
Abstract
Glycolytic reprogramming is one of the most important features of cancer and plays an integral role in the progression of cancer. In cancer cells, changes in glucose metabolism meet the needs of self-proliferation, angiogenesis and lymphangiogenesis, metastasis, and also affect the immune escape, prognosis evaluation and therapeutic effect of cancer. The n6-methyladenosine (m6A) modification of RNA is widespread in eukaryotic cells. Dynamic and reversible m6A modifications are widely involved in the regulation of cancer stem cell renewal and differentiation, tumor therapy resistance, tumor microenvironment, tumor immune escape, and tumor metabolism. Lately, more and more evidences show that m6A modification can affect the glycolysis process of tumors in a variety of ways to regulate the biological behavior of tumors. In this review, we discussed the role of glycolysis in tumor genesis and development, and elaborated in detail the profound impact of m6A modification on different tumor by regulating glycolysis. We believe that m6A modified glycolysis has great significance and potential for tumor treatment.
Similar content being viewed by others


Introduction
Cancer cells have the potential to replicate indefinitely, which is inseparable from a continuous supply of energy and the support of macromolecular raw materials. In order to maintain their proliferative ability, cancer cells alter their metabolic state through various pathways. This makes the metabolism of tumor cells significantly different from that of normal cells [1]. As early as 1920s, Warburg and his colleagues found a special phenomenon in the process of glycolysis of cancer cells: even in the presence of oxygen, cancer cells mainly rely on glycolysis to provide energy. This is known as the Warburg effect [2]. Until 2011, the concept of cancer metabolism reprogramming, as one of the main characteristics of cancer, has attracted widespread attention [3]. Tumor cells’ metabolic reprogramming primarily involves glucose, fatty acid, amino acid, and mitochondrial metabolism. Among these, glycolysis, situated at the core of the tumor cell metabolic network, plays an irreplaceable role in tumor initiation and progression. For example, during the proliferation of tumor cells, glycolysis can provide enough energy and metabolic intermediates to ensure the smooth progress of cell biosynthesis [4]. The lactate produced by glycolysis of tumor cells has also been proved to inhibit the function of various immune cells, and is also related to the resistance of tumor immunotherapy [5, 6]. Given the crucial role of tumor glycolysis in its genesis, development, and treatment, it is necessary to explore the glycolysis of tumor cells at a deeper level.
Methylation of m6A is an epigenetic regulatory mechanism that has become increasingly familiar in recent years, and is also a widespread RNA modification in mammalian eukaryotic cells [7]. The so-called m6A modification is a methylation substitution reaction, which occurs at the N6-position of adenosine, and is most common in the 3’UTR region and RRACH ([G > A] m6AC [U > A > C]) sequence of RNA [8,9,10]. This dynamic reversible process is regulated by the interaction between the “writers” (methyltransferases that catalyzes methylation), the “erasers” (demethylases that removes methylation) and the “readers” (RNA-binding proteins that recognizes methylation) [11]. In tumor cells, m6A modification is widely involved in gene expression by regulating processes such as RNA splicing, maturation, stability, translation, and localization, thus controlling the progression of cancer [12]. Compelling evidence indicates that m6A regulates tumor metabolism through multiple pathways, significantly impacting tumorigenesis and tumor development [13, 14]. For example, m6A modifications can modulate fatty acid metabolic enzymes and pathways, thereby regulating lipid metabolism in tumor cells [15, 16]. Additionally, by influencing glutamine metabolism, a pivotal component of tumor amino acid metabolism, m6A modification can indirectly affect nucleotide synthesis [17, 18]. Moreover, m6A participates in tumor chemotherapy resistance by regulating mitochondrial metabolism [19]. However, beyond the aforementioned metabolic pathways, the most prominent area of research on m6A modification in tumor metabolism is tumor glycolysis. Over the past few years, studies on m6A in tumor glycolysis have experienced explosive growth. Numerous investigations have unveiled the molecular mechanisms through which m6A modification regulates tumor glycolysis and its pivotal role in this metabolic process.
In this review, we provide a brief introduction to the regulation of m6A modification. Then, we systematically describe the effects of glycolysis on tumor proliferation, angiogenesis and lymphangiogenesis, metastasis, immune escape, prognosis evaluation and treatment. Finally, we analyze the potential mechanism by which m6A affects the development of various tumors through the regulation of glycolysis, providing a theoretical basis for the future targeting of m6A modification and glycolysis for tumor treatment.
The m6A modification
m6A modification has been identified as the most common chemical modification in eukaryotic cell mRNA and lncRNA. It is almost involved in the whole process of RNA metabolism, such as RNA processing, nuclear export, translation and degradation [20,21,22,23]. The m6A modification of RNA is dynamic and reversible. This complex reaction cannot be separated from the interaction of the “writers”, “erasers” and “readers”. Specifically, the “writers” and “erasers” of m6A are responsible for regulating the methylation level of related RNA. Then, various m6A “readers” can recognize and combine with m6A modified RNA to regulate gene expression (Fig. 1; Table 1).

Regulation of m6A modification. The writers and erasers of m6A are responsible for regulating the methylation level of related RNA. Then, various m6A readers can recognize and combine with m6A modified RNA to regulate gene expression. METTL3, Methyltransferase-like 3; METTL14, Methyltransferase-like 14; WTAP, Wilms tumor 1-associated protein; RBM15, RNA binding motif protein 15; VIRMA, Vir-like m6A methyltransferase associated; ZC3H13, zinc finger CCCH-type containing 13; METTL16, Methyltransferase-like 16; METTL5, Methyltransferase-like 5; ZCCHC4, zinc finger CCHC-type containing 4; FTO, Fat mass and obesity-associated protein; ALKBH3, ALKB homologue 3; ALKBH5, ALKB homologue 5; YTHDF1, YTH N6-Methyladenosine RNA Binding Protein 1; YTHDF2, YTH N6-Methyladenosine RNA Binding Protein 2; YTHDF3, YTH N6-Methyladenosine RNA Binding Protein 3; YTHDC1, YTH Domain Containing 1; YTHDC2, YTH Domain Containing 2; HNRNPA2B1, Heterogeneous nuclear ribonucleoprotein A2B1; HNRNPAC/G, Heterogeneous nuclear ribonucleoprotein C/G; HNRNPR, Heterogeneous nuclear ribonucleoprotein R; IGF2BP1-3, Insulin Like Growth Factor Binding Protein 1–3; NKAP, NF-κB activating protein; eIF3, Eukaryotic translation initiation factor 3
m6A writers
As the m6A “writer”, the methyltransferase complex is composed of multiple components, which are mutually supportive and jointly regulate the methylation of nucleic acid. As is known to all, METTL3 and METTL14 are S-adenosylmethionine (SAM) dependent methyltransferase from the same family. The heterodimer formed by them plays a vital role in the regulation of m6A and is also related to the repair of DNA damage in vitro [24, 25]. Here, we mainly discuss the role of METTL3 and METTL14 as m6A “writers”. Wang et al. showed that that METTL3 folded to form a positively charged groove at the molecular level and contained a large amount of SAM, while no SAM was found in METTL14 [26]. This shows that METTL3 is the core force of catalytic methylation modification, but its catalytic ability is inhibited after the modification of small ubiquitin-like modifier on the lysine residue of METTL3 [27]. Besides, the function of METTL3 is also inseparable from the structural basis of Cys-Cys-Cys-His (CCCH)-type zinc-binding motif [28]. As another component of METTL3-METTL14 dimer complex, METTL14 can combine with RNA through its terminal RGG repeat sequence, and can also form a wide range of connections with METTL3 at the molecular level to improve the biological activity of METTL3 [26, 29]. METTL14 can also avoid the ubiquitination degradation mediated by U-box-containing protein 1 under the protection of METTL3 [30]. Wilms tumor 1-associated protein (WTAP) is one of the components of the core complex that catalyzes the modification of m6A. It exists as a regulatory subunit of METTL3-METTL14 complex, and its expression is also regulated by METTL3 [31, 32]. RNA binding motif protein 15 (RBM15) is mainly used to bind with METTL3 and WTAP. It also improves the nucleating efficiency of mRNA by promoting the binding of RNA helicase DBP5 with mRNA [33]. VIRMA plays a role in recruiting the catalytic core complex composed of METTL3, METTL14 and WTAP. It mainly plays its catalytic function in the 3’UTR region of RNA and near the termination codon [34]. Hakai is a ring finger E3 ubiquitin ligase, which plays an irreplaceable role in maintaining the stability of m6A methyltransferase complex [35]. ZC3H13, like a rivet, connects RBM15, WTAP and VIRMA, and is also related to the nuclear positioning of WTAP, VIRMA and Hakai [36, 37].
In addition to the above mentioned, there are also some methyltransferases that cannot be ignored. For example, METTL16, which is a methyltransferase gradually known in recent years, regulates the expression of MAT2A mRNA and U6 snRNA through its N-terminal domain and C-terminal VCRs, maintains the homeostasis of SAM in cells, and is also related to the repair of DNA after damage [38,39,40,41]. Moreover, METTL5 and ZCCHC4 are responsible for the methylation of 18 S rRNA and 28 S rRNA in cells [42, 43]. The specific mechanism of their function needs further study.
m6A erasers
Fat mass and obesity-associated protein (FTO), ALKB homolog 5 (ALKBH5) and ALKB homolog 3 (ALKBH3) are all members of the dioxygenase ALKB protein family. They rely on α-ketoglutarate and iron (II) to reduce the level of m6A modification of RNA [44, 45]. At the microstructure level, ALKBH3, ALKBH5 and FTO all have shallow gaps, so they prefer to combine with single-stranded nucleic acids such as mRNA [46]. Besides, the demethylation activity of FTO is affected by the sequence and tertiary structure of RNA, and it also has SFPQ (an RNA-binding protein) as its chaperone protein. SFPQ can combine with the CUGUG sequence on RNA and recruit FTO to promote proximal demethylation [47, 48].
m6A readers
Unlike “writers” and “erasers”, m6A “readers” do not directly change the methylation level, but recognize and combine the methylation sites of RNA. In this process, various “readers” combine with various m6A sites, affecting the fate of RNA.
Proteins YTHDF1-3 containing the YT521-B homology (YTH) domain are common “readers” of m6A modification. It is generally believed that YTHDF1 can induce mRNA translation; YTHDF2 accelerates the degradation of mRNA; YTHDF3 has the above two functions [49]. But recently, a new model of YTHDF proteins showed that YTHDF1/2/3 acted on the same subset of mRNAs, resulting in its degradation, but did not promote translation [50, 51]. The model also indicates that there is a certain functional compensation among them [52]. Another type of m6A “readers” with YTH domain are YTH Domain Containing 1 (YTHDC1) and YTHDC2, the former is related to RNA splicing and nuclear export, and the latter is related to RNA translation [53, 54]. Heterogeneous nuclear ribonucleoprotein (HNRNP) family is another m6A “reader”. Among them, heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1) can combine with miRNA containing RGm6AC sequence, recruit microRNA microprocessor complex protein DGCR8 to splice the precursor of miRNA, and promote the formation of mature miRNA [55]. Both HNRNPC and HNRNPG are related to the special mechanism of m6A switch. The so-called m6A switch refers to the process that the structure of m6A-modified RNA changes, affecting the binding of RNA-binding proteins to corresponding sites, so as to regulate the relevant life activities in cells [56, 57]. The latest research shows that HNPNPR is also a member of HNRNPs protein family, which has strong correlation with m6A modification and participates in tumor glycolysis [58]. Additionally, the insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) are mainly involved in regulating the stability and translation of mRNA [59]. NF-κB activating protein (NKAP) promotes the splicing and maturation of mRNA [60]. Eukaryotic translation initiation factor 3 (eIF3) is also considered as a “reader” of m6A and plays a central role in the initiation process of eukaryotic translation [61]. It can be seen that the role played by m6A “readers” is very complex and diverse, including but not limited to the regulation of RNA splicing, maturation, stability, translation, and localization. In this respect, there are still many unknown “readers” waiting to be discovered.
Glycolysis process
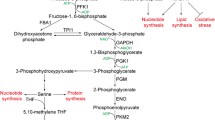
Glycolysis is the process of decomposing one molecule of glucose into two molecules of pyruvate, and then producing two molecules of ATP [62]. In this process, extracellular glucose is transferred to the cell fluid with the help of glucose transporters (GLUT). Then glucose is decomposed into mutually convertible glyceraldehyde 3-phosphate and dihydroxyacetone phosphate under the action of hexokinase (HK), glucose-6-phosphate isomerase (GPI), phosphofructokinase-1 (PFK) and aldolase (ALD) [63,64,65]. So far, glucose containing six carbon atoms has been decomposed into two three-carbon units. After that, glyceraldehyde 3-phosphate is finally converted into two molecules of pyruvate under the catalysis of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase (PGK), phosphoglycerate mutase (PGAM), enolase (ENO) and pyruvate kinase (PK) [62, 66].
