
Abstract
Given that hypoxia is a persistent physiological feature of many different solid tumors and a key driver for cancer malignancy, it is thought to be a major target in cancer treatment recently. Tumor-associated macrophages (TAMs) are the most abundant immune cells in the tumor microenvironment (TME), which have a large impact on tumor development and immunotherapy. TAMs massively accumulate within hypoxic tumor regions. TAMs and hypoxia represent a deadly combination because hypoxia has been suggested to induce a pro-tumorigenic macrophage phenotype. Hypoxia not only directly affects macrophage polarization, but it also has an indirect effect by altering the communication between tumor cells and macrophages. For example, hypoxia can influence the expression of chemokines and exosomes, both of which have profound impacts on the recipient cells. Recently, it has been demonstrated that the intricate interaction between cancer cells and TAMs in the hypoxic TME is relevant to poor prognosis and increased tumor malignancy. However, there are no comprehensive literature reviews on the molecular mechanisms underlying the hypoxia-mediated communication between tumor cells and TAMs. Therefore, this review has the aim to collect all recently available data on this topic and provide insights for developing novel therapeutic strategies for reducing the effects of hypoxia.
Similar content being viewed by others


Introduction
As a prominent feature of solid tumors, hypoxia is thought to be a common cause of poor patient prognosis and therapeutic outcomes [1,2,3]. There is an increasing number of hypoxia-related publications highlighting its importance in tumors [4]. Studies have shown that long-term hypoxia is the main driving force of cancer development [5, 6]. According to the in vitro and tumor xenograft studies [7], even minutes of exposure of tumor cells to ambient air is enough to induce signaling alterations that affect their biology. Most preclinical studies collect and process tumor tissues in normoxia rather than physioxia, which contributes to therapy failure in clinic despite promising preclinical results [7]. Hypoxia contributes to various critical aspects of cancer, including genome instability [8], autophagy [9], metabolic reprogramming [10, 11], angiogenesis [12], migration, invasion [13], extracellular matrix remodeling [14], epithelial mesenchymal transition (EMT) [15], stem cell maintenance [16], immune evasion [17] and therapy resistance [18] (Fig. 1). Furthermore, in response to hypoxic stress, intercellular communication becomes more frequent and complex [19]. Therefore, hypoxia is thought to be a big obstacle to overcome in the treatment of malignancies [20].

Overview of the effects of hypoxia on tumor cells. Hypoxia contributes to many critical aspects of cancer, including genome instability, autophagy, metabolic reprogramming, angiogenesis, migration, invasion, extracellular matrix remodeling, epithelial mesenchymal transition (EMT), stem cell maintenance, immune evasion and therapy resistance
Increasing evidence supports the vital role of TME during cancer development [21]. Complex TME is composed of various cells, including stromal cells and immune cells. Macrophages are centered in the innate immune system [22] and can acquire distinct functional properties in response to environmental stimuli [23]. A macrophage spectrum is a popular model for describing the properties of macrophage activation, with M1 and M2 macrophages at opposite ends and other types of macrophages in between [23, 24]. Within the cancerous tissue, macrophages can be referred as TAMs, that involve in every part of the tumorigenesis [22, 25, 26]. M1-like macrophages have tumor-suppressing properties, whereas M2-like macrophages have tumor-promoting properties [27]. Thus, TAMs are important regulators of tumor immunity and immunotherapy [28]. Recent literature suggested that higher proximity of tumor cells to M2-like TAMs correlates to lower overall survival rates [29]. Notably, hypoxia can drive macrophages to polarize into immune-suppressive [28] or angiogenic phenotypes [30]. For example, hypoxia directly drives macrophage polarization through the unfolded protein response pathways [31]. TAMs are preferentially presented in the hypoxic region [29, 32, 33]. The attraction of macrophages by various chemoattractants and the hampered mobility of macrophages in the hypoxic region are plausible mechanisms accounting for the accumulation of TAMs within the hypoxic niche [34, 35]. Once entrapped, macrophages will gradually polarize into M2 subtypes and serve protumoral functions [22]. Together with intra-tumor hypoxia, M2 phenotype TAMs can drive tumor aggressiveness [36] and severely restrict to the efficacy of immunotherapy [37]. Thus, in order to better understand the functional roles of TAMs in tumor progression, the effects of hypoxia should be taken into account.
It is well known that the crosstalk between TAMs and tumor cells plays a fundamental role in driving cancer progression [38]. The interaction of cancer cells with TAMs in the hypoxic TME, in particular, plays a significant role in tumorigenesis and may be a novel therapeutic target in cancer [34]. According to the most recent single-cell RNA-Seq data, hypoxia is the most important factor influencing cell communication [39]. Chemokines and exosomes are both crucial mediators of the crosstalk between TAMs and tumor cells. Recent in vivo and in vitro studies have shown that hypoxia can alter the secretion of chemokines and exosomes [40, 41]. The attraction of macrophages to tumor cells can be increased by exposing tumor cells to hypoxia [42]. In addition to the direct effects of hypoxia on macrophage polarization, hypoxia can also indirectly affect this process by altering the communication of tumor cells with macrophages [43]. Interestingly, some studies showed that blocking the CD47-SIRPα “don’t eat me signal” to promote macrophage phagocytosis of cancer cells may be ineffective in hypoxic colorectal cancer [44]. Despite the fact that the importance of hypoxia in oncology is now widely recognized, understanding the many complex interactions of hypoxia and related TME stresses with cancer biology and therapy remains a work in progress [45]. Additionally, there are some excellent comprehensive reviews on the interplay between tumor cells and macrophages [46,47,48,49], but there are few reviews on the crosstalk mediated by hypoxia. Therefore, in order to identify new therapeutic targets, it is urgently necessary to have a thorough understanding of the intricate mechanisms underlying the hypoxia-mediated interaction between TAMs and tumor cells.
In this review, we provided a piece of detailed information on pathophysiological features of tumor hypoxia and its mechanism in terms of sensing oxygen. Besides, we also summarized recent results in the experiments that focused on hypoxia-driven interaction between TAMs and tumor cells. Lastly, we analyzed current clinical strategies for limiting hypoxia-induced responses.
