
Abstract
Ovarian cancer is the leading cause of gynecological cancer-related death. Drug resistance is the bottleneck in ovarian cancer treatment. The increasing use of novel drugs in clinical practice poses challenges for the treatment of drug-resistant ovarian cancer. Continuing to classify drug resistance according to drug type without understanding the underlying mechanisms is unsuitable for current clinical practice. We reviewed the literature regarding various drug resistance mechanisms in ovarian cancer and found that the main resistance mechanisms are as follows: abnormalities in transmembrane transport, alterations in DNA damage repair, dysregulation of cancer-associated signaling pathways, and epigenetic modifications. DNA methylation, histone modifications and noncoding RNA activity, three key classes of epigenetic modifications, constitute pivotal mechanisms of drug resistance. One drug can have multiple resistance mechanisms. Moreover, common chemotherapies and targeted drugs may have cross (overlapping) resistance mechanisms. MicroRNAs (miRNAs) can interfere with and thus regulate the abovementioned pathways. A subclass of miRNAs, “epi-miRNAs”, can modulate epigenetic regulators to impact therapeutic responses. Thus, we also reviewed the regulatory influence of miRNAs on resistance mechanisms. Moreover, we summarized recent phase I/II clinical trials of novel drugs for ovarian cancer based on the abovementioned resistance mechanisms. A multitude of new therapies are under evaluation, and the preliminary results are encouraging. This review provides new insight into the classification of drug resistance mechanisms in ovarian cancer and may facilitate in the successful treatment of resistant ovarian cancer.
Similar content being viewed by others
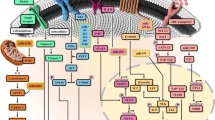

Introduction
Ovarian cancer (OC) is the third most common and the most lethal malignancy of the female reproductive system. Seventy percent of patients are diagnosed at an advanced stage (FIGO stage III and IV) with distant metastasis [1]. Despite receiving standard-of-care therapy (optimal cytoreductive surgery followed by adjuvant chemotherapy), most patients develop recurrent disease, which is resistant to chemotherapy, resulting in a 5-year survival rate of approximately 30–40% worldwide [2]. Although maintenance therapy with poly (adenosine diphosphate-ribose) polymerase (PARP) inhibitors (PARPis) has prolonged progression-free survival (PFS) and 5-year overall survival (OS) [3,4,5], unfortunately, many patients do not respond to PARPi treatment due to intrinsic or acquired resistance. Drug resistance is a formidable challenge in the treatment of ovarian cancer and is the primary contributor to poor prognosis.
According to the National Comprehensive Cancer Network (NCCN) guidelines (version 1.2023), there are many therapeutic regimens for resistant ovarian cancer, including some novel agents. However, the objective remission rate is still low, and the median survival time is less than 12 months due to complicated resistance mechanisms. In resistant ovarian cancer, the classical mechanisms of action of common drugs can be disrupted or altered, possibly resulting in impaired therapeutic effects. Thus, treatment regimens should not rely only on empirical options. In addition to traditional drugs, novel compounds are being investigated and tested in early clinical trials [6]. As the number of categories of agents increases, after the development of multidrug resistance (MDR), the decision of appropriate later-line therapeutic regimens is very challenging. This issue prompted us to consider the interactions of drug resistance mechanisms among different agents.
Even if resistance can develop to different drugs, the underlying mechanisms may be similar. Thus, instead of simply distinguishing resistance by agent, we attempted to classify drug resistance by mechanism. We summarized four major mechanisms (Fig. 1) from the published literature: 1) abnormalities in transmembrane transport, 2) alterations in DNA damage repair (DDR), 3) dysregulation of cancer-associated signaling pathways, and 4) epigenetic modifications. MicroRNAs (miRNAs) post-transcriptionally regulate the expression of target genes and affect a variety of biological processes, including cancer cell proliferation, metastasis, and therapeutic resistance [7]. miRNAs significantly regulate drug resistance by acting on molecules or/and pathways related to the four abovementioned mechanisms. Abnormal miRNA expression can lead to dysregulation of drug transporters, which control drug influx and efflux [8, 9]. The expression of some components of DDR mechanisms, such as homologous recombination repair (HRR) and nonhomologous end joining (NHEJ), is modulated by miRNAs [10]. In addition, miRNAs can interfere with multiple cancer-associated signaling pathways by targeting their components, thereby promoting tumor resistance to therapy [11].

The summery of miRNA-mediated resistance mechanisms (a) Abnormal transmembrane transport; (b) Alterations of DNA damage repair; (c) Dysregulation of cancer-associated signal pathway; (d) Epigenetic modification
Based on the abovementioned findings, we retrieved phase I/II clinical trials (Table 1, Figure S1 and S2) of novel drugs for resistant ovarian cancer. Understanding the underlying resistance mechanisms is expected to contribute to the identification of new clinical options for reversing resistance and improving the prognosis of ovarian cancer patients.
Mechanisms of drug resistance in ovarian cancer
Abnormal transmembrane transport
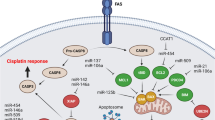
Decreased influx and increased efflux are two forms of abnormal transmembrane transport that reduce the intracellular drug concentration and result in resistance (Fig. 2). Moreover, in platinum-resistant ovarian cancer (PROC), the expression of the related genes and transporters is decreased. Thus, the intracellular concentration of platinum is insufficient, and platinum resistance subsequently develops [12,13,14,15,16]. miRNAs can directly target transmembrane transporters, thereby regulating cellular resistance to drugs [17]. They directly bind to the 3'-untranslated region (3'-UTR) of a targeted transporter gene to regulate its transcription, leading to abnormalities in drug influx and efflux [18].

Abnormal transmembrane transport. The SLC31A1, SLC22A1/2/3, as members of SLC superfamily, are significant transporters in charge of drug inflow. Downregulation of SLC transporters reduce platinum uptake, leading to chemoresistance in ovarian cancer. The role of miRNA in SLC expression lacks sufficient evidence. The ABC transporter family include ABCB1, ABCG2, ABCC1, which are responsible for drug efflux and then reduce intracellular concentration of platinum. miR130a/b, miR-186, miR-495 can directly bind with the 3'-UTR of ABCB1 mRNA or regulate PTEN, XIPA, and PI3K, leading to decreased ABCB1 transcription or translation level. miR-21-5p and miR-212-3p also have a regulatory factor of ABCB1 and ABCG2, respectively. miR-185-5p, miR-326, miR-508-3p and miR-134 can regulate the expression of ABCC1. ATP7A/7B are another contributor of drug efflux. miR-139 can directly bind to the 3'-UTR of ATP7A/7B, leading to apoptosis induction and increasing the chemosensitivity of ovarian cancer. MT can bind to cisplatin and deactivates it, which decreases drug efficacy and induces drug resistance. GST catalyzes glutathione to bind platinum and causes drug inactivation, which is associated with platinum resistance in ovarian cancer. (SLC, solute carrier superfamily; GST, Glutathione transferase; MT, Metallothionein)
Reduced drug influx
Sodium pumps, copper ion transporters, and organic cation transporters on the cell membrane or plasma membrane, such as the drug-transporting solute carrier (SLC) superfamily transporters (e.g., SLC31A1, SLC22A1/2/3), are key transporters controlling drug influx. SLC31A1 has been convincingly demonstrated to transport cisplatin and its analogs carboplatin and oxaliplatin, leading to intracellular accumulation of platinum [19]. The low expression of SLC22A2 in ovarian cancer may correlate with platinum drug resistance via a reduction in platinum uptake [20]. miRNAs play pivotal roles in the expression of drug-transporting SLC transporters and may influence treatment responses in prostate cancer, hepatocellular carcinoma and colorectal cancer [9]. However, the association and interaction mechanisms of miRNAs and SLC transporters in drug resistance in ovarian cancer have not been investigated, and further research is warranted.
Increased drug efflux
The ABC transporter family is mainly responsible for drug efflux. Abnormal expression of miRNAs (e.g., the miR-200 family, let-7 family and miR-130a/b) plays a role in ABC transporter regulation, thereby inducing resistance in ovarian cancer [21]. The characterized efflux transporters in the ABC family include ABCB1, ABCG2 and ABCCs [8, 22]. The abovementioned miRNAs can bind to the 3'-UTRs of ABC transporter-encoding mRNAs, or participate in imperfect base pairing with genes encoding nuclear receptors, transcription factors (TFs), and signaling molecules associated with ABC transporters. Through this action, the mRNAs of ABC transporters are degraded or the translation of the corresponding proteins is inhibited [8]. In addition, the vault protein lung drug resistance-related protein (LRP) can transport cytostatic drugs from intracellular targets, conferring drug resistance [23].
Whole-genome microarray analysis revealed that ABCB1 was the only drug transporter with increased expression in resistant ovarian cancer cells, while the expression of several other ABC transporters was significantly decreased [24]. The membrane transporter P-glycoprotein (P-gp) is encoded by ABCB1 and is an ATP-dependent drug efflux pump. Its overexpression in resistant cell lines is considered the crucial mechanism of resistance to paclitaxel, doxorubicin, sorafenib [25], and PARPis [26]. Notably, dysregulated miRNAs can mediate the overexpression of ABCB1, resulting in MDR. For instance, miR130a/b, miR-186, and miR-495 can directly bind to the 3'-UTR of ABCB1 mRNA or regulate the expression of other targets (e.g., PTEN, XIAP, and PI3K) [11, 27], leading to ABCB1 mRNA degradation or translational inhibition. A strong increase in ABCB1 expression was found to correlate with decreased expression of miR-21-5p, but the regulatory mechanism involved remains unknown [21]. In addition, upregulation of ABCB1 is associated with the transcriptional fusion of ABCB1 and SLC25A40, which was identified through whole-genome analysis in patients with high-grade serous ovarian cancer (HGSOC) who underwent prior chemotherapy and targeted therapy [28]. These findings indicate that ABCB1 upregulation frequently induces cross-resistance to chemotherapeutics and targeted drugs. Therefore, PARPis that are not dependent on the P-gp transporter might show greater therapeutic efficacy in patients who have received chemotherapy [24]. ABCC1 is associated with poor survival and chemoresistance in HGSOC. miR-185-5p and miR-326 both target the ABCC1 3'-UTR to regulate the expression of ABCC1 [2]. miR-508-3p [29] and miR-134 [30], which are sponged by CircETDB1 and LINC01118, respectively, can posttranscriptionally regulate the expression of ABCC1. ABCG2 is involved in topotecan resistance in ovarian cancer, which is associated with miR-212-3p downregulation [31].
In addition, upregulation of the copper efflux transporters ATP7A and ATP7B contributes to chemoresistance in ovarian cancer [32]. miR-139 can directly bind to the 3'-UTR of ATP7A/B, contributing to apoptosis induction and increasing the chemosensitivity of ovarian cancer cells [33].
Drug inactivation
Metallothionein (MT) and glutathione (GSH) are two major thiol-containing proteins that bind to platinum-based drugs. Detoxification of cisplatin by intracellular thiol-containing proteins is considered a major hurdle to overcome. MT binding to cisplatin can induce drug resistance, which can be reversed by short hairpin MT (shMT) [34]. GSH reacts with cisplatin to form a GS-platinum complex, reducing the available intracellular platinum content [35]. Glutathione S-transferase (GST) catalyzes the binding of GSH to platinum and causes drug inactivation, which is associated with platinum resistance in ovarian cancer [36, 37].
Alterations in DDR
If DNA damage is not repaired promptly, cellular senescence or apoptotic signals are activated, while abnormal activation of DDR maintains the viability of cancer cells, significantly inducing resistance to chemotherapeutic drugs and PARPis and affecting therapeutic efficacy [38]. DDR generally consists of seven pathways (Fig. 3): the HRR, NHEJ, base excision repair (BER), nucleotide excision repair (NER), mismatch repair (MMR), translesion DNA synthesis (TLS), and Fanconi anemia (FA) pathways. Interactions among the DNA damage response, DNA repair components and miRNAs have been reported [39]. The ectopic expression of miRNAs, as regulatory factors, can interfere with the activity of DNA repair mechanisms, which have been implicated in multiple types of resistance [40]. Some miRNAs can reverse drug resistance by targeting genes encoding DDR-related enzymes [41].

Alterations of DNA damage repair. DDR generally consists of HRR, NHEJ, Replication fork, BER, NER, MMR, TLS, and FA. The repair of DSBs occurs predominately through NHEJ repair pathway in conjunction with HRR pathway. NHEJ are initiated by binding of Ku70–Ku80 heterodimer to DNA ends. The subsequent recruitment and autophosphorylation of DNA-PKcs bring the DNA ends together and allow their ligation by XRCC4–LIG4. MRN complex (MRE11-RAD50-NBS1), an important repair factor of HRR, detects the DNA damage firstly and activates downstream signaling. Besides, it exerts nuclease activity to resect DNA end, guiding to HRR. Further, DYNLL1 binds directly to MRE11 to limit its end-resection activity. Decreased DYNLL1 restores HR-mediated double-strand DNA breaks repair. Replication fork protection is a modality independent of DSBs, which contributes to gene stabilization, leading to chemoresistance and PARPi resistance. Additionally, down-expression of 53BP1 protein is another mechanism to restore DNA end resection. Shieldin (SHLD1, SHLD2, SHLD3 and REV7), as an effector complex of 53BP1, can mediate 53BP1 dependent DNA repair in a BRCA-independent manner. The kinases ATR and ATM have crucial roles in DDR pathway, such as maintaining replication fork stability and regulating CHK1 and CHK2.CHK1 can activate the G2/M inhibiting kinase WEE1 to maintain genomic integrity. Some miRNAs were shown to regulate the expression of components involved in HRR, NHEJ, Replication fork protection, TLS, and FA, but the interaction between miRNA and BER/ NER/ MMR lack sufficient evidence. (SLC, solute carrier superfamily; GST, Glutathione transferase; MT, Metallothionein)
HRR
HR deficiency is characteristic of many HGSOC cases (approximately 50%) and is considered a predictive biomarker of sensitivity to platinum agents and PARPis [42]. Restoration of HR pathway activity likely results in acquired resistance to platinum agents and PARPis in ovarian cancer patients with HR deficiency [43]. Notably, miRNAs have been revealed to impede DDR by directly targeting components of the DDR response, leading to reduced drug resistance [44]. miR-146 targets BRCA1 and is associated with the response to double-strand breaks (DSBs) [45]. Overexpressed miR-182 and miR-9 mediate the downregulation of BRCA1 and increase sensitivity to cisplatin and PARPis in ovarian cancer [46, 47]. miR-96 directly targets the coding region of RAD51 and the 3'-UTR of REV1 and decreases the efficiency of HRR [43]. miR-1255b, miR-193b*, and miR-148b* (“*” indicates minor products at low concentrations) can target the transcripts of the HR-mediated DSB repair factors BRCA1, BRCA2, and RAD51, respectively, thereby regulating PARPi sensitivity [48]. miR-506, miR-103 and miR-107 are robust clinical markers for the chemotherapy response and survival in patients with ovarian cancer and can sensitize cancer cells to DNA damage by directly targeting RAD51 and inhibiting the formation of RAD51 foci [49, 50]. Importantly, reversion mutations in BRCA1/2, RAD51C, and PALB have been identified during prolonged exposure to platinum agents and PARPis and in post-progression biopsies. The restoration of the open reading frame by these mutations leads to the functional restoration of HRR [51, 52]. Furthermore, HSP90 was found to mediate the stabilization of BRCA1, which interacts with the PALB2-BRCA2-RAD51 complex. This interaction was found to be essential for RAD51 focus formation and for conferring PARPi and cisplatin resistance [53]. Combination therapy with an HSP90 inhibitor and platinum is an innovative antitumor strategy that has the potential to reverse platinum resistance in ovarian cancer [54, 55].