When the cell is aerobic and has mitochondria, pyruvate usually enters mitochondria for tricarboxylic acid cycle, producing a large amount of ATP to provide energy for cells; However, pyruvate is reduced to lactate if the cells are in hypoxia or lack of mitochondria [67]. In these processes, metabolic intermediates can also enter other biochemical reaction processes, so that various intracellular reactions can be coordinated and orderly. However, biochemist Otto Warburg found a special phenomenon: in cancer cells, even if they have normal mitochondria and sufficient oxygen, they rarely undergo oxidative phosphorylation (OXPHOS). On the contrary, cancer cells mainly rely on glycolysis to provide energy. This characteristic is the expression that cancer cells can tolerate hypoxic environment, and also increases the level of lactate in tumor cell microenvironment, stimulating tumor growth and development [68].
Glycolysis and tumor
For most normal cells, they prefer OXPHOS to provide energy for themselves. However, in tumor cells, glycolysis holds a unique position [69]. This is because compared to OXPHOS, glycolysis produces less ATP but at a faster rate. Therefore, at the same time, glycolysis can produce more ATP to provide energy for the life activities of tumor cells [70]. Besides, various intermediate products produced during the glycolysis process are crucial for the synthesis of biological macromolecules such as lipids and nucleic acids within cells, contributing to tumor cell proliferation [71]. Lactate, one of the products of glycolysis, can effectively inhibit the function of a variety of immune cells and contribute to the immune escape of tumor cells [5]. In recent years, accumulating studies have confirmed that tumor glycolysis plays an indispensable role in tumor progression, including tumor proliferation, angiogenesis and lymphangiogenesis, metastasis, immune escape, and has a certain correlation with tumor prognosis evaluation and treatment (Fig. 2; Table 2).

The relationship between glycolysis and tumors. In the upper part of the figure, the glycolysis pathway is depicted for both normal and tumor cells, with a focus on the involvement of m6A regulators in glucose metabolism. The lower part of the figure illustrates how glycolysis impacts various aspects of tumor biology, including proliferation, angiogenesis and lymphangiogenesis, metastasis, immune escape, and its relevance to tumor prognosis evaluation and treatment. (I) Changes in glycolysis enzymes within tumor cells influence tumor proliferation by affecting glycolysis. (II) Enhanced tumor glycolysis contributes to angiogenesis and lymphangiogenesis. (III) Elevated tumor glycolysis promotes tumor metastasis. (IV) Increased tumor glycolysis inhibits the activity of T and NK cells, affects PD-L1 expression, and promotes the generation of MDSCs, thereby inducing tumor immune escape. (V) The substantial uptake of 18 F-FDG by tumor cells leads to the accumulation of FDG-6-phosphate, which can be imaged using PET/CT. (VI) Attenuation of tumor glycolysis inhibits proliferation, enhances chemosensitivity, and promotes apoptosis in cancer cells
Glycolysis and tumor proliferation
The life activities of cells cannot be separated from energy. Even tenacious tumor cells cannot survive without the energy provided by glycolysis. In pancreatic cancer, TRIM47 can ubiquitinate fructose-1,6-diphosphatase (FBP1) and accelerate the degradation of FBP1, so as to promote the glycolysis and proliferation of pancreatic cancer cells [72].
LDH is the key enzyme that catalyzes the forward reaction of pyruvate reduction to lactate, and LDHB is a subunit of LDH. The serine/threonine kinase Aurora-A can directly bind to lactate dehydrogenase B (LDHB) and phosphorylate it at serine 162. Phosphorylated LDHB reduces the inhibition of substrates in the reduction reaction, significantly accelerates the conversion of pyruvate to lactate, and promotes the glycolysis and proliferation of tumor cells [73]. In addition to LDHB, LDHA, as a component of LDH, also plays an important role in tumor proliferation. In thyroid cancer, lncRNA LINC00671 is negatively correlated with LDHA levels. Under hypoxia condition, the transcription factor STAT3 is activated, which inhibits the expression of LINC00671. This increases the LDHA content in tumor cells and promotes glycolysis and proliferation of thyroid cancer [74].
Fructose-2,6-diphosphatase 3 (PFKFB3) is a regulating enzyme in glycolysis. Under the effect of LINC00930, more RBBP5-GCN5 complexes combine with PFKFB3 promoter. Therefore, H3K4 trimethylation and H3K9 acetylation in the PFKFB3 promoter region increased, resulting in a significant increase in the expression of PFKFB3. Over-expressed PFKFB3 can catalyze the production of lactate, regulate the expression of cell cycle related proteins, and promote glycolysis and proliferation of nasopharyngeal carcinoma cells [75].
As a key enzyme in the glycolysis process, PGK1 catalyzes the conversion of 1,3-bisphosphoglycerate to 3-phosphoglycerate and produces a molecule of ATP. In colorectal cancer, the O-linked N-acetylglucosamine (O-GlcNAc) in PGK1 modifies the glycosylation site at threonine 255. The activity of PGK1 modified by O-GlcNAcylation has been greatly improved and can enter the mitochondria with the help of TOM20 (a translocation enzyme on the outer membrane of mitochondria). PKG1 entering mitochondria can inhibit OXPHOS in cancer cells, but improve glycolysis activity and lactate production. Therefore, colon cancer cells promote tumor proliferation by increasing the level of PGK1 O-GlcNAcylation [76].
ENO1 can catalyze 2-Phosphoglycerate to produce phosphoenolpyruvate. Studies have shown that the overexpression of CD47 is associated with poor prognosis of colorectal cancer. Mechanically, CD47 prevents ENO1 from ubiquitin-mediated degradation by inhibiting FBXW7. This can enhance the glycolysis of tumor cells, activate the ERK signal pathway and ultimately accelerate the proliferation of tumor cells [77]. In lung adenocarcinoma, circ-ENO1 up-regulates the expression of ENO1 gene by interacting with miR-22-3p, resulting in an increase in ATP level, glucose uptake and lactate production of tumor cells, and promoting tumor proliferation [78].
Glycolysis and tumor angiogenesis and lymphangiogenesis
Tumor angiogenesis and lymphangiogenesis is a complicated process. Abnormality in this process is one of the important characteristics of tumor metastasis [79]. Dickkopf associated protein 2 (DKK2) can cooperate with lipoprotein receptor-related protein 6 (LRP6) and activate downstream AKT/mTOR signal pathway to accelerate aerobic glycolysis of colorectal cancer cells. At this time, a large amount of lactate produced by glycolysis accumulates in the tumor microenvironment, further stimulating the growth of endothelial cells, accelerating tumor angiogenesis and metastasis of colorectal cancer [80].
Tumor lymphangiogenesis and metastasis to sentinel lymph node are critical for tumor progression. Li et al. found that the expression of S100A4 in mice can enhance the movement ability of lymphatic endothelial cells (LECs) and promote lymphangiogenesis and lymph node metastasis of melanoma by studying the mouse model of melanoma popliteal lymph node metastasis. Under hypoxic conditions, S100A4 expression in LECs is up-regulated, which activates AMPK-dependent glycolysis, and regulates the budding of tumor lymphatic vessels by affecting the position and movement of tip cells in the new lymphatic vessels [81].
Glycolysis and tumor metastasis
In the tumor microenvironment, cancer-associated fibroblasts (CAF) are a large number of stromal cells that can secrete a variety of cancer cell regulatory factors [82]. The regulatory factors secreted by CAF can promote the genesis and development of tumor. CAF also has strong glycolysis ability and releases a large amount of lactate outside the cell. After that, this part of lactate will be absorbed by the cancer cells, converted into pyruvate in the cancer cell plasma, and then provide continuous energy for the proliferation of cancer cells through the OXPHOS of mitochondria. This process is called “reverse Warburg effect” [83]. Ataxia-telangiectasia mutant protein kinase (ATM) in cancer-associated fibroblasts can be induced to oxidize under hypoxia. Oxidized ATM can phosphorylate GLUT1 and increase PKM2 expression to promote glycolysis. In this process, a large amount of accumulated lactate can activate TGFβ1/p38 MAPK/MMP2/9 signal axis and stimulate the mitochondrial activity of cancer cells to improve the invasion and metastasis ability of breast cancer cells [84].
The animal model by Li et al. showed that circRPN2 inhibits glycolysis and metastasis of hepatocellular carcinoma. Specifically, circRPN2 inhibits aerobic glycolysis of HCC by accelerating ENO1 degradation and regulating the miR-183-5p/FOXO1 axis, ultimately inhibiting liver cancer metastasis [85]. In addition, Zhou and colleagues analyzed data from the TCGA database and found that the ZEB1-PFKM-glycolysis axis was significantly associated with tumorigenesis and intrahepatic metastasis of HCC. Moreover, the same results were obtained in the follow-up experiment of in-situ HCC xenograft assays [86].
Glycolysis and tumor immune escape
Tumor “immune escape” refers to the phenomenon that cancer cells escape the surveillance and attack of the immune system, so as to survive and proliferate in the body [87]. Considerable evidences show that tumor glycolysis is strongly related to immune escape of various cancer cells [88, 89].
It has been found that the positive feedback loop formed by Notch1 signaling and TAZ has the effect of promoting the expression of glycolytic genes and accelerating lactate production in cancer cells. Lactic acid accumulated in the extracellular fluid can inhibit T and NK cell activity and induce immune escape from lung cancer [90]. In breast cancer, increased glycolysis activity can up regulate IL-17 signaling pathway, recruit macrophages and other immune cells, but reduce the aggregation of tumor killer cells. High glycolysis can also induce the expression of immune checkpoints such as PD-L1 and CTLA-4, so that breast cancer can escape the surveillance of the immune system [91]. Furthermore, tumor glycolysis can also promote the expression of granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage colony-stimulating factor (GM-CSF) in triple-negative breast cancer (TNBC) cells, induce the generation of tumor derived myeloid-derived suppressor cells (MDSCs), and affect tumor immune escape and tumor growth [92].
In glioblastoma, high levels of aerobic glycolysis transfer HK2 from mitochondria to cytoplasm. Then, HK2 acts as a protein kinase to phosphorylate IκBα and induce PD-L1 expression, leading to tumor immune escape [93]. Moreover, it has been found that tumor-associated macrophages (TAM) participate in the glycolysis and immune escape of non-small cell lung cancer by reducing the oxygen content in the tumor immune microenvironment, secreting TNF to promote tumor glycolysis, and inhibiting the expression of PD-L1 in tumor cells [94].
Glycolysis and tumor prognosis evaluation
18 F-FDG PET/CT imaging is a non-invasive tool for evaluating the stage, recurrence or treatment response of various malignant tumors [95, 96]. The most commonly used imaging agent for this examination is 18 F-FDG, which is a glucose analogue. After being injected into the human body, it can enter the cells like glucose and be phosphorylated to form FDG-6-phosphate. However, it cannot be further metabolized and can only accumulate in cells. Tumor cells are characterized by increased glucose metabolism, which is manifested by increased glucose uptake and metabolic rate. Therefore, the imaging principle of PET/CT is to use the high metabolism, high uptake and accumulation of 18 F-FDG in various malignant tumors to find the primary and metastatic lesions.
At present, 18 F-FDG PET/CT parameters that combine tumor volume and tumor metabolic activity, such as metabolic tumor volume and total lesion glycolysis, are prognostic imaging biomarkers for predicting multiple cancers, such as oropharyngeal squamous cell carcinoma [97], epithelial ovarian carcinoma [98], renal cell carcinoma [99] and colorectal cancer [100].
Glycolysis and tumor treatment
In order to adapt to the harsh tumor microenvironment, tumor cells provide energy support for their various life activities by regulating aerobic glycolysis. Therefore, the destruction of tumor cell glycolysis is considered as a potential tumor treatment strategy. At present, several inhibitors targeting glycolysis have been developed, but further research is still needed to make them play a more comprehensive role in tumor inhibition [101].
DHNQ is a new inhibitor of phosphatidylinositol 3-kinase (PI3K) signal pathway. DHNQ blocks the glycolysis of cancer cells by reducing the expression of glycolytic enzyme genes such as PFK1 and PKM2, so as to inhibit the proliferation, migration and angiogenesis of colorectal cancer [102]. In castration-resistant prostate cancer (CRPC) cells, androgen receptors directly bind to the promoter of the GLUT1 gene, promoting GLUT1 transcription to meet the large glucose demand of CRPC. Inhibition of GLUT1 can significantly reduce the glycolysis and proliferation rate of CRPC cells, make CRPC more sensitive to the treatment of enzalutamide and reduce drug resistance [103].
Metformin is a drug that selectively inhibits the activity of HK2. It can destroy the glycolysis of tumor cells by activating AMP-AMPK pathway and p53 pathway. Glucose oxidase reduces the content of glucose in tumor tissue by consuming accumulated glucose, which improves the efficacy of starvation treatment. The combination of the two drugs has better tumor inhibition effect than the single drug [104].
