingolipids (% of control WT-Sham) in the brain of WT-Sham (WT-0) and WT-Blast (WT-50) mice one day after TBI. B Ceramide-1-phosphate (C1P), Sphingosine (Sph), Sphingosine-1-phosphate (S1P), and Sphinganine (Sa) level (% of control WT-Sham) in the brain of four groups of animals as shown in A. (n = 6; p < 0.05; ANOVA)
TBI-induced changes in individual SPL species in the brain
Although there were no major changes in the total levels of SPL and its major classes, such as Cer, MHC, and SM, compositional changes in those classes from an alteration in many individual species were found. At 1 day post-TBI, there were significantly elevated mol% levels of Cer C18:0 and SM C18:0 and decreased levels of Cer C22:0, Cer C24:1, MHC C22:0, and SM C26:1 in the Blast-exposed mice compared to the Sham-exposed mice brains (Table 1). After correction for multiple testing, the increase of Cer C18:0 and decrease in C24:1 remained highly significant (p < 0.0001 and p < 0.001, respectively) (Table 1). At 3 days post-TBI, there were significantly decreased mol% levels of Cer C14:0 and increased levels of MHC C20:0 in Blast-exposed brains compared to Sham (Table 1). After correction for multiple testing, the increase of MHC C20:0 remained significant (p < 0.05) (Table 1). At 7 days post-TBI, there were significant changes in many SPL species that included increased levels of Cer C18:1, Cer C26:0; MHC C18:0, MHC C26:0; SM C24:0, SM C26:1, and SM C26:0 and decreased levels of Cer C22:0 in the Blast-exposed brains compared to Sham (Table 1). After correction for multiple testing, the increase of MHC (C18:0, C24:1) and SM (C18:0, C24:0) remained significant p < 0.05 for all) (Table 1).
At 1 day post-TBI, there was elevation in total ceramide-1-phosphate (C1P) (Fig. 2D). Among CIP species, measurable levels of C14:0, C16:0, C18:0, C22:0, C24:1, C24:0, and C26:0 were detected in mice brains. There was a significant increase in the levels of C1P and C18:0 (0.34 ± 0.04 vs. 0.22 ± 0.02 pmol/mg; p = 0.01) in the brains of Blast-exposed mice at 1-day post-TBI.
This study attempted to determine the temporal changes in the species of Cer, MHC, and SM over 1 day, 3 days, and 7 days post-TBI brains. The species that showed significant changes in their mole % either with time or between the groups (control vs. TBI) are shown in Supplemental Fig. S1. Most species showed concordance between control vs. TBI in their levels as a function of time but a small number showed differences in their mole % either at day 1 or day 7 (Cer C18:0, C18:1, C22:0, C24:1; MHC C18:0, C20:0, C22:0, C26:0; and SM C18:0 (Supplemental Fig. S1). The observed changes that include increases in the most abundant saturated Cer species C18:0 at 1 day post-TBI and very-long-chain Cer, MHC, and SM species at 7 days post-TBI may have implications with the TBI pathology.
Expression of SPL metabolic marker genes in the brain tissue
qRT-PCR was used to quantify changes in gene expression of SPL metabolic markers in the brain tissue of mice exposed to Sham and Blast at 1, 3, and 7 days post-TBI. At 1 day post-TBI, out of 14 genes acquired, there was a significant elevation (fivefold) in the expression of de novo Cer biosynthetic gene, Spt1 (Serine palmitoyltransferase 1) in the Blast-exposed brains compared to Sham (Fig. 3A). Moreover, there were significant increases in the expression of Cers2 (Ceramide synthase 2), Cerk (Ceramide kinase), Smpd1 (Sphingomyelin phosphodiesterase 1), and Sphk1 (Sphingosine kinase 1) in the Blast-exposed mice brains compared to Sham (Fig. 3A). At 3 days post-TBI, Blast-exposed mice showed a significant increase in Spt1 but significant decreases in Gcs (Glucosylceramide synthase), Smpd1, and Asah1 (Acylsphingosine amidohydrolase 1 or acid-Ceramidase) expression compared to Sham mice (Fig. 3B). At 7 days post-TBI, there were no differences in the expression of 14 SPL genes investigated between the Blast-exposed and Sham-exposed brains (Fig. 3C).

Expression analysis of sphingolipid metabolic genes in the brain tissues of wild type (WT) mice A) 1 day after treatment with 0-psi (sham) or 50-psi (blast) TBI, B) 3 days after treatment with 0-psi (sham) or 50-psi (blast) TBI, C) 7 days after treatment with 0-psi (sham) or 50-psi (blast) TBI. mRNA expression levels are presented over wild type sham (WT-0) (= 1.0) after normalization with two housekeeping genes, Ribosomal protein L19 (Rpl19) and Glyceraldehyde-3-phosphate dehydrogenase (Gapdh) (n = 6; p < 0.05; ANOVA). Spt1, Serine palmitoyl transferase 1; Spt2; CerS1, Ceramide synthase 1; CerS2; CerS5; CerS6; GCS, Glucosyl-ceramide-synthase; Asah1, Acyl-sphingosine amido-hydrolase-1(acid-Ceramidase); Asah2; Smpd1, Sphingomyelin phosphodiesterase 1; Smpd2; Cerk, Ceramide kinase; Sphk1, Sphingosine kinase 1; Sphk2
Changes of SPL profile in the plasma
Plasma SPL has been investigated as a biomarker for various neurodegeneration, neuroinflammation, and psychiatric diseases [37,38,39,40,41,42,43,44]. To determine if there is any effect of traumatic head injury on the plasma profile of sphingolipids, the profiles of SPL in the plasma from both Blast-exposed and Sham-exposed mice were analyzed at days 1, 3, and 7 post-TBI. At 1 day post-TBI, there were no significant changes in total SPL as well in the major SPL classes, including total Cer, total MHC, and total SM (Fig. 4A). However, there was a significant increase in the levels of sphingosine (Sph) and a significant decrease in sphingosine-1-phosphate (S1P) in the Blast-exposed mice compared to Sham-exposed mice (Fig. 4D). At 3 days post-TBI, there were no differences in the total SPL and various SPL classes as well as in the levels of bioactive C1P, Sph, S1P and Sa between the Blast- and Sham exposed mice (Fig. 4B, E). At 7 days post-TBI, like day 1 and day 3, no changes in the total levels of major sphingolipids in the plasma were detected; however, there was significantly decreased Sph levels in Blast-exposed mice compared to Sham-exposed mice (Fig. 4C, F). There were also.