The MRE11-RAD50-NBS1 (MRN) complex, an important factor of HRR, first detects DNA damage and then activates signaling molecules [56]. In addition, it exerts nuclease activity to resect DNA ends, guiding HRR. Furthermore, recombinant human cytoplasmic dynein light chain 1 (DYNLL1) was found to bind directly to MRE11 to limit its end resection activity. Thus, downregulation of DYNLL1 restores HR-mediated DNA DSB repair, thereby inducing chemoresistance and PARPi resistance in ovarian cancer [57]. Additionally, loss-of-function mutations in the TP53BP1 gene result in decreased 53BP1 protein expression and facilitate BRCA1-independent DNA end resection, which accounts for platinum and PARPi resistance [58].
Given the expanding role of immune checkpoint inhibitors as therapeutic agents, the interaction of tumor DNA damage and repair with the immune response has recently come into focus. HGSOC patients with BRCA mutation and homologous recombination deficiency (HRD) were found to exhibit increases in CD3 + /CD8 + tumor-infiltrating lymphocytes (TILs), immunohistochemical staining for PD-1/PD-L1, and neoantigen load. Moreover, wild-type BRCA1/2 ovarian tumors with mutations in RAD51, ATM, and ATR had higher predicted neoantigen levels than HR-proficient tumors [59, 60]. Mu Chen et al. showed that DNA damage resulted in the production of many DNA fragments in the cytoplasm, leading to increased antigen presentation on the cell surface and activation of the immune response [61]. However, a clinical trial of avelumab did not show an improved response in patients with BRCA1/2-mutated ovarian cancer (NCT01772004). Thus, additional clinical trials are warranted to determine the complexities of the interactions between DNA damage and immunomodulatory agents.
NHEJ
NHEJ repairs DNA DSBs by competing with HRR during the repair process, and its machinery includes TP53BP1, DNA-PK, etc. [62] miRNAs play important roles in regulating the expression of these NHEJ-related genes [39]. miR-136 overexpression downregulates DNA-PK, cell cycle-related genes, and antiapoptotic genes, resensitizing ovarian cancer cells to paclitaxel [63]. miR-622 suppresses NHEJ and facilitates HR-mediated DSB repair by targeting the Ku complex. Therefore, high expression of miR-622 in BRCA1-deficient HGSOC cells induces platinum and PARPi resistance [64]. DNA-PK, composed of DNA-PKcs and the DNA end-binding Ku70/80 heterodimer, has emerged as an intriguing therapeutic target within the NHEJ pathway [65, 66]. This heterodimer can recognize DSBs and form the Ku-DNA complex, which can recruit DNA-PKs to DSB sites [67]. DNA-PKcs plays a major role in promoting NHEJ through autophosphorylation and recruitment of downstream effectors, such as endonucleases (Artemis) [68] and polymerases (DNA POLM (Pol µ) and POLL (Pol λ)) [69, 70]. DNA-PK inhibition was found to induce restoration of HR function and resulted in resistance to PARPis in patient-derived ovarian cancer xenografts [71]. Ectopic expression of XRCC5/Ku80 [66] and XRCC6/Ku70 [65] induces platinum and PARPi resistance. Crucially, TP53BP1 can promote NHEJ and reduce BRCA1-mediated HRR by restricting DSB resection and antagonizing BRCA2/RAD51 loading in BRCA1-deficient cells [72]. The shieldin complex (comprising SHLD1, SHLD2, and SHLD3), an effector complex of 53BP1, regulates 53BP1-dependent NHEJ in various settings and impacts resistance to PARPis in HRD-defective cells [73, 74]. Finally, XRCC4, DNA ligase IV (LIG4) and XLF are central components of end ligation.
Replication fork protection
Replication fork protection contributes to genome stability in a manner independent of DSB-induced HR, leading to chemoresistance and PARPi resistance [75]. PARP1, BRCA1 and BRCA2 play key roles in protecting the replication fork under replication stress (RS) conditions [76, 77]. PTIP, PARP1 and CHD4 deficiency in BRCA-deficient cells prevent the recruitment of the MRE11 nuclease to stall replication forks and subsequently protect nascent DNA from degradation, thus conferring chemoresistance and PARPi resistance [78]. In both cells and patients with BRCA2 mutation, EZH2 downregulation leads to inhibition of the MUS81 nuclease, which restores DNA replication fork protection, leading to PARPi resistance [79]. miRNA-493-5p significantly preserves replication fork stability in BRCA2-mutant ovarian cancer cells through downregulation of MRE11 and CHD4, conferring platinum and PARPi resistance [10]. However, restoration of miR223-3p expression, which delays the repair of the replication fork, leads to genomic instability and enhances drug sensitivity in BRCA1-deficient OC [80].
NER and BER
NER is responsible for repairing single-stranded DNA damage, and 8% of HGSOC patients exhibit alterations in some NER genes, according to The Cancer Genome Atlas (TCGA) database [81]. The NER signaling pathway can repair platinum-induced adducts, therefore, upregulation of NER genes, including ERCC1, ERCC2-XPD, ERCC3-XPB, ERCC4-XPF, ERCC5-XPG, ERCC6, ERCC8 and XPA, might mediate chemoresistance [63]. Indeed, overexpression of ERCC1 or XPF not only increased platinum resistance but also decreased the toxicity of olaparib [82]. Although certain NER gene mutations (ERCC6-Q524* and ERCC4-A583T) were found to be functionally associated with platinum sensitivity in vitro, these NER alterations did not affect HR or confer sensitivity to PARPis.
BER is accelerated by PARPs and the scaffold protein XRCC1. Currently, it has been reported that the BER pathway has both positive and negative associations with platinum resistance. Although BER pathway intermediates underlie the efficacy of PARPis, they mediate the activity of PARP family proteins (especially PARP1) to initiate repair, resulting in PARPi resistance.
MMR deficiency
MMR defects in OC are relatively underinvestigated, although they are the most common cause of hereditary ovarian cancer after BRCA1/2 mutations. The MMR pathway contains seven proteins (MSH2, MSH3, MSH6, MLH1, MLH3, PMS1, and PMS2) [66]. The frequency of MMR deficiency (loss of any protein) reportedly ranges from 2 to 29% in patients with ovarian cancer [67]. A small number of studies have suggested that MMR deficiency is associated with drug resistance, but the results were inconclusive [83,84,85,86]. The possible role of MMR defects in drug resistance in ovarian cancer deserves further investigation. Currently, MMR deficiency is proposed to occur due to loss of ineffective MMR activity, replication fork stalling, the inability to recognize DNA damage, an increase in the net replicative bypass of cisplatin adducts and modulation of the level of recombination-dependent bypass [87, 88].
Other DDR pathways
The FA core complex consists of at least 10 FA-associated proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, FAAP100, FAAP20 and FAAP24) [89]. Inhibition of components of the FA repair pathway such as FA complementation group D2 (FANCD2) and FANCI, can increase sensitivity to chemotherapeutic agents [90]. miR-15a-5p, miR-494-3p and miR-544a potentially inhibit the entire FA/HR pathway [91].
TLS is mediated by DNA polymerases (e.g., Pol η and REV1). It increases the tolerance of tumor cells to platinum-induced DNA adducts and results in platinum resistance [92]. Pol η and REV1 are translesion DNA polymerases [93]. Upregulation of miR-93 might reverse resistance through targeting of DNA Pol η [92]. It has been reported that miR-96 can prevent the emergence of chemoresistance by inhibiting REV1-mediated TLS.
Dysregulated cancer-associated signaling pathways
A series of signaling pathways (Fig. 4) collectively regulate biological processes in human malignancies and are associated with the proliferation, invasion and therapeutic resistance of cancer cells [94]. The expression of signaling pathway components can be modulated by miRNAs through miRNA–mRNA binding, typically to miRNA target sites in the mRNA 3’-UTR [40, 95]. Although cancer-associated signaling pathways are complex, the identification of potential therapeutic targets is promising.

Dysregulation of cancer-associated signal pathway. A series of signal pathways collectively regulates the biological process in human malignancies, which is associated with the proliferation, invasion and therapeutic resistance. The signaling pathways mainly include NFκB, PI3K/Akt, JAK/STAT, Notch, GAS6/AXL, TGF-β, MAPK, Hippo/YAP patwhay. Some miRNAs have ability to regulate the key members of these mentioned pathway, including JAK/STAT, GAS/AXL, MAPK, PI3K/Akt, NFκB,, TGF-β, Hippo/YAP, but there are no investigations about the interaction between miRNAs and Notch in ovarian cancer. The dysregulated cancer-associated signal pathway interfere with apoptosis, cell cycle, and immune status, resulting in multidrug resistance. Molecule targets in these pathway may provide a new approach for drug resistance in OC. The γ-secretase inhibitor DAPT, c-Myc targeting small molecule JQ1, an inhibitor of NFκB DHMEQ suppress the proliferation and induce apoptosis to reversing drug resistance in OC. (JQ1, novel cell-permeable small molecule; BAD, Bcl-2 antagonist of death; IKKα, inhibitor of nuclear factor-κB subunit-α; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor-κB; DHMEQ, Dehydroxymethylepoxyquinomicin; MDSCs, Myeloid-derived suppressor cells; CSCs, cancer stem cells;BEZ235,a dual PI3K/mTOR inhibitor; DAPT, γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester)
NFκB signaling pathway
NFκB can perform a biphasic function in ovarian cancer. It plays an anticarcinoma role in ovarian cancer cells and renders them sensitive to apoptosis induced by carboplatin and paclitaxel, but it also has carcinogenic effects on promoting aggressiveness and chemoresistance in ovarian cancer cells and confers resistance to these therapeutic agents [96]. Common chemotherapeutic drugs, including taxanes, platinum agents, vinca alkaloids and erlotinib, activate NFκB and its prosurvival downstream targets, which contribute to chemoresistance [97]. Activation of the NFκB pathway is correlated with platinum resistance and leads to poor prognosis in patients with ovarian cancer [98]. Mechanistically, increased nuclear translocation of the p65 subunit and phosphorylation of inhibitor of IκB kinase subunits alpha and beta are markers of NFκB activation, which promotes chemoresistance [99]. Moreover, NF-κB p65 increases miR-200b/c expression by binding to its promoter, subsequently sensitizing ovarian cancer cells to cisplatin [100]. It also regulates the downstream miRNAs miR-452-5p and miR-335-5p through the NF-κB TFs RelA and RelB, preventing the recurrence of OC [101]. Moreover, the NF-κB signaling pathway has been implicated in immunosuppression and immune evasion in ovarian cancer cells partly via NFκB-dependent production of IL-6, which impairs DCs but generates and recruits immunosuppressive MDSCs, and IL-8, which increases the expression of the immunosuppressive enzyme arginase [102]. Dehydroxymethylepoxyquinomicin (DHMEQ), an inhibitor of NFκB, induces apoptosis, increases the response to platinum-based drugs and reverses immunosuppression in ovarian cancer cells [102, 103].
PI3K/Akt pathway
The PI3K/Akt pathway is frequently upregulated in ovarian cancer, and activated PI3K/Akt signaling contributes to increased cancer cell chemoresistance [104, 105]. Many miRNAs have been found to modulate the PI3K/Akt pathway, influencing ovarian cancer chemosensitivity [106]. miR-337-3p directly targets PIK3CA and PIK3CB, suppresses the proliferation of epithelial ovarian cancer cells and reverses resistance [107]. The let-7 miRNA family deregulates this pathway by governing PI3K and Akt1 phosphorylation and activity [108]. However, miR-20a and miR-200c activate and upregulate this pathway, contributing to paclitaxel resistance [109]. The aberrant PI3K-Akt signaling in tumor cells is attributed to the platinum-resistant phenotype, and the combination of cisplatin and LY-294002 (a PI3K-Akt dual kinase inhibitor) was found to prevent 3D spheroid formation and sensitize cells to cisplatin [110]. Furthermore, mTOR is a key downstream signaling kinase in the PI3K/Akt pathway [111]. Activated mTOR signaling can trigger epithelial–mesenchymal transition (EMT) and promote the maintenance of cancer stem cells (CSCs), resulting in chemoresistance in ovarian cancer patients, and treatment with BEZ235 (a dual PI3K/mTOR inhibitor) might be a promising approach for reversing chemoresistance [112]. In addition, miR-497 and miR-199a were found to quantitatively control mTOR expression to induce apoptosis in ovarian cancer cells [106].