Hypoxic tumor microenvironment
Pathophysiologic features of tumor hypoxia
The majority of solid tumors, like normal tissues, require effective clearance of produced cellular metabolic wastes in addition to regular oxygen and nutrient supplies. The host blood vessels surrounding tumors are unable to meet the above demands of tumors due to the rapid proliferation of tumor cells. To compensate, tumors generate their own vasculature. Unfortunately, the tumor neo-vasculature is abnormal in structure and function. This situation makes the hostile tumor microenvironment to be characterized by poor perfusion, insufficient oxygen, nutritional deprivation, low pH, and elevated interstitial fluid pressure [50]. The oxygen level of tumor tissue lower than 10 mmHg (1.3 kPa) is defined as hypoxia [51].
Generally, tumor tissues harbor three regions in terms of different oxygen levels: normoxic region (with functional blood vessels nearby), hypoxic region (100 μm away from functional blood vessels), and necrotic region (150 μm away from functional blood vessels) [19]. Due to different mechanisms and duration of hypoxia, tumor hypoxia can be roughly divided into chronic hypoxia and acute hypoxia. Within each class, it can be further categorized into different subtypes according to the involved pathogenetic processes [50, 52,53,54,55]. Chronic hypoxia is caused primarily by diffusion limitations due to increased diffusion distances and adverse diffusion geometries. Uncontrolled development of tumors can cause some tumor cells located far away from blood vessels and thus be deprived from sufficient oxygen. A small proportion of chronic hypoxia is attributed to hypoxemia (e.g. the formation of HbCO in anemic patients and heavy smokers) and compromised perfusion of microvessels (e.g. disturbed starling force or solid-phase stress in tumor). On the other hand, acute hypoxia, also known as cyclic, intermittent, transient, repetitive, or fluctuating hypoxia, is primarily caused by temporal flow blocking in microvessels (e.g. blockage of blood vessels by cell aggregates and fibrin plugs) and transient hypoxemia (e.g. fluctuating red blood cell fluxes).
Of note, other studies have described three types of hypoxia: chronic hypoxia, acute hypoxia, and cyclic hypoxia [56]. The first type of hypoxia—chronic hypoxia is caused by over-proliferation of cancer cells with a key character of prolonged timescales (> 24 h). The second type of hypoxia—acute hypoxia arises is resulting from sudden blockages of small blood vessels, and it may last from few minutes to few hours (< 24 h). The third type of hypoxia—cyclic hypoxia (also referred as intermittent hypoxia or IH) is due to the short-term shutdown of immature tumor vasculature ranging from several minutes to days, which can be reversed by restoring blood flow [56, 57]. In this classification, cyclic hypoxia is characterized by the presence of cycles of hypoxia and reoxygenation (H-R cycles), whereas acute hypoxia is not followed by reoxygenation.
The total duration of hypoxia, oxygen concentration, and frequency of H-R cycles are three indicators that have a significant impact on the regulation of molecular mechanisms. Oxygen levels are believed to be correlated with tumor types [4, 58, 59]. There is no unambiguous and uniform classification system. One of the possible explanations is that there is currently no agreement on the methods for studying tumor hypoxia in vitro or in vivo [56, 60]. Therefore, there is an urgent need to standardize methods to recreate intratumoral hypoxia in the laboratory and detect intratumoral hypoxia in the clinic. A variety of hypoxia-mimicking model systems and technologies for quantification of hypoxia levels emerge as time requires [1, 61,62,63], which is expected to help us gain deeper insights in pathophysiological hallmarks of tumors and the mechanisms for adaptation to hypoxia.
Oxygen sensing mechanisms
The oxygen level in hypoxic tumor region dynamically changes as tumor progression [64], thus understanding the molecular mechanism by which cells dominate the oxygen regulation is of great significance for cancer treatment. Drs. William G. Kaelin, Jr., Peter Ratcliffe,and Gregg Semenza won the 2019 Nobel Prize in Physiology or Medicine for their outstanding discoveries of cellular oxygen-sensing mechanisms.
Studies have revealed that cellular responses to hypoxia are mediated by hypoxia-inducible factor (HIF)-dependent pathways and histone lysine demethylases (KDMs), as shown in Fig. 2. KDMs are oxygen-dependent enzymes that regulate histone methylation [65], which are novel oxygen sensors beyond HIF [66]. Certain histone demethylases, such as KDM6A and KDM5A, directly sense oxygen to regulate gene expression by controlling chromatin structure [66]. For example, hypoxia-induced KDM6A inactivation leads to the persistence of histone-3 lysine-27 trimethylation (H3K27me3), eventually blocking cellular differentiation [65].

Oxygen sensing mechanisms. In presence of oxygen, HIF α is hydroxylated by prolyl hydroxylase (PHD) and FIH (factor inhibiting HIF), leading to rapid proteosomal degradation mediated by von Hippel–Lindau (VHL) protein and failure of recruiting transcriptional coactivators. The absence of oxygen leads to the stabilization and translocation of HIF-α to the nucleus where it heterodimerizes with HIF-1β to form the HIF–α/1β complex. Then, this complex recruits transcriptional coactivator and regulates target gene expression. Histone lysine demethylases (KDMs) can directly sense oxygen to control cell fate by regulating the chromatin structure in a HIF-independent manner. For example, KDM6A and KDM5A are inactivated during hypoxia, causing hypermethylation of H3K27 (KDM6A target) and H3K4 (KDM5A target)
With the ability to regulate the expression of hundreds of target genes, the HIF pathway plays a central role in coordinating cellular responses to oxygen deprivation. HIF consists of two distinct subunits: HIF-α (HIF-1α, HIF-2α, HIF-3α) and HIF-1β (also called ARNT, aryl hydrocarbon receptor nuclear translocator). Under hypoxic conditions, HIF-α proteins are stable and can be translocated into the nucleus, where they heterodimerize with HIF-1β proteins to form the functional HIF transcription factor complex. Following the recruitment of transcriptional coactivators, the HIF-α/HIF-1β complex regulates the expression of responsive genes by binding to the HRE located on the promoter regions of a large number of target genes [67]. In contrast, under normoxic conditions, HIF-α proteins are quickly degraded, failing to exert its functions [68].