In addition to the inhibitors mentioned above, there are also some natural compounds that can regulate tumor glycolysis and proliferation. Xu et al. found that chrysin, a bio active flavone, slowed down the glycolysis rate of liver cancer cells and accelerated the apoptosis of liver cancer cells by reducing the expression of HK2 [105]. Piperlongumine is another natural compound with anti-tumor effects. In non-small cell lung cancer, Piperlongumine can reduce the expression of HK2, inhibit the glycolysis of cancer cells, and accelerate cancer cell apoptosis to achieve anti-tumor effects [106].
m6A regulates tumor glycolysis
Mounting evidence suggests that m6A modification regulates tumor glycolysis through a variety of ways, which is of great significance for tumor proliferation, metastasis and treatment (Fig. 3; Table 3).

m6A regulates tumor glycolysis. Writers, erasers and readers of m6A participate in tumor glycolysis by regulating glycolysis related enzymes or pathways in a series of direct or indirect ways, and ultimately affect the genesis and development of tumors
Colorectal cancer
Colorectal cancer (CRC) is the second leading cause of cancer death in the world [107]. In colorectal cancer cells, METTL3 directly acts on HK2 and GLUT1 mRNA, and the stability of HK2 and GLUT1 mRNA modified by m6A increases. In the subsequent process, IGF2BP2 combines with 5’/3’UTR regions of HK2. IGF2BP2/3 combine with the 3’UTR region of GLUT1. This promotes the expression of HK2 and GLUT1, and improve the glycolysis and proliferation of colorectal cancer cells [108]. Another methyltransferase VIRMA increases the methylation level of HK2 mRNA in an m6A-dependent manner, up-regulates the level of HK2 mRNA and improves its mRNA stability, which can ultimately accelerate the aerobic glycolysis of cancer cells and improve the degree of malignancy [109]. Liu et al.’s analysis shows that m6A can modify the GLUT1 gene to enhance the stability of GLUT1 mRNA, thereby promoting the glycolysis and cell proliferation of CRC [110]. Pinin (PNN) is a desmosome associated protein that is involved in the development of some tumors. He et al. found that PNN mRNA undergoes m6A modification under the action of METTL3 and improves stability by binding to YTHDF1, ultimately promoting glycolysis and proliferation of colon cancer [111]. Zhang et al. found that WTAP and YTHDF1 enhance the stability of FOXP3 mRNA in an m6A-dependent manner. High expression of FOXP3 can directly promote downstream glycolysis processes, and can also play a role in promoting glycolysis and proliferation of colon adenocarcinoma by increasing the expression of SMARCE1 [112].
In addition to directly regulating the mRNA of enzymes involved in glycolysis, m6A modification can also act on some lncRNAs and miRNAs to regulate tumor glycolysis. Wang et al. found that LINRIS (Long Intergenic Noncoding RNA for IGF2BP2 Stability) was at a high level in colorectal cancer cells and was associated with poor prognosis of patients. LINRIS can induce the K139 ubiquitination of IGF2BP2, thus preventing the degradation of IGF2BP2 by lysosomes, which keeps the content of IGF2BP2 protein at a certain level, and promotes the glycolysis and proliferation of cancer cells through the IGF2BP2/MYC/glycolysis axis [113]. Zheng et al. showed that the expression of lncRNA PTTG3P is closely related to the prognosis of patients with colorectal cancer. PTTG3P was methylated under the action of METTL3 and combined with IGF2BP2 to obtain higher stability. After that, the glycolysis and proliferation of colorectal cancer were improved under the synergistic effect of transcription regulator YAP1 [114]. As the m6A “reader”, IMP2 interacts with the m6A modified lncRNA ZFAS1 through the KH3–4 domain and increases the stability of ZFAS1. The highly expressed ZFAS1 can combine with Obg-like ATPase 1 (OLA1) to improve its ability to hydrolyze ATP and activate glycolysis [115]. Through the study of CRC cells expressing wild-type p53, Hou and his colleagues found that wild-type p53 acts on the promoter region of METTL14 and induces the expression of METTL14. Next, METTL14, with the help of YTHDF2, promoted the biological maturation of pri-miR-6769b and pri-miR-499a. Finally, the miR-6769b-3p/GLUT3 axis and miR-499a-3p/PGAM1 axis can inhibit aerobic glycolysis and malignant phenotype of p53 WT colorectal cancer cells by decreasing the expression of GLUT3 and PGAM1 [116].
Lung cancer
Lung cancer is the second most common cancer in the world [107]. In non-small cell lung cancer, lncRNA ABHD11-AS1 is more stable after being modified with m6A mediated by METTL3, and promotes aerobic glycolysis of NSCLC through ABHD11‐AS1/EZH2/KLF4 axis [117]. Zhang and his colleagues found that METTL3 can also increase the methylation level of lncRNA DLGAP1-AS2 and improve its stability. Next, DLGAP1-AS2 and YTHDF1 work together to promote the expression of c-MYC mRNA, accelerate the growth of cancer cells and enhance glycolysis [118].
In lung adenocarcinoma (LUAD), ENO1 promotes tumor development in an m6A-dependent manner. The up-regulation of METTL3 and down-regulation of ALKBH5 increased the overall m6A level and ENO1 mRNA methylation level in lung adenocarcinoma. The combination of YTHDF1, a m6A “reader”, with methylated ENO1 mRNA leads to increased expression of ENO1 and promotes glycolysis of lung adenocarcinoma [119]. Liu and colleagues found that NPM1 is associated with m6A modification and glycolysis through analysis of the TCGA and GEO datasets. They also believe that m6A modification may enhance the glycolytic capacity and development of LUAD by enhancing the stability of NPM1 to promote the expression of glycolytic enzymes such as ENO1, HK2, LDHA, LDHB, and GLUT1 in LUAD [120]. Yang et al. showed that the WNT/β-catenin axis can act on the promoter region of FTO to inhibit the expression of FTO. This can increase the modification of m6A in c-MYC mRNA and recruit YTHDF1 to promote the translation of c-MYC mRNA, ultimately accelerating the glycolysis and proliferation of lung adenocarcinoma cells [121].
Breast cancer
So far, breast cancer has surpassed lung cancer to become the most common cancer in the world [107]. In breast cancer, C5aR1 positive neutrophils can enhance the glycolysis of breast cancer cells. Mechanically, IL1β and TNFα secreted by C5aR1 positive neutrophils can act on the downstream ERK1/2-WTAP-ENO1 signal axis, increase the m6A modification level of ENO1 mRNA, and promote ENO1 expression and glycolysis in breast cancer cells [122]. Xu and his colleagues found that m6A can also regulate glycolysis in breast cancer through Hippo pathway. Specifically, METTL3/LATS1/YTHDF2 axis promotes glycolysis and tumorigenesis of breast cancer by inhibiting YAP/TAZ in Hippo pathway [123]. Yao et al. found that under hypoxia, the transcription of HIF-1α in tumor cells increased, and the expression of miR-16-5p was inhibited, resulting in the decrease of the binding of miR-16-5p and YTHDF1 mRNA, and promoting the expression of YTHDF1. YTHDF1 then binds to PKM2 mRNA to increase PKM2 expression and glycolysis in cancer cells, accelerating the genesis and metastasis of breast cancer [124]. In addition, bioinformatics analysis shows that GPI is closely related to m6A modification in breast cancer and may be a new prognostic marker for breast cancer [125].
Esophageal cancer
At present, the overall mortality and incidence rate of esophageal cancer rank sixth and seventh respectively [107]. A TCGA ESCA cohort analysis showed that the key glycolytic enzyme HK2 was closely related to m6A modification in esophageal cancer [126]. Functional studies show that HLA complex P5 (HCP5) can enhance the binding of YTHDF1 and m6A-modified HK2 mRNA, thus improving the stability of HK2 mRNA, promoting the aerobic glycolysis of ESCC cells, and increasing the malignant phenotype of ESCC [127].
APC is a common tumor suppressor gene. In esophageal cancer, METTL3 and METTL14 act synergistically on APC mRNA, and the APC mRNA modified by m6A degrades after binding with YTHDF. Therefore, the regulatory effect of APC on WNT/β-catenin pathway is weakened, and the expression of cyclin D1, c-MYC and PKM2 is increased, which promotes the aerobic glycolysis and development of tumor [128].
Liver cancer
Liver cancer is the sixth most common cancer in the world, but it is the third leading cause of cancer death [107]. In hepatocellular carcinoma, YTHDF3, a m6A “reader”, inhibits the degradation of PFKL mRNA by m6A modification, and promotes the expression of PFKL and aerobic glycolysis. Surprisingly, PFKL positively regulates the expression of YTHDF3 protein, forming a positive feedback loop of YTHDF3-PFKL-YTHDF3, promoting the growth and lung metastasis of liver cancer [129]. The research of Zhao et al. shows that ubiquitin protein ligase E3 component N-recognin 7 (UBR7) activates the Keap1/Nrf2/Bach1/HK2 axis to reduce the content of HK2 in hepatoma cells and inhibit the glycolysis and proliferation of hepatoma cells. However, the m6A “eraser” ALKBH5 promotes the expression of UBR7 in an m6A-dependent manner [130]. Du et al. found that METTL14/USP48/SIRT6 axis also has the function of inhibiting liver cancer. This is because the ubiquitin-specific peptidase 48 (USP48) in hepatoma cells can combine with SIRT6, a histone deacetylase, to improve the stability of SIRT6, which plays a role in weakening the glycolysis of hepatoma cells. USP48 is also regulated by METTL14 [131].
LNCAROD is a lncRNA associated with the glycolysis and poor prognosis of HCC. Under the action of METTL3 and IGF2BP1, the expression and stability of LNCAROD were improved. The high expression of LNCAROD can promote the conversion of PKM to PKM2 mediated by SRSF3, and can also act as a ceRNA of miR-145-5p to maintain PKM2 levels in the cytoplasm. These two pathways synergistically increase the level of PKM2 and induce the aerobic glycolysis, proliferation and invasion of liver cancer cells [132]. Yang and his colleagues found in their research on hepatitis B virus X interacting protein (HBXIP) that the expression of HBXIP can promote glycolysis of liver cancer cells and improve the malignancy. This is because HBXIP can induce the expression of methyltransferase METTL3. In HCC cells overexpressed with METTL3, the m6A modification level of HIF-1α increased, activating downstream glycolytic enzymes and increasing the invasive ability of HCC [133].
Gastric cancer
The mortality and incidence rate of gastric cancer are the fourth and fifth respectively [107]. In gastric cancer, HDGF mRNA was modified by m6A under the action of METTL3, and its stability and expression increased after binding with IGF2BP3. HDGF in the nucleus can combine with the promoters of GLUT4 and ENO2, increase the content of glycolytic enzymes in cells, and promote glycolysis, proliferation and liver metastasis of gastric cancer [134]. METTL3 can also catalyze m6A modification at the 3’UTR of NADH dehydrogenase-1α sub-complex 4 (NDUFA4) mRNA and recruit IGF2BP1 to increase the stability of NDUFA4 mRNA and promote NDUFA4 expression. As a component of mitochondrial electron transfer chain complex, NDUFA4 promotes the proliferation of gastric cancer cells and tumor growth by promoting glycolysis of GC cells [135]. WTAP, another methyltransferase, catalyzes m6A modification at 3’UTR of HK2 mRNA, prolongs the half-life of HK2 mRNA, and promotes glycolysis and proliferation of GC cells [136].
LncRNA and WNT pathway also play an important role in glycolysis of gastric cancer. For example, LINC00958 can promote aerobic glycolysis of GC cells. In mechanism, m6A methyltransferase VIRMA induced m6A modification on the methylation site of LINC00958. The degradation of LINC00958 was reduced after modification with m6A, and it combined with GLUT1 mRNA to improve the stability of GLUT1 mRNA, thus accelerating the aerobic glycolysis of GC [137]. Xu et al. found that the RNA-binding proteins IGF2BP3 and LIN28B can recognize and bind the m6A site of c-MYC mRNA. LncRNA LOC101929709 acts as a scaffold to support the combination of the above three. The overexpression of c-MYC in turn promotes the expression of LOC101929709 and LIN28B and forms a positive feedback loop that promotes the proliferation, migration and glycolysis of gastric cancer [138]. Besides, Luo et al. found that overexpression of IGF2BP1 is associated with poor prognosis in gastric cancer. Mechanically, the stability of c-MYC mRNA modified with m6A is greatly improved after binding to IGF2BP1. Overexpression of c-MYC promotes the development of gastric cancer through the c-MYC/glycolysis axis [139].
Different from the way described above, the stability of phospholysine phosphohistidine inorganic pyrophosphate phosphotase (LHPP) mRNA is enhanced after METTL14-mediated methylation and plays a role in inhibiting the glycolysis of gastric cancer. After that, the acetylation of LHPP can inhibit the phosphorylation of GSK3b and then inhibit the WNT pathway, so as to inhibit the glycolysis and proliferation of gastric cancer cells [140]. In another study by Zhang et al., it was found that FTO removes the m6A modification in the 3’UTR region of AMPK catalytic subunit α1 (PRKAA1) mRNA, preventing YTHDF2 from recognizing and binding to PRKAA1 mRNA. As a result, the stability of PRKAA1 mRNA increases, ultimately promoting glycolysis in gastric cancer cells and inhibiting apoptosis [141].