Analysis of different sphingolipids in plasma of wild type (WT) mice 1 to 7 days after treatment with 0-psi (sham) or 50-psi (blast) TBI. A Ceramide (Cer), Monohexosylceramides (MHC), Sphingomyelins (SM), and total level of these sphingolipids (% of control WT-Sham) in plasma of WT-Sham (WT-0) and WT-Blast (WT-50) mice one day after TBI. B Ceramide-1-phosphate (C1P), Sphingosine (Sph), Sphingosine-1-phosphate (S1P), and Sphinganine (Sa) level (% of control WT-Sham) in plasma of four groups of animals as shown in A. (n = 6; p < 0.05; ANOVA)
TBI-induced changes in individual SPL species in plasma
At the individual species level, considerable changes in the plasma SPL in the relative mol% composition of chain-length variants in all classes of SPL were detected. At 1 day post-TBI, the Blast-exposed mice showed significant increases in Cer C16:0, C18:1, C18:0, and MHC C16:0 compared to Sham (Table 2). After correction for multiple testing, the decrease of Cer C22:0 and MHC C22:0 and increase in MHC C16:0 remained significant (p < 0.05 for all) (Table 2). At 3 days post-TBI, Blast-exposed mice had elevated mol% levels of Cer C24:1, MHC C14:0, and SM C14:0 in their plasma but decreased relative levels of MHC C26:1, and SM C20:0 and C22:0 (Table 2). After correction for multiple testing, the increase of Cer C24:1 and decrease in Cer C24:0, MHC C22:0, and SM C22:0 remained highly significant (p < 0.001, p < 0.05, p < 0.05, and p < 0.05, respectively) (Table 2). At 7 days post-TBI, Blast-exposed mice had significantly elevated levels of Cer C22:0, MHC C22:0, and SM C22:0 and decreased levels of Cer C24:1 and MHC C14:0, C24:1, and C24:0 compared to Sham-exposed mice (Table 2). After correction for multiple testing, the increase of Cer C22:0, SM C22:0, MHC C22:0 and decrease in Cer C24:1, MHC 24:1, and MHC C24:0 remained significant (p < 0.05 for all) (Table 2).
This study also analyzed the temporal changes in the species of Cer, MHC, and SM over 1 day, 3 days, and 7 days post-TBI plasma. The species that showed significant changes in their mole % either with time or between the groups (control vs. TBI) are shown in Supplemental Fig. S2. The SPL species that showed discordance between control vs. TBI in their levels as a function of time include Cer C24:0; MHC C14:0, C16:0, C22:0, C24:0, C24:1, C26:1; and SM C14:0, C20:0 and C22:0 (Supplemental Fig. S2).
Comparison of ratios of sphingolipid species
Historically, specific ratios SPL species in the plasma have been documented as biomarkers for systemic or neurodegenerative diseases [68,69,70]. This study analyzed the ratios of individual species in all classes of SPL in various combinations and examined if they were different between Blast-exposed mice and Sham mice. At 1 day post-TBI, after multiple comparison adjustments in P-values, plasma ratios of Cer C24:1/C18:1 and MHC C24:1/C26:1 were significantly reduced in the Blast-exposed mice compared to the Sham-exposed plasma (Table 3). Very interestingly, a complete reversal (significantly increased) of those two ratios of Cer and MHC occurred at 3 days post-TBI plasma, suggesting their association with the exposure of TBI (Table 3). At 7 days post-TBI, there was a decrease in the plasma ratio of MHC C24:0/C22:0 in the Blast-exposed mice compared to the Sham-exposed plasma (Table 3).
A comparison of changes among the SPL species in plasma and brain tissues following mTBI is presented in Supplemental Table 1. The changes in the plasma profile of SPL species, some of their particular ratios, and their temporal variability indicate their association with exposure of the brain to TBI and may serve as biomarkers for the prognosis of traumatic head injury.
Discussion
Associations of sphingolipid signaling with neuroinflammation pathology have been well documented and predicted to be a promising target for therapeutic management as well as a potential prognostic marker in neurodegenerative diseases [71, 72]. In a similar pathological context, TBI involves neuroinflammation-mediated neurodegeneration where aberrations in sphingolipid metabolism have been demonstrated [49, 73]. Our previous study with a mouse model of mild TBI (mTBI) revealed changes in brain SPL profiles even after 30 days post-TBI [52]. In this study, brain and plasma SPL profile changes were observed within the first seven days following exposure to mTBI, which could be associated with the early pathophysiology of the TBI process.
The major findings in the brain tissue included significant elevation of 1) acid sphingomyelinase (aSMase) at 1 and 3 days post-TBI but no changes in nSMase levels (Fig. 1); 2) elevation of nSmase levels at 7 days post-TBI (Fig. 1); 3) C1P levels at 1 day post-TBI (Fig. 2); 4) monohexosylceramide (MHC) and sphingosine at 7 days post-TBI (Fig. 2); 5) relative mole% of Cer C18:0 at 1 day (Table 1), MHC C20:0 at 3 days (Table 1), and MHC (C18:0, C24:1) and sphingomyelins (SM) (C18:0, C24:0) at 7 days (Table 1); and 6) significant increase of several SPL metabolic genes at 1-day, reduced numbers at 3-days and none at 7-days post-TBI (Fig. 3). The major findings in plasma included 1) a significant increase in MHC C16:0 as well as a decrease in C22:0 in Cer and MHC classes at 1 day (Table 2); 2) a significant increase in long-chain Cer (C24:1) accompanied by decreases in Cer C24:0 and C22:0 in MHC and SM classes at 3 days (Table 2); 3) a significant increase of C22:0 in all classes of SPL (Cer, MHC and SM) as well as a decrease in Cer C24:1, MHC C24:1 and MHC C24:0 at day 7 (Table 2); and 4) a significant elevation of Sph and reduction of S1P at 1 day; and 5) a significant reduction of Sph at day 7 (Fig. 4).
This study also explored the relationship between SPL metabolites in plasma and brain tissues following mTBI (Supplemental Table 1). Most significant metabolites demonstrated an inverse relationship between brain and plasma. Notably, Cer C24:1, MHC C20:0, and MHC C24:1 exhibited significant alterations in both brain and plasma. Interestingly, there was a significant elevation of MHC C24:1 in plasma and reduction in brain at 7 days post-TBI. This observed inverse relationship may be attributable to the complex interplay of central and systemic inflammatory responses following a mTBI. After a TBI, chemokines and other inflammatory molecules can leak through a compromised blood–brain barrier into the systemic circulation, attracting cells of the peripheral immune system to the site of injury and potentially activating an overactive inflammatory response known as systemic immune response syndrome [74]. Thus, negative feedback to systemic inflammation is often provided by the hypothalamus–pituitary–adrenal axis [74]. This adaptive response may aim to mitigate inflammatory damage, highlighting a sophisticated interplay between central and peripheral immune responses post-TBI.