Under normoxia, HIF-α is hydroxylated by prolyl hydroxylases (PHDs) and then recognized by E3-ubiquitin ligase von Hippel-Lindau (VHL), resulting in the rapid degradation of HIF-α protein [69, 70]. The activity of PHDs is related to Fe(II), 2-oxoglutarate (2OG), and oxygen. Under normoxia, dioxygen is delivered to the active site in the PHD2.Fe(II)0.2OG.HIF substrate complex through a single hydrophobic tunnel. This reversible binding of dioxygen is central to the hypoxia-sensing capacity of the PHDs, influencing the extent of HIF-α substrate prolyl hydroxylation [71]. The PHDs family has three canonical members: PHD1, PHD2, and PHD3. Each isoform has a differential function in regulating HIF-α activity [72]. In addition, studies in vitro have shown that the negative regulation of HIF-1α and HIF-2α by VHL is functionally distinct. Compared to HIF-2α, HIF-1α has a stronger affinity for VHL [73]. Additionally, different sites of proline hydroxylation play different roles in HIF-1α-pVHL interactions [74]. An additional oxygen-dependent hydroxylase involved in the regulation of the HIF pathway is the factor-inhibiting HIF (FIH). FIH is an asparagine hydroxylase that suppresses the transcriptional activity of HIF-α by preventing the recruitment of transcriptional coactivators without affecting the stability of HIF-α protein [75].
Notably, recent studies have revealed that the activation of HIF-α is not always correlated with hypoxia. In the study by Xiaotong Diao et al. [76], oleoylethanolamide (OEA) could selectively bind to the Per-ARNT-Sim-B (PAS-B) pocket of HIF-3α, resulting in the enhanced activity of HIF-3α. OEA is an oleic acid derivative that regulates food intake and metabolism. The identification of OEA provides evidence that endogenous small-molecule ligands can control the HIF pathways directly. It's interesting to note that some small-molecule compounds (described in detail below) can similarly bind to the PAS-B pocket, but they function as antagonists to suppress HIF-α activity, which may be related to their particular allosteric effects. In the study by Andrea L Casillas et al. [77], PIM1 kinase directly phosphorylated HIF-1α regardless of oxygen tension to prevent PHDs from binding to and hydroxylating HIF-1α, hence interrupting its degradation pathway. Dong Zhao et al. [78] discovered that the oncogene iASPP (inhibitor of apoptosis-simulating protein of p53) bound directly to VHL and prevented HIF-1α from degrading without affecting the PHD-mediated HIF-1α hydroxylation. In conclusion, the fact that the HIF pathway is regulated by a variety of cellular conditions highlights the importance of this pathway in numerous biological processes.
Hypoxia-driven crosstalk between tumor cells and TAMs
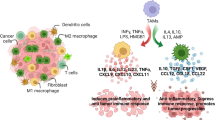
Extracellular vesicles, cytokines, growth factors, and proteins mediate reciprocal interactions between cells within TME to fulfill the growing demands of tumor cells [79]. The expression and release of these mediators are greatly affected by hypoxia. Thus, a comprehensive understanding of the mediators and pathways involved in the hypoxia-induced macrophage-cancer cell crosstalk should be helpful in finding accurate biomarkers and therapeutic targets. This section will summary the mediators and signaling pathways participating in macrophage-cancer cell crosstalk under hypoxia (Fig. 3) (Table 1).

Hypoxia-driven crosstalk between tumor cells and tumor-associated macrophages (TAMs). The complex interplay between tumor cells and TAMs under hypoxia conditions may have tumor-promoting and tumor-suppressive consequences. The mediators that are responsible for tumor cell-to-TAM communication under hypoxia include exosomes, cytokines, growth factors, cellular debris, and oncometabolites. In addition, hypoxia can regulate the expression of cell surface ligands and receptors mediating cell signaling transduction
Exosomes
Exosomes are extracellular particles with diameters ranging from 40 to 160 nm that can be released into the cell's surrounding environment. Exosomes contain a variety of constituents, depending on their cellular origins, such as nucleic acids, proteins, lipids, amino acids, metabolites, and cytosolic [110,111,112]. Exosomes have an impact on tumor growth, metastasis, paraneoplastic syndromes and provide resistance to therapy, making them a research focus in the field of oncology [110]. Plenty of evidence suggests that hypoxic effects in the TME are mediated by exosomes that carry information in cell-to-cell communication [19]. Hypoxia exerts its effects on cancer-derived exosomes in many ways, including increasing exosome release [91], elevating exosomal heterogeneity (for example, size and cargo), and enhancing exosome target cell recognition and internalization [19]. The results of comprehensive proteomics [91] showed that exosomes secreted from hypoxic tumors contain elevated protein levels of: (1) chemokines, such as colony-stimulating factor 1 (CSF1), C–C motif chemokine 2 (CCL2), and endothelial monocyte-activating polypeptide 2 (EMAP2); (2) pro-tumorigenic molecules, including matrix metalloproteinases 2 (MMP2), procollagen-lysine, 2-oxoglutarate 5-dioxygenase 1 gene (PLOD1) and annexin A4 (ANXA4); (3) soluble inhibitory factors, like transforming growth factor beta (TGFβ), macrophage migration inhibitory factor (MIF) and ferritin heavy/light chain (FTH, FTL); (4) microRNAs processing proteins and growth factors, for example, argonaute 1 (AGO1), AGO3, hepatoma‑derived growth factor (HDGF). Some of these proteins, such as CSF1, CCL2, and EMAP2, can mediate macrophage recruitment and M2 polarization. The content of this section demonstrates that the key cargos in exosomes mediated the interaction between tumor cells and TAMs under hypoxia are microRNAs (miRNAs), followed by long noncoding RNAs (lncRNAs), circular RNA (circRNAs), and interleukins (ILs).
Exosomal MiRNAs and ILs
MiRNAs, a class of regulatory non-coding RNAs (ncRNAs), are frequently found in different exosomes and are involved in tumor proliferation, angiogenesis, metastasis and chemoresistance [113]. Tumor-derived exosomes generally contain one or more miRNAs, which are involved in different signaling pathways. The production of miRNAs in tumor-derived exosomes can be regulated by HIF-1α or HIF-2α [86].
MiR-1246 targets telomeric repeat binding factor 2 interacting protein (TERF2IP) and markedly promotes M2 macrophage polarization by activating the STAT3 pathway and inhibiting the NF-κB pathway, and ultimately leading to tumor proliferation, migration and invasion [81]. Moreover, miR-1246-rich exosomes derived from hypoxic tumor cells are delivered to normoxic tumor cells for inducing tumor migration and invasion [114]. TERF2IP (also known as RAP1), a member of the shelterin complex, plays a crucial part in protecting telomeric function and maintaining chromosome stability. It also acts as an essential modulator to enhance NF-kB signaling and attenuate STAT3 signaling [81, 115]. Of note, activating the NF-κB pathway and the STAT3 pathway can induce M1 and M2 gene expression, respectively [22, 116, 117]. In addition, NF-κB is be a crucial transcription factor that regulates the release of ILs from TAMs. For example, an in vitro study illustrated that macrophages transfected with NF-κB (p50) siRNA exhibited decreased expression levels of IL-10, VEGF, and matrix metalloproteinase-9 (MMP-9), whereas increased expression levels of IL-12, tumor necrosis factor-α (TNF-α), and IL-6 [118].