Pancreatic cancer
Pancreatic cancer is a highly malignant tumor, and its death toll is almost the same as the number of patients [107]. In pancreatic cancer, lncRNA DICER1-AS1 can inhibit glycolysis and proliferation of pancreatic cancer. By identifying the m6A modification site on DICER1-AS1, YTHDF3 binds to and destabilizes DICER1-AS1, which reduces maturation of miR-5586-5p. Therefore, the expression of glycolytic genes such as LDHA, HK2, PGK1 and GLUT1 increases, promoting glycolysis, proliferation and metastasis of pancreatic cancer [142]. Another lncRNA linc-UROD was modified with m6A under the action of METTL3, and its stability was enhanced after binding with IGF2BP3. Linc-UROD can prevent ENO1 and PKM from being degraded by proteasome by reducing the ubiquitination of ENO1 and PKM proteins, and finally enhance the glycolysis and invasion ability of pancreatic cancer [143]. miR-30d is a kind of miRNA related to glycolysis and is positively correlated with YTHDC1. miR-30d acts directly with the transcription factor RUNX1 to reduce the expression of GLUT1 and HK1 genes, and inhibits the genesis of pancreatic tumors by inhibiting aerobic glycolysis [144]. These reports collectively suggest that alterations in RNA m6A modifications, mediated by “writers” and “erasers”, can modulate RNA stability by influencing the binding of different “readers” to m6A-modified RNA, and finally impact tumor glycolysis through downstream pathways.
In addition, Li et al. found that glycolysis of pancreatic ductal adenocarcinoma (PDAC) can promote its perineural invasion (PNI). PNI is a special way of cancer metastasis, which can make PDAC spread to the distance along the nerve fibers. This is because glutamate secreted by nerve cells can bind to NMDAR receptors on PDAC cells, causing calcium influx, and promoting the expression of METTL3 through the downstream Ca2+ dependent protein kinase CaMKII/ERK-MAPK pathway. Later, METTL3 promoted the expression of HK2 in an m6A-dependent manner, and enhanced the glycolysis and transfer of PDAC cells [145].
Cervical carcinoma
Cervical cancer is the fourth most common cancer and the fourth leading cause of cancer death in women [107]. At present, there are many studies on the glycolysis of cervical cancer by METTL3. METTL3 modifies HK2 mRNA with m6A at the 3’UTR of HK2 mRNA, and also recruit YTHDF1 to combine with HK2 mRNA to improve the stability of HK2 mRNA and promote the aerobic glycolysis of cervical cancer [146]. METTL3 can also catalyze m6A modification at the 5’UTR of PDK4 mRNA. After that, PDK4 and IGF2BP3 combine to obtain stronger stability, or combine with YTHDF1/eEF-2 complex to improve translation efficiency and ultimately promote glycolysis and proliferation of cancer cells [147].
Persistent infection of human papillomavirus type 16 and 18 (HPV16/18) is considered to be a high-risk factor for cervical cancer [148]. The E6/E7 protein produced by HPV promotes the expression of IGF2BP2, allowing it to recognize and bind more m6A-MYC mRNA to promote the proliferation and aerobic glycolysis of cervical cancer cells [149].
Renal carcinoma
In renal cell carcinoma, the transcription factor BPTF can enhance the glycolysis of cancer cells to drive the distant metastasis of renal cell carcinoma. However, this process is inhibited by METTL14. The stability and expression of BPTF mRNA catalyzed by METTL14 decreased. The enhancer action of low levels of BPTF is weakened, leading to reduced activation of downstream enolase 2 and other glycolytic related enzymes, and ultimately, the glycolytic level of renal cell carcinoma is reduced and distant metastasis is inhibited [150]. There is also a mitochondrial enzyme called MTHFD2 in renal cell carcinoma, which has a positive regulatory effect on METTL3-induced HIF-2α mRNA methylation. The expression of m6A modified HIF-2α mRNA increased, which accelerated the aerobic glycolysis and proliferation of cancer cells [151]. In addition, Yuan et al. found that the expression of m6A “reader” IGF2BP1 in renal clear cell carcinoma cells increased compared with normal renal histiocyte. The highly expressed IGF2BP1 directly binds to the m6A modification site on LDHA mRNA, thereby enhancing the stability of LDHA mRNA and promoting glycolysis and malignant phenotype of clear cell renal cell carcinoma [152].
Osteosarcoma
In osteosarcoma, circ-CTNNB1 directly binds to RBM15 and modifies the mRNA of glycolytic enzymes by m6A. The expression of methylated glycolytic enzymes such as HK2, GPI and PGK1 increased to promote glycolysis and osteosarcoma development [153]. Liu and colleagues found that the stability of m6A-modified PGK1 mRNA was improved after binding to YTHDF3. Afterwards, high expression of PGK1 can promote glycolysis and proliferation of osteosarcoma cells [154]. Besides, Bi et al. analyzed seven prognostic m6A related lncRNAs in osteosarcoma and found that they play an important role in the glycolysis process of osteosarcoma [155].
Other tumors
Oral squamous cell carcinoma (OSCC) is the most common malignant tumor in the head and neck. The expression of circular RNA circFOXK2 was significantly increased after m6A modification, and was related to the malignant degree of OSCC. In mechanism, m6A-circFOXK2 and IGF2BP3 act synergistically on GLUT1 mRNA to improve its stability and promote glycolysis and transfer of OSSC [156]. In glioblastoma (GBM), lncRNA CASC9 can promote glycolysis and is related to the poor prognosis of GBM. The m6A “reader” IGF2BP2 can recognize and combine methylated CASC9 to enhance the stability of CASC9. The complex can also bind to the methylation site on HK2 mRNA to improve the stability of HK2 mRNA and promote GBM glycolysis [157]. In cholangiocarcinoma, AKR1B10 in aldosterone reductase family is associated with glycolysis and malignant phenotype of cancer cells. Under the action of METTL3, the stability and expression of AKR1B10 mRNA were improved, which promoted the growth and glycolysis of cholangiocarcinoma [158]. In thyroid papillary carcinoma, apolipoprotein E (APOE) induces the expression of GLUT1 and other glycolytic enzymes through the IL-6/JAK2/STAT3 signal pathway to promote the genesis and development of cancer. However, FTO can erase the m6A modification level of APOE mRNA, reduce its binding with IGF2BP2 and reduce its stability, and ultimately inhibit the glycolysis and proliferation of thyroid papillary carcinoma [159]. In prostate cancer, lncRNA SNHG7 is modified with m6A under the action of METTL3 and its stability is improved. High expression SNHG7 can recruit SRSF1, which can improve the stability of c-MYC mRNA and ultimately promote the glycolysis and development of prostate cancer [160]. In ovarian cancer, HIF-1α can promote the expression of WTAP in hypoxic environments. WTAP not only contributes to the recognition of pri-miR-200 by DGCR8, a key component of the canonical microprocessor complex of microRNA biogenesis, but also induces m6A methylation of pri-miR-200 to promote its maturation. Finally, miR-200 enhances the glycolysis and proliferation of ovarian cancer by promoting the expression of HK2 [161].
Clinical significance of m6A-regulated tumors glycolysis
The aforementioned information demonstrates that m6A-regulated tumor glycolysis plays an important role in tumor genesis and development. This also aroused our curiosity about whether targeting m6A-regulated tumor glycolysis is beneficial to tumor treatment. Therefore, here, we further discuss the mechanism of m6A-regulated tumor glycolysis in cancer pathogenesis, drug treatment response and drug resistance, and the therapeutic potential of targeting m6A-regulated tumor glycolysis (Fig. 4; Table 4).

Clinical significance of m6A-regulated tumors glycolysis. m6A regulation impacts the efficacy of tumor chemotherapy, immunotherapy, and targeted therapy by influencing glycolysis. Additionally, m6A inhibitors exert anti-tumor effects by inhibiting glycolysis
In tumor chemotherapy
Chemotherapy of cancer is a method of treating cancer by using certain chemical drugs to destroy or inhibit the proliferation of cancer cells.
Some studies have shown that the m6A modification in tumor glycolysis is related to the response to chemotherapy. Cisplatin can inhibit the division of cancer cells and induce cancer cell death. Therefore, it has become an effective chemotherapy drug for the treatment of multiple solid tumors [162]. In bladder cancer tissues, the low expression of ALKBH5 cannot reduce the m6A modification level of CK2α mRNA, which increases the stability of CK2α mRNA, prolongs its half-life, ultimately accelerates the glycolysis, proliferation, migration and invasion of bladder cancer cells, and reduces the sensitivity of bladder cancer cells to cisplatin chemotherapy [163]. Wang et al. found in the study of liver cancer that the m6A modification induced by methyltransferase ZC3H13 can reduce the stability of PKM2 mRNA, inhibit the glycolysis of liver cancer cells and reduce the degree of malignancy, and also make liver cancer more sensitive to cisplatin treatment [164].
5-FU is produced by replacing hydrogen with fluorine at C-5 position of uracil, and exerts its anti-tumor effect by inhibiting thymidylate synthase [165]. Jia et al. found that LNCAROD can promote the glycolysis and malignancy of HCC and induce the resistance of HCC to 5-FU in an m6A-dependent manner [132]. In colorectal cancer, METTL3 can induce HIF-1α mRNA methylation and recruit IGF3BP3 to improve its stability, resulting in an increase in the transcription level of LDHA; On the other hand, METTL3 catalyzes m6A modification in the CDS region of LDHA mRNA and recruits YTHDF1 to bind with m6A-LDHA mRNA. Through the above two pathways, the expression of LDHA in colorectal cancer cells increased significantly, and the rate of glycolysis increased, which also led to the resistance of CRC cells to 5-FU [166].
Temozolomide (TMZ) is a first-line chemotherapy drug for GBM, which can effectively penetrate the blood-tumor barrier of GBM, and play an anti-tumor role by inducing DNA double-strand breaks in cancer cells [167]. Li et al. found that the high expression of long non-coding RNA just proximally to the X-inactive specific transcript (JPX) can promote the aerobic glycolysis of GBM cells and is related to the chemoresistance of GBM to temozolomide. In GBM cells, JPX can directly bind to PDK1 mRNA and can also enhance FTO-mediated demethylation of PDK1 mRNA, both of which improve the stability of PDK1 mRNA. In summary, JPX promote GBM aerobic glycolysis to accelerate tumor development and TMZ resistance in an m6A-dependent manner [168]. Other studies have shown that aerobic glycolysis induces the release of exosomal circ_0072083, thus promoting the expression of NANGO mediated by ALKBH5 and making GBM resistant to TMZ [169].
Doxorubicin is a commonly used drug in transcatheter arterial chemoembolization, with broad-spectrum anti-tumor activity [170]. WW domain containing protein 2 (WWP2) is a ubiquitin E3 ligase. The study by Qin et al. suggests that WWP2 regulates glycolysis and proliferation of liver cancer cells in an m6A-dependent manner, and is also associated with the resistance of liver cancer to doxorubicin therapy. Mechanically, the stability of WWP2 mRNA was improved under the combined action of METTL3 and IGF2BP2. Overexpression of WWP2 inhibits the anti-liver cancer effect of doxorubicin through WWP2/AKT/glycolysis axis [171].
Numerous evidence suggests that sevoflurane can inhibit the growth of cancer cells and inhibit platelet induced invasion of cancer cells [172]. Sun et al. found that the stability of HK2 mRNA in lung cancer cell lines decreased after m6A modification. However, lncRNA HOTAIR can collaborate with FTO to remove the methylation modification of HK2 mRNA, thereby increasing the expression of HK2 and promoting glycolysis and proliferation of lung cancer. This process is inhibited by sevoflurane [173].
In tumor immunotherapy
Tumor immunotherapy is to restore the normal anti-tumor immune response of the body by restarting and maintaining the tumor-immune cycle. At present, the effect of m6A modified tumor glycolysis on the treatment of tumor immune checkpoints inhibitors (anti-PD-L1/anti-PD-1 and anti-CTLA-4) has attracted people’s attention.