In order to design therapeutic interventions for TBI, it is imperative to understand the pathophysiology of TBI by designing animal models. The procedure used in this study is an over-pressure air blast using a device developed and standardized by our group to induce TBI in mice using a small horizontally mounted air cannon system [17, 51,52,53, 75, 76]. This model simulates a closed-head primary blast injury with forces similar to those seen in an explosion or blunt force-mediated human mild-TBI (mTBI). Over the past decade, our research team has performed an extensive neurological and behavioral characterization of the mice exposed to this TBI procedure. This TBI caused by a single 50–60 psi blast in mice results in diffuse axonal injury in white matter tracts, characterized by swollen axonal bulbs, during the first few days [51, 75,76,77,78], followed by degeneration of injured axons. These mice exhibit clear motor deficiency in the early phase of recovery (1–2 weeks) and demonstrate persistent microglial activation in the major fiber tracts and degeneration of neurons in various brain regions at 2–3 months post-TBI. These pathologic characteristics are consistent with human mTBI; therefore, this procedure can act as a model for mTBI in mice.
Although several mechanisms in TBI have been proposed, a growing consensus favors the critical role of brain lipids and lipid pathway enzymes, including SMase activation and glia-mediated neuroinflammation [16, 79]. Activation and signaling of the enzymes involved in the metabolism of SPLs, especially that of bioactive SPLs, such as neutral and acid sphingomyelinase (nSMase, aSMase), ceramidase, and sphingosine kinase (SphK), have been linked to the development of various inflammatory and neurological diseases [80,81,82,83,84]. SMase hydrolyzes SM to produce phosphocholine, and Cer is a key step in SPL signaling. Like in many other neurodegenerative conditions, activation of SMases and Cer-induced neuroinflammatory pathology have been documented in TBI [85,86,87,88]. This study indicates activation of aSMase in the acute damage phase but nSMase in the chronic phase (Fig. 1). Our previous study also supports this notion of no changes in aSMase at 30-day post-TBI, but nSMase increased significantly [52]. This suggests that TBI’s initial insult impacts or is associated with aSMase activity, whereas chronic inflammatory effects are associated with nSMase activity. Prior studies have reported that aSMase-Cer axis regulates the pathogenesis of Major Depressive Disorder (MDD) which can occur after TBI [89]. Niziloket al., observed a similar pathology in relation to aSMase-Cer axis and accumulation of phosphorylated-tau protein in the mouse model of TBI [87]. Moreover, inhibition of aSMase activity by a single injection of amitriptyline, an inhibitor of aSMase, 1 h after TBI decreased accumulation of p-tau in the hippocampus one month after injury and decreased levels of depression [87]. Genetically ablated Smpd1−/− (aSMase) also reproduced similar results, indicating a major role of aSMase activation in the initiation of TBI neuropathology [87]. In another study modeling TBI in mice, inhibiting aSMase by intraperitoneal imipramine significantly reduced levels of Cer and aSMase activity in the brain tissue, neuronal death, and cognitive dysfunction [90]. In the context of these prior studies, these findings align well with other experimental models of Cer-regulated neuroinflammation-mediated neurodegeneration in TBI and suggests aSMase could be a potential therapeutic target in the acute phases of TBI, whereas nSMase for chronic phases of TBI.
Another important finding was an elevation of ceramide-1-phosphate (C1P) in the brains of blast-exposed mice at 1-day post-TBI (Fig. 2). Phosphorylation of Cer by ceramide kinase (CK) generates C1P, a process known to activate cytosolic phospholipase A2 (cPLA2), which is crucial in producing proinflammatory eicosanoids from arachidonic acid (AA) by COX and LOX enzymes [66, 91,92,93]. The data suggests potential roles of C1P in generating proinflammatory lipids (eicosanoids) and acute inflammation in TBI.
This study also found significantly elevated level of Cer C18:0 in the blast-exposed brain at 1 day post-TBI (Table 1). Pathological elevation of Cer C18:0 has been observed in the cerebrospinal fluid of stroke patients [94]. In ischemic brain injury in mice, Cer C16:0 and Cer C18:0 have been found to be increased at 3 h post-injury, and de novo synthesis of Cer has been proposed [95]. Moreover, a study by Nielson et al. reported an increased level of Cer C18:1 in their model on focal cerebral ischemia at 5-days of ischemic injury and simultaneous elevation of C1P and S1P 7 days post-injury [96]. The elevation of Cer and Cer-mediated upregulation of proinflammatory factors are well known for inducing apoptosis, parthanatos, and neurodegeneration [97, 98]. Cer C18:0 can be converted to CIP by ceramide kinases, and the present study found that ceramide-1-phosphate (C1P) was elevated in the brains of blast-exposed mice at 1-day post-TBI (Fig. 2).
Elevation of Cer precursor dihydrosphingosine (Sa) at 1, 2 and 7 days post-injury in animal models suggests de novo synthesis of Cer [99]; however, the present study did not find significantly different levels of Sa between Blast-exposed and Sham-exposed mice. In this study, there were no significant alterations in the total levels of Cer detected; however, there was a noteworthy increase in the total MHC species at 7 days post TBI in Blast-induced brains compared to the sham condition (Fig. 2C). To maintain cellular homeostasis, elevated Cer get quickly converted to MHC to reduce its adverse effects, which may explain this study’s findings of increasing MHC levels in the brains of 7 days post-TBI [100]. Cer can also be generated from SMase activation as well as from de novo synthesis [101]. Significant elevation of de novo pathway genes (Spt1) at both 1 and 3 days, CerS2 at 1 day may indicate activation of de novo pathway and at the same time elevation of Smpd1 may associate with aSMase activation, Cerk with C1P generation, and Sphk1 for S1P (Fig. 3).