JAK/STAT pathway
Following the phosphorylation of JAK, STAT is phosphorylated and activated, after which its nuclear translocation induces the transcription of its target genes involved in growth and apoptosis. M Koti et al. reported that STAT1 was the most significantly differentially expressed gene between chemoresistant and chemosensitive HGSOC. Upregulation of STAT1 is associated with platinum resistance [113]. c-Myc is a downstream target of the JAK/STAT signaling pathway and is linked with the malignancy and chemotherapeutic response of OC [114]. The novel cell-permeable small molecule JQ1 can target c-Myc to suppress the proliferation and induce the apoptosis of OC cells. Along with chemotherapeutic agents and PARPis, JQ1 warrants further investigation regarding its ability to reverse drug resistance in OC patients through interaction with the JAK-STAT signaling pathway [115]. This pathway is also regulated by miRNAs, and miRNA interactions are linked to drug resistance. Restoration of miR-503-5p expression can block the downstream JAK2/STAT3 pathway through the binding of this miRNA to the 3’-UTR of the mediator CD97 [116]. miR-340 can also directly target LGR5, FHL2, CTNNB1, and BAG3 to inhibit the JAK/STAT3, Wnt/β-catenin, Notch and PI3K/Akt pathways, respectively [117]. miR-637 is regulated by competing endogenous RNAs (ceRNAs) and is involved in five signaling pathways, including the JAK/STAT3, Wnt/β-catenin, and PI3K/Akt signaling pathways, in OC [118]. Additionally, the JAK/STAT pathway can exert effects on ovarian cancer by shaping immune cell infiltration. Interferon-mediated activation of STAT1 leads to the expression of the downstream target CXCL10, which is key to the trafficking and differentiation of effector Th1 CD4 + cells, natural killer (NK) cells and CD8 + cells [113]. Moreover, attenuation of the JAK/STAT3 signaling pathway mediated by overexpression of miR-217 can suppress M2 macrophage polarization and regulate the immune status [119].
Notch signaling pathway
The Notch signaling pathway is activated by the binding of ligands to Notch receptors. Following proteolytic cleavage of Notch by γ-secretase (an instrumental proteolytic enzyme in the Notch pathway), the active NICD fragment is translocated to the nucleus, where it induces the transcription of Notch target genes through interaction with CSL transcriptional regulators [120]. Aberrant Notch pathway can cause drug resistance in ovarian cancer cells, whereas Notch knockdown can increase platinum sensitivity through downregulation of ABCC1 and ABCB1 [121, 122]. In addition, inhibition of the Notch signaling pathway can induce apoptosis and reverse drug resistance. The γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT) can induce apoptosis by downregulating Notch signaling, in turn reversing platinum resistance in ovarian cancer cells [123, 124]. In addition, suppression of Notch signaling can increase apoptosis in ovarian cancer cells in animal models and reverse resistance to cisplatin and paclitaxel [121, 125].
GAS6/AXL pathway
GAS6 binding to AXL leads to AXL dimerization and autophosphorylation at tyrosine residues, which results in intracellular signal transduction [126]. The GAS6/AXL pathway influences drug resistance through interactions with other signals and regulation of the tumor microenvironment (TME). For instance, AXL-related EMT mediates resistance to chemotherapy and targeted therapy [127, 128]. The GAS6/AXL pathway also confers resistance through interactions with other signaling pathways, such as the PI3K, JAK/STAT and MAPK pathways, in ovarian cancer [129]. Moreover, the role of the GAS/AXL pathway in DDR has gradually been revealed in ovarian cancer. Inhibition of AXL (via bemcentinib or MYD1-72) resensitizes ovarian cancer cells to platinum, ATR inhibitors (ATRis) and PARPis by increasing DNA damage and inducing RS [130,131,132]. Furthermore, GAS6/AXL signaling promotes the generation of an immunosuppressive TME by modulating the expression of MHC and PD-L1 in neoplastic cells, increasing the secretion of immunosuppressive chemokines, and interfering with the infiltration of immune cells [133]. Although miR-515-3p regulates oxaliplatin sensitivity in mucinous ovarian cancer, in part by targeting AXL [134], there is still a lack of sufficient evidence demonstrating the roles of miRNAs in regulating the GAS6/AXL pathway.
Transforming growth factor-beta (TGF-β) pathway
Activation of the TGF-β signaling pathway occurs via the interaction of the dimeric TGF-β ligand with its specific transmembrane receptors [135]. TGF-β signaling is transduced via downstream SMAD effectors and non-SMAD proteins, such as AKT and MAPK [136]. miRNAs can target the components of the TGF-β signaling pathway to mediate drug resistance in ovarian cancer. For instance, miR-33a-5p influences the expression of SMAD2/4 by targeting carnitine O-octanoyl transferase (CROT), which induces paclitaxel resistance in ovarian cancer [137]. Decreased miR-30a expression can result in upregulation of TGF-β and SMAD4 to ultimately activate autophagy, mediating cisplatin resistance in ovarian cancer [138]. However, miR-181a plays an unappreciated role in mediating resistance in HGSOC via the activation of TGF-β signaling by directly targeting SMAD7 [139].
The TGF-β pathway has biphasic effects and acts as a tumor suppressor at early stages but later stimulates cancer progression by impacting tumor cells and their microenvironment [135]. Aberrant activation of this pathway blocks apoptosis and confers chemoresistance on ovarian cancer cells [140]. In addition, the TGF-β pathway plays a vital role in platinum resistance via canonical downstream EMT-related molecules [141]. The TGF-β pathway also suppresses immunity within the TME and contributes to chemoresistance. Daniel Newsted et al. developed an inhibitory antibody (anti-TGFBR2) to block TGF-β signaling and showed that this antibody improved the efficacy of chemotherapy and the limited antitumor immune response [142]. Moreover, the immunosuppressive effects of the TGF-β signaling pathway can be induced via CRISPR/Cas9-mediated knockout of TGF-β receptor 2 (TGFBR2) in TILs [143].
MAPK pathway
RAS/RAF/MEK/ERK are the classical and key signaling mediators in the MAPK pathway, and low-grade serous carcinoma (LGSC) of the ovary and peritoneum are characterized by MAPK pathway alterations and chemoresistance [144]. Excessive activation of Ras and Erk1/2 is positively and significantly correlated with chemoresistance in ovarian cancer [145]. Both the PI3K/Akt and Ras/MAPK signaling pathways can mediate the phosphorylation of the proapoptotic protein BAD, which leads to increased platinum resistance by inhibiting apoptosis [146]. miRNAs also play regulatory roles in the MAPK pathway by interfering with its components. For example, miR-634 can directly repress GRB2, ERK2 and RSK2, hence, inhibition of the Ras-MAPK pathway restores chemosensitivity in ovarian cancer cells [147]. Low levels of miR-508/miR-18a and increased expression of MAPK1 and ERK were identified in ovarian cancer, while miR-508 mimics were found to repress MAPK1 and ERK, resulting in suppression of EMT and the malignant progression of cancer cells [148, 149].
Hippo/yes-associated protein (YAP) pathway
The Hippo pathway confers resistance to therapeutic agents that are commonly used to treat ovarian cancer [150, 151]. YAP and its paralog TAZ are the main downstream effectors of the Hippo–YAP pathway and act as transcriptional coactivators, and their signaling has emerged as key mechanism of drug resistance [152, 153]. YAP and TAZ mediate gene transcription by binding to TFs, such as the TEA domain family (TEAD) proteins, to promote tumor progression and resistance [153, 154]. miRNAs can regulate the expression of YAP1 and modulate the Hippo pathway, but the regulatory mechanism involved remains vague. miR-509-3p, miR-509–3-5p [155] and miR-141 [156] are associated with cisplatin resistance via YAP1 and the Hippo signaling pathway. It is hypothesized that miR-509–3-5p can directly regulate YAP1 expression by targeting its coding region [155].
Epigenetic modifications
Epigenetic regulation refers to the effects of heritable changes in gene expression without DNA sequence changes. DNA methylation, histone modification and noncoding RNA (ncRNA) activity (Fig. 5) are common epigenetic regulatory mechanisms [157]. Increasing evidence shows that abnormal epigenetic regulation leads to tumor drug resistance.

Epigenetic modification. Epigenetic processes regulate gene expression through DNA methylation, histone modification, and non-coding RNA (ncRNAs) without altered DNA sequences. Hypermethylation of ABCB1 and demethylation of ABCG2 promoter lead to chemoresistance in ovarian cancer. The loss of RAD51C promoter methylation and the downregulation of BRCA1 methylation have been verified to cause drug resistance. The specific H3K27 methyltransferase EZH2 confers chemoresistance on ovarian cancer cells through H3K27 methylation. A subclass of miRNAs, “epi-miRNAs”, can modulate epigenetic regulators to impact therapeutic responses. miR-152 and miR-185 co-contribute to the cisplatin resistance by directly targeting DNMT1, miR-15a and miR-16 directly target the Bmi-1 (a member of Polycomb complexes). They may serve as potential epigenetic therapeutic targets. Epigenetic therapy including DNMTi and HDACi can increase the number of CD45 + immune cells, active CD8 + T and NK cells in TME, reducing immunosuppression. Thus, the epigenetic therapy combined with immunotherapy may be a promising therapeutic strategy for resistant OC. (HDACs, histone deacetylases; H3K27, histone H3 lysine 27; EZH2, enhancer of zeste homolog 2; DNMTis, DNA methyltransferase inhibitors; HDACis, histone deacetylase inhibitors; Bmi-1: a member of Polycomb complexes)
DNA methylation can affect therapeutic responses through various mechanisms, including affecting membrane transport, DNA repair, signaling pathway activity and apoptosis [158]. For instance, hypermethylation of ABCB1 and demethylation of the ABCG2 promoter may affect therapeutic efficacy and lead to chemoresistance in ovarian carcinoma, effects attributed to upregulation of P-gp [159, 160]. Abnormal methylation of genes involved in the PI3K-AKT, MAPK, and Wnt pathways and in EMT confers resistance on HGSOC cells [161,162,163]. In addition, loss of RAD51C promoter methylation and a low level of BRCA1 methylation have been verified to cause drug resistance. Homozygous RAD51C methylation and hypermethylation of BRCA1 could be predictive biomarkers for the treatment response in HGSOC [164]. Epigenetic alterations in the docking protein 2 (DOK2) gene can induce carboplatin resistance in ovarian cancer via suppression of apoptosis [165].
Histone modifications mainly include histone methylation and acetylation [166]. Min-Gyun Kim et al. confirmed the correlation between overexpression of histone deacetylases (HDACs) and cisplatin resistance in the ovarian cancer cell lines SKOV3 and OVCAR3 [167]. Recent data have provided novel insight into the role of histone H3 lysine 27 (H3K27) methylation in resistance mechanisms [168]. The specific H3K27 methyltransferase enhancer of zeste homolog 2 (EZH2) confers chemoresistance on ovarian cancer cells through H3K27 methylation [169]. In addition, Yujie Fang et al. revealed the roles of histone acetylation in a weak immune response and chemoresistance in ovarian cancer based on analysis of the TCGA and Gene Expression Omnibus (GEO) databases [170]. In terms of treatments, epigenetic therapy, including treatment with DNA methyltransferase and histone deacetylase inhibitors (DNMTis and HDACis, respectively), can increase the numbers of CD45 + immune cells, active CD8 + T cells and NK cells in the TME, reducing immunosuppression and the tumor burden through activation of type I interferon signaling in murine ovarian cancer [171, 172].
NcRNAs, comprising long ncRNAs (lncRNAs), small ncRNAs (sncRNAs) and circular RNAs (circRNAs), can regulate gene expression via epigenetic modification [173]. Most commonly, lncRNAs and circRNAs play roles in drug resistance by acting as miRNA sponges to regulate downstream gene expression [174]. “Epi-miRNAs” exert their effects by directly targeting epigenetic regulators, such as DNMTs and HDACs, or components of polycomb repressor complexes [175]. miRNAs affect mRNA transcription by binding to mRNA 3'-UTRs, leading to restoration of the expression of hypermethylated tumor suppressor genes [176]. Downregulated miR-152 and miR-185 contribute cooperatively to cisplatin resistance by directly targeting DNMT1 and may thus serve as epigenetic therapeutic targets [177]. miR-15a and miR-16 directly target the 3'-UTR of Bmi-1 (a component of Polycomb complexes), and their expression levels are significantly correlated with the Bmi-1 protein level in ovarian cancer [178].
Other mechanisms
Indeed, determining the complex mechanisms of resistance in ovarian cancer remains highly challenging. The resistance mechanisms cross-talk with each other and may interfere by generating an immunosuppressive environment, thus resulting in drug resistance, including immunotherapy resistance. An imbalance of Treg/Th17 cells [179], M2 polarization of macrophages [180], NK-cell exhaustion [181], and aberrant expression of IFNγ [182] and PD-L1 [183, 184] mediate immunosuppression, promoting tumor progression and resistance. miRNAs, such as miR-29a-3p, miR-21-5p, miR-1246, miR-29c, and miR-424, can modulate the expression of immune-related molecules to influence the immune status. Conversely, the TME or immunotherapy can regulate the expression of many miRNAs to promote drug resistance [185, 186]. The Hedgehog (Hh) and Wnt/β-catenin pathways can also promote T-cell exclusion and checkpoint inhibitor resistance [187, 188]. However, monotherapy with the Hh pathway inhibitor vismodegib did not show any significant antitumor activity in patients with ovarian cancer in a phase II clinical trial (NCT00739661) [189]. Interestingly, although Wnt signaling is a driver of resistance in ovarian cancer, the genetic driver of Wnt signaling is largely unknown [190].
In addition to the above mechanisms, aberrations in apoptosis, ferroptosis, autophagy, and endoplasmic reticulum stress (ER stress) act simultaneously or sequentially to enable cancer cells to survive treatment with antitumor agents. miR-130a [191] and miR-142-5p [192] have been reported to modulate apoptosis by targeting XIAP. An in-depth study of ferroptosis revealed that ferroptosis played a pivotal role in acquired resistance to sorafenib [193], EGFR tyrosine kinase inhibitors [194], and immunotherapy tolerance [195]. Intriguingly, autophagic flux can be driven by paclitaxel to promote paclitaxel resistance in ovarian cancer [196] and can be regulated by miR-30a [138], miR-200c [197], and miR-133a [198]. Furthermore, as a popular research topic, ER stress has a considerable impact on drug resistance in ovarian cancer [199]. The IRE1α/XBP1s pathway activates the unfolded protein response (UPR) during ER stress, resulting in microenvironment remodeling or resistance to treatment [199, 200].
Strategies for overcoming drug resistance
Clinical trials targeting transmembrane transport
Overexpression of ABCB1 (also known as p-gp/MDR1) mediates increased drug efflux. Increased drug efflux makes attaining a sufficient intracellular concentration of drugs challenging, thus resulting in drug resistance [201, 202]. The ABCB1 inhibitors (verapamil and elacridar) can reverse MDR through reducing the efflux of many drugs, including paclitaxel, olaparib, doxorubicin and rucaparib [24]. Moreover, PARPi resistance was evaluated in a mouse model and was found to be reversed by coadministration of tariquidar (a P-gp inhibitor) [26]. Although preclinical studies of the response to P-gp inhibitors have been performed, clinical trials of P-gp inhibitors are limited and outdated due to the severe toxic effects of these drugs [203]. For instance, P-gp inhibition increases the intracellular accumulation of paclitaxel, leading to paclitaxel-induced peripheral neuropathy [204]. NcRNAs play key roles in the regulation of ABC transporters and their clinical implications for MDR [8]. Thus, novel strategies for post-resistance therapy include delivering ncRNA mimics or antisense oligonucleotides of ncRNAs to interfere with ncRNA-ABC transporter axes. Moreover, codelivery of miR-129-5p and doxorubicin via polypeptide nanoparticles was found to effectively overcome MDR by directly inhibiting P-gp, thereby increasing intracellular doxorubicin accumulation and enhancing chemosensitivity [205].