In liver cancer, circRHBDD1 can recruit YTHDF1 and combine with m6A modified PIK3R1 mRNA, improve the translation level of PIK3R1 mRNA, promote the glycolysis of liver cancer cells through subsequent PI3K/AKT signal transduction pathway and make liver cancer resistant to anti-PD-1 treatment. Therefore, the combined treatment of glycolytic inhibitors and immunosuppressants for liver cancer deserves our attention [174]. Chen et al. found that non-smc lectin I complex subunit H (NCAPH) has a strong correlation with glycolysis and immune tolerance of clear cell renal cell carcinoma. During the expression of NCAPH, IGF2BP3 and YTHDC1 regulate the stability and nuclear export of NCAPH mRNA in an m6A-dependent manner. The highly expressed NCAPH can promote the glycolysis of cancer cells, increase the expression of PD-L1 and induce resistance to anti-PD-1 therapy by stabilizing the β-catenin protein [175]. In colorectal cancer, circQSOX1 is methylated under the action of METTL3, and its stability is improved after binding with IGF2BP2. m6A modified circQSOX1 can recruit miR-326 and miR-330-5p to increase the expression of PGAM1, enhance the glycolysis of CRC, increase the production of lactate, promote the immune escape of CRC and reduce the therapeutic effect of anti-CTLA-4. It can be seen that the combined treatment of sh-circQSOX1 and anti-CTLA-4 is of great value in avoiding the drug resistance of CRC immunotherapy [176]. In addition, GNRa-CSP12 is a potential immunosuppressive agent for the treatment of leukemia. It can destroy the balance of intracellular Fe2+/Fe3+, inactivate the Fe2+-dependent demethylase FTO and ALKBH5, and increase the level of intracellular m6A. This can reduce the stability of GLUT3 and PKM transcripts that respectively mediate glycolysis and immune checkpoint pathway, so as to inhibit the proliferation of AML cells and enhance the therapeutic effect of PD-L1 checkpoint blockade [177].
In tumor-targeted therapy
In addition to chemotherapy and immunotherapy, m6A modifications may reduce the efficacy of HER2-targeted therapies for breast cancer by modulating glycolysis. Specifically, ALKBH5 removes the m6A modification of GLUT4 mRNA in cancer cells, and recruits YTHDF2 to combine with GLUT4 mRNA to enhance its stability, leading to increased glycolysis of breast cancer cells and resistance to HER2 targeted therapy. Therefore, inhibiting the ALKBH5/GLUT4 axis is of great significance in improving the efficacy of HER2 targeted therapy for breast cancer, including trastuzumab and lapatinib [178].
Combining m6A and glycolysis inhibitors for tumor therapy
As mentioned above, tumor glycolysis regulated by m6A has an immeasurable role in chemotherapy, immunotherapy, and targeted therapy of tumors. It also raises the question of whether the combination of m6A and glycolysis inhibitors could contribute to better anti-tumor therapy.
In order to fulfill the glucose demand, the vast majority of cancer cells have a high expression of GLUT1 on the cell membrane. GLUT1 specific inhibitors, such as STF-31 and WZB117, have been developed to block glucose uptake by cancer cells, thereby inhibiting tumor growth [179, 180]. Additionally, several inhibitors targeting key enzymes in the glycolysis process have been investigated, including 2-deoxy-D-glucose (2-DG), PFK158, 3-BrPA, Shikonin, and Galloflavin. These inhibitors act on HK2, PFKFB3, GAPDH, PKM2, and LDH, respectively, effectively inhibiting glycolysis and tumor proliferation [181,182,183,184,185,186]. Among them, 2-DG, a glycolysis inhibitor targeting HK2, has advanced to clinical trials.
With the advancement of high-throughput screening technology and continuous exploration of the molecular mechanisms underlying m6A modification, several m6A regulatory inhibitors with anti-cancer potential have emerged. For instance, STM2457 and Elvitegravir are specific inhibitors of METTL3, while CWI1-2 and JX5 selectively inhibit IGF2BP2 [187,188,189,190]. BTYNB functions as an inhibitor of IGF2BP1, and MV1035 targets ALKBH5 [191, 192]. However, the most significant results have been observed with FTO inhibitors. Rhein, FB23, FB23-2, MO-I-500, MA2, CS1 and CS2 have all demonstrated promising anti-tumor effects by inhibiting FTO’s m6A demethylase activity [193,194,195,196,197,198,199,200]. Notably, we highlight two FTO inhibitors, R-2-hydroxyglutarate (R-2HG) and Dac51. R-2HG competitively inhibits FTO’s enzymatic activity, resulting in increased m6A modifications on PFKP and LDHB mRNA in AML cells. Subsequently, YTHDF2 identifies and binds to these transcripts, reducing their stability and expression, and ultimately hindering AML proliferation by inhibiting the glycolytic pathway [201]. In melanoma, Dac51 enhances m6A modification levels in JunB and C/EBPβ mRNA by inhibiting FTO, which facilitates the binding of YTHDF2 to both mRNAs, leading to significant reductions in JunB and C/EBPβ expression. JunB and C/EBPβ are transcription factors that activate genes involved in glycolysis, such as PFKP, PGAM1, and HK1, thereby promoting glycolysis in cancer cells and limiting the activation and effector states of CD8+ T cells, promoting tumorigenesis and progression. By targeting these processes, the FTO inhibitor Dac51 effectively suppresses melanoma. Interestingly, overexpression of JunB in FTO-KD cells was sufficient to inhibit CD8+ T cell activation. Blocking tumor glycolysis with the HK2 inhibitor 2-DG significantly restored impaired CD8+ T cell activation [202].
The available evidence strongly suggests that m6A can modulate the sensitivity of various cancer cell types to anti-cancer therapies by regulating tumor glycolysis. Therefore, the combination of m6A inhibitors with tumor glycolysis inhibitors holds promise in enhancing anti-tumor efficacy. We eagerly anticipate early breakthroughs in the clinical application of these inhibitors, as they bring hope for the future of cancer treatment.
Conclusion and future perspectives
The story between tumor and glycolysis has been explored for decades. The most typical example is the discovery of Warburg effect, which reveals differences between tumor cells and normal cells from the perspective of glucose metabolism. With the rapid development of m6A detection technologies such as MeRIP-seq, miCLIP and SELECT-m6A, there are more and more studies on the relationship between m6A and tumor glycolysis [203,204,205]. We found that m6A modification regulates glycolysis and proliferation of tumor cells through a series of direct or indirect ways. For example, m6A can directly modify the mRNA of key glycolysis enzymes such as HK2, GLUT1, ENO1 and PDK4, and regulate tumor glycolysis by influencing the expression of these enzymes [108, 119, 147]. m6A can also modulate lncRNA and circRNA to regulate tumor glycolysis by controlling their downstream pathway. Examples include the circFOXK2/IGF2BP3/GLUT1 axis in OSSC and the lncRNA CASC9/IGF2BP2/HK2 axis in glioblastoma [156, 157]. From the current research, we can predict that m6A regulates tumor glycolysis through an extremely complex interaction network, and thereby affects the genesis, development and treatment of tumor. Further study of the potential link between m6A and tumor glycolysis will help us gain a deeper understanding of tumor metabolism and have important implications for the future development of new tumor diagnostic methods and treatment options.
Although the strategy of targeting m6A-modified tumor glycolysis for tumor therapy shows great promise, there are still challenges in related research. As early as the 1950s, m6A modification was detected in organisms, but the research on m6A regulation of tumor glycolysis began to be carried out in recent years [206]. Therefore, the relevant research is still in the initial stage, and there are many potential mechanisms that have not been discovered. From a metabolic point of view, the heterogeneity of tumor cells makes different cells in tumor tissues have different metabolic states, which poses a serious challenge for targeted glycolysis in cancer therapy [207]. Currently, preclinical studies have demonstrated the anti-tumor properties of certain glycolytic inhibitors, such as metformin and natural compounds chrysin and piperlongumine [104,105,106]. Besides, in vitro and animal studies have shown that inhibiting METTL3 in colorectal cancer cells can reduce glycolysis levels and restore therapeutic sensitivity to 5-FU [166]. In drug-resistant breast cancer cells, specific knockdown of ALKBH5 inhibited glycolysis and restored therapeutic response to trastuzumab and lapatinib in vitro and in vivo experiments [178]. It can be predicted that the combined action of m6A inhibitors and tumor glycolysis inhibitors will open a new window of hope for the treatment of tumors in the future. However, the existing specific drugs and small molecule inhibitors of m6A regulators and tumor glycolysis are in the development stage, and have not yet entered the clinical trial stage. There is an urgent need to identify specific inhibitors that have strong anti-tumor efficacy, high bioavailability and few drug-related side effects in order to be applied in clinic as soon as possible [71, 200, 208, 209].
In summary, glucose metabolism is closely related to tumor proliferation, angiogenesis and lymphangiogenesis, metastasis, immune escape, prognostic evaluation and treatment. In different types of tumors, m6A modification affects the biological behavior, therapeutic response and prognosis of tumors by altering the mRNA stability of key glycolytic enzymes and activating or inhibiting glycolytic-related signaling pathways. Therefore, further exploration of the complex relationship and mechanism of m6A methylation regulation of tumor glycolysis provides a new idea for future tumor therapy. Developing specific inhibitors targeting m6A regulators and tumor glycolysis will be a new direction for tumor treatment in the future.
Data availability
Not applicable.
Abbreviations
- ALKBH:
-
ALKB homolog
- APOE:
-
Apolipoprotein E
- ATM:
-
Ataxia-telangiectasia mutant protein kinase
- ATP:
-
Adenosine-triphosphate
- CAFs:
-
Cancer-associated fibroblasts
- CCCH:
-
Cys-Cys-Cys-His
- CRC:
-
Colorectal cancer
- CRPC:
-
Castration-resistant state cancer
- DKK2:
-
Dickkopf associated protein 2
- ENO:
-
Enolase
- FBP1:
-
Fructose-1,6-diphosphatase
- FTO:
-
Fat mass and obesity-associated protein
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GLUT:
-
Glucose transporters
- GPI:
-
Glucose-6-phosphate isomerase
- G-CSF:
-
Granulocyte colony-stimulating factor
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- HBXIP:
-
Hepatitis B virus X interacting protein
- HCP5:
-
HLA complex P5
- HNRNP:
-
Heterogeneous nuclear ribonucleoprotein
- HNRNPA2B1:
-
Heterogeneous nuclear ribonucleoprotein A2/B1
- HK:
-
Hexokinase
- JPX:
-
Just proximally to the X-inactive specific transcript
- LDH:
-
Lactate dehydrogenase
- LECs:
-
Lymphatic endothelial cells
- LHPP:
-
Phospholysine phosphohistidine inorganic pyrophosphate phosphotase
- LINRIS:
-
Long Intergenic Noncoding RNA for IGF2BP2 Stability
- LRP6:
-
Receptor-related protein 6
- LUAD:
-
Lung adenocarcinoma
- m6A:
-
n6-methyladenosine
- MAT2A:
-
Methionine adenosyltransferase 2 A
- MDSCs:
-
Myeloid-derived suppressor cells
- METTL:
-
Methyltransferase Like
- NCAPH:
-
Non-smc lectin I complex subunit H
- NDUFA4:
-
NADH dehydrogenase-1α sub-complex 4
- NKAP:
-
NF-κB activating protein
- OLA1:
-
Obg-like ATPase 1
- OSCC:
-
Oral squamous cell carcinoma
- O-GlcNAc:
-
O-linked N-acetylglucosamine
- OXPHOS:
-
Oxidative phosphorylation
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PFK:
-
Phosphofructokinase-1
- PFKFB3:
-
Fructose-2,6-diphosphatase 3
- PGAM:
-
Phosphoglycerate mutase
- PGK:
-
Phosphoglycerate kinase
- PI3K:
-
Phosphatidylinositol 3-kinase
- PK:
-
Pyruvate kinase
- PNI:
-
Perineural invasion
- RBM15:
-
RNA binding motif protein 15
- SAM:
-
S-adenosylmethionine
- SFPQ:
-
Splicing factor, proline- and glutamine-rich
- TMZ:
-
Temozolomide
- TNBC:
-
Triple-negative breast cancer
- UBR7:
-
Ubiquitin protein ligase E3 component N-recognin 7
- USP48:
-
Ubiquitin-specific peptidase 48
- UTR:
-
Untranslated region
- VCRs:
-
Vertebrate-conserved regions
- VIRMA:
-
Vir like m6A methyltransferase associated protein
- WTAP:
-
Wilms tumor 1-associated protein
- YTHDC:
-
YTH Domain Containing
- YTHDF:
-
YT521-B homology Domain-containing Family
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- ZCCHC4:
-
Zinc finger CCHC-type containing 4
References
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–9.
Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11(5):325–37.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated Lactic Acid Production blunts Tumor Immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657–71.
Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in Low-Glucose, high-lactate environments. Cell Metab. 2017;25(6):1282–93. e7.
Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)a RNA methylation. Nat Rev Genet. 2014;15(5):293–306.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149(7):1635–46.
Wang S, Sun C, Li J, Zhang E, Ma Z, Xu W, et al. Roles of RNA methylation by means of N(6)-methyladenosine (m(6)A) in human cancers. Cancer Lett. 2017;408:112–20.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–6.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19(1):88.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74.
Han X, Wang L, Han Q. Advances in the role of m(6)a RNA modification in cancer metabolic reprogramming. Cell Biosci. 2020;10:117.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21(1):14.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Fang R, Chen X, Zhang S, Shi H, Ye Y, Shi H, et al. EGFR/SRC/ERK-stabilized YTHDF2 promotes cholesterol dysregulation and invasive growth of glioblastoma. Nat Commun. 2021;12(1):177.