Exosomes released from hypoxic epithelial ovarian cancer cells deliver miR-21–3p, miR-125 b-5p and miR-181 d-5p to macrophages and induce M2 macrophage polarization, ultimately leading to tumor proliferation and migration [83]. Among these three miRNAs, miR-21–3p and miR-125 b-5p bind to SOCS4, whereas miR-21-3p and miR-181 d-5p bind to SOCS5, resulting in the decrease of SOCS4/5 expression and the increase of phosphorylated STAT3 [83]. To be clear, SOCS4 and SOCS5, members of suppressor of cytokine signaling (SOCS) families, are critical negative regulators of the JAK-STAT pathway.
Under hypoxia, total expression of let-7a miRNA (a well-known epigenetic tumor suppressor) in tumor cells is only about 30% of that in normoxia, whereas exosomal let-7a miRNA is increased by almost 25 times. These observations indicate that let-7a miRNA is extruded from tumor cells via exosomes [91]. Exosomal let-7a miRNA is transferred to TAMs and downregulates the expression of insulin-like growth factor 1 receptor (IGF1R), insulin receptor (INSR), insulin receptor substrate-1 (IRS-1) and IRS-2. The inhibition of insulin signaling-related genes can negatively regulate the insulin AKT-mTOR signaling pathway, leading to a metabolic shift from glycolysis to oxidative phosphorylation (OXPHOS) and eventually M2-like polarization in macrophages [91].
In addition, tumor-derived exosomal miRNAs can regulate inflammatory cytokine secretion in macrophages. Hypoxia has no influences in regulating IL1A or IL6 expression in macrophages, but instead dramatically promotes their expression in the co-culture with tumor cells. The reason for this could be that the tumor inhibitor miR101 is disturbed in tumor-derived exosomes under hypoxic stress, leading to the upregulation of cyclin-dependent kinase 8 (CDK8) in macrophages and the stimulation of IL1A and IL6 secretion in macrophages [80]. As a tumor inhibitor miRNA, miR101 is participating in various cancer-related biological processes by targeting multiple oncogenes. Thus, miR101 is considered to be a potentially novel approach for cancer therapy [119, 120]. CDK8 is an oncogene that functions as a transcriptional coactivator for several oncogenic transcription factors. [121]. IL1A and IL6 are two crucial inflammatory cytokines in macrophages, playing important roles in tumor development. IL-6 has multiple functions the activation of pro-oncogenic STAT3 signaling, the enhancement of cell motility, the reduction of cell–cell adhesion, the promotion of EMT, and the stimulation of cell proliferation [122]. Given that IL-1A has both pro- and anti-tumor effects, its role in cancer development is controversial. Results of in vivo experiments revealed that IL-1A overexpression in mice suppressed liver metastasis of lymphoma, which was related to an increase in CD8+ T-cells [123]. An in vivo and in vitro investigation on hepatocellular carcinoma (HCC) illustrated that tumor-derived IL-1A promoted tumor growth by increasing tumoral infiltration of myeloid-derived suppressor cells (MDSCs), which suppressed T and NK cell activation. In contrast, systemic administration of recombinant IL-1A protein exerted an anti-tumor effect by directly activating T cells. The location of released IL-1A is therefore crucial to understanding how it contributes to tumor growth [124]. Furthermore, the detailed function of IL-1A may be related to the type of cancer [125].
Hypoxic glioma-derived exosomes can induce macrophage autophagy and M2 polarization through their highly expressed IL-6 and miR-155-3p. IL-6 facilitates M2-like macrophage polarization either directly or indirectly through the IL-6-pSTAT3 pathway and the IL-6-autophagy-pSTAT3 pathway, respectively. Overexpression of miR-155-3p downregulates CREBRF gene expression and promotes autophagy in macrophages [87]. CREB3 regulatory factor (CREBRF), a negative regulator of CREB3 (cAMP responsive element binding protein 3), contributes an important part in hypoxia-induced autophagy in glioma cells [126]. Additionally, IL-6 can upregulate miR-155-3p expression by activating STAT3 in TAMs [87]. Autophagy plays a pivotal role in promoting M2-like macrophage polarization by activating the STAT3 pathway [127,128,129].
Tumor–derived exosomal miR-301a-3p, which is regulated by HIF-1α and HIF-2α, can be transferred to TAMs, promoting tumor cell EMT, migration, invasion, and metastatic potential. Exosomal miR-301a-3p mediates macrophages M2 polarization via downregulating PTEN (phosphatase and tensin homolog deleted on chromosome ten) expression and activating the PI3Kγ signaling pathway [86]. PTEN is a widely known tumor suppressor gene with phosphatase activity and regulates cell growth, proliferation, apoptosis, adhesion, migration, invasion, and genomic integrity [130]. PTEN can inhibit the PI3K/AKT pathway by dephosphorylation of phosphatidylinositol 3,4,5-trisphosphate (PIP3) [130]. It is worth noting that even a tiny decrease in PTEN levels can contribute to obvious cancer susceptibility and tumor progression [131]. Furthermore, miR-1305 [82], miR-21 [84], and miR-940 [88] in tumor-derived exosomes can promote M2 macrophage phenotype, but mechanisms of these miRNAs have not been thoroughly studied.
Under hypoxic conditions, macrophages can also secrete miRNAs via exosomes to regulate tumor biological functions. Exosomal miR223 derived from hypoxic TAMs is internalized into co-cultured tumor cells, resulting in the decreased apoptosis rate, increased cell viability, and enhanced drug resistance. Specifically, miR-223 down-regulates expression of PTEN and gradually increases PI3K/AKT signal activation [85]. However, inhibition of miR-223 expression cannot completely eliminate the promotion of chemoresistance by hypoxic macrophage-derived exosomes [85], indicating that the communication between tumor cells and macrophages under hypoxia is quite complex. Taken together, exosomal miRNA is a well-investigated mediator of tumor-macrophage communication under hypoxia.