Recently, antibody‒drug conjugates (ADCs), which can directly deliver potent cytotoxic drugs to cancer cells with appropriate target antigens while avoiding toxic effects on healthy cells, have gained increasing attention. Currently, the only FDA-approved ADC, namely, mirvetuximab soravtansine, has attracted widespread attention in the context of ovarian cancer drug resistance. A phase III clinical trial, MIRASOL (NCT04209855), is underway to compare the efficacy of chemotherapy and mirvetuximab soravtansine in FRα-positive, platinum-resistant HGSOC. The novel ADC BA3011 can target the Axl receptor on cancer cells through conditionally active biologics technology. A phase II clinical trial is underway to evaluate the combination of BA3011 and durvalumab in patients with platinum-resistant HGSOC (NCT04918186). MUC16 is another common target for platinum-resistant ovarian cancer treatment evaluated in two completed phase I trials (NCT01335958 [206] and NCT02146313 [207]). The results showed that the anti-MUC16 ADC had a tolerable safety profile and encouraging antitumor activity in patients with platinum-resistant ovarian cancer with high MUC16 expression. Additionally, down-regulation of some miRNAs could lead to abnormal MUC16 levels in OC. Thus, their up-regulation or mimics could be potential options along with anti-MUC16 for OC patients [208]. Mesothelin is an glycoprotein overexpressed on the surface of cancer cells. Two phase I clinical trials (NCT01469793/NCT02751918) evaluated a novel anti-mesothelin ADC in platinum-resistant ovarian cancer. Conclusions drawn from these trials indicated the tolerability and promising clinical activity of anetumab ravtansine combined with PEGylated liposomal doxorubicin [209], although the results of previous trials were inconsistent. Another ADC drug Zilovertamab Vedotin, targeting ROR1, was applicated in the II-phase clinical trials (NCT04504916). ROR1 also can be targeted by miR‑382, which might serve as another option for OC [210]. Additionally, HER2, TROP2, DLL3, and Nectin-4 are major targets of ADCs. Combination strategies with ADCs have shown considerable promise as emerging therapies in further investigations and clinical trials [211].
Clinical trials targeting DDR
The HRR pathway contributes to a key mechanism of acquired platinum and PARPi resistance in ovarian cancer. DNA repair-targeted therapy is a promising precision medicine strategy for ovarian cancer. Many clinical trials, including trials evaluating drugs targeting ATR, ATM, WEE1, checkpoint kinase 1/2 (CHK1/2), BRCA1/2 and RAD51, have been designed to evaluate interference with DDR pathways to overcome platinum and PARPi resistance in ovarian cancer.
ATR/ATM kinase inhibitors
ATR/ATM kinases, key molecules in DDR, are potential therapeutic targets for overcoming drug resistance in ovarian cancer. miR-203a-3p mimics and ATMis were reported to synergistically hinder OC progression, which could serve as a potential therapeutic option for OC [212]. It has been reported that ATRis can reverse PARPi resistance by blocking RAD51 loading onto DSBs and disrupting fork protection in human-derived cell lines [213]. An increasing number of clinical trials have evaluated the efficacy of ATRi or ATMi in combination with chemotherapeutic agents or PARPis. An interventional and crossover phase II randomized clinical trial (NCT02595892) was the first randomized clinical trial of an ATRi and demonstrated the benefit of adding berzosertib to gemcitabine for the treatment of platinum-resistant HGSOC [214]. M4344 enhances the activity of clinical DNA-damaging agents, including topoisomerase inhibitors, gemcitabine, cisplatin, and talazoparib, in advanced solid tumors [215]. Recently, another single-group interventional phase I trial (NCT04149145) in patients with PARPi-resistant HGSOC was just announced, in which a combination regimen of M4344 (an ATRi) plus niraparib will be evaluated.
WEE1 inhibitors
WEE1 is a vital target in the HRR pathway, and WEE1 inhibitors have been widely evaluated in combination with chemotherapeutic agents or PARPis in many ongoing clinical trials. A phase Ib nonrandomized, multicenter study (NCT04516447) in patients with platinum-resistant ovarian cancer evaluated the preclinical activity of ZN-c3 in combination with carboplatin, PLD, paclitaxel, and gemcitabine individually. The WEE1 inhibitor MK-1775 in combination with carboplatin or gemcitabine hydrochloride was tested in two phase II trials (NCT01164995 and NCT02272790). Adavosertib combined with chemotherapy showed preliminary therapeutic efficacy in platinum-resistant ovarian cancer, but the hematologic toxicity of this combination may limit its application [216, 217]. In addition, a phase I/II clinical trial of the WEE1 inhibitor ZN-c3 combined with niraparib was conducted in patients with platinum-resistant ovarian cancer (NCT05198804), but no results have been published. In addition, a conference abstract (ASCO 2021) reported that adavosertib alone or in combination with olaparib demonstrated efficacy in patients with PARPi resistance. Although grade 3 and 4 toxicities could be managed, they led to dose interruption and reduction (NCT03579316).
CHK1/2 inhibitors
Investigations of CHK1/2 inhibitors have been limited until recently. CHK1 inhibitors play a preliminary role in the clinical treatment of PARPi-resistant HGSOC by inducing DNA damage and RS [218]. miRNA‑199b‑3p suppressed CHK1 expression and EMT transition, which may represent a promising therapeutic target for ovarian cancer [219]. A phase Ia dose-escalation trial, the combination of PHI-101 (a selective CHK2 inhibitor) with a PARPi showed good safety and tolerability, and is a potential therapeutic regimen for platinum-resistant recurrent ovarian cancer [220]. In summary, the therapeutic efficacy and underlying mechanisms of CHK1/2 inhibitors are unknown, and further studies are attractive and needed.
Downregulation of BRCA1/2 and RAD51
The reactivation of the HRD genes BRCA1/2 and RAD51 is the genetic mechanism of PARPi resistance and confers a dismal prognosis [221]. Cediranib can potentially reverse PARPi resistance by downregulating BRCA1/2 and RAD51 and ultimately resensitizing cells to PARPis [222]. However, this combination regimen showed activity in patients with ovarian cancer who progressed on PARPi therapy in another phase II trial (EVOLVE) [221]. However, in a randomized phase II trial (BAROCCO), the combination of a PARPi and cediranib did not improve PFS in platinum-resistant ovarian cancer patients compared with chemotherapy alone [223]. The underlying mechanisms of these combination strategies have not been thoroughly elucidated.
Clinical trials targeting signaling pathways
Targeting the PI3K/AKT pathway
The PI3K/AKT pathway is regarded as a common oncogenic signaling pathway. Approximately 70% of ovarian cancer patients have aberrations in the PI3K/AKT signaling pathway, and mutations in the gene encoding the catalytic subunit PIK3CA occur in 6–12% of patients [224, 225]. CYH33, a PI3Kα inhibitor, exhibited a manageable safety profile and preliminary antitumor efficacy in patients with PI3KCA-mutant ovarian cancer (NCT03544905). In addition, a phase I clinical trial (NCT04586335) is underway to further evaluate the therapeutic efficacy of CYH33 in combination with olaparib in platinum-resistant ovarian cancer. In addition, a combination regimen of PARPi and copanlisib (a PI3K inhibitor) was tested in phase I/II trials (NCT03586661 and NCT05295589) in patients with BRCA-mutated, resistant ovarian cancer. PI3K inhibition is believed to lead to downregulation of the BRCA1/2 proteins, which enhances HRR deficiency and the efficacy of PARPis. In addition, the Akt inhibitor afuresertib is under assessment in an interventional randomized clinical trial (NCT04374630) in patients with platinum-resistant ovarian, fallopian tube, or peritoneal cancer.
Targeting the GAS6-AXL pathway
The GAS6-AXL signaling pathway is another crucial player in drug resistance in ovarian cancer. Carboplatin/olaparib plus AVB-500, a selective inhibitor of GAS6-AXL, can increase DNA damage and RAD51 focus formation and slow replication fork progression, resulting in rapid death of ovarian cancer cells in vitro and decreased tumor burden in vivo [131]. A phase 1b trial (NCT03639246) evaluated AVB-S6-500 in combination with paclitaxel or PEGylated liposomal doxorubicin. PROC patients may derive the greatest benefit from AVB-500 treatment [226]. Another phase I/II clinical trial (NCT04019288) was designed and was commenced in 2019 to evaluate the safety and clinical benefit of durvalumab plus AVB-S6-500 (an AXL inhibitor) in platinum-resistant ovarian cancer patients. It was reported that the combination of AVB-S6-500 and durvalumab was tolerable in PROC patients [227]. Moreover, a humanized anti-AXL monoclonal antibody, tilvestamab, blocks GAS6-mediated AXL receptor activation and has been tested in platinum-resistant HGSOC patients (NCT04893551), but no results have been published.
Targeting the MAPK pathway
The RAS/RAF/MEK/ERK kinase pathway, also known as the MAPK pathway, participates in cancerogenesis, metastasis and resistance. Although VS-6766 (a RAF/MEK inhibitor) exhibited antitumor activity in platinum-resistant low-grade serous ovarian cancer and endometrial adenocarcinoma with RAF–RAS–MEK pathway mutations, patients later experienced progression. Thus, the use of VS-6766 in combination regimens warrants further evaluation. The combination of defactinib (a FAK inhibitor) and VS-6766 was evaluated for its pharmacodynamic activity in PROC patients (NCT03875820). In addition, combined PI3K/mTOR and ERK inhibition can reverse therapeutic resistance in ovarian cancer cell lines, but the clinical efficacy of these agents requires further preclinical determination [228]. ONC201, a dual inhibitor of Akt and ERK, is being evaluated in combination with paclitaxel for the treatment of platinum-resistant ovarian cancer in an ongoing phase II trial (NCT04055649). The unpublished results of this trial are likely to provide strong evidence for the development of novel treatment strategies.
Targeting the Notch pathway
The Notch pathway is linked to the proliferation, migration, and drug resistance of ovarian cancer cells [229]. Pretreatment with the γ-secretase inhibitor DAPT increased the sensitivity of PROC to platinum by downregulating the Notch pathway, suggesting a promising approach for treating patients with PROC [123, 230]. The SIERRA open-label phase Ib trial (NCT01952249) was conducted to observe the safety and efficacy of demcizumab (potent inhibitor of the Notch pathway) combined with paclitaxel for the treatment of platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer. The results indicated that this combination had a manageable toxicity profile and showed a clinical benefit rate of 42% in patients with heavily pretreated platinum-resistant ovarian cancer [231].
Targeting the NF-κB pathway
Activation of the NF-κB pathway contributes to aggressive behaviors, mediating the oncogenic activity of DDR-related genes [232]. Furthermore, the scientific literature supports the interaction and colocalization of NF-κB and BRCA1 [233]. Denosumab, an inhibitor of RANKL (an NF-κB ligand) and NF-κB signaling, was evaluated in ovarian cancer patients with BRCA1 mutations. However, the pilot study (NCT03382574), which compared growth and metastatic spread between the denosumab and control groups, was terminated early due to the inability to enroll participants [234].
In addition, components of the cell cycle and apoptosis machineries, including topoisomerase I (NCT04029909), P53 (NCT03113487), and CDK2 (NCT05252416), could be promising treatment targets. An increasing number of early-phase clinical trials involving the glucocorticoid receptor (GR), FAK, and HER2 are underway. Although the results are pending, these studies could provide sufficient rationale for the involvement of these signaling pathways. The restoration of miR-206 expression represented a potential anti-FAK strategy to control ovarian cancer progression in EOC lines [221]. Some miRNAs were designed to target 3’-UTR of HER2 to inhibit HER2 protein expression [235]. However, the miRNA targeting drugs lacks application in clinical trials. Emerging peptide vaccines aimed to elicit a host immune response against tumor-specific antigens, such as p53, HER2, NY-ESO-1, and FRα, are being evaluated [236]. However, cancer vaccines have had limited clinical success, and research on most peptide vaccines for gynecological malignancies is still at an exploratory stage.
Clinical trials targeting epigenetic modifications
Increased DNA methylation and histone modifications can alter the transcription of tumor suppressors and genes related to the apoptotic response to chemotherapy [224, 237]. An increasing number of trials have provided insight into the role of epigenetic modifications in the drug resistance of ovarian cancer. Researchers have attempted to overcome platinum resistance by coadministration of hypomethylating agents. For instance, guadecitabine plus carboplatin was tolerable and resulted in a detectable clinical response in patients with PROC in a phase I clinical trial [238]. However, in the phase II trial, the guadecitabine plus carboplatin group did not show any superior effect compared with the traditional chemotherapy group [239]. Furthermore, combination regimens of hypomethylating agents with PARPis or immune checkpoint inhibitors are increasingly being developed. Talazoparib and ZEN003694 (a BET inhibitor) are being evaluated in an ongoing phase II clinical trial (NCT05327010) for recurrent PARPi-resistant cancer. This series of novel therapeutic regimens has spurred the development of triplet regimens. In an ongoing phase I trial (NCT04840589), ZEN003694 and nivolumab alone or combined with ipilimumab were assessed in PROC patients. In addition, another combination therapy comprising CDX-1401 (a vaccine), atezolizumab, and guadecitabine was evaluated in a clinical trial (NCT03206047) to improve clinical efficacy. These innovative clinical trials are anticipated to provide therapeutic opportunities for drug-resistant patients.
Conclusion
With the increasing use of novel therapeutic drugs for ovarian cancer, the development of later-line treatments has been under enormous pressure. Recently, resistance to a variety of therapeutic drugs, such as PARPis, angiogenesis inhibitors, and immune checkpoint inhibitors, has been found to occur. In the past, therapeutic agents for ovarian cancer have been limited, and researchers have usually described the underlying mechanism and explored therapeutic strategies for overcoming resistance based on the drug classification. However, as increasing numbers of new agents are applied in clinical practice, the resistance mechanisms of these various new drugs must be identified, and these mechanisms may be similar or even identical to those of other drugs. Thus, the classification of drug resistance should not be confined to the drug category, and we should attempt to obtain insight into classification of resistance based on molecular mechanisms. The concept of drug resistance classification provides a sound basis for further research to develop more precise reversal strategies.