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40(9):1609–27.
Xiao Y, Thakkar KN, Zhao H, Broughton J, Li Y, Seoane JA, et al. The m(6)a RNA demethylase FTO is a HIF-independent synthetic lethal partner with the VHL tumor suppressor. Proc Natl Acad Sci U S A. 2020;117(35):21441–9.
Liu X, Gonzalez G, Dai X, Miao W, Yuan J, Huang M, et al. Adenylate kinase 4 modulates the resistance of breast Cancer cells to tamoxifen through an m6A-Based epitranscriptomic mechanism. Mol Ther. 2020;28(12):2593–604.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Tan B, Zhou K, Liu W, Prince E, Qing Y, Li Y, et al. RNA N(6) -methyladenosine reader YTHDC1 is essential for TGF-beta-mediated metastasis of triple negative breast cancer. Theranostics. 2022;12(13):5727–43.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N(6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20(3):285–95.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20(10):608–24.
Yu D, Horton JR, Yang J, Hajian T, Vedadi M, Sagum CA, et al. Human MettL3-MettL14 RNA adenine methyltransferase complex is active on double-stranded DNA containing lesions. Nucleic Acids Res. 2021;49(20):11629–42.
Woodcock CB, Yu D, Hajian T, Li J, Huang Y, Dai N, et al. Human MettL3-MettL14 complex is a sequence-specific DNA adenine methyltransferase active on single-strand and unpaired DNA in vitro. Cell Discov. 2019;5:63.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, et al. Structural basis of N(6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534(7608):575–8.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46(10):5195–208.
Wang P, Doxtader KA, Nam Y. Structural basis for Cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63(2):306–17.
Yoshida A, Oyoshi T, Suda A, Futaki S, Imanishi M. Recognition of G-quadruplex RNA by a crucial RNA methyltransferase component, METTL14. Nucleic Acids Res. 2022;50(1):449–57.
Zeng ZC, Pan Q, Sun YM, Huang HJ, Chen XT, Chen TQ, et al. METTL3 protects METTL14 from STUB1-mediated degradation to maintain m(6) a homeostasis. EMBO Rep. 2023;24(3):e55762.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Sorci M, Ianniello Z, Cruciani S, Larivera S, Ginistrelli LC, Capuano E, et al. METTL3 regulates WTAP protein homeostasis. Cell Death Dis. 2018;9(8):796.
Zolotukhin AS, Uranishi H, Lindtner S, Bear J, Pavlakis GN, Felber BK. Nuclear export factor RBM15 facilitates the access of DBP5 to mRNA. Nucleic Acids Res. 2009;37(21):7151–62.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m(6)a mRNA methylation in 3’UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Bawankar P, Lence T, Paolantoni C, Haussmann IU, Kazlauskiene M, Jacob D, et al. Hakai is required for stabilization of core components of the m(6)a mRNA methylation machinery. Nat Commun. 2021;12(1):3778.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)a machinery component Wtap/Fl(2)d. Genes Dev. 2018;32(5–6):415–29.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)a methylation and mouse embryonic stem cell Self-Renewal. Mol Cell. 2018;69(6):1028–38. e6.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, et al. The U6 snRNA m(6)a methyltransferase METTL16 regulates SAM synthetase Intron Retention. Cell. 2017;169(5):824–35. e14.
Perez-Pepe M, Alarcon CR. An RNA link for METTL16 and DNA repair in PDAC. Nat Cancer. 2022;3(9):1018–20.
Ruszkowska A. METTL16, Methyltransferase-Like protein 16: current insights into structure and function. Int J Mol Sci. 2021;22(4).
Doxtader KA, Wang P, Scarborough AM, Seo D, Conrad NK, Nam Y. Structural basis for regulation of METTL16, an S-Adenosylmethionine homeostasis factor. Mol Cell. 2018;71(6):1001–11e4.
van Tran N, Ernst FGM, Hawley BR, Zorbas C, Ulryck N, Hackert P, et al. The human 18S rRNA m6A methyltransferase METTL5 is stabilized by TRMT112. Nucleic Acids Res. 2019;47(15):7719–33.
Pinto R, Vagbo CB, Jakobsson ME, Kim Y, Baltissen MP, O’Donohue MF, et al. The human methyltransferase ZCCHC4 catalyses N6-methyladenosine modification of 28S ribosomal RNA. Nucleic Acids Res. 2020;48(2):830–46.
Wei J, Liu F, Lu Z, Fei Q, Ai Y, He PC, et al. Differential m(6)A, m(6)A(m), and m(1)a demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol Cell. 2018;71(6):973–85. e5.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Xu B, Liu D, Wang Z, Tian R, Zuo Y. Multi-substrate selectivity based on key loops and non-homologous domains: new insight into ALKBH family. Cell Mol Life Sci. 2021;78(1):129–41.
Song H, Wang Y, Wang R, Zhang X, Liu Y, Jia G, et al. SFPQ is an FTO-Binding protein that facilitates the demethylation substrate preference. Cell Chem Biol. 2020;27(3):283–91. e6.
Zhang X, Wei LH, Wang Y, Xiao Y, Liu J, Zhang W, et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc Natl Acad Sci U S A. 2019;116(8):2919–24.
Zou Z, Sepich-Poore C, Zhou X, Wei J, He C. The mechanism underlying redundant functions of the YTHDF proteins. Genome Biol. 2023;24(1):17.
Zaccara S, Jaffrey SR. A unified model for the function of YTHDF Proteins in regulating m(6)A-Modified mRNA. Cell. 2020;181(7):1582–95. e18.
Xia Z, Tang M, Ma J, Zhang H, Gimple RC, Prager BC, et al. Epitranscriptomic editing of the RNA N6-methyladenosine modification by dCasRx conjugated methyltransferase and demethylase. Nucleic Acids Res. 2021;49(13):7361–74.
Lasman L, Krupalnik V, Viukov S, Mor N, Aguilera-Castrejon A, Schneir D, et al. Context-dependent functional compensation between Ythdf m(6)a reader proteins. Genes Dev. 2020;34(19–20):1373–91.
Li S, Qi Y, Yu J, Hao Y, He B, Zhang M, et al. Nuclear Aurora kinase A switches m(6)a reader YTHDC1 to enhance an oncogenic RNA splicing of tumor suppressor RBM4. Signal Transduct Target Ther. 2022;7(1):97.
Kim GW, Siddiqui A. N6-methyladenosine modification of HCV RNA genome regulates cap-independent IRES-mediated translation via YTHDC2 recognition. Proc Natl Acad Sci U S A. 2021;118(10).
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m(6)A-Dependent Nuclear RNA Processing events. Cell. 2015;162(6):1299–308.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518(7540):560–4.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–63.
Liu XY, Gao Y, Kui XY, Liu XS, Zhang YH, Zhang Y, et al. High expression of HNRNPR in ESCA combined with (18)F-FDG PET/CT metabolic parameters are novel biomarkers for preoperative diagnosis of ESCA. J Transl Med. 2022;20(1):450.
Ramesh-Kumar D, Guil S. The IGF2BP family of RNA binding proteins links epitranscriptomics to cancer. Semin Cancer Biol. 2022;86(Pt 3):18–31.
Sun S, Gao T, Pang B, Su X, Guo C, Zhang R, et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m(6)A-dependent manner. Cell Death Dis. 2022;13(1):73.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Chandel NS, Glycolysis. Cold Spring Harb Perspect Biol. 2021;13(5).
DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. 2018;9(1):446.
Kathagen-Buhmann A, Maire CL, Weller J, Schulte A, Matschke J, Holz M, et al. The secreted glycolytic enzyme GPI/AMF stimulates glioblastoma cell migration and invasion in an autocrine fashion but can have anti-proliferative effects. Neuro Oncol. 2018;20(12):1594–605.
Houles T, Gravel SP, Lavoie G, Shin S, Savall M, Meant A, et al. RSK regulates PFK-2 activity to promote metabolic rewiring in Melanoma. Cancer Res. 2018;78(9):2191–204.
Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, et al. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science. 2018;360(6387):449–53.
Li X, Yang Y, Zhang B, Lin X, Fu X, An Y, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther. 2022;7(1):305.
Bononi G, Masoni S, Di Bussolo V, Tuccinardi T, Granchi C, Minutolo F. Historical perspective of tumor glycolysis: a century with Otto Warburg. Semin Cancer Biol. 2022;86(Pt 2):325–33.
Paul S, Ghosh S, Kumar S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin Cancer Biol. 2022;86(Pt 3):1216–30.
Pfeiffer T, Schuster S, Bonhoeffer S. Cooperation and competition in the evolution of ATP-producing pathways. Science. 2001;292(5516):504–7.
Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152.
Li L, Yu Y, Zhang Z, Guo Y, Yin T, Wu H, et al. TRIM47 accelerates aerobic glycolysis and tumor progression through regulating ubiquitination of FBP1 in pancreatic cancer. Pharmacol Res. 2021;166:105429.
Cheng A, Zhang P, Wang B, Yang D, Duan X, Jiang Y, et al. Aurora-A mediated phosphorylation of LDHB promotes glycolysis and tumor progression by relieving the substrate-inhibition effect. Nat Commun. 2019;10(1):5566.
Huo N, Cong R, Sun ZJ, Li WC, Zhu X, Xue CY, et al. STAT3/LINC00671 axis regulates papillary thyroid tumor growth and metastasis via LDHA-mediated glycolysis. Cell Death Dis. 2021;12(9):799.
He B, Pan H, Zheng F, Chen S, Bie Q, Cao J, et al. Long noncoding RNA LINC00930 promotes PFKFB3-mediated tumor glycolysis and cell proliferation in nasopharyngeal carcinoma. J Exp Clin Cancer Res. 2022;41(1):77.
Nie H, Ju H, Fan J, Shi X, Cheng Y, Cang X, et al. O-GlcNAcylation of PGK1 coordinates glycolysis and TCA cycle to promote tumor growth. Nat Commun. 2020;11(1):36.
Hu T, Liu H, Liang Z, Wang F, Zhou C, Zheng X, et al. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics. 2020;10(9):4056–72.
Zhou J, Zhang S, Chen Z, He Z, Xu Y, Li Z. CircRNA-ENO1 promoted glycolysis and tumor progression in lung adenocarcinoma through upregulating its host gene ENO1. Cell Death Dis. 2019;10(12):885.
Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. 2007;8(6):464–78.
Deng F, Zhou R, Lin C, Yang S, Wang H, Li W, et al. Tumor-secreted dickkopf2 accelerates aerobic glycolysis and promotes angiogenesis in colorectal cancer. Theranostics. 2019;9(4):1001–14.
Li A, Zhu L, Lei N, Wan J, Duan X, Liu S, et al. S100A4-dependent glycolysis promotes lymphatic vessel sprouting in tumor. Angiogenesis. 2023;26(1):19–36.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19(1):43.
Marin-Hernandez A, Rodriguez-Enriquez S, Moreno-Sanchez R. Oxidized ATM protein kinase is a new signal transduction player that regulates glycolysis in CAFs as well as tumor growth and metastasis. EBioMedicine. 2019;41:24–5.
Sun K, Tang S, Hou Y, Xi L, Chen Y, Yin J, et al. Oxidized ATM-mediated glycolysis enhancement in breast cancer-associated fibroblasts contributes to tumor invasion through lactate as metabolic coupling. EBioMedicine. 2019;41:370–83.
Li J, Hu ZQ, Yu SY, Mao L, Zhou ZJ, Wang PC, et al. CircRPN2 inhibits aerobic glycolysis and metastasis in Hepatocellular Carcinoma. Cancer Res. 2022;82(6):1055–69.
Zhou Y, Lin F, Wan T, Chen A, Wang H, Jiang B, et al. ZEB1 enhances Warburg effect to facilitate tumorigenesis and metastasis of HCC by transcriptionally activating PFKM. Theranostics. 2021;11(12):5926–38.
Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):10.
Ganapathy-Kanniappan S. Linking tumor glycolysis and immune evasion in cancer: emerging concepts and therapeutic opportunities. Biochim Biophys Acta Rev Cancer. 2017;1868(1):212–20.
Jiang Z, Liu Z, Li M, Chen C, Wang X. Increased glycolysis correlates with elevated immune activity in tumor immune microenvironment. EBioMedicine. 2019;42:431–42.
Xie M, Fu XG, Jiang K. Notch1/TAZ axis promotes aerobic glycolysis and immune escape in lung cancer. Cell Death Dis. 2021;12(9):832.
Li W, Xu M, Li Y, Huang Z, Zhou J, Zhao Q, et al. Comprehensive analysis of the association between tumor glycolysis and immune/inflammation function in breast cancer. J Transl Med. 2020;18(1):92.
Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and Tumor Immunity via a specific CEBPB isoform in Triple-Negative breast Cancer. Cell Metab. 2018;28(1):87–103. e6.
Guo D, Tong Y, Jiang X, Meng Y, Jiang H, Du L, et al. Aerobic glycolysis promotes tumor immune evasion by hexokinase2-mediated phosphorylation of IkappaBalpha. Cell Metab. 2022;34(9):1312–24. e6.
Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-Associated Macrophages enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019;79(4):795–806.
Suzuki Y, Okabayashi K, Hasegawa H, Tsuruta M, Shigeta K, Murakami K, et al. Metabolic tumor volume and total lesion glycolysis in PET/CT correlate with the pathological findings of Colorectal Cancer and allow its Accurate Staging. Clin Nucl Med. 2016;41(10):761–5.
Nakajima R, Matsuo Y, Kondo T, Abe K, Sakai S. Prognostic value of metabolic tumor volume and total lesion glycolysis on preoperative 18F-FDG PET/CT in patients with renal cell carcinoma. Clin Nucl Med. 2017;42(4):e177–e82.
Lim R, Eaton A, Lee NY, Setton J, Ohri N, Rao S, et al. 18F-FDG PET/CT metabolic tumor volume and total lesion glycolysis predict outcome in oropharyngeal squamous cell carcinoma. J Nucl Med. 2012;53(10):1506–13.
Lee JW, Cho A, Lee JH, Yun M, Lee JD, Kim YT, et al. The role of metabolic tumor volume and total lesion glycolysis on (1)(8)F-FDG PET/CT in the prognosis of epithelial ovarian cancer. Eur J Nucl Med Mol Imaging. 2014;41(10):1898–906.
Hwang SH, Cho A, Yun M, Choi YD, Rha SY, Kang WJ. Prognostic value of pretreatment metabolic tumor volume and total lesion glycolysis using 18F-FDG PET/CT in patients with metastatic renal cell carcinoma treated with anti-vascular endothelial growth factor-targeted agents. Clin Nucl Med. 2017;42(5):e235–e41.
Woff E, Hendlisz A, Ameye L, Garcia C, Kamoun T, Guiot T, et al. Validation of metabolically active tumor volume and total lesion glycolysis as 18F-FDG PET/CT-derived prognostic biomarkers in Chemorefractory Metastatic Colorectal Cancer. J Nucl Med. 2019;60(2):178–84.
Ganapathy-Kanniappan S. Targeting tumor glycolysis by a mitotropic agent. Expert Opin Ther Targets. 2016;20(1):1–5.
Hussain A, Qazi AK, Mupparapu N, Guru SK, Kumar A, Sharma PR, et al. Modulation of glycolysis and lipogenesis by novel PI3K selective molecule represses tumor angiogenesis and decreases colorectal cancer growth. Cancer Lett. 2016;374(2):250–60.
Wang J, Xu W, Wang B, Lin G, Wei Y, Abudurexiti M, et al. GLUT1 is an AR target contributing to tumor growth and glycolysis in castration-resistant and enzalutamide-resistant prostate cancers. Cancer Lett. 2020;485:45–55.
Meng X, Lu Z, Lv Q, Jiang Y, Zhang L, Wang Z. Tumor metabolism destruction via metformin-based glycolysis inhibition and glucose oxidase-mediated glucose deprivation for enhanced cancer therapy. Acta Biomater. 2022;145:222–34.
Xu D, Jin J, Yu H, Zhao Z, Ma D, Zhang C, et al. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J Exp Clin Cancer Res. 2017;36(1):44.
Zhou L, Li M, Yu X, Gao F, Li W. Repression of Hexokinases II-Mediated glycolysis contributes to Piperlongumine-Induced Tumor suppression in Non-Small Cell Lung Cancer cells. Int J Biol Sci. 2019;15(4):826–37.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. M(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19(1):72.
Li Y, He L, Wang Y, Tan Y, Zhang F. N(6)-methyladenosine methyltransferase KIAA1429 elevates colorectal cancer aerobic glycolysis via HK2-dependent manner. Bioengineered. 2022;13(5):11923–32.
Liu XS, Yang JW, Zeng J, Chen XQ, Gao Y, Kui XY, et al. SLC2A1 is a diagnostic biomarker involved in Immune Infiltration of Colorectal Cancer and Associated with m6A modification and ceRNA. Front Cell Dev Biol. 2022;10:853596.
He M, Jiang D, Xun A, Yang J, Luo Q, Wu H. Methyltransferase like 3 enhances pinin mRNA stability through N(6) -methyladenosine modification to augment tumourigenesis of colon adenocarcinoma. Exp Physiol. 2022;107(11):1283–97.
Zhang Y, Tian X, Bai Y, Liu X, Zhu J, Zhang L, et al. WTAP mediates FOXP3 mRNA stability to promote SMARCE1 expression and augment glycolysis in colon adenocarcinoma. Mamm Genome. 2022;33(4):654–71.
Wang Y, Lu JH, Wu QN, Jin Y, Wang DS, Chen YX, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18(1):174.
Zheng Y, Wang Y, Liu Y, Xie L, Ge J, Yu G, et al. N6-Methyladenosine modification of PTTG3P contributes to Colorectal Cancer Proliferation via YAP1. Front Oncol. 2021;11:669731.
Lu S, Han L, Hu X, Sun T, Xu D, Li Y, et al. N6-methyladenosine reader IMP2 stabilizes the ZFAS1/OLA1 axis and activates the Warburg effect: implication in colorectal cancer. J Hematol Oncol. 2021;14(1):188.
Hou Y, Zhang X, Yao H, Hou L, Zhang Q, Tao E et al. METTL14 modulates glycolysis to inhibit colorectal tumorigenesis in p53-wild-type cells. EMBO Rep. 2023:e56325.
Xue L, Li J, Lin Y, Liu D, Yang Q, Jian J, et al. M(6) a transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J Cell Physiol. 2021;236(4):2649–58.
Zhang Q, Zhang Y, Chen H, Sun LN, Zhang B, Yue DS, et al. METTL3-induced DLGAP1-AS2 promotes non-small cell lung cancer tumorigenesis through m(6)A/c-Myc-dependent aerobic glycolysis. Cell Cycle. 2022;21(24):2602–14.
Ma L, Xue X, Zhang X, Yu K, Xu X, Tian X, et al. The essential roles of m(6)a RNA modification to stimulate ENO1-dependent glycolysis and tumorigenesis in lung adenocarcinoma. J Exp Clin Cancer Res. 2022;41(1):36.
Liu XS, Zhou LM, Yuan LL, Gao Y, Kui XY, Liu XY, et al. NPM1 is a prognostic biomarker involved in Immune Infiltration of Lung Adenocarcinoma and Associated with m6A modification and glycolysis. Front Immunol. 2021;12:724741.
Yang X, Shao F, Guo D, Wang W, Wang J, Zhu R, et al. WNT/beta-catenin-suppressed FTO expression increases m(6)a of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 2021;12(5):462.
Ou B, Liu Y, Yang X, Xu X, Yan Y, Zhang J. C5aR1-positive neutrophils promote breast cancer glycolysis through WTAP-dependent m6A methylation of ENO1. Cell Death Dis. 2021;12(8):737.
Xu Y, Song M, Hong Z, Chen W, Zhang Q, Zhou J, et al. The N6-methyladenosine METTL3 regulates tumorigenesis and glycolysis by mediating m6A methylation of the tumor suppressor LATS1 in breast cancer. J Exp Clin Cancer Res. 2023;42(1):10.
Yao X, Li W, Li L, Li M, Zhao Y, Fang D, et al. YTHDF1 upregulation mediates hypoxia-dependent breast cancer growth and metastasis through regulating PKM2 to affect glycolysis. Cell Death Dis. 2022;13(3):258.
Zeng J, Yi J, Tan S, Zeng Y, Zou L, Zhang C, et al. GPI: an indicator for immune infiltrates and prognosis of human breast cancer from a comprehensive analysis. Front Endocrinol (Lausanne). 2022;13:995972.
Liu XS, Liu JM, Chen YJ, Li FY, Wu RM, Tan F, et al. Comprehensive analysis of hexokinase 2 Immune infiltrates and m6A related genes in human esophageal carcinoma. Front Cell Dev Biol. 2021;9:715883.
Wang Y, Yu Z, Shi W, Shen J, Guan Y, Ni F. HLA complex P5 upregulation is correlated with poor prognosis and tumor progression in esophageal squamous cell carcinoma. Bioengineered. 2022;13(4):9301–11.
Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y, et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat Commun. 2021;12(1):3803.
Zhou R, Ni W, Qin C, Zhou Y, Li Y, Huo J, et al. A functional loop between YTH domain family protein YTHDF3 mediated m(6)a modification and phosphofructokinase PFKL in glycolysis of hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):334.
Zhao L, Kang M, Liu X, Wang Z, Wang Y, Chen H, et al. UBR7 inhibits HCC tumorigenesis by targeting Keap1/Nrf2/Bach1/HK2 and glycolysis. J Exp Clin Cancer Res. 2022;41(1):330.
Du L, Li Y, Kang M, Feng M, Ren Y, Dai H, et al. USP48 is upregulated by Mettl14 to Attenuate Hepatocellular Carcinoma via regulating SIRT6 stabilization. Cancer Res. 2021;81(14):3822–34.
Jia G, Wang Y, Lin C, Lai S, Dai H, Wang Z, et al. LNCAROD enhances hepatocellular carcinoma malignancy by activating glycolysis through induction of pyruvate kinase isoform PKM2. J Exp Clin Cancer Res. 2021;40(1):299.
Yang N, Wang T, Li Q, Han F, Wang Z, Zhu R, et al. HBXIP drives metabolic reprogramming in hepatocellular carcinoma cells via METTL3-mediated m6A modification of HIF-1alpha. J Cell Physiol. 2021;236(5):3863–80.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m(6)a modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–205.
Xu W, Lai Y, Pan Y, Tan M, Ma Y, Sheng H, et al. m6A RNA methylation-mediated NDUFA4 promotes cell proliferation and metabolism in gastric cancer. Cell Death Dis. 2022;13(8):715.
Yu H, Zhao K, Zeng H, Li Z, Chen K, Zhang Z, et al. N(6)-methyladenosine (m(6)A) methyltransferase WTAP accelerates the Warburg effect of gastric cancer through regulating HK2 stability. Biomed Pharmacother. 2021;133:111075.
Yang D, Chang S, Li F, Ma M, Yang J, Lv X, et al. M(6) a transferase KIAA1429-stabilized LINC00958 accelerates gastric cancer aerobic glycolysis through targeting GLUT1. IUBMB Life. 2021;73(11):1325–33.
Xu TP, Yu T, Xie MY, Fang Y, Xu TT, Pan YT, et al. LOC101929709 promotes gastric cancer progression by aiding LIN28B to stabilize c-MYC mRNA. Gastric Cancer. 2023;26(2):169–86.
Luo F, Lin K. N(6)-methyladenosine (m(6)A) reader IGF2BP1 accelerates gastric cancer aerobic glycolysis in c-Myc-dependent manner. Exp Cell Res. 2022;417(1):113176.
Lin JX, Lian NZ, Gao YX, Zheng QL, Yang YH, Ma YB, et al. m6A methylation mediates LHPP acetylation as a tumour aerobic glycolysis suppressor to improve the prognosis of gastric cancer. Cell Death Dis. 2022;13(5):463.
Zhang Y, Zhou X, Cheng X, Hong X, Jiang X, Jing G, et al. PRKAA1, stabilized by FTO in an m6A-YTHDF2-dependent manner, promotes cell proliferation and glycolysis of gastric cancer by regulating the redox balance. Neoplasma. 2022;69(6):1338–48.
Hu Y, Tang J, Xu F, Chen J, Zeng Z, Han S, et al. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J Exp Clin Cancer Res. 2022;41(1):69.
He Y, Liu Y, Wu D, Chen L, Luo Z, Shi X, et al. Linc-UROD stabilizes ENO1 and PKM to strengthen glycolysis, proliferation and migration of pancreatic cancer cells. Transl Oncol. 2023;27:101583.
Hou Y, Zhang Q, Pang W, Hou L, Liang Y, Han X, et al. YTHDC1-mediated augmentation of miR-30d in repressing pancreatic tumorigenesis via attenuation of RUNX1-induced transcriptional activation of Warburg effect. Cell Death Differ. 2021;28(11):3105–24.
Li F, He C, Yao H, Zhao Y, Ye X, Zhou S, et al. Glutamate from nerve cells promotes perineural invasion in pancreatic cancer by regulating tumor glycolysis through HK2 mRNA-m6A modification. Pharmacol Res. 2023;187:106555.
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M, et al. N(6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 2020;11(10):911.
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J, et al. N(6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat Commun. 2020;11(1):2578.
Force USPST, Curry SJ, Krist AH, Owens DK, Barry MJ, Caughey AB, et al. Screening for cervical Cancer: US Preventive Services Task Force Recommendation Statement. JAMA. 2018;320(7):674–86.
Hu C, Liu T, Han C, Xuan Y, Jiang D, Sun Y, et al. HPV E6/E7 promotes aerobic glycolysis in cervical cancer by regulating IGF2BP2 to stabilize m(6)A-MYC expression. Int J Biol Sci. 2022;18(2):507–21.