Exosomal CircRNAs
CircRNAs are a novel class of nc-RNAs that have stronger stability than linear RNAs due to their covalently closed loops and have become a hotspot in recent years [132]. As a competitive endogenous RNA (ceRNA), circRNAs regulate gene expression through sponging miRNAs. For example, Hsa-circ-0048117 can be used as a ceRNA to inhibit the activity of miR-140. Tumor-derived exosomal Has-circ-0048117 inhibits miR-140 expression, upregulates the TLR4 expression, and promotes M2 polarization. Subsequently, Arg1, IL-10 and TGF-β secreted by M2 macrophages facilitate tumor cell invasion and migration [89]. TLR4 is a typical receptor from the Toll-like receptors (TLRs) family that is expressed on both immune cells and tumor cells, and its overexpression may lead to cancer progression [133, 134]. Paradoxically, TLR4 can cause macrophage polarization towards M1 [135]or M2 [136, 137].
Exosomal LncRNAs
LncRNAs are nc-RNAs that have more than 200 nucleotides and perform a variety of functions in the nucleus and cytoplasm. In the nucleus, they are involved in regulating chromosome architecture, modulating inter- and intrachromosomal interactions, remodeling chromatin, and directly regulating transcription. In cytoplasm, they can modulate mRNA stability, translation, and post-translation [138]. The newly discovered lncRNA hyaluronan-mediated motility receptor antisense RNA 1 (HMMR-AS1) [139] is found in tumor cytoplasm and is involved in cell proliferation, cell migration, and EMT [90]. Hypoxic condition can enhance the transcription of HMMR-AS1 by introducing the binding of promoter regions and HIF-1α. Tumor-derived exosomal HMMR-AS1 regulates M2-shifted polarization by miR-147a/A-T rich interacting domain 3a (ARID3A) axis [90]. Concretely, HMMR-AS1 functions as a ceRNA of miR-147a, limiting ARID3A degradation while increasing inhibition of M1 type polarization and promoting M2 type polarization [90].
Other exosomes
Exosomes released from intermittently hypoxic tumor cells also promote PD-L1 expression in macrophages, providing biological plausibility for explaining the underlying mechanisms of poor prognosis observed in patients with cancer and obstructive sleep apnea (OSA) [92]. Unfortunately, this study did not fully elucidate the specific exosome components that dominate this effect [92]. PD-L1 commonly expresses on TAMs in high grade serous ovarian cancers (HGSOC) at both the original and metastatic locations [140]. The binding of PD-L1 expressed on macrophages to PD-1 expressed on T-cells inhibits T cell cytotoxicity [141].
Cytokines and growth factors
Cytokines, a class of diverse low-molecular weight proteins, include IL, colony-stimulating factors, chemokines, and tumor necrosis factors [142]. Chemokines can be categorized into four subclasses regarding to their amino acid motif at N-termini: CXC, CC, C, or CX3C, where C and X stand for cysteine and non-cysteine residues, respectively [143].
The inflammatory cytokine oncostatin M (OSM), which belongs to the IL-6 superfamily, is an essential part of the secretome of hypoxic cancer cells. OSM can enhance the expression of M2 macrophage surface markers (viz. CD206 and CD163) as well as functional markers (viz. arginase-1, and cyclooxygenase-2) in macrophages, which are involved in activation of mTOR signaling complex 2 (mTORC2) pathway [93]. Additional research showed that activated mTORC2 leads to M2 polarization by relaying signals through its effector kinases Akt, particularly Akt1, rather than PKCα. IL-4 is another classic mediator of M2 polarization by activating the mTORC2 pathway [93].
IL-8 (also known as CXCL8) is mainly produced by macrophages and plays a controversial role in regulating cancer progression [144]. Hypoxia increases IL-8 secretion significantly in macrophages but only slightly in gastric cancer (GC) cells [94]. Macrophage-derived CXCL8 induced by hypoxia can activate the JAK/STAT1 signaling pathway through binding to CXCR1/2 expressed on GC cells, leading to GC invasion and proliferation. The activation of STAT1 directly upregulates the expression of IL-10, stimulating M2 polarization of macrophages through the NF-κB signaling pathway. CXCL8 production is further encouraged by NF-κB activation [94].
The expression level of C–C chemokine ligand 8 (CCL8) is increased by zinc finger E-box binding homeobox 1 (Zeb1) via directly interacting with the CCL8 promoter in the hypoxic TME. Subsequently, CCL8 promotes TAMs infiltration via CCR2–NF-κB pathway, and this result is typically associated with a poor prognosis in cervical cancer [95]. Zeb1 is a typical transcription factor that is widely expressed in carcinomas and has a significant role in the development of cancer by promoting EMT and chemoresistance in cancer cells [145]. Monocyte chemoattractant protein-1 (MCP-1), also known as CCL2, is a key chemokine controlling macrophage migration and invasion [146]. Under hypoxic stress, NF-κB/HIF-1α activation encourages lung cancer cells to secrete MCP-1, which furthers the accumulation of macrophages [96].
Under a hypoxic microenvironment, vascular endothelial growth factor (VEGF) and IL-6, generated by head and neck squamous cell carcinoma (HNSCC) attract macrophages and polarize them to the M2 type. Later, M2-type TAMs release CCL15 through the HIF-2α pathway, which leads to gefitinib resistance in HNSCC via CCL15-CCR1-NF-κB pathway [97]. Moreover, VEGF can also be secreted from macrophages, and both its mRNA and protein levels rise in response to hypoxia in a time-dependent manner. The upregulation of VEGF increases the phosphorylation of Akt and p38, contributing to the proliferation and invasion of tumor cells [98]. Under hypoxic conditions, TAMs-derived VEGF binds to its receptor VEGFR on tumor cells, activating the PI3K-Akt and p38 MAP kinase pathways to promote tumor cell proliferation and invasion [98].