Although the resistance mechanisms of different agents are complicated, we classified miRNA-mediated mechanisms into four categories: abnormalities in transmembrane transport, dysregulation of DDR, dysregulation of signaling pathways and epigenetic modification. On the basis of the above four mechanisms, clinical trials of new agents are underway to overcome drug resistance. Notably, ADCs, a current research hotspot, hold promise for overcoming resistance in patients with ovarian cancer. The FDA's approval of mirvetuximab soravtansine-gynx for FRα-positive, platinum-resistant HGSOC was based on Study 0417 (SORAYA, NCT04296890) [240]. Thus, many additional ADCs against various targets, including NaPi2b, HER2/3, mesothelin, and MUC16, which are expressed in ovarian cancer, are under investigation [241]. Future innovative studies and targeted therapies with ADCs will provide opportunities for reversing drug resistance in ovarian cancer. In addition, another potential approach for reversing resistance is based on miRNAs [242]. Codelivery of miRNAs with chemotherapeutic agents is a promising option for overcoming resistance, but further investigations of the underlying mechanism and the clinical application of this strategy are needed [243]. Polypeptide nanoparticles carrying doxorubicin and miR-129-5p could be a promising and synergistic strategy to overcome drug resistance in ovarian cancer [205].
In the context of the increasing number of novel agents, our summary of the four resistance mechanisms of ovarian cancer provides a new concept for resistance classification by molecular mechanism, not by drug category. Given the intersections between drug resistance mechanisms, this concept is likely to result in the realization of “two birds with one stone” effects on the reversal of drug resistance in ovarian cancer. Furthermore, these findings are anticipated to have broad implications for the development of precise therapeutic approaches for reversing drug resistance in ovarian cancer. On this basis, umbrella trials can be carried out to explore the diagnostic and therapeutic targets of the four resistance mechanisms, and this may be a direction of future researches on drug resistance in ovarian cancer.
Availability of data and materials
No datasets were generated or analysed during the current study.
Abbreviations
- ADC:
-
Antibody–drug conjugate
- ATRi:
-
Ataxia telangiectasia and Rad3-related protein inhibitor
- ATMi:
-
Ataxia telangiectasia mutated protein inhibitor
- BCRP:
-
Breast cancer drug resistance protein
- BER:
-
Base excision repair
- ceRNA:
-
Competitive endogenous RNA
- CHK1/2:
-
Checkpoint kinase 1/2
- CSC:
-
Cancer stem cell
- DAPT:
-
N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester
- DDR:
-
DNA damage repair
- DHMEQ:
-
Dehydroxymethylepoxyquinomicin
- DNMTi:
-
DNA methyltransferase inhibitor
- DOK2:
-
Docking protein 2
- DYNLL1:
-
Dynein light chain 1
- EAH2:
-
Zeste homologue 2
- EMT:
-
Epithelial mesenchymal transformation
- ER:
-
Endoplasmic Reticulum
- FANC:
-
Fanconi anemia complementation group
- GST:
-
Glutathione transferase
- HDACs:
-
Histone deacetylase
- Hh:
-
Hedgehog
- HRD:
-
Homologous recombination deficiency
- HRR:
-
Homologous recombination repair
- HGSOC:
-
High-grade serous ovarian cancer
- HSP:
-
Heat shock protein
- LGSC:
-
Low grade serous carcinoma
- LRP:
-
Lung drug resistance-related protein
- MMR:
-
Mismatch repair
- MRP:
-
Multidrug resistance-related protein
- NCCN:
-
National Comprehensive Cancer Network
- ncRNA:
-
Non-coding RNA
- NHEJ:
-
Non-homologous end junction
- NER:
-
Nucleotide excision repair
- OS:
-
Overall survival
- PARPi:
-
Poly ADP-ribose polymerase inhibitor
- PFS:
-
Progression-free survival
- P-gp:
-
P-glycoproteins
- SHLD:
-
Shieldin
- TGF-β:
-
Transforming growth factor-beta
- TIL:
-
Tumor-infiltrating lymphocytes
- TLS:
-
Translesion DNA synthesis
- TEAD:
-
TEA domain family
- UCA1:
-
Urothelial carcinoma-associated 1
- 3’-UTR:
-
3’-Untranslated region
- XIAP:
-
X-linked inhibitor of apoptosis
- YAP:
-
Yes-associated protein
References
Cortez AJ, Tudrej P, Kujawa KA, Lisowska KM. Advances in ovarian cancer therapy. Cancer Chemother Pharmacol. 2018;81:17–38.
Ovarian Cancer — Cancer Stat Facts. [cited 2023 Aug 10]. Available from: https://seer.cancer.gov/statfacts/html/ovary.html.
Ray-Coquard I, Leary A, Pignata S, Cropet C, González-Martin A, Marth C, et al. Olaparib plus bevacizumab first-line maintenance in ovarian cancer: final overall survival results from the PAOLA-1/ENGOT-ov25 trial. Ann Oncol Off J Eur Soc Med Oncol. 2023;S0923–7534(23):00686–95.
DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim B-G, Oaknin A, et al. Overall Survival With Maintenance Olaparib at a 7-Year Follow-Up in Patients With Newly Diagnosed Advanced Ovarian Cancer and a BRCA Mutation: The SOLO1/GOG 3004 Trial. J Clin Oncol Off J Am Soc Clin Oncol. 2023;41:609–17.
González-Martín A, Pothuri B, Vergote I, Graybill W, Lorusso D, McCormick CC, et al. Progression-free survival and safety at 3.5years of follow-up: results from the randomised phase 3 PRIMA/ENGOT-OV26/GOG-3012 trial of niraparib maintenance treatment in patients with newly diagnosed ovarian cancer. Eur J Cancer Oxf Engl. 1990;2023(189):112908.
Richardson DL, Eskander RN, O’Malley DM. Advances in Ovarian Cancer Care and Unmet Treatment Needs for Patients With Platinum Resistance: A Narrative Review. JAMA Oncol. 2023;9:851.
Kandettu A, Adiga D, Devi V, Suresh PS, Chakrabarty S, Radhakrishnan R, et al. Deregulated miRNA clusters in ovarian cancer: Imperative implications in personalized medicine. Genes Dis. 2022;9:1443–65.
Wang Y, Wang Y, Qin Z, Cai S, Yu L, Hu H, et al. The role of non-coding RNAs in ABC transporters regulation and their clinical implications of multidrug resistance in cancer. Expert Opin Drug Metab Toxicol. 2021;17:291–306.
Yi C, Yu A-M. MicroRNAs in the Regulation of Solute Carrier Proteins Behind Xenobiotic and Nutrient Transport in Cells. Front Mol Biosci. 2022;9: 893846.
Meghani K, Fuchs W, Detappe A, Drané P, Gogola E, Rottenberg S, et al. Multifaceted Impact of MicroRNA 493–5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018;23:100–11.
Mihanfar A, Fattahi A, Nejabati HR. MicroRNA-mediated drug resistance in ovarian cancer. J Cell Physiol. 2019;234:3180–91.
Yonezawa A, Masuda S, Yokoo S, Katsura T, Inui K. Cisplatin and oxaliplatin, but not carboplatin and nedaplatin, are substrates for human organic cation transporters (SLC22A1-3 and multidrug and toxin extrusion family). J Pharmacol Exp Ther. 2006;319:879–86.
Fu S, Naing A, Fu C, Kuo MT, Kurzrock R. Overcoming platinum resistance through the use of a copper-lowering agent. Mol Cancer Ther. 2012;11:1221–5.
Hsu KF, Shen MR, Huang YF, Cheng YM, Lin SH, Chow NH, et al. Overexpression of the RNA-binding proteins Lin28B and IGF2BP3 (IMP3) is associated with chemoresistance and poor disease outcome in ovarian cancer. Br J Cancer. 2015;113:414–24.
Sun S, Zhao S, Yang Q, Wang W, Cai E, Wen Y, et al. Enhancer of zeste homolog 2 promotes cisplatin resistance by reducing cellular platinum accumulation. Cancer Sci. 2018;109:1853–64.
Kalayda GV, Wagner CH, Jaehde U. Relevance of copper transporter 1 for cisplatin resistance in human ovarian carcinoma cells. J Inorg Biochem. 2012;116:1–10.
Rashid K, Ahmad A, Liang L, Liu M, Cui Y, Liu T. Solute carriers as potential oncodrivers or suppressors: their key functions in malignant tumor formation. Drug Discov Today. 2021;26:1689–701.
Barbier RH, McCrea EM, Lee KY, Strope JD, Risdon EN, Price DK, et al. Abiraterone induces SLCO1B3 expression in prostate cancer via microRNA-579-3p. Sci Rep. 2021;11:10765.
Kuo MT, Chen HHW, Song I-S, Savaraj N, Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metastasis Rev. 2007;26:71–83.
Burger H, Zoumaro-Djayoon A, Boersma AWM, Helleman J, Berns EMJJ, Mathijssen RHJ, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2). Br J Pharmacol. 2010;159:898–908.
Kazmierczak D, Jopek K, Sterzynska K, Nowicki M, Rucinski M, Januchowski R. The Profile of MicroRNA Expression and Potential Role in the Regulation of Drug-Resistant Genes in Cisplatin- and Paclitaxel-Resistant Ovarian Cancer Cell Lines. Int J Mol Sci. 2022;23:526.
Beretta GL, Benedetti V, Cossa G, Assaraf YG, Bram E, Gatti L, et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem Pharmacol. 2010;79:1108–17.
Izquierdo MA, van der Zee AG, Vermorken JB, van der Valk P, Beliën JA, Giaccone G, et al. Drug resistance-associated marker Lrp for prediction of response to chemotherapy and prognoses in advanced ovarian carcinoma. J Natl Cancer Inst. 1995;87:1230–7.
Vaidyanathan A, Sawers L, Gannon A-L, Chakravarty P, Scott AL, Bray SE, et al. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel- and olaparib-resistant ovarian cancer cells. Br J Cancer. 2016;115:431–41.
Zhang B, Kang Z, Zhang J, Kang Y, Liang L, Liu Y, et al. Simultaneous binding mechanism of multiple substrates for multidrug resistance transporter P-glycoprotein. Phys Chem Chem Phys PCCP. 2021;23:4530–43.
Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AOH, Zander SAL, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84.
Zou Z, Zou R, Zong D, Shi Y, Chen J, Huang J, et al. miR-495 sensitizes MDR cancer cells to the combination of doxorubicin and taxol by inhibiting MDR1 expression. J Cell Mol Med. 2017;21:1929–43.
Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol. 2021;18:773–91.
Huang C, Qin L, Chen S, Huang Q. CircSETDB1 contributes to paclitaxel resistance of ovarian cancer cells by sponging miR-508-3p and regulating ABCC1 expression. Anticancer Drugs. 2023;34:395–404.
Shi C, Wang M. LINC01118 Modulates Paclitaxel Resistance of Epithelial Ovarian Cancer by Regulating miR-134/ABCC1. Med Sci Monit Int Med J Exp Clin Res. 2018;24:8831–9.
Stasiak P, Kaźmierczak D, Jopek K, Nowicki M, Rucinski M, Januchowski R. The Profile of MicroRNA Expression and Potential Role in the Regulation of Drug-Resistant Genes in Doxorubicin and Topotecan Resistant Ovarian Cancer Cell Lines. Int J Mol Sci. 2022;23:5846.
Lukanović D, Herzog M, Kobal B, Černe K. The contribution of copper efflux transporters ATP7A and ATP7B to chemoresistance and personalized medicine in ovarian cancer. Biomed Pharmacother Biomedecine Pharmacother. 2020;129: 110401.
Xiao F, Li Y, Wan Y, Xue M. MircroRNA-139 sensitizes ovarian cancer cell to cisplatin-based chemotherapy through regulation of ATP7A/B. Cancer Chemother Pharmacol. 2018;81:935–47.
Lee J-H, Chae J-W, Kim JK, Kim HJ, Chung JY, Kim Y-H. Inhibition of cisplatin-resistance by RNA interference targeting metallothionein using reducible oligo-peptoplex. J Control Release Off J Control Release Soc. 2015;215:82–90.
Ishikawa T, Ali-Osman F. Glutathione-associated cis-diamminedichloroplatinum(II) metabolism and ATP-dependent efflux from leukemia cells. Molecular characterization of glutathione-platinum complex and its biological significance. J Biol Chem. 1993;268:20116–25.
Zhang J, Xie S, Zhou L, Tang X, Guan X, Deng M, et al. Up-regulation of GSTT1 in serous ovarian cancer associated with resistance to TAXOL / carboplatin. J Ovarian Res. 2021;14:122.
Boušová I, Skálová L. Inhibition and induction of glutathione S-transferases by flavonoids: possible pharmacological and toxicological consequences. Drug Metab Rev. 2012;44:267–86.
Pilié PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol. 2019;16:81–104.
Peraza-Vega RI, Valverde M, Rojas E. Interactions between miRNAs and Double-Strand Breaks DNA Repair Genes, Pursuing a Fine-Tuning of Repair. Int J Mol Sci. 2022;23:3231.
Hu X-Y, Song Z, Yang Z-W, Li J-J, Liu J, Wang H-S. Cancer drug resistance related microRNAs: recent advances in detection methods. Analyst. 2022;147:2615–32.
Si W, Shen J, Zheng H, Fan W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin Epigenetics. 2019;11:25.
González-Martín A, Desauw C, Heitz F, Cropet C, Gargiulo P, Berger R, et al. Maintenance olaparib plus bevacizumab in patients with newly diagnosed advanced high-grade ovarian cancer: Main analysis of second progression-free survival in the phase III PAOLA-1/ENGOT-ov25 trial. Eur J Cancer Oxf Engl. 1990;2022(174):221–31.
Burdett NL, Willis MO, Alsop K, Hunt AL, Pandey A, Hamilton PT, et al. Multiomic analysis of homologous recombination-deficient end-stage high-grade serous ovarian cancer. Nat Genet. 2023;55:437–50.
He M, Zhou W, Li C, Guo M. MicroRNAs, DNA Damage Response, and Cancer Treatment. Int J Mol Sci. 2016;17:2087.
Zhang X, Wan G, Berger FG, He X, Lu X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol Cell. 2011;41:371–83.
Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell. 2011;41:210–20.
Sun C, Li N, Yang Z, Zhou B, He Y, Weng D, et al. miR-9 regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and PARP inhibition. J Natl Cancer Inst. 2013;105:1750–8.