Plasma sphingolipids have been increasingly recognized as potential biomarkers for neurodegenerative diseases, which prompted this investigation into their role in TBI. In animal models of stroke, an elevation of plasma long-chain Cer and long-chain SM has been demonstrated at 24 and 48 h post-injury, respectively [102]. In this study’s TBI model, plasma long-chain Cer C24:1 increased significantly after three days post-TBI (Table 2). In Parkinson’s disease, higher plasma Cer C16, C18, C20, C22, and C24:1 have been associated with worse cognition [103]. Similar changes in plasma Cer have also been reported in dementia, with increasing level of C18:1 and C24:1 MHC predicting severe pathology in Alzheimer’s Disease [104]. These pathological alterations of plasma SPL metabolites in neurodegenerative disorders and stroke suggest its potential role as a TBI biomarker in predicting outcomes of patients and TBI severity. To search for the potential metabolites in association with TBI severity, one study group recruited a cohort of 716 patients with TBI and compared it with non-TBI reference patients. They observed a significant increase in Cer levels but a significant decrease in SM levels between groups [105]. Moreover, increasing severity of TBI was associated with a decrease in SM, which was supported by clinicopathological parameters such as head CT [105]. With an abundance of research suggesting a strong association of lipid markers with the severity of TBI patients, lipids show promise as potential biomarkers for predicting TBI severity and merit further investigation for their clinical applicability.
Recently, US Food and Drug Administration (FDA) has approved a combination of two disease-specific biomarkers, one is the ubiquitin C-terminal hydrolase-L1 (UCH-L1) for neurons, and the other one is a glial fibrillary acidic protein (GFAP) for astrocytes for effective diagnosis of mild TBI having a brain lesion [106,107,108]. Although protein biomarkers effectively measure tissue damage, they often lack disease specificity and do not provide insights into metabolic disruption, one of the most significant events in TBI-associated energy failure/crisis [109]. As lipids are one of the major constituents of the brain and have the ability to cross the blood–brain barrier, evaluating lipid metabolites is a strength in this study and could be one of the best approaches in biomarker identification that addresses metabolic disruption. TBI pathology is a protracted form of neuroinflammation-mediated neurodegeneration, and lipids could act as either pan-inflammatory markers (ceramide, arachidonic acid), pan-anti-inflammatory markers (eicosapentaenoic acid and docosahexaenoic acid), and other bioactive intermediates (eicosanoids, prostaglandins). As with all studies, it is important to consider this study within the context of its limitations. Limitations of the TBI model exist as it negates certain variables, such as hypoxia from blast-induced pulmonary injury, which may independently contribute to neuronal stress and damage [17, 52, 53, 75, 76]. Additionally, this study ensured meticulous pre-analytical stability of samples with a standardized protocol, and the results demonstrate consistent levels of sphingolipids and their enzymes across the sample groups, indicating uniformity in sample handling and processing. However, despite these precautions, subtle variations in pre-analytical procedures could still potentially impact the results. Moreover, analysis was limited by a small sample size, preventing advanced statistical techniques such as ternary plots or logistic regression [110]. It is important to validate these findings and undertake more comprehensive analyses with larger sample sizes.
Conclusions
The present study aimed to elucidate the status of inflammatory SPL metabolites in the acute stage of TBI and explore the potential of SPL metabolites as biomarkers for TBI. In this study, TBI was induced under controlled conditions, and further research in other TBI animal models and human subjects is necessary to assess the effect of the types of TBI and their severity on the brain and circulating lipids to understand their pathophysiological connections. In conclusion, SPL metabolites in the plasma hold immense promise as biomarkers in TBI, and harnessing their capabilities may prove useful in clinical settings. The goal is to give providers the ability to objectively assess the severity and progression of TBI one day, allowing for a more accurate diagnosis, prognosis, and treatment monitoring. It is imperative to continue conducting more studies to determine the usefulness and accuracy of SPL biomarkers in TBI.
Availability of data and materials
All materials and data will be available from the corresponding author following University of Tennessee’s policy of sharing research materials and data.
References
Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM, et al. Estimating the global incidence of traumatic brain injury. J Neurosurg. 2018;130:1080–97.
Rabinowitz AR, Levin HS. Cognitive sequelae of traumatic brain injury. Psychiatr Clin North Am. 2014;37:1–11.
Shangkuan WC, Lin HC, Shih CP, Cheng CA, Fan HC, Chung CH, Lin FH, Tsao CH, Chien WC. Increased long-term risk of hearing loss in patients with traumatic brain injury: a nationwide population-based study. Laryngoscope. 2017;127:2627–35.
Armstrong RA. Visual problems associated with traumatic brain injury. Clin Exp Optom. 2018;101:716–26.
Brett BL, Gardner RC, Godbout J, Dams-O’Connor K, Keene CD. Traumatic brain injury and risk of neurodegenerative disorder. Biol Psychiatry. 2022;91:498–507.
Bombardier CH, Fann JR, Temkin NR, Esselman PC, Barber J, Dikmen SS. Rates of major depressive disorder and clinical outcomes following traumatic brain injury. JAMA. 2010;303:1938–45.
Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–41.
Magnuson J, Leonessa F, Ling GS. Neuropathology of explosive blast traumatic brain injury. Curr Neurol Neurosci Rep. 2012;12:570–9.
Masel BE, DeWitt DS. Traumatic brain injury: a disease process, not an event. J Neurotrauma. 2010;27:1529–40.
Agimi Y, Marion D, Schwab K, Stout K. Estimates of long-term disability among US Service Members With Traumatic Brain Injuries. J Head Trauma Rehabil. 2021;36:1–9.
Goldstein LE, Fisher AM, Tagge CA, Zhang XL, Velisek L, Sullivan JA, Upreti C, Kracht JM, Ericsson M, Wojnarowicz MW, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra160.
Jorge RE, Robinson RG, Moser D, Tateno A, Crespo-Facorro B, Arndt S. Major depression following traumatic brain injury. Arch Gen Psychiatry. 2004;61:42–50.
Jorge RE, Starkstein SE. Pathophysiologic aspects of major depression following traumatic brain injury. J Head Trauma Rehabil. 2005;20:475–87.
Kinoshita K. Traumatic brain injury: pathophysiology for neurocritical care. J Intensive Care. 2016;4:29.
Abdul-Muneer PM, Chandra N, Haorah J. Interactions of oxidative stress and neurovascular inflammation in the pathogenesis of traumatic brain injury. Mol Neurobiol. 2015;51:966–79.
Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, van Noort JM. Inflammation in neurodegenerative diseases–an update. Immunology. 2014;142:151–66.
Bu W, Ren H, Deng Y, Del Mar N, Guley NM, Moore BM, Honig MG, Reiner A. Mild traumatic brain injury produces neuron loss that can be rescued by modulating microglial activation using a CB2 receptor inverse agonist. Front Neurosci. 2016;10:449.