Semaphorin3A (Sema3A) is a membrane-bound protein that has been shown to be a prognostic marker for patients with metastatic colorectal cancer (mCRC) [147]. In vivo studies have revealed that Sema3A is an endogenous inhibitor of angiogenesis that counteracts angiogenic factors like VEGF-A. Sema3A has the ability to regulate tumor blood vessels, alleviate tumor hypoxia, and inhibit tumor growth [148]. Sema3A expression is higher in hypoxic tumor single cell suspensions than in normoxic conditions [99]. In vivo and in vitro studies have documented that Sema3A drives TAMs toward hypoxic niches via the Sema3A–neuropilin-1 (Nrp1) pathway. Following macrophage localization in the hypoxic environment, Nrp1 is downregulated, and Sema3A captures TAMs locally via Nrp1-independent plexinA1-plexinA4-mediated stop signals [99]. Finally, hypoxic TAMs acquire protumoral phenotypes. The absence of Sema3A leads to a more M1-like phenotype and a reduced tumor growth [99]. Nrp-1 is a pleiotropic single-pass transmembrane protein that functions as a co-receptor to many extracellular ligands [149]. In vitro study showed that the expression of Nrp-1 in tumor cells was upregulated under hypoxic situation, which resulted in recruiting more macrophages and educating them into M2-phenotype [105]. However, more research is required to fully understand how Nrp-1, which is produced by cancer cells, promotes M2 macrophage polarization under hypoxic conditions.
Myeloid-derived growth factor (MYDGF), which is generated when tissue is damaged, exerts a crucial role in regulating neutrophil interstitial motility and inflammation in a way that is HIF-1α dependent [150]. Hepatocellular carcinoma contains hypoxia-induced MYDGF in its cytoplasm and cell membrane, which can stimulate tumor angiogenesis and boost the potential of cancer stem cells to self-renew. Additionally, tumor-derived MYDGF promotes the release of inflammatory cytokines including TNF-α and IL-6 and increases macrophage infiltration, all of which ultimately aid in the growth of tumors [100]. However, the molecular mechanism of MYDGF action in tumor progression is still unclear, and it merits further research.
Binding proteins and protease inhibitors
Galectin-3 (Gal‐3), a member of the β-galactoside binding protein family [151], is both a prognostic indicator and a potential target for cancer treatment [152,153,154]. Gal‐3 is also expressed in tumor tissues in a HIF-1α-dependent manner [155], causing an increase in PD‐L1 level via STAT3 phosphorylation in carcinomas [156]. Gal‐3 secreted by TAMs during hypoxia promotes tumor metastasis and angiogenesis, which is highly dependent on the degree and duration of hypoxia [101]. Although the expression level of HIF-1α is elevated in hypoxic TAMs, HIF-1α inhibitors have no effect on the expression of Gal-3 there, suggesting that HIF-1α may not be involved in Gal-3 expression in hypoxic TAMs. Interestingly, HIF-1α inhibitor 2ME2 can upregulate Gal-3 expression in normoxia but not in hypoxia. Further research revealed that the upregulation of Gal‐3 expression in hypoxic TAMs is associated with an increase in intracellular reactive oxygen species (ROS) level via activation of NF-κB nucleation. In addition, Gal-3 overexpression enhances VEGFA secretion and glucose consumption in TAMs [101].
As a nuclear DNA-binding protein, high-mobility group box 1 (HMGB1) can be robustly upregulated via HIF-1α signaling after prolonged exposure to hypoxia in tumors. Increased HMGB1 levels encourage macrophage infiltration and cytokine expression(i.e. IL-6). Subsequently, macrophage-derived IL-6 activates STAT3 signaling and promotes EMT in tumor cells [102].
However, other studies noticed that tumor-infiltrating macrophages in the hypoxic microenvironment may have tumor suppressing effects. Under hypoxic conditions, HIF-2α highly expressed in TAMs induces the secretion of the serine protease inhibitor Spint1. Spint1 is then released into TME to block the serine protease HGF activator (HGFA), preventing the cleavage of pro-HGF into active hepatocyte growth factor (HGF) [103]. When activated HGF binds to the c-Met receptor on tumor cells, it activates several signaling pathways, including MAPK, PI3K/AKT, and STAT3, which promotes tumor growth and therapeutic resistance [157]. Thus, TAM-secreted Spint1 can reduce tumor cell proliferation.
Ligand-receptor interaction between tumor cells and TAMs
The binding of CD47 (cluster of differentiation 47) on tumor cells to SIRPα (signal-regulatory protein α) ligand on macrophages is a typical tumor escape mechanism. The "don't eat me" signal is released by the CD47 receptor when it binds to SIRPα, which impairs the phagocytic activity of macrophages [158]. HIF has been shown to activate highly expressed CD47 in various cancer types [158]. However, hypoxia may have beneficial effects on cancer therapy via SIRPα-CD47 axis. Colon cancer has a better prognosis than other cancer types because it has higher levels of macrophage infiltration and HIF-1α expression [104]. Hypoxia can decrease SIRPα expression in macrophages while simultaneously increasing CD47 expression in colon cancer cells. The heightened signal of "don't eat me" is countered by the reduced SIRP expression level, increasing the phagocytic capacity of macrophages [104]. Therefore, HIF-1α does have the ability to enhance phagocytosis of macrophages, which may be dependent on cancer types [158].
Tumor cell debris
Tumor necrotic debris caused by hypoxia can release different signals leading to cancer progression [159]. Macrophages can accumulate in perivascular and perinecrotic niches in tumors [160] where they can operate as immune scavengers to sweep away cellular debris [161]. An interesting study demonstrated that the necrotic debris from severely hypoxic cancer cells modulates the communication between tumors and TAMs [106]. Under conditions of moderate hypoxia, HIF-1α facilitates IL-1β secretion in macrophages. When exposed to severe hypoxia, necrotic cancer cell debris can stimulate IL-1β secretion in macrophages via TLR4/TRIF/NF-κB signaling. Specifically, necrotic debris enhances TLR4 signaling by attracting more TLR4 receptors to the macrophage membrane and activating TIR domain-containing adapter-inducing interferon-β (TRIF). Following then, phosphorylated NF-κB is up-regulated, resulting in macrophage M2 polarization and IL-1β secretion. Macrophage-derived IL-1β activates the IL-1β/HIF-1α/COX-2 axis, enhancing tumor cell EMT and promoting tumor invasion and metastasis [106].