Choi YE, Pan Y, Park E, Konstantinopoulos P, De S, D’Andrea A, et al. MicroRNAs down-regulate homologous recombination in the G1 phase of cycling cells to maintain genomic stability. eLife. 2014;3:e02445.
Liu G, Yang D, Rupaimoole R, Pecot CV, Sun Y, Mangala LS, et al. Augmentation of response to chemotherapy by microRNA-506 through regulation of RAD51 in serous ovarian cancers. J Natl Cancer Inst. 2015;107:djv108.
Huang J-W, Wang Y, Dhillon KK, Calses P, Villegas E, Mitchell PS, et al. Systematic Screen Identifies miRNAs that Target RAD51 and RAD51D to Enhance Chemosensitivity. Mol Cancer Res MCR. 2013;11(12):1564–73.
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI, et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017;7:984–98.
Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20.
Johnson N, Johnson SF, Yao W, Li Y-C, Choi Y-E, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A. 2013;110:17041–6.
Lombardi R, Sonego M, Pucci B, Addi L, Iannelli F, Capone F, et al. HSP90 identified by a proteomic approach as druggable target to reverse platinum resistance in ovarian cancer. Mol Oncol. 2021;15:1005–23.
Wang Y, Chen Q, Wu D, Chen Q, Gong G, He L, et al. Lamin-A interacting protein Hsp90 is required for DNA damage repair and chemoresistance of ovarian cancer cells. Cell Death Dis. 2021;12:786.
Altan B, Yokobori T, Ide M, Bai T, Yanoma T, Kimura A, et al. High Expression of MRE11-RAD50-NBS1 Is Associated with Poor Prognosis and Chemoresistance in Gastric Cancer. Anticancer Res. 2016;36:5237–47.
He YJ, Meghani K, Caron M-C, Yang C, Ronato DA, Bian J, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563:522–6.
Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013;3:68–81.
McAlpine JN, Porter H, Köbel M, Nelson BH, Prentice LM, Kalloger SE, et al. BRCA1 and BRCA2 mutations correlate with TP53 abnormalities and presence of immune cell infiltrates in ovarian high-grade serous carcinoma. Mod Pathol Off J U S Can Acad Pathol Inc. 2012;25:740–50.
Clarke B, Tinker AV, Lee C-H, Subramanian S, van de Rijn M, Turbin D, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol Off J U S Can Acad Pathol Inc. 2009;22:393–402.
Chen M, Huang B, Zhu L, Wang Q, Pang Y, Cheng M, et al. DNA Damage Response Evaluation Provides Novel Insights for Personalized Immunotherapy in Glioma. Front Immunol. 2022;13: 875648.
Dev H, Chiang TW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–65.
Zhao M, Li S, Zhou L, Shen Q, Zhu H, Zhu X. Prognostic values of excision repair cross-complementing genes mRNA expression in ovarian cancer patients. Life Sci. 2018;194:34–9.
Choi YE, Meghani K, Brault M-E, Leclerc L, He YJ, Day TA, et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Rep. 2016;14:429–39.
Sakamoto M, Kondo A, Kawasaki K, Goto T, Sakamoto H, Miyake K, et al. Analysis of gene expression profiles associated with cisplatin resistance in human ovarian cancer cell lines and tissues using cDNA microarray. Hum Cell. 2001;14:305–15.
Li G-M. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98.
Xiao X, Melton DW, Gourley C. Mismatch repair deficiency in ovarian cancer – molecular characteristics and clinical implications. Gynecol Oncol. 2014;132:506–12.
Riballo E, Kühne M, Rief N, Doherty A, Smith GCM, Recio M-J, et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol Cell. 2004;16:715–24.
Bebenek K, Pedersen LC, Kunkel TA. Structure-function studies of DNA polymerase λ. Biochemistry. 2014;53:2781–92.
Moon AF, Pryor JM, Ramsden DA, Kunkel TA, Bebenek K, Pedersen LC. Sustained active site rigidity during synthesis by human DNA polymerase μ. Nat Struct Mol Biol. 2014;21:253–60.
McCormick A, Donoghue P, Dixon M, O’Sullivan R, O’Donnell RL, Murray J, et al. Ovarian Cancers Harbor Defects in Nonhomologous End Joining Resulting in Resistance to Rucaparib. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23:2050–60.
Dev H, Chiang T-WW, Lescale C, de Krijger I, Martin AG, Pilger D, et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat Cell Biol. 2018;20:954–65.
Noordermeer SM, Adam S, Setiaputra D, Barazas M, Pettitt SJ, Ling AK, et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–21.
Gupta R, Somyajit K, Narita T, Maskey E, Stanlie A, Kremer M, et al. DNA Repair Network Analysis Reveals Shieldin as a Key Regulator of NHEJ and PARP Inhibitor Sensitivity. Cell. 2018;173:972–988.e23.
Liptay M, Barbosa JS, Rottenberg S. Replication Fork Remodeling and Therapy Escape in DNA Damage Response-Deficient Cancers. Front Oncol. 2020;10:670.
Classen S, Rahlf E, Jungwirth J, Albers N, Hebestreit LP, Zielinski A, et al. Partial Reduction in BRCA1 Gene Dose Modulates DNA Replication Stress Level and Thereby Contributes to Sensitivity or Resistance. Int J Mol Sci. 2022;23:13363.
Kanev P-B, Atemin A, Stoynov S, Aleksandrov R. PARP1 roles in DNA repair and DNA replication: The basi(c)s of PARP inhibitor efficacy and resistance. Semin Oncol. 2023;S0093–7754(23):00061–71.
Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee J-E, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–7.
Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19:1371–8.
Srinivasan G, Williamson EA, Kong K, Jaiswal AS, Huang G, Kim H-S, et al. MiR223-3p promotes synthetic lethality in BRCA1-deficient cancers. Proc Natl Acad Sci U S A. 2019;116:17438–43.
Ovejero-Sánchez M, González-Sarmiento R, Herrero AB. DNA Damage Response Alterations in Ovarian Cancer: From Molecular Mechanisms to Therapeutic Opportunities. Cancers. 2023;15:448.
Mesquita KA, Alabdullah M, Griffin M, Toss MS, Fatah TMAA, Alblihy A, et al. ERCC1-XPF deficiency is a predictor of olaparib induced synthetic lethality and platinum sensitivity in epithelial ovarian cancers. Gynecol Oncol. 2019;153:416–24.
Ercoli A, Ferrandina G, Raspaglio G, Marone M, Maggiano N, Del Mastro P, et al. hMSH2 and GTBP expression in advanced stage epithelial ovarian cancer. Br J Cancer. 1999;80:1665–71.
Marcelis CL, van der Putten HW, Tops C, Lutgens LC, Moog U. Chemotherapy resistant ovarian cancer in carriers of an hMSH2 mutation? Fam Cancer. 2001;1:107–9.
Samimi G, Fink D, Varki NM, Husain A, Hoskins WJ, Alberts DS, et al. Analysis of MLH1 and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2000;6:1415–21.
Helleman J, van Staveren IL, Dinjens WNM, van Kuijk PF, Ritstier K, Ewing PC, et al. Mismatch repair and treatment resistance in ovarian cancer. BMC Cancer. 2006;6:201.
Durant ST, Morris MM, Illand M, McKay HJ, McCormick C, Hirst GL, et al. Dependence on RAD52 and RAD1 for anticancer drug resistance mediated by inactivation of mismatch repair genes. Curr Biol CB. 1999;9:51–4.
Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, et al. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–85.
Burkitt K, Ljungman M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol Cancer. 2008;7:24.
Yuan M, Wu Q, Zhang M, Lai M, Chen W, Yang J, et al. Disulfiram enhances the antitumor activity of cisplatin by inhibiting the Fanconi anemia repair pathway. J Zhejiang Univ Sci B. 2023;24:207–20.
Hater N, Iwaniuk KM, Leifeld C, Grüten P, Wiek C, Raba K, et al. Identification of new RAD51D-regulating microRNAs that also emerge as potent inhibitors of the Fanconi anemia/homologous recombination pathways. Hum Mol Genet. 2022;31:4241–54.
Srivastava AK, Han C, Zhao R, Cui T, Dai Y, Mao C, et al. Enhanced expression of DNA polymerase eta contributes to cisplatin resistance of ovarian cancer stem cells. Proc Natl Acad Sci U S A. 2015;112:4411–6.
Wang Y, Huang J-W, Calses P, Kemp CJ, Taniguchi T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res. 2012;72:4037–46.
You M, Xie Z, Zhang N, Zhang Y, Xiao D, Liu S, et al. Signaling pathways in cancer metabolism: mechanisms and therapeutic targets. Signal Transduct Target Ther. 2023;8:196.
Pasquinelli AE. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet. 2012;13:271–82.
Yang G, Xiao X, Rosen DG, Cheng X, Wu X, Chang B, et al. The Biphasic Role of NF-κB in Progression and Chemoresistance of Ovarian Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:2181–94.
Devanaboyina M, Kaur J, Whiteley E, Lin L, Einloth K, Morand S, et al. NF-κB Signaling in Tumor Pathways Focusing on Breast and Ovarian Cancer. Oncol Rev. 2022;16:10568.
Harrington BS, Annunziata CM. NF-κB Signaling in Ovarian Cancer Cancers. 2019;11:1182.
Shen H, Liao B, Wan Z, Zhao Y, You Z, Liu J, et al. PTOV1 promotes cisplatin-induced chemotherapy resistance by activating the nuclear factor kappa B pathway in ovarian cancer. Mol Ther Oncolytics. 2021;20:499–507.
Huang X, Yan Y, Gui A, Zhu S, Qiu S, Chen F, et al. A Regulatory Loop Involving miR-200c and NF-κB Modulates Mortalin Expression and Increases Cisplatin Sensitivity in an Ovarian Cancer Cell Line Model. Int J Mol Sci. 2022;23:15300.
Kamdar RD, Harrington BS, Attar E, Korrapati S, Shetty J, Zhao Y, et al. NF-κB Signaling Modulates miR-452-5p and miR-335-5p Expression to Functionally Decrease Epithelial Ovarian Cancer Progression in Tumor-Initiating Cells. Int J Mol Sci. 2023;24:7826.
Nishio H, Yaguchi T, Sugiyama J, Sumimoto H, Umezawa K, Iwata T, et al. Immunosuppression through constitutively activated NF-κB signalling in human ovarian cancer and its reversal by an NF-κB inhibitor. Br J Cancer. 2014;110:2965–74.
Michalak M, Lach MS, Borska S, Nowakowski B, Umezawa K, Suchorska WM. DHMEQ enhances the cytotoxic effect of cisplatin and carboplatin in ovarian cancer cell lines. Am J Cancer Res. 2021;11:6024–41.
Huang T-T, Lampert EJ, Coots C, Lee J-M. Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat Rev. 2020;86: 102021.
Hoarau-Véchot J, Blot-Dupin M, Pauly L, Touboul C, Rafii S, Rafii A, et al. Akt-Activated Endothelium Increases Cancer Cell Proliferation and Resistance to Treatment in Ovarian Cancer Cell Organoids. Int J Mol Sci. 2022;23:14173.
Gasparri ML, Besharat ZM, Farooqi AA, Khalid S, Taghavi K, Besharat RA, et al. MiRNAs and their interplay with PI3K/AKT/mTOR pathway in ovarian cancer cells: a potential role in platinum resistance. J Cancer Res Clin Oncol. 2018;144:2313–8.
Zhang Z, Zhang L, Wang B, Wei R, Wang Y, Wan J, et al. MiR-337-3p suppresses proliferation of epithelial ovarian cancer by targeting PIK3CA and PIK3CB. Cancer Lett. 2020;469:54–67.
Frederick MI, Siddika T, Zhang P, Balasuriya N, Turk MA, O’Donoghue P, et al. miRNA-Dependent Regulation of AKT1 Phosphorylation. Cells. 2022;11:821.
Luo X, Dong Z, Chen Y, Yang L, Lai D. Enrichment of ovarian cancer stem-like cells is associated with epithelial to mesenchymal transition through an miRNA-activated AKT pathway. Cell Prolif. 2013;46:436–46.
Parashar D, Geethadevi A, Mittal S, McAlarnen LA, George J, Kadamberi IP, et al. Patient-Derived Ovarian Cancer Spheroids Rely on PI3K-AKT Signaling Addiction for Cancer Stemness and Chemoresistance. Cancers. 2022;14:958.
Musa F, Alard A, David-West G, Curtin JP, Blank SV, Schneider RJ. Dual mTORC1/2 Inhibition as a Novel Strategy for the Resensitization and Treatment of Platinum-Resistant Ovarian Cancer. Mol Cancer Ther. 2016;15:1557–67.
Santiskulvong C, Konecny GE, Fekete M, Chen K-YM, Karam A, Mulholland D, et al. Dual targeting of phosphoinositide 3-kinase and mammalian target of rapamycin using NVP-BEZ235 as a novel therapeutic approach in human ovarian carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2011;17:2373–84.
Koti M, Siu A, Clément I, Bidarimath M, Turashvili G, Edwards A, et al. A distinct pre-existing inflammatory tumour microenvironment is associated with chemotherapy resistance in high-grade serous epithelial ovarian cancer. Br J Cancer. 2015;112:1215–22.
Iba T, Kigawa J, Kanamori Y, Itamochi H, Oishi T, Simada M, et al. Expression of the c-myc gene as a predictor of chemotherapy response and a prognostic factor in patients with ovarian cancer. Cancer Sci. 2004;95:418–23.
Bagratuni T, Mavrianou N, Gavalas NG, Tzannis K, Arapinis C, Liontos M, et al. JQ1 inhibits tumour growth in combination with cisplatin and suppresses JAK/STAT signalling pathway in ovarian cancer. Eur J Cancer Oxf Engl. 1990;2020(126):125–35.
Park GB, Kim D. MicroRNA-503-5p Inhibits the CD97-Mediated JAK2/STAT3 Pathway in Metastatic or Paclitaxel-Resistant Ovarian Cancer Cells. Neoplasia N Y N. 2019;21:206–15.
Huang Z, Xu Y, Wan M, Zeng X, Wu J. miR-340: A multifunctional role in human malignant diseases. Int J Biol Sci. 2021;17:236–46.
Shen J, Liang C, Su X, Wang Q, Ke Y, Fang J, et al. Dysfunction and ceRNA network of the tumor suppressor miR-637 in cancer development and prognosis. Biomark Res. 2022;10:72.
Jiang B, Zhu S-J, Xiao S-S, Xue M. MiR-217 Inhibits M2-Like Macrophage Polarization by Suppressing Secretion of Interleukin-6 in Ovarian Cancer. Inflammation. 2019;42:1517–29.
Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol. 2016;17:722–35.
Zhou Y, Chen Q, Qin R, Zhang K, Li H. MicroRNA-449a reduces cell survival and enhances cisplatin-induced cytotoxicity via downregulation of NOTCH1 in ovarian cancer cells. Tumour Biol J Int Soc Oncodevelopmental Biol Med. 2014;35:12369–78.
Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, et al. Notch3 overexpression as potential therapeutic target in advanced stage chemoresistant ovarian cancer. Am J Clin Pathol. 2012;138:535–44.
Wang M, Ma X, Wang J, Wang L, Wang Y. Pretreatment with the γ-secretase inhibitor DAPT sensitizes drug-resistant ovarian cancer cells to cisplatin by downregulation of Notch signaling. Int J Oncol. 2014;44:1401–9.
Akbarzadeh M, Akbarzadeh S, Majidinia M. Targeting Notch signaling pathway as an effective strategy in overcoming drug resistance in ovarian cancer. Pathol Res Pract. 2020;216: 153158.
Zou W, Ma X, Hua W, Chen B, Cai G. Caveolin-1 mediates chemoresistance in cisplatin-resistant ovarian cancer cells by targeting apoptosis through the Notch-1/Akt/NF-κB pathway. Oncol Rep. 2015;34:3256–63.
Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, et al. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271:30022–7.
Wilson C, Ye X, Pham T, Lin E, Chan S, McNamara E, et al. AXL inhibition sensitizes mesenchymal cancer cells to antimitotic drugs. Cancer Res. 2014;74:5878–90.
Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19:279–90.
Antony J, Tan TZ, Kelly Z, Low J, Choolani M, Recchi C, et al. The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci Signal. 2016;9:ra97.
Yeo XH, Sundararajan V, Wu Z, Phua ZJC, Ho YY, Peh KLE, et al. The effect of inhibition of receptor tyrosine kinase AXL on DNA damage response in ovarian cancer. Commun Biol. 2023;6:660.
Mullen MM, Lomonosova E, Toboni MD, Oplt A, Cybulla E, Blachut B, et al. GAS6/AXL Inhibition Enhances Ovarian Cancer Sensitivity to Chemotherapy and PARP Inhibition through Increased DNA Damage and Enhanced Replication Stress. Mol Cancer Res MCR. 2022;20:265–79.
Kariolis MS, Miao YR, Diep A, Nash SE, Olcina MM, Jiang D, et al. Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J Clin Invest. 2017;127:183–98.
Tanaka M, Siemann DW. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers. 2020;12:1850.
Hisamatsu T, McGuire M, Wu SY, Rupaimoole R, Pradeep S, Bayraktar E, et al. PRKRA/PACT Expression Promotes Chemoresistance of Mucinous Ovarian Cancer. Mol Cancer Ther. 2019;18:162–72.
Colak S, ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends Cancer. 2017;3:56–71.
Ali S, Rehman MU, Yatoo AM, Arafah A, Khan A, Rashid S, et al. TGF-β signaling pathway: Therapeutic targeting and potential for anti-cancer immunity. Eur J Pharmacol. 2023;947: 175678.
Li X, Gao X, Yuan J, Wang F, Xu X, Wang C, et al. The miR-33a-5p/CROT axis mediates ovarian cancer cell behaviors and chemoresistance via the regulation of the TGF-β signal pathway. Front Endocrinol. 2022;13: 950345.
Cai Y, An B, Yao D, Zhou H, Zhu J. MicroRNA miR-30a inhibits cisplatin resistance in ovarian cancer cells through autophagy. Bioengineered. 2021;12:10713–22.
Parikh A, Lee C, Joseph P, Marchini S, Baccarini A, Kolev V, et al. microRNA-181a has a critical role in ovarian cancer progression through the regulation of the epithelial–mesenchymal transition. Nat Commun. 2014;5:2977.
Liang S, Liu Y, He J, Gao T, Li L, He S. Family with sequence similarity 46 member a confers chemo-resistance to ovarian carcinoma via TGF-β/Smad2 signaling. Bioengineered. 2022;13:10629–39.
Marchini S, Fruscio R, Clivio L, Beltrame L, Porcu L, Fuso Nerini I, et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer Oxf Engl. 1990;2013(49):520–30.
Newsted D, Banerjee S, Watt K, Nersesian S, Truesdell P, Blazer LL, et al. Blockade of TGF-β signaling with novel synthetic antibodies limits immune exclusion and improves chemotherapy response in metastatic ovarian cancer models. Oncoimmunology. 2018;8: e1539613.
Fix SM, Forget M-A, Sakellariou-Thompson D, Wang Y, Griffiths TM, Lee M, et al. CRISPR-mediated TGFBR2 knockout renders human ovarian cancer tumor-infiltrating lymphocytes resistant to TGF-β signaling. J Immunother Cancer. 2022;10: e003750.
Musacchio L, Valsecchi AA, Salutari V, Valabrega G, Camarda F, Tuninetti V, et al. MEK inhibitor as single agent in low grade serous ovarian and peritoneal cancer: a systematic review and meta-analysis. Cancer Treat Rev. 2022;110: 102458.
Lee S, Yoon S, Kim D-H. A high nuclear basal level of ERK2 phosphorylation contributes to the resistance of cisplatin-resistant human ovarian cancer cells. Gynecol Oncol. 2007;104:338–44.
Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, et al. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 2000;60:5988–94.
van Jaarsveld MTM, van Kuijk PF, Boersma AWM, Helleman J, van IJcken WF, Mathijssen RHJ, et al. miR-634 restores drug sensitivity in resistant ovarian cancer cells by targeting the Ras-MAPK pathway. Mol Cancer. 2015;14:196.
Hong L, Wang Y, Chen W, Yang S. MicroRNA-508 suppresses epithelial-mesenchymal transition, migration, and invasion of ovarian cancer cells through the MAPK1/ERK signaling pathway. J Cell Biochem. 2018;119:7431–40.
Zhao Y, Liu X-L, Huang J-H, Yin A-J, Zhang H. MicroRNA-18a suppresses ovarian carcinoma progression by targeting CBX7 and regulating ERK/MAPK signaling pathway and epithelial-to-mesenchymal transition. Eur Rev Med Pharmacol Sci. 2020;24:5292–302.
Hall CA, Wang R, Miao J, Oliva E, Shen X, Wheeler T, et al. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 2010;70:8517–25.
Zhang X, George J, Deb S, Degoutin JL, Takano EA, Fox SB, et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene. 2011;30:2810–22.
Omori H, Nishio M, Masuda M, Miyachi Y, Ueda F, Nakano T, et al. YAP1 is a potent driver of the onset and progression of oral squamous cell carcinoma. Sci Adv. 2020;6:eaay3324.
Zhang H, Liu C-Y, Zha Z-Y, Zhao B, Yao J, Zhao S, et al. TEAD transcription factors mediate the function of TAZ in cell growth and epithelial-mesenchymal transition. J Biol Chem. 2009;284:13355–62.
Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–71.
Yoshida K, Yokoi A, Sugiyama M, Oda S, Kitami K, Tamauchi S, et al. Expression of the chrXq27.3 miRNA cluster in recurrent ovarian clear cell carcinoma and its impact on cisplatin resistance. Oncogene. 2021;40:1255–68.
Mo Y, Leung LL, Mak CSL, Wang X, Chan W-S, Hui LMN, et al. Tumor-secreted exosomal miR-141 activates tumor-stroma interactions and controls premetastatic niche formation in ovarian cancer metastasis. Mol Cancer. 2023;22:4.
Borley J, Brown R. Epigenetic mechanisms and therapeutic targets of chemotherapy resistance in epithelial ovarian cancer. Ann Med. 2015;47:359–69.
Balch C, Matei DE, Huang TH-M, Nephew KP. Role of epigenomics in ovarian and endometrial cancers. Epigenomics. 2010;2:419–47.
Vaclavikova R, Klajic J, Brynychova V, Elsnerova K, Alnaes GIG, Tost J, et al. Development of high-resolution melting analysis for ABCB1 promoter methylation: Clinical consequences in breast and ovarian carcinoma. Oncol Rep. 2019;42:763–74.
Bram EE, Stark M, Raz S, Assaraf YG. Chemotherapeutic drug-induced ABCG2 promoter demethylation as a novel mechanism of acquired multidrug resistance. Neoplasia N Y N. 2009;11:1359–70.
Su H-Y, Lai H-C, Lin Y-W, Liu C-Y, Chen C-K, Chou Y-C, et al. Epigenetic silencing of SFRP5 is related to malignant phenotype and chemoresistance of ovarian cancer through Wnt signaling pathway. Int J Cancer. 2010;127:555–67.
Alghamian Y, Soukkarieh C, Abbady AQ, Murad H. Investigation of role of CpG methylation in some epithelial mesenchymal transition gene in a chemoresistant ovarian cancer cell line. Sci Rep. 2022;12:7494.
Chan DW, Lam WY, Chen F, Yung MMH, Chan YS, Chan WS, et al. Genome-wide DNA methylome analysis identifies methylation signatures associated with survival and drug resistance of ovarian cancers. Clin Epigenetics. 2021;13:142.
Nesic K, Kondrashova O, Hurley RM, McGehee CD, Vandenberg CJ, Ho G-Y, et al. Acquired RAD51C Promoter Methylation Loss Causes PARP Inhibitor Resistance in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2021;81:4709–22.
Lum E, Vigliotti M, Banerjee N, Cutter N, Wrzeszczynski KO, Khan S, et al. Loss of DOK2 induces carboplatin resistance in ovarian cancer via suppression of apoptosis. Gynecol Oncol. 2013;130:369–76.
Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–6.
Vriezen ER, Moscovitch M. Memory for temporal order and conditional associative-learning in patients with Parkinson’s disease. Neuropsychologia. 1990;28:1283–93.
Abbosh PH, Montgomery JS, Starkey JA, Novotny M, Zuhowski EG, Egorin MJ, et al. Dominant-negative histone H3 lysine 27 mutant derepresses silenced tumor suppressor genes and reverses the drug-resistant phenotype in cancer cells. Cancer Res. 2006;66:5582–91.
Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010;10:788–95.
Fang Y, Zhao J, Guo X, Dai Y, Zhang H, Yin F, et al. Establishment, immunological analysis, and drug prediction of a prognostic signature of ovarian cancer related to histone acetylation. Front Pharmacol. 2022;13: 947252.
Silva R, Glennon K, Metoudi M, Moran B, Salta S, Slattery K, et al. Unveiling the epigenomic mechanisms of acquired platinum-resistance in high-grade serous ovarian cancer. Int J Cancer. 2023;153:120–32.
Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci U S A. 2017;114:E10981–90.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482:339–46.
Qin M, Zhang C, Li Y. Circular RNAs in gynecologic cancers: mechanisms and implications for chemotherapy resistance. Front Pharmacol. 2023;14:1194719.
Papadimitriou M-A, Panoutsopoulou K, Pilala K-M, Scorilas A, Avgeris M. Epi-miRNAs: Modern mediators of methylation status in human cancers. Wiley Interdiscip Rev RNA. 2023;14: e1735.
Zhang L, Volinia S, Bonome T, Calin GA, Greshock J, Yang N, et al. Genomic and epigenetic alterations deregulate microRNA expression in human epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2008;105:7004–9.
Xiang Y, Ma N, Wang D, Zhang Y, Zhou J, Wu G, et al. MiR-152 and miR-185 co-contribute to ovarian cancer cells cisplatin sensitivity by targeting DNMT1 directly: a novel epigenetic therapy independent of decitabine. Oncogene. 2014;33:378–86.
Bhattacharya R, Nicoloso M, Arvizo R, Wang E, Cortez A, Rossi S, et al. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 2009;69:9090–5.
Zhou J, Li X, Wu X, Zhang T, Zhu Q, Wang X, et al. Exosomes Released from Tumor-Associated Macrophages Transfer miRNAs That Induce a Treg/Th17 Cell Imbalance in Epithelial Ovarian Cancer. Cancer Immunol Res. 2018;6:1578–92.
Kanlikilicer P, Bayraktar R, Denizli M, Rashed MH, Ivan C, Aslan B, et al. Exosomal miRNA confers chemo resistance via targeting Cav1/p-gp/M2-type macrophage axis in ovarian cancer. EBioMedicine. 2018;38:100–12.
Deng M, Wu D, Zhang Y, Jin Z, Miao J. MiR-29c downregulates tumor-expressed B7–H3 to mediate the antitumor NK-cell functions in ovarian cancer. Gynecol Oncol. 2021;162:190–9.
Miyamoto T, Murakami R, Hamanishi J, Tanigaki K, Hosoe Y, Mise N, et al. B7–H3 Suppresses Antitumor Immunity via the CCL2-CCR2-M2 Macrophage Axis and Contributes to Ovarian Cancer Progression. Cancer Immunol Res. 2022;10:56–69.
Li S, Wu Y, Zhang J, Sun H, Wang X. Role of miRNA-424 in Cancers. OncoTargets Ther. 2020;13:9611–22.
Nguyen HT, Phung CD, Tran TH, Pham TT, Pham LM, Nguyen TT, et al. Manipulating immune system using nanoparticles for an effective cancer treatment: Combination of targeted therapy and checkpoint blockage miRNA. J Control Release Off J Control Release Soc. 2021;329:524–37.
Guyon N, Garnier D, Briand J, Nadaradjane A, Bougras-Cartron G, Raimbourg J, et al. Anti-PD1 therapy induces lymphocyte-derived exosomal miRNA-4315 release inhibiting Bim-mediated apoptosis of tumor cells. Cell Death Dis. 2020;11:1048.
Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q, et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res CR. 2019;38:81.
Doo DW, Meza-Perez S, Londoño AI, Goldsberry WN, Katre AA, Boone JD, et al. Inhibition of the Wnt/β-catenin pathway enhances antitumor immunity in ovarian cancer. Ther Adv Med Oncol. 2020;12:1758835920913798.
Cascio S, Chandler C, Zhang L, Sinno S, Gao B, Onkar S, et al. Cancer-associated MSC drive tumor immune exclusion and resistance to immunotherapy, which can be overcome by Hedgehog inhibition. Sci Adv. 2021;7:eabi5790.
Kaye SB, Fehrenbacher L, Holloway R, Amit A, Karlan B, Slomovitz B, et al. A Phase II, Randomized, Placebo-Controlled Study of Vismodegib as Maintenance Therapy in Patients with Ovarian Cancer in Second or Third Complete Remission. Clin Cancer Res. 2012;18:6509–18.