Desai A, Park T, Barnes J, Kevala K, Chen H, Kim HY. Reduced acute neuroinflammation and improved functional recovery after traumatic brain injury by alpha-linolenic acid supplementation in mice. J Neuroinflammation. 2016;13:253.
Mayeux JP, Teng SX, Katz PS, Gilpin NW, Molina PE. Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behav Brain Res. 2015;279:22–30.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet. 1974;2:81–4.
Li A, Atem FD, Venkatachalam AM, Barnes A, Stutzman SE, Olson DM. Admission Glasgow Coma Scale score as a predictor of outcome in patients without traumatic brain injury. Am J Crit Care. 2021;30:350–5.
Matis GK, Birbilis TA. Poor relation between Glasgow coma scale and survival after head injury. Med Sci Monit. 2009;15:Cr62–65.
Mena JH, Sanchez AI, Rubiano AM, Peitzman AB, Sperry JL, Gutierrez MI, Puyana JC. Effect of the modified Glasgow Coma Scale score criteria for mild traumatic brain injury on mortality prediction: comparing classic and modified Glasgow Coma Scale score model scores of 13. J Trauma. 2011;71:1185–92. discussion 1193.
Sinke MRT, Otte WM, Meerwaldt AE, Franx BAA, Ali MHM, Rakib F, van der Toorn A, van Heijningen CL, Smeele C, Ahmed T, et al. Imaging markers for the characterization of gray and white matter changes from acute to chronic stages after experimental traumatic brain injury. J Neurotrauma. 2021;38:1642–53.
Ghaith HS, Nawar AA, Gabra MD, Abdelrahman ME, Nafady MH, Bahbah EI, Ebada MA, Ashraf GM, Negida A, Barreto GE. A literature review of traumatic brain injury biomarkers. Mol Neurobiol. 2022;59:4141–58.
Hier DB, Obafemi-Ajayi T, Thimgan MS, Olbricht GR, Azizi S, Allen B, Hadi BA, Wunsch DC 2nd. Blood biomarkers for mild traumatic brain injury: a selective review of unresolved issues. Biomark Res. 2021;9:70.
Passos IC, Vasconcelos-Moreno MP, Costa LG, Kunz M, Brietzke E, Quevedo J, Salum G, Magalhães PV, Kapczinski F, Kauer-Sant’Anna M. Inflammatory markers in post-traumatic stress disorder: a systematic review, meta-analysis, and meta-regression. Lancet Psychiatry. 2015;2:1002–12.
Dadas A, Washington J, Diaz-Arrastia R, Janigro D. Biomarkers in traumatic brain injury (TBI): a review. Neuropsychiatr Dis Treat. 2018;14:2989–3000.
Sastry PS. Lipids of nervous tissue: composition and metabolism. Prog Lipid Res. 1985;24:69–176.
Cermenati G, Mitro N, Audano M, Melcangi RC, Crestani M, De Fabiani E, Caruso D. Lipids in the nervous system: from biochemistry and molecular biology to patho-physiology. Biochim Biophys Acta. 2015;1851:51–60.
Tracey TJ, Kirk SE, Steyn FJ, Ngo ST. The role of lipids in the central nervous system and their pathological implications in amyotrophic lateral sclerosis. Semin Cell Dev Biol. 2021;112:69–81.
Adibhatla RM, Hatcher JF, Dempsey RJ. Lipids and lipidomics in brain injury and diseases. Aaps j. 2006;8:E314–321.
Wenk MR. The emerging field of lipidomics. Nat Rev Drug Discov. 2005;4:594–610.
Horn A, Jaiswal JK. Structural and signaling role of lipids in plasma membrane repair. Curr Top Membr. 2019;84:67–98.
Adibhatla RM, Hatcher JF. Role of lipids in brain injury and diseases. Future Lipidol. 2007;2:403–22.
Ayub M, Jin HK, Bae JS. Novelty of sphingolipids in the central nervous system physiology and disease: focusing on the sphingolipid hypothesis of neuroinflammation and neurodegeneration. Int J Mol Sci. 2021;22:7353.
van Kruining D, Luo Q, van Echten-Deckert G, Mielke MM, Bowman A, Ellis S, Oliveira TG, Martinez-Martinez P. Sphingolipids as prognostic biomarkers of neurodegeneration, neuroinflammation, and psychiatric diseases and their emerging role in lipidomic investigation methods. Adv Drug Deliv Rev. 2020;159:232–44.
Di Pietro P, Izzo C, Abate AC, Iesu P, Rusciano MR, Venturini E, Visco V, Sommella E, Ciccarelli M, Carrizzo A, Vecchione C. The dark side of sphingolipids: searching for potential cardiovascular biomarkers. Biomolecules. 2023;13:168.
Bhadwal P, Dahiya D, Shinde D, Vaiphei K, Math RGH, Randhawa V, Agnihotri N. LC-HRMS based approach to identify novel sphingolipid biomarkers in breast cancer patients. Sci Rep. 2020;10:4668.
Lim GB. Sphingolipids are biomarkers of coronary disease. Nat Rev Cardiol. 2020;17:200.
Sedic M, Grbcic P, Pavelic SK. Bioactive sphingolipids as biomarkers predictive of disease severity and treatment response in cancer: current status and translational challenges. Anticancer Res. 2019;39:41–56.
Othman A, Saely CH, Muendlein A, Vonbank A, Drexel H, von Eckardstein A, Hornemann T. Plasma 1-deoxysphingolipids are predictive biomarkers for type 2 diabetes mellitus. BMJ Open Diabetes Res Care. 2015;3:e000073.
Fox TE, Bewley MC, Unrath KA, Pedersen MM, Anderson RE, Jung DY, Jefferson LS, Kim JK, Bronson SK, Flanagan JM, Kester M. Circulating sphingolipid biomarkers in models of type 1 diabetes. J Lipid Res. 2011;52:509–17.
Oizumi H, Sugimura Y, Totsune T, Kawasaki I, Ohshiro S, Baba T, Kimpara T, Sakuma H, Hasegawa T, Kawahata I, et al. Plasma sphingolipid abnormalities in neurodegenerative diseases. PLoS ONE. 2022;17:e0279315.
Meikle PJ, Wong G, Barlow CK, Kingwell BA. Lipidomics: potential role in risk prediction and therapeutic monitoring for diabetes and cardiovascular disease. Pharmacol Ther. 2014;143:12–23.
Zhao YY, Wu SP, Liu S, Zhang Y, Lin RC. Ultra-performance liquid chromatography-mass spectrometry as a sensitive and powerful technology in lipidomic applications. Chem Biol Interact. 2014;220:181–92.