It is worth noting that IL-1β in the TME can be an important driver of immune suppression. For instance, in mouse models of spontaneous breast cancer metastasis, IL1β stimulates IL17 expression from γδ T cells, leading to neutrophil accumulation via systemic induction of G-CSF. Neutrophils inhibit CD8+ T cell activation, allowing cancer cells to spread [162]. Interestingly, the results of IL1β mRNA expression in diverse cell populations separated from the transplanted tumors indicate that macrophages are the most abundant IL1β-expressing cell type [162]. These findings also highlight the importance of cross-talk between immune cells in influencing immune responses in tumors. Similarly, Máté Kiss et al. [163] also observed that IL1β exerted an immune-suppressive function in TME in two distinct mouse models. The researchers found that increased IL1β production within tumors was released mainly by neutrophils, monocytes, and macrophages. A noteworthy finding in that study was that the immunostimulatory major histocompatibility complex (MHC)-IIhigh TAMs produced large amounts of IL1β.
The NF-κB pathway has been identified as a critical regulator of macrophage behavior in the TME. Interestingly, NF-κB pathway exerts dual effects on macrophage polarization—both promotion and inhibition of M1 polarization. Studies in vitro showed that transfection of NF-κB (p50) siRNA into M2-like macrophages leaded to the anti-tumorigenic M1 phenotype [118]. Similarly, an in vivo study suggested that specific blockade of NF‐κB signaling in macrophages could switch macrophages from a M2 to a M1 phenotype [164]. However, some studies in vivo showed that the increased NF-κB activity in macrophages resulted in reduced tumor burden and persistent macrophage M1 polarization [165]. There are five members of the NF-κB family of transcription factors: p65 (RELA), p50 (NFKB1), p52 (NFKB2), c-REL, and RELB [166]. These members can couple to form different homo- or heterodimers, which have opposing effects on macrophage polarization, depending on the source of macrophage populations and the way that macrophages are activated [167]. Lipopolysaccharide, for example, promotes the overexpression of p50-p50 homodimers, allowing M1 to M2 macrophage reprogramming [168,169,170]. In contrast, Bufalin promotes the overexpression of p65-p50 heterodimers, leading to the transition of macrophage from M2 to M1 [171]. Therefore, in order to better understand the multifaceted role that NF-κB plays in regulating TAMs function, it may be useful to investigate the exact functions of various NF-κB dimers.
Oncometabolites
Under hypoxic conditions, tumor cells may undergo metabolic reprogramming allowing them to shift from oxidative phosphorylation to anaerobic glycolysis. Succinate, an intermediate of the tricarboxylic acid (TCA) cycle, and lactate, an end product of glycolysis, are two examples of tumor metabolites produced by hypoxic tumor cells that can influence macrophage activity [172].
Lactic acid shuttles among different cells within the TME, acting not only as a stromal cell energy supplier, but also as a signaling molecule to intensify crosstalk between tumor cells and adjacent cells [173]. In vivo and in vitro study showed that lactate induces VEGF expression and M2-like polarization of TAMs, both of which are mediated by HIF-1α [107]. Lactate targets the protein-coupled receptors on the surface of the TAMs membrane and induces M2-type polarization via the PKA/CREB pathway [174]. Additionally lactic acid produced under hypoxia is believed to be a weapon for activating pro-angiogenic TAMs and increasing PD-L1 protein expression in TAMs [172, 175, 176]. It's important to note that in vitro research indicated that lactate differentially influenced TAM subgroup metabolism [108]. These subsets are known to reside in different intratumoral locations, with MHC-IIlo TAMs being enriched in hypoxic tumor areas. Lactate promotes oxidative metabolism in MHC-IIlo TAMs while inhibiting it in MHC-IIhi TAMs. Furthermore, in the presence of lactate, MHC-IIlo TAMs showed an improved ability to inhibit T cells.
Extensive evidences showed a positive synergistic relationship between hypoxia and lactate [177]. When normoxic and hypoxic macrophages are treated with different lactate doses, the protein levels of ARG1 increase concomitantly in hypoxic macrophages but not in normoxic ones, indicating that the combination of low oxygen and lactate is already sufficient enough to trigger Arg-1 expression [177]. Moreover, a high concentration of lactic acid causes medium acidification, which kills macrophages rather than causing Arg-1 expression. Macrophages have the ability to detect the presence of hypoxia and lactate. These signals can then be integrated with phenotypic responses by MAPK signaling, which results in the release of pro-angiogenic cytokines like VEGFA [177]. In-depth bioinformatics analysis of macrophages transcriptome data indicated that lactate has only mild impacts on macrophages under normoxic conditions. When lactate is combined with hypoxia, macrophages become significantly more M2-polarized via the HIF-1, Hedgehog and mTOR pathways is observed [178]. However, it is important to note that most of the studies on the effect of lactate on macrophages were conducted under normoxic conditions, rather than hypoxic conditions. The information in this section may serve as a reminder of the importance of conducting research under hypoxic conditions.
Succinate is a typical TCA cycle intermediate that promotes inflammation and can accumulate in macrophages in response to lipopolysaccharide (LPS). Succinate accumulation robustly boosts HIF-1α protein levels, leading to increased secretion of IL-1β from macrophages [179]. Specifically, succinate produced by tumors is released into extracellular milieu and interacts to succinate receptor (SUCNR1) on the membrane of macrophages. As a result of this binding, the PI3K-HIF-1 axis is activated, which causes macrophage recruitment, migration, and an M2-skewed phenotype. M2 polarized macrophages secret IL-6 to enhance cancer cell migration. Meanwhile, tumor-derived succinate also activates SUCNR1 on the membrane of tumor cells to induce cancer cell migration and EMT through the PI3K/HIF-1α pathway [109]. These findings demonstrated that the tumor-derived succinate has great potential to be a novel target for anti-tumor therapy because of its ability to control TAM polarization and tumorigenic pathway.
HIF-1α/2α inhibitors for cancer treatment in clinical studies
As previously mentioned, hypoxia not only affects the biological function of tumor cells and macrophages but also the communication between them, triggering a series of signal pathways to support tumor survival. The activation of HIF-1α/2α is one of the main initiators of macrophage-cancer cell interaction. Inhibiting the activity of HIF-1α/2α can therefore open up new possibilities for tumor treatment approaches. This section will provide insights for therapeutic development by summarizing those pharmaceuticals that successfully entered clinical trials or the market for inhibiting HIF (Fig. 4) (Table 2).