Nagaraj AB, Knarr M, Sekhar S, Connor RS, Joseph P, Kovalenko O, et al. The miR-181a-SFRP4 axis regulates Wnt activation to drive stemness and platinum resistance in ovarian cancer. Cancer Res. 2021;81:2044–55.
Zhang X, Huang L, Zhao Y, Tan W. Downregulation of miR-130a contributes to cisplatin resistance in ovarian cancer cells by targeting X-linked inhibitor of apoptosis (XIAP) directly. Acta Biochim Biophys Sin. 2013;45:995–1001.
Li X, Chen W, Jin Y, Xue R, Su J, Mu Z, et al. miR-142-5p enhances cisplatin-induced apoptosis in ovarian cancer cells by targeting multiple anti-apoptotic genes. Biochem Pharmacol. 2019;161:98–112.
Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatol Baltim Md. 2016;64:488–500.
Zhang T, Sun B, Zhong C, Xu K, Wang Z, Hofman P, et al. Targeting histone deacetylase enhances the therapeutic effect of Erastin-induced ferroptosis in EGFR-activating mutant lung adenocarcinoma. Transl Lung Cancer Res. 2021;10:1857–72.
Jiang Z, Lim S-O, Yan M, Hsu JL, Yao J, Wei Y, et al. TYRO3 induces anti-PD-1/PD-L1 therapy resistance by limiting innate immunity and tumoral ferroptosis. J Clin Invest. 2021;131(e139434): 139434.
Zhang S-F, Wang X-Y, Fu Z-Q, Peng Q-H, Zhang J-Y, Ye F, et al. TXNDC17 promotes paclitaxel resistance via inducing autophagy in ovarian cancer. Autophagy. 2015;11:225–38.
Vescarelli E, Gerini G, Megiorni F, Anastasiadou E, Pontecorvi P, Solito L, et al. MiR-200c sensitizes Olaparib-resistant ovarian cancer cells by targeting Neuropilin 1. J Exp Clin Cancer Res CR. 2020;39:3.
Zhou Y, Wang C, Ding J, Chen Y, Sun Y, Cheng Z. miR-133a targets YES1 to reduce cisplatin resistance in ovarian cancer by regulating cell autophagy. Cancer Cell Int. 2022;22:15.
Urra H, Dufey E, Avril T, Chevet E, Hetz C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer. 2016;2:252–62.
Mo J, Ruan S, Yang B, Jin Y, Liu K, Luo X, et al. A novel defined risk signature of endoplasmic reticulum stress-related genes for predicting the prognosis and immune infiltration status of ovarian cancer. J Zhejiang Univ Sci B. 2023;24:64–77.
Morosi L, Matteo C, Ceruti T, Giordano S, Ponzo M, Frapolli R, et al. Quantitative determination of niraparib and olaparib tumor distribution by mass spectrometry imaging. Int J Biol Sci. 2020;16:1363–75.
Wang N, Yang Y, Jin D, Zhang Z, Shen K, Yang J, et al. PARP inhibitor resistance in breast and gynecological cancer: Resistance mechanisms and combination therapy strategies. Front Pharmacol. 2022;13: 967633.
Karthika C, Sureshkumar R, Zehravi M, Akter R, Ali F, Ramproshad S, et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life. 2022;12:897.
Stage TB, Mortensen C, Khalaf S, Steffensen V, Hammer HS, Xiong C, et al. P-glycoprotein Inhibition Exacerbates Paclitaxel Neurotoxicity in Neurons and Cancer Patients. Clin Pharmacol Ther. 2020;108:671–80.
Yi H, Liu L, Sheng N, Li P, Pan H, Cai L, et al. Synergistic Therapy of Doxorubicin and miR-129-5p with Self-Cross-Linked Bioreducible Polypeptide Nanoparticles Reverses Multidrug Resistance in Cancer Cells. Biomacromol. 2016;17:1737–47.
Liu JF, Moore KN, Birrer MJ, Berlin S, Matulonis UA, Infante JR, et al. Phase I study of safety and pharmacokinetics of the anti-MUC16 antibody-drug conjugate DMUC5754A in patients with platinum-resistant ovarian cancer or unresectable pancreatic cancer. Ann Oncol Off J Eur Soc Med Oncol. 2016;27:2124–30.
Liu J, Burris H, Wang JS, Barroilhet L, Gutierrez M, Wang Y, et al. An open-label phase I dose-escalation study of the safety and pharmacokinetics of DMUC4064A in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2021;163:473–80.
Zhang S, Lu Z, Unruh AK, Ivan C, Baggerly KA, Calin GA, et al. Clinically relevant microRNAs in ovarian cancer. Mol Cancer Res MCR. 2015;13:393–401.
Santin AD, Vergote I, González-Martín A, Moore K, Oaknin A, Romero I, et al. Safety and activity of anti-mesothelin antibody-drug conjugate anetumab ravtansine in combination with pegylated-liposomal doxorubicin in platinum-resistant ovarian cancer: multicenter, phase Ib dose escalation and expansion study. Int J Gynecol Cancer Off J Int Gynecol Cancer Soc. 2023;33:562–70.
Tan H, He Q, Gong G, Wang Y, Li J, Wang J, et al. miR-382 inhibits migration and invasion by targeting ROR1 through regulating EMT in ovarian cancer. Int J Oncol. 2016;48:181–90.
Qi X, Li Y, Liu W, Wang Y, Chen Z, Lin L. Research Trend of Publications Concerning Antibody-Drug Conjugate in Solid Cancer: A Bibliometric Study. Front Pharmacol. 2022;13: 921385.
Liu H-Y, Zhang Y-Y, Zhu B-L, Feng F-Z, Zhang H-T, Yan H, et al. MiR-203a-3p regulates the biological behaviors of ovarian cancer cells through mediating the Akt/GSK-3β/Snail signaling pathway by targeting ATM. J Ovarian Res. 2019;12:60.
Yazinski SA, Comaills V, Buisson R, Genois M-M, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017;31:318–32.
Konstantinopoulos PA, Cheng S-C, Wahner Hendrickson AE, Penson RT, Schumer ST, Doyle LA, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21:957–68.
Jo U, Senatorov IS, Zimmermann A, Saha LK, Murai Y, Kim SH, et al. Novel and Highly Potent ATR Inhibitor M4344 Kills Cancer Cells With Replication Stress, and Enhances the Chemotherapeutic Activity of Widely Used DNA Damaging Agents. Mol Cancer Ther. 2021;20:1431–41.
Moore KN, Chambers SK, Hamilton EP, Chen L-M, Oza AM, Ghamande SA, et al. Adavosertib with Chemotherapy in Patients with Primary Platinum-Resistant Ovarian, Fallopian Tube, or Peritoneal Cancer: An Open-Label, Four-Arm, Phase II Study. Clin Cancer Res Off J Am Assoc Cancer Res. 2022;28:36–44.
Embaby A, Kutzera J, Geenen JJ, Pluim D, Hofland I, Sanders J, et al. WEE1 inhibitor adavosertib in combination with carboplatin in advanced TP53 mutated ovarian cancer: A biomarker-enriched phase II study. Gynecol Oncol. 2023;174:239–46.
Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A, et al. Phase 1 Combination Study of the CHK1 Inhibitor Prexasertib and the PARP Inhibitor Olaparib in High-grade Serous Ovarian Cancer and Other Solid Tumors. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27:4710–6.
Wei L, He Y, Bi S, Li X, Zhang J, Zhang S. miRNA-199b-3p suppresses growth and progression of ovarian cancer via the CHK1/E-cadherin/EMT signaling pathway by targeting ZEB1. Oncol Rep. 2021;45:569–81.
Park SJ, Chang S-J, Suh DH, Kong TW, Song H, Kim TH, et al. A phase IA dose-escalation study of PHI-101, a new checkpoint kinase 2 inhibitor, for platinum-resistant recurrent ovarian cancer. BMC Cancer. 2022;22:28.
Lheureux S, Oaknin A, Garg S, Bruce JP, Madariaga A, Dhani NC, et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib Plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26:4206–15.
An D, Banerjee S, Lee J-M. Recent advancements of antiangiogenic combination therapies in ovarian cancer. Cancer Treat Rev. 2021;98: 102224.
Colombo N, Tomao F, Benedetti Panici P, Nicoletto MO, Tognon G, Bologna A, et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol Oncol. 2022;164:505–13.
Barton CA, Clark SJ, Hacker NF, O’Brien PM. Epigenetic markers of ovarian cancer. Adv Exp Med Biol. 2008;622:35–51.
Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11:2875–8.
Fuh KC, Bookman MA, Liu JF, Coleman RL, Herzog TJ, Thaker PH, et al. Phase 1b study of AVB-500 in combination with paclitaxel or pegylated liposomal doxorubicin platinum-resistant recurrent ovarian cancer. Gynecol Oncol. 2021;163:254–61.
Phase Ib study of AVB-S6-500 (axl inhibition) in combination with durvalumab (MEDI4736) in patients with platinum-resistant, recurrent epithelial ovarian cancer (NCT04019288) (320) - ScienceDirect [cited 2023 Aug 18]. Available from: https://www.sciencedirect.com/science/article/pii/S0090825822015426?via%3Dihub.
Dunn E, Chitcholtan K, Sykes P, Garrill A. The Anti-Proliferative Effect of PI3K/mTOR and ERK Inhibition in Monolayer and Three-Dimensional Ovarian Cancer Cell Models. Cancers. 2022;14:395.
Diao B, Sun C, Yu P, Zhao Z, Yang P. LAMA5 promotes cell proliferation and migration in ovarian cancer by activating Notch signaling pathway. FASEB J Off Publ Fed Am Soc Exp Biol. 2023;37: e23109.
Muñoz-Galván S, Felipe-Abrio B, García-Carrasco M, Domínguez-Piñol J, Suarez-Martinez E, Verdugo-Sivianes EM, et al. New markers for human ovarian cancer that link platinum resistance to the cancer stem cell phenotype and define new therapeutic combinations and diagnostic tools. J Exp Clin Cancer Res CR. 2019;38:234.
Coleman RL, Handley KF, Burger R, Molin GZD, Stagg R, Sood AK, et al. Demcizumab combined with paclitaxel for platinum-resistant ovarian, primary peritoneal, and fallopian tube cancer: The SIERRA open-label phase Ib trial. Gynecol Oncol. 2020;157:386–91.
Li Y, Wang S, Li P, Li Y, Liu Y, Fang H, et al. Rad50 promotes ovarian cancer progression through NF-κB activation. J Cell Mol Med. 2021;25:10961–72.
Morrone M da S, Somensi N, Franz L, Ramos V de M, Gasparotto J, da Rosa HT, et al. BRCA-1 depletion impairs pro-inflammatory polarization and activation of RAW 264.7 macrophages in a NF-κB-dependent mechanism. Mol Cell Biochem. 2019;462:11–23.
Trivedi MS, Arber N, Friedman E, Garber JE, Holcomb K, Horowitz NS, et al. Lessons from the Failure to Complete a Trial of Denosumab in Women With a Pathogenic BRCA1/2 Variant Scheduling Risk-Reducing Salpingo-Oophorectomy. Cancer Prev Res Phila Pa. 2022;15:721–6.
Tsuda N, Kawano K, Efferson CL, Ioannides CG. Synthetic microRNA and double-stranded RNA targeting the 3’-untranslated region of HER-2/neu mRNA inhibit HER-2 protein expression in ovarian cancer cells. Int J Oncol. 2005;27:1299–306.
Tang M, Cai J-H, Diao H-Y, Guo W-M, Yang X, Xing S. The progress of peptide vaccine clinical trials in gynecologic oncology. Hum Vaccines Immunother. 2022;18:2062982.
Balch C, Fang F, Matei DE, Huang TH-M, Nephew KP. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009;150:4003–11.
Matei D, Ghamande S, Roman L, Alvarez Secord A, Nemunaitis J, Markham MJ, et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin Cancer Res Off J Am Assoc Cancer Res. 2018;24:2285–93.
Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26:1009–16.
Dilawari A, Shah M, Ison G, Gittleman H, Fiero MH, Shah A, et al. FDA Approval Summary: Mirvetuximab Soravtansine-Gynx for FRα-Positive, Platinum-Resistant Ovarian Cancer. Clin Cancer Res. 2023;29(19):OF1–6.
Tolcher A, Hamilton E, Coleman RL. The evolving landscape of antibody-drug conjugates in gynecologic cancers. Cancer Treat Rev. 2023;116: 102546.
Gandhi NS, Tekade RK, Chougule MB. Nanocarrier mediated delivery of siRNA/miRNA in combination with chemotherapeutic agents for cancer therapy: current progress and advances. J Control Release Off J Control Release Soc. 2014;194:238–56.
Pereira DM, Rodrigues PM, Borralho PM, Rodrigues CMP. Delivering the promise of miRNA cancer therapeutics. Drug Discov Today. 2013;18:282–9.
Acknowledgements
We thank our colleagues for the critical reading of this manuscript, their valuable suggestions, as well as their useful comments for the preparation of this manuscript. Figures were created with Biorender.com.
Funding
This work was funded by the National Natural Science Foundation of China (No. 82073129), Chongqing Science and Technology Bureau (No. CSTB2023NSCQ-MSX1030, No. cstc2022jxjl120039), Chongqing Health Commission (No. 2023ZDXM029, No. 2023MSXM043), Special Project for Improving Scientific Research Ability of Chongqing University Cancer Hospital (2023nlts005, 2023nlts009).
Author information
Authors and Affiliations
Contributions
Ling Wang and Xin Wang summerized the related papers and originally drafted the manuscript. Xin Wang prepared the figures and Ling Wang made the tables. Xueping Zhu and Lin Zhong searched the clinical trials about drug resistance of ovarian cancer. Qingxiu Jiang and Ya Wang summarized the targets involved in these clinical trials. Qin Tang, Qiaoling Li, and Cong Zhang searched literatures about drug resistance in ovarian cancer. Haixia Wang and Dongling Zou initiated the study, revised and finalized the manuscript.
All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is a review article; data is not related to ethics approval.
Consent for publication
All authors agree with the content of the manuscript and consent to publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1/S2.
The summary of flow charts of clinical trials in I/II phase. The components in these flow chart include inclusion criteria, sample size, study duration, study arms, and study endpoints of these I-phase clinical trials about resistant ovarian cancer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, L., Wang, X., Zhu, X. et al. Drug resistance in ovarian cancer: from mechanism to clinical trial. Mol Cancer 23, 66 (2024). https://doi.org/10.1186/s12943-024-01967-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-01967-3