Hyotylainen T, Oresic M. Optimizing the lipidomics workflow for clinical studies–practical considerations. Anal Bioanal Chem. 2015;407:4973–93.
Sun N, Keep RF, Hua Y, Xi G. Critical role of the sphingolipid pathway in stroke: a review of current utility and potential therapeutic targets. Transl Stroke Res. 2016;7:420–38.
Novgorodov SA, Riley CL, Yu J, Borg KT, Hannun YA, Proia RL, Kindy MS, Gudz TI. Essential roles of neutral ceramidase and sphingosine in mitochondrial dysfunction due to traumatic brain injury. J Biol Chem. 2014;289:13142–54.
Abe T, Niizuma K, Kanoke A, Saigusa D, Saito R, Uruno A, Fujimura M, Yamamoto M, Tominaga T. Metabolomic analysis of mouse brain after a transient middle cerebral artery occlusion by mass spectrometry imaging. Neurol Med Chir (Tokyo). 2018;58:384–92.
Heldt SA, Elberger AJ, Deng Y, Guley NH, Del Mar N, Rogers J, Choi GW, Ferrell J, Rex TS, Honig MG, Reiner A. A novel closed-head model of mild traumatic brain injury caused by primary overpressure blast to the cranium produces sustained emotional deficits in mice. Front Neurol. 2014;5:2.
Mondal K, Takahashi H, Cole J 2nd, Del Mar NA, Li C, Stephenson DJ, Allegood J, Cowart LA, Chalfant CE, Reiner A, Mandal N. Systemic elevation of n-3 polyunsaturated fatty acids (n-3-PUFA) is associated with protection against visual, motor, and emotional deficits in mice following closed-head mild traumatic brain injury. Mol Neurobiol. 2021;58:5564–80.
Reiner A, Heldt SA, Presley CS, Guley NH, Elberger AJ, Deng Y, D’Surney L, Rogers JT, Ferrell J, Bu W, et al. Motor, visual and emotional deficits in mice after closed-head mild traumatic brain injury are alleviated by the novel CB2 inverse agonist SMM-189. Int J Mol Sci. 2014;16:758–87.
Airola MV, Hannun YA. Sphingolipid metabolism and neutral sphingomyelinases. Handb Exp Pharmacol. 2013;(215):57–76. PMID: 23579449.
Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, Molotkovsky JG, Malinina L, Hinchcliffe EH, Chalfant CE, et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013;500:463–7.
Shaner RL, Allegood JC, Park H, Wang E, Kelly S, Haynes CA, Sullards MC, Merrill AH Jr. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009;50:1692–707.
MacKnight HP, Stephenson DJ, Hoeferlin LA, Benusa SD, DeLigio JT, Maus KD, Ali AN, Wayne JS, Park MA, Hinchcliffe EH, et al. The interaction of ceramide 1-phosphate with group IVA cytosolic phospholipase A(2) coordinates acute wound healing and repair. Sci Signal. 2019;12:eaav5918.
Vu NT, Kim M, Stephenson DJ, MacKnight HP, Chalfant CE. Ceramide kinase inhibition drives ferroptosis and sensitivity to cisplatin in mutant KRAS lung cancer by dysregulating VDAC-mediated mitochondria function. Mol Cancer Res. 2022;20:1429–42.
Nelson AJ, Stephenson DJ, Cardona CL, Lei X, Almutairi A, White TD, Tusing YG, Park MA, Barbour SE, Chalfant CE, Ramanadham S. Macrophage polarization is linked to Ca(2+)-independent phospholipase A(2)β-derived lipids and cross-cell signaling in mice. J Lipid Res. 2020;61:143–58.
Nelson AJ, Stephenson DJ, Bone RN, Cardona CL, Park MA, Tusing YG, Lei X, Kokotos G, Graves CL, Mathews CE, et al. Lipid mediators and biomarkers associated with type 1 diabetes development. JCI Insight. 2020;5:e138034.
Stephenson DJ, MacKnight HP, Hoeferlin LA, Washington SL, Sawyers C, Archer KJ, Strauss JF 3rd, Walsh SW, Chalfant CE. Bioactive lipid mediators in plasma are predictors of preeclampsia irrespective of aspirin therapy. J Lipid Res. 2023;64:100377.
Paranjpe V, Tan J, Nguyen J, Lee J, Allegood J, Galor A, Mandal N. Clinical signs of meibomian gland dysfunction (MGD) are associated with changes in meibum sphingolipid composition. Ocul Surf. 2019;17:318–26.
Galor A, Sanchez V, Jensen A, Burton M, Maus K, Stephenson D, Chalfant C, Mandal N. Meibum sphingolipid composition is altered in individuals with meibomian gland dysfunction-a side by side comparison of Meibum and Tear Sphingolipids. Ocul Surf. 2022;23:87–95.
Gary AA, Prislovsky A, Tovar A, Locatelli E, Felix ER, Stephenson D, Chalfant CE, Lai J, Kim C, Mandal N, Galor A. Lipids from ocular meibum and tears may serve as biomarkers for depression and post-traumatic stress disorder. Clin Exp Ophthalmol. 2023. https://doi.org/10.1111/ceo.14343.
Sugano E, Edwards G, Saha S, Wilmott LA, Grambergs RC, Mondal K, Qi H, Stiles M, Tomita H, Mandal N. Overexpression of acid ceramidase (ASAH1) protects retinal cells (ARPE19) from oxidative stress. J Lipid Res. 2019;60:30–43.
Nixon GF. Sphingolipids in inflammation: pathological implications and potential therapeutic targets. Br J Pharmacol. 2009;158:982–93.
Pavoine C, Pecker F. Sphingomyelinases: their regulation and roles in cardiovascular pathophysiology. Cardiovasc Res. 2009;82:175–83.
Hammad SM, Harden OC, Wilson DA, Twal WO, Nietert PJ, Oates JC. Plasma sphingolipid profile associated with subclinical atherosclerosis and clinical disease markers of systemic lupus erythematosus: potential predictive value. Front Immunol. 2021;12:694318.
Chua XY, Torta F, Chong JR, Venketasubramanian N, Hilal S, Wenk MR, Chen CP, Arumugam TV, Herr DR, Lai MKP. Lipidomics profiling reveals distinct patterns of plasma sphingolipid alterations in Alzheimer’s disease and vascular dementia. Alzheimer’s Research & Therapy. 2023;15:214.