Zhang C, Chen L, Liu Y, Huang J, Liu A, Xu Y, et al. Downregulated METTL14 accumulates BPTF that reinforces super-enhancers and distal lung metastasis via glycolytic reprogramming in renal cell carcinoma. Theranostics. 2021;11(8):3676–93.
Green NH, Galvan DL, Badal SS, Chang BH, LeBleu VS, Long J, et al. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene. 2019;38(34):6211–25.
Yuan B, Zhou J. N(6)-methyladenosine (m(6)A) reader IGF2BP1 facilitates clear-cell renal cell carcinoma aerobic glycolysis. PeerJ. 2023;11:e14591.
Yang F, Liu Y, Xiao J, Li B, Chen Y, Hu A, et al. Circ-CTNNB1 drives aerobic glycolysis and osteosarcoma progression via m6A modification through interacting with RBM15. Cell Prolif. 2023;56(1):e13344.
Liu D, Li Z, Zhang K, Lu D, Zhou D, Meng Y. N6-methyladenosine reader YTHDF3 contributes to the aerobic glycolysis of osteosarcoma through stabilizing PGK1 stability. J Cancer Res Clin Oncol. 2022.
Bi Y, Meng D, Wan M, Xu N, Xu Y, Yuan K, et al. m6A-Related lncRNAs predict overall survival of patients and regulate the Tumor Immune Microenvironment in Osteosarcoma. Comput Intell Neurosci. 2022;2022:9315283.
Cui Y, Liu J, Liu L, Ma X, Gui Y, Liu H, et al. M(6)A-modified circFOXK2 targets GLUT1 to accelerate oral squamous cell carcinoma aerobic glycolysis. Cancer Gene Ther. 2023;30(1):163–71.
Liu H, Qin S, Liu C, Jiang L, Li C, Yang J, et al. M(6)a reader IGF2BP2-stabilized CASC9 accelerates glioblastoma aerobic glycolysis by enhancing HK2 mRNA stability. Cell Death Discov. 2021;7(1):292.
Cai J, Cui Z, Zhou J, Zhang B, Lu R, Ding Y, et al. METTL3 promotes glycolysis and cholangiocarcinoma progression by mediating the m6A modification of AKR1B10. Cancer Cell Int. 2022;22(1):385.
Huang J, Sun W, Wang Z, Lv C, Zhang T, Zhang D, et al. FTO suppresses glycolysis and growth of papillary thyroid cancer via decreasing stability of APOE mRNA in an N6-methyladenosine-dependent manner. J Exp Clin Cancer Res. 2022;41(1):42.
Liu J, Yuan JF, Wang YZ. METTL3-stabilized lncRNA SNHG7 accelerates glycolysis in prostate cancer via SRSF1/c-Myc axis. Exp Cell Res. 2022;416(1):113149.
Lyu Y, Zhang Y, Wang Y, Luo Y, Ding H, Li P, et al. HIF-1alpha regulated WTAP overexpression promoting the Warburg Effect of Ovarian Cancer by m6A-Dependent manner. J Immunol Res. 2022;2022:6130806.
Tang C, Livingston MJ, Safirstein R, Dong Z. Cisplatin nephrotoxicity: new insights and therapeutic implications. Nat Rev Nephrol. 2023;19(1):53–72.
Yu H, Yang X, Tang J, Si S, Zhou Z, Lu J, et al. ALKBH5 inhibited cell proliferation and sensitized bladder Cancer cells to cisplatin by m6A-CK2alpha-Mediated glycolysis. Mol Ther Nucleic Acids. 2021;23:27–41.
Wang Q, Xie H, Peng H, Yan J, Han L, Ye G. ZC3H13 inhibits the progression of Hepatocellular Carcinoma through m(6)A-PKM2-Mediated glycolysis and enhances Chemosensitivity. J Oncol. 2021;2021:1328444.
Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther. 2020;206:107447.
Zhang K, Zhang T, Yang Y, Tu W, Huang H, Wang Y, et al. N(6)-methyladenosine-mediated LDHA induction potentiates chemoresistance of colorectal cancer cells through metabolic reprogramming. Theranostics. 2022;12(10):4802–17.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–96.
Li XD, Wang MJ, Zheng JL, Wu YH, Wang X, Jiang XB. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021;112(11):4543–52.
Ding C, Yi X, Chen X, Wu Z, You H, Chen X, et al. Warburg effect-promoted exosomal circ_0072083 releasing up-regulates NANGO expression through multiple pathways and enhances temozolomide resistance in glioma. J Exp Clin Cancer Res. 2021;40(1):164.
Yu Z, Guo J, Hu M, Gao Y, Huang L. Icaritin exacerbates Mitophagy and Synergizes with Doxorubicin to induce immunogenic cell death in Hepatocellular Carcinoma. ACS Nano. 2020;14(4):4816–28.
Qin Y, Wang CJ, Ye HL, Ye GX, Wang S, Pan DB, et al. WWP2 overexpression inhibits the antitumor effects of doxorubicin in hepatocellular carcinoma. Cell Biol Int. 2022;46(10):1682–92.
Liang H, Yang CX, Zhang B, Zhao ZL, Zhong JY, Wen XJ. Sevoflurane attenuates platelets activation of patients undergoing lung cancer surgery and suppresses platelets-induced invasion of lung cancer cells. J Clin Anesth. 2016;35:304–12.
Sun X, Li Q, Yang L. Sevoflurane inhibits lncRNA HOTAIR-Modulated Stability of HK2 mRNA in a m6A-Dependent manner to Dampen Aerobic glycolysis and proliferation in Lung Cancer. Biomed Res Int. 2022;2022:4668774.
Cai J, Chen Z, Zhang Y, Wang J, Zhang Z, Wu J, et al. CircRHBDD1 augments metabolic rewiring and restricts immunotherapy efficacy via m(6)a modification in hepatocellular carcinoma. Mol Ther Oncolytics. 2022;24:755–71.
Chen Z, Ruan W, Guo C, Chen K, Li L, Tian J et al. Non-SMC condensin I complex subunit H participates in anti-programmed cell death-1 resistance of clear cell renal cell carcinomas. Cell Prolif. 2023:e13400.
Liu Z, Zheng N, Li J, Li C, Zheng D, Jiang X, et al. N6-methyladenosine-modified circular RNA QSOX1 promotes colorectal cancer resistance to anti-CTLA-4 therapy through induction of intratumoral regulatory T cells. Drug Resist Updat. 2022;65:100886.
Du Y, Han M, Cao K, Li Q, Pang J, Dou L, et al. Gold nanorods exhibit intrinsic therapeutic activity via Controlling N6-Methyladenosine-based epitranscriptomics in Acute myeloid leukemia. ACS Nano. 2021;15(11):17689–704.
Liu H, Lyu H, Jiang G, Chen D, Ruan S, Liu S, et al. ALKBH5-Mediated m6A demethylation of GLUT4 mRNA promotes glycolysis and resistance to HER2-Targeted therapy in breast Cancer. Cancer Res. 2022;82(21):3974–86.
Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70.
Goodwin J, Neugent ML, Lee SY, Choe JH, Choi H, Jenkins DMR, et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat Commun. 2017;8:15503.
Zhang D, Li J, Wang F, Hu J, Wang S, Sun Y. 2-Deoxy-D-glucose targeting of glucose metabolism in cancer cells as a potential therapy. Cancer Lett. 2014;355(2):176–83.
Mondal S, Roy D, Sarkar Bhattacharya S, Jin L, Jung D, Zhang S, et al. Therapeutic targeting of PFKFB3 with a novel glycolytic inhibitor PFK158 promotes lipophagy and chemosensitivity in gynecologic cancers. Int J Cancer. 2019;144(1):178–89.
Xiao Y, Jin L, Deng C, Guan Y, Kalogera E, Ray U, et al. Inhibition of PFKFB3 induces cell death and synergistically enhances chemosensitivity in endometrial cancer. Oncogene. 2021;40(8):1409–24.
Ganapathy-Kanniappan S, Kunjithapatham R, Torbenson MS, Rao PP, Carson KA, Buijs M, et al. Human hepatocellular carcinoma in a mouse model: assessment of tumor response to percutaneous ablation by using glyceraldehyde-3-phosphate dehydrogenase antagonists. Radiology. 2012;262(3):834–45.
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C, et al. PKM2 inhibitor shikonin overcomes the cisplatin resistance in bladder Cancer by inducing necroptosis. Int J Biol Sci. 2018;14(13):1883–91.
Han X, Sheng X, Jones HM, Jackson AL, Kilgore J, Stine JE, et al. Evaluation of the anti-tumor effects of lactate dehydrogenase inhibitor galloflavin in endometrial cancer cells. J Hematol Oncol. 2015;8:2.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593(7860):597–601.
Liao L, He Y, Li SJ, Zhang GG, Yu W, Yang J, et al. Anti-HIV drug Elvitegravir suppresses Cancer Metastasis via increased proteasomal degradation of m6A methyltransferase METTL3. Cancer Res. 2022;82(13):2444–57.
Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y, et al. The m(6)a reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40(12):1566–82e10.
Feng P, Chen D, Wang X, Li Y, Li Z, Li B, et al. Inhibition of the m(6)a reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia. Leukemia. 2022;36(9):2180–8.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl Oncol. 2017;10(5):818–27.
Malacrida A, Rivara M, Di Domizio A, Cislaghi G, Miloso M, Zuliani V, et al. 3D proteome-wide scale screening and activity evaluation of a new ALKBH5 inhibitor in U87 glioblastoma cell line. Bioorg Med Chem. 2020;28(4):115300.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134(43):17963–71.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, et al. A dynamic N(6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28(11):1062–76.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in Acute myeloid leukemia. Cancer Cell. 2019;35(4):677–91. e10.
Zheng G, Cox T, Tribbey L, Wang GZ, Iacoban P, Booher ME, et al. Synthesis of a FTO inhibitor with anticonvulsant activity. ACS Chem Neurosci. 2014;5(8):658–65.
Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, Lucci A. Important role of FTO in the survival of Rare Panresistant Triple-Negative inflammatory breast Cancer cells facing a severe metabolic challenge. PLoS ONE. 2016;11(7):e0159072.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43(1):373–84.
Xiao L, Li X, Mu Z, Zhou J, Zhou P, Xie C, et al. FTO Inhibition enhances the Antitumor Effect of Temozolomide by targeting MYC-miR-155/23a Cluster-MXI1 Feedback Circuit in Glioma. Cancer Res. 2020;80(18):3945–58.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses Cancer Stem Cell maintenance and Immune Evasion. Cancer Cell. 2020;38(1):79–96e11.
Qing Y, Dong L, Gao L, Li C, Li Y, Han L, et al. R-2-hydroxyglutarate attenuates aerobic glycolysis in leukemia by targeting the FTO/m(6)A/PFKP/LDHB axis. Mol Cell. 2021;81(5):922–39. e9.
Liu Y, Liang G, Xu H, Dong W, Dong Z, Qiu Z, et al. Tumors exploit FTO-mediated regulation of glycolytic metabolism to evade immune surveillance. Cell Metab. 2021;33(6):1221–33e11.
Zheng Y, Nie P, Peng D, He Z, Liu M, Xie Y, et al. m6AVar: a database of functional variants involved in m6A modification. Nucleic Acids Res. 2018;46(D1):D139–d45.
Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–72.
Xiao Y, Wang Y, Tang Q, Wei L, Zhang X, Jia G. An elongation- and ligation-based qPCR amplification method for the Radiolabeling-Free detection of locus-specific N(6) -Methyladenosine modification. Angew Chem Int Ed Engl. 2018;57(49):15995–6000.
Dunn DB, Smith JD. Occurrence of a new base in the deoxyribonucleic acid of a strain of Bacterium coli. Nature. 1955;175(4451):336–7.
Kondo H, Ratcliffe CDH, Hooper S, Ellis J, MacRae JI, Hennequart M, et al. Single-cell resolved imaging reveals intra-tumor heterogeneity in glycolysis, transitions between metabolic states, and their regulatory mechanisms. Cell Rep. 2021;34(7):108750.
Zhou X, Li C, Chen T, Li W, Wang X, Yang Q. Targeting RNA N6-methyladenosine to synergize with immune checkpoint therapy. Mol Cancer. 2023;22(1):36.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172(1–2):90–105. e23.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 81874189, 81202300), the National Key Research and Development Program of China (2018YFA0208904).
Author information
Authors and Affiliations
Contributions
The authors contributed as follows: writing–original draft preparation, SWY, HLL; writing–review & editing, SWY, HLL; figure preparation and editing, SWY, HFS; supervision, HFL, BXZ, WZ; project administration, HFL, BXZ, WZ. All authors reviewed the article and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yue, SW., Liu, HL., Su, HF. et al. m6A-regulated tumor glycolysis: new advances in epigenetics and metabolism. Mol Cancer 22, 137 (2023). https://doi.org/10.1186/s12943-023-01841-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01841-8