Mantovani A, Dugo C. Ceramides and risk of major adverse cardiovascular events: a meta-analysis of longitudinal studies. J Clin Lipidol. 2020;14:176–85.
Alaamery M, Albesher N, Aljawini N, Alsuwailm M, Massadeh S, Wheeler MA, Chao CC, Quintana FJ. Role of sphingolipid metabolism in neurodegeneration. J Neurochem. 2021;158:25–35.
Alessenko AV, Albi E. Exploring sphingolipid implications in neurodegeneration. Front Neurol. 2020;11:437.
Novgorodov SA, Voltin JR, Wang W, Tomlinson S, Riley CL, Gudz TI. Acid sphingomyelinase deficiency protects mitochondria and improves function recovery after brain injury. J Lipid Res. 2019;60:609–23.
Das M, Mohapatra S, Mohapatra SS. New perspectives on central and peripheral immune responses to acute traumatic brain injury. J Neuroinflammation. 2012;9:236.
Guley NH, Rogers JT, Del Mar NA, Deng Y, Islam RM, D’Surney L, Ferrell J, Deng B, Hines-Beard J, Bu W, et al. A novel closed-head model of mild traumatic brain injury using focal primary overpressure blast to the cranium in mice. J Neurotrauma. 2016;33:403–22.
Guley NM, Del Mar NA, Ragsdale T, Li C, Perry AM, Moore BM, Honig MG, Reiner A. Amelioration of visual deficits and visual system pathology after mild TBI with the cannabinoid type-2 receptor inverse agonist SMM-189. Exp Eye Res. 2019;182:109–24.
Honig MG, Dorian CC, Worthen JD, Micetich AC, Mulder IA, Sanchez KB, Pierce WF, Del Mar NA, Reiner A. Progressive long-term spatial memory loss following repeat concussive and subconcussive brain injury in mice, associated with dorsal hippocampal neuron loss, microglial phenotype shift, and vascular abnormalities. Eur J Neurosci. 2021;54(5):5844–79.
Honig MG, Del Mar NA, Henderson DL, Ragsdale TD, Doty JB, Driver JH, Li C, Fortugno AP, Mitchell WM, Perry AM, et al. Amelioration of visual deficits and visual system pathology after mild TBI via the cannabinoid Type-2 receptor inverse agonism of raloxifene. Exp Neurol. 2019;322:113063.
Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154–69.
Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19:175–91.
Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60.
Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67.
Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002;1585:114–25.
Pettus BJ, Chalfant CE, Hannun YA. Sphingolipids in inflammation: roles and implications. Curr Mol Med. 2004;4:405–18.
Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, Balaban CD, Barbacci D, Schultz JA, Gouty S, et al. Mass spectrometry imaging of rat brain lipid profile changes over time following traumatic brain injury. J Neurosci Methods. 2016;272:19–32.
Barbacci DC, Roux A, Muller L, Jackson SN, Post J, Baldwin K, Hoffer B, Balaban CD, Schultz JA, Gouty S, et al. Mass Spectrometric Imaging of Ceramide Biomarkers Tracks Therapeutic Response in Traumatic Brain Injury. ACS Chem Neurosci. 2017;8:2266–74.
Niziolek GM, Boudreau RM, Baker J, Friend LA, Makley AT, Edwards MJ, Gulbins E, Goodman MD. Acid sphingomyelinase inhibition mitigates histopathological and behavioral changes in a murine model of traumatic brain injury. J Neurotrauma. 2020;37:1902–9.
Niziolek GM, Hoehn RS, Seitz AP, Jernigan PL, Makley AT, Gulbins E, Edwards MJ, Goodman MD. The role of acid sphingomyelinase inhibition in repetitive mild traumatic brain injury. J Surg Res. 2021;259:296–304.
Lin C-H, Kornhuber J, Zheng F, Alzheimer C. Tonic control of secretory acid sphingomyelinase over ventral hippocampal synaptic transmission and neuron excitability. Front Cell Neurosci. 2021;15:660561.
Lee SH, Kho AR, Lee SH, Hong DK, Kang BS, Park MK, Lee CJ, Yang HW, Woo SY, Park SW. Acid sphingomyelinase inhibitor, imipramine, reduces hippocampal neuronal death after traumatic brain injury. Int J Mol Sci. 2022;23:14749.
Pettus BJ, Bielawska A, Subramanian P, Wijesinghe DS, Maceyka M, Leslie CC, Evans JH, Freiberg J, Roddy P, Hannun YA, Chalfant CE. Ceramide 1-phosphate is a direct activator of cytosolic phospholipase A2. J Biol Chem. 2004;279:11320–6.
Ghidoni R, Caretti A, Signorelli P. Role of sphingolipids in the pathobiology of lung inflammation. Mediators Inflamm. 2015;2015:487508.
Schutze S, Potthoff K, Machleidt T, Berkovic D, Wiegmann K, Kronke M. TNF activates NF-kappa B by phosphatidylcholine-specific phospholipase C-induced “acidic” sphingomyelin breakdown. Cell. 1992;71:765–76.
Testai FD, Hillmann M, Amin-Hanjani S, Gorshkova I, Berdyshev E, Gorelick PB, Dawson G. Changes in the cerebrospinal fluid ceramide profile after subarachnoid hemorrhage. Stroke. 2012;43:2066–70.
Chao HC, Lee TH, Chiang CS, Yang SY, Kuo CH, Tang SC. Sphingolipidomics investigation of the temporal dynamics after ischemic brain injury. J Proteome Res. 2019;18:3470–8.
Nielsen MM, Lambertsen KL, Clausen BH, Meyer M, Bhandari DR, Larsen ST, Poulsen SS, Spengler B, Janfelt C, Hansen HS. Mass spectrometry imaging of biomarker lipids for phagocytosis and signalling during focal cerebral ischaemia. Sci Rep. 2016;6:39571.
Mondal K, Takahashi H, Cole J, Del Mar NA, Li C, Stephenson DJ, Allegood J, Cowart LA, Chalfant CE, Reiner A. Systemic elevation of n-3 polyunsaturated fatty acids (n-3-PUFA) is associated with protection against visual, motor, and emotional deficits in mice following closed-head mild traumatic brain injury. Mol Neurobiol. 2021;58:5564–80.
Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin K-M. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–17.
Nessel I, Michael-Titus AT. Lipid profiling of brain tissue and blood after traumatic brain injury: A review of human and experimental studies. Semin Cell Dev Biol. 2021;112:145–56. https://doi.org/10.1016/j.semcdb.2020.08.004.
Stiles M, Qi H, Sun E, Tan J, Porter H, Allegood J, Chalfant CE, Yasumura D, Matthes MT, LaVail MM, Mandal NA. Sphingolipid profile alters in retinal dystrophic P23H–1 rats and systemic FTY720 can delay retinal degeneration. J Lipid Res. 2016;57:818–31.
Stancevic B, Kolesnick R. Ceramide-rich platforms in transmembrane signaling. FEBS Lett. 2010;584:1728–40.
Sheth SA, Iavarone AT, Liebeskind DS, Won SJ, Swanson RA. Targeted lipid profiling discovers plasma biomarkers of acute brain injury. PLoS ONE. 2015;10:e0129735.
Mielke MM, Maetzler W, Haughey NJ, Bandaru VV, Savica R, Deuschle C, Gasser T, Hauser A-K, Gräber-Sultan S, Schleicher E. Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS ONE. 2013;8:e73094.
Savica R, Murray ME, Persson X-M, Kantarci K, Parisi JE, Dickson DW, Petersen RC, Ferman TJ, Boeve BF, Mielke MM. Plasma sphingolipid changes with autopsy-confirmed Lewy body or Alzheimer’s pathology. Alzheimers Dement. 2016;3:43–50.
Thomas I, Dickens AM, Posti JP, Czeiter E, Duberg D, Sinioja T, Kråkström M, Retel Helmrich IR, Wang KK, Maas AI. Serum metabolome associated with severity of acute traumatic brain injury. Nat Commun. 2022;13:2545.
Czeiter E, Amrein K, Gravesteijn BY, Lecky F, Menon DK, Mondello S, Newcombe VF, Richter S, Steyerberg EW, Vyvere TV. Blood biomarkers on admission in acute traumatic brain injury: relations to severity, CT findings and care path in the CENTER-TBI study. EBioMedicine. 2020;56:102785.
Bazarian JJ, Biberthaler P, Welch RD, Lewis LM, Barzo P, Bogner-Flatz V, Brolinson PG, Büki A, Chen JY, Christenson RH. Serum GFAP and UCH-L1 for prediction of absence of intracranial injuries on head CT (ALERT-TBI): a multicentre observational study. Lancet Neurol. 2018;17:782–9.
Wang KK, Kobeissy FH, Shakkour Z, Tyndall JA. Thorough overview of ubiquitin C-terminal hydrolase-L1 and glial fibrillary acidic protein as tandem biomarkers recently cleared by US Food and Drug Administration for the evaluation of intracranial injuries among patients with traumatic brain injury. Acute Med Surg. 2021;8:e622.
Timofeev I, Carpenter KL, Nortje J, Al-Rawi PG, O’Connell MT, Czosnyka M, Smielewski P, Pickard JD, Menon DK, Kirkpatrick PJ. Cerebral extracellular chemistry and outcome following traumatic brain injury: a microdialysis study of 223 patients. Brain. 2011;134:484–94.
Chen X, Song Y, Song W, Han J, Cao H, Xu X, Li S, Fu Y, Ding C, Lin F, et al. Multi-omics reveal neuroprotection of Acer truncatum Bunge Seed extract on hypoxic-ischemia encephalopathy rats under high-altitude. Commun Biol. 2023;6:1001.
Acknowledgements
This work was supported by National Eye Institute grants [EY022071, R01 EY031316] (NM); US Department of Defense office of the Congressionally Directed Medical Research Programs (CDMRP), Vision Research Program grant (W81XWH-20-1-0900) (NM); the Veterans’ Administration (VA Merit Review Award I01BX004893 (NM); the Veterans’ Administration (VA Merit Review, I BX001792 (CEC) and a Senior Research Career Scientist Award, IK6BX004603 (CEC)); National Institute of Allergy and Infectious Diseases grant (R01 AI139072 (CEC), National Institute of General Medical Sciences grant (R01 GM137578 (CEC) and R01 GM137394 (CEC), grants from Research to Prevent Blindness Inc., USA (NM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health, the Department of Veterans Affairs, or the U.S. government. UTHSC Plough Center funding (NM) and Neuroscience Institute for Postdoctoral Award (KM) are duly acknowledged.
Animals
Animal studies approved by UTHSC IACUC committee. Approval # 19–0104.
Author consent
All the authors have seen the manuscript and approved for publication.
Funding
1. US Department of Defense office of the Congressionally Directed Medical Research Programs (CDMRP), Vision Research Program grant (W81XWH-20–1-0900) (NM)
2. US Veterans’ Administration (VA Merit Review Award I01BX004893) (NM)
3. US Veterans’ Administration (VA Merit Review, I BX001792 (CEC) and a Senior Research Career Scientist Award, IK6BX004603 (CEC)
4. National Eye Institute grants [EY022071, R01 EY031316] (NM)
5. National Institute of Allergy and Infectious Diseases grant (R01 AI139072 (CEC)
6. National Institute of General Medical Sciences grant (R01 GM137578 (CEC) and R01 GM137394 (CEC)
7. Research to Prevent Blindness Inc., USA (NM)
Author information
Authors and Affiliations
Contributions
K.M. conducted experiments, performed data analysis, and contributed to manuscript writing. N.D.M. conducted experiments. A.A.G. contributed to data analysis, manuscript writing, and reviewing. R.C.G. contributed to data analysis and figure construction. M.Y. contributed to data analysis. F.T. contributed to data tabulation. B.S. contributed to methods and experiment execution. D.J. contributed to methods and experiment execution. C.E.C. contributed to study design, analysis, and provided funding. A.R. contributed to study design and analysis. N.M. contributed to study design, analysis, manuscript writing, and provided research support and funding.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12944_2024_2186_MOESM1_ESM.pdf
Supplementary Material 1: Supplemental Table 1. Comparative Analysis of Significant Metabolite Changes in Brain and Plasma Following Traumatic Brain Injury (TBI). Supplemental Figure S1. Longitudinal Variability of Brain Sphingolipid Species: Temporal changes in the levels of Ceramide (Cer), Monohexosylceramide (MHC), and Sphingomyelin (SM) species. Supplemental Figure S2. Longitudinal Variability of Plasma Sphingolipid Species: Temporal changes in the levels of Ceramide (Cer), Monohexosylceramide (MHC), and Sphingomyelin (SM) species.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mondal, K., Del Mar, N.A., Gary, A.A. et al. Sphingolipid changes in mouse brain and plasma after mild traumatic brain injury at the acute phases. Lipids Health Dis 23, 200 (2024). https://doi.org/10.1186/s12944-024-02186-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-024-02186-x