Mechanisms of action of the HIF inhibitors currently on the market or under clinical trials. HIF inhibitors target HIF on different levels, ranking from transcription, translation, protein stabilization, transcriptional coactivators recruitment, and dimerization. The clearance of ROS leads to decreased HIF-α stabilization and accumulation
Small-molecule inhibitors
The PAS-B binding pocket on the HIF-2α contains a unique hydrophilic cavity that can accommodate a small molecule, which may result in a conformation change in HIF-2α and disruption of its interaction with ARNT [180, 181]. Accordingly, HIF-2α small-molecule inhibitors are discovered, such as Belzutifan (Welireg™, MK-6482) [182] and PT2385 [183]. Despite the high sequence identity between HIF-2α and HIF-1α, these small-molecule inhibitors are highly selective in dissociating the HIF-2α/ARNT heterodimer while having no effect on HIF-1 function [181, 184].
Belzutifan is the first FDA-approved treatment for Von Hippel-Lindau (VHL) disease in patients with renal cell carcinoma (RCC), central nervous system (CNS) hemangioblastomas, or pancreatic neuroendocrine tumors (pNET) without the request of immediate surgery [185,186,187]. VHL disease is a rare autosomal dominantly inherited tumor syndrome caused by germline mutation or deletion of VHL gene [188, 189]. The incidence of RCC patients with VHL disease is high due to VHL gene inactivation and constitutive activation of the transcription factor HIF-2α [190]. Belzutifan is also expected to be used in the treatment of polycythemia and multiple paragangliomas (the Pacak–Zhuang syndrome), which are caused by somatic mosaicism for an activating mutation in EPAS1 [191]. Several ongoing clinical trials are currently focusing on the evaluation of the efficacy of belzutifan in combination with other medicines, such as pembrolizumab, Lenvatinib, and cabozantinib (ClinicalTrials.gov Identifier: NCT04976634, NCT05239728, NCT03634540, NCT04736706, NCT05030506, NCT04626518, NCT04586231, NCT04626479). There are also some clinical trials of belzutifan used alone for other tumors (Table 2). It is hoped that these studies will yield excellent patient survival data.
PT2385, a small molecule drug, is the first HIF-2α antagonist progressed into clinical trials [192]. PT2385 can inhibit HIF-2 dimerization in healthy tissue and ccRCC metastases [193, 194]. PT2385 can significantly alleviate the undesirable adverse effects of sorafenib through inhibiting HIF-2α, increasing androgen receptor (AR) and suppressing downstream pSTAT3/pAKT/pERK pathways [195].
PX-478 is an active HIF-1α small-molecule inhibitor with potent antitumor activities [196, 197]. PX-478 can inhibit HIF-1α protein levels, transactivating activity, and deubiquitination. In addition, PX-478 prevents the synthesis of VEGF that is generated by hypoxia in various cancer cell lines [198]. By inhibiting the HIF-1α/lysyl oxidase-like 2 (LOXL2) signaling pathway, PX-478 can enhance immunotherapeutic effectiveness and reduce the EMT phenotypes induced by hypoxia [199]. PX-478 can drastically reduce the expression level of granulocyte–macrophage-colony-stimulating factors (GM-CSF) and the incidence of perineural invasion (PNI) in pancreatic ductal adenocarcinoma (PDAC) [200].
Cycling hypoxia increases the production of ROS, which promotes HIF-1α and NF-κB activation in tumor cells [201]. ROS is an important mediator of HIF-stability by inhibiting the activity of PHD and FIH in the cytoplasm [201]. MBM-02, also known as Tempol, is a dual-specific HIF-1 and HIF-2 inhibitor (ClinicalTrials.gov Identifier: NCT04876755). As a well-known antioxidant, MBM-02 promotes the clearance of ROS and inhibits cycling hypoxia-induced chemoresistance [202]. In addition, DFF332 (ClinicalTrials.gov Identifier: NCT04895748) and NKT2152 (ClinicalTrials.gov Identifier: NCT05119335) are also small molecules that inhibit HIF2α.
Nucleic acid therapeutics
Due to their high target-specificity, nucleic acid therapeutics, such as miRNA-based molecules, lncRNAs, small interfering RNAs (siRNAs), antisense oligonucleotides (ASOs), mRNA therapeutics, and nucleic acid aptamers, have recently been successful in emerging into a highly attractive class of medicines [203].
RO7070179 (EZN-2968) is an ASO specifically targeting HIF-1α in a synthetic locked nucleic acid (LNA) form, which can reduce HIF-1α mRNA levels [204]. ASOs typically contain < 20 mER DNA or RNA nucleotides and can target mRNAs that are largely degraded through RNAse H-mediated cleavage. ASOs also inhibit the interaction between its targeted mRNAs and their paired enzymes, which blocks the transcription or translation of target genes [205, 206]. LNA-based oligonucleotides offer the advantages of remarkable stability, low off-target events, and high target-mRNA binding affinity [207, 208].
ARO-HIF2 is a synthetic double-stranded RNA interference (RNAi) trigger with an αvβ3 targeting ligand designed to silence HIF-2α expression. RNAi is a natural protective mechanism induced by double stranded RNAs (dsRNAs), leading to efficient and specific degradation of homologous mRNA [209]. Integrins αvβ3, which is related to tumor progression and metastasis, is frequently overexpressed in ccRCC and can be selectively bound by ARO-HIF2 [210]. The results of the phase I clinical trial provides preliminary evidence for the safety and efficacy of ARO-HIF2 in patients with advanced ccRCC [211].
Drug repurposing
Compared with traditional de novo drug application, drug repurposing has become an effective alternative drug therapeutic strategy due to its lower risk of failure, reduce costs, and higher efficiency [212]. As shown in Table 2, there are some examples of drug repurposing to target hypoxia signaling in cancer.
Camptothecin (CPT) and its analogs (including SN-38, topotecan, and irinotecan) are important Topoisomerase I inhibitors that can block HIF-1α expression [213]. CPT has been approved for its ability to decrease the number of cancer stem cells (CSCs) and inhibit the accumulation of HIF-1α [214]. CPT can initiate the transcription of long noncoding antisense RNAs at the 5′ and 3′ ends of the HIF-1α, leading to posttranscriptional regulation of gene expression. Interestingly, low camptothecin concentrations have an effect on miR expression profiles, particularly increasing miR-17-5p and miR-155, which are two important players in reducing of HIF-1α protein accumulation and activity [215]. CRLX101 (NLG207) is a nanoparticle-drug conjugate (NDC) of CPT designed to overcome the poor physicochemical properties of CPT and allow more of CPT to accumulate at tumor sites. CRLX101 plus enzalutamide has been shown to be effective in preclinical prostate cancer models with enzalutamide resistance, and clinical trials are currently underway (NCT03531827) [216]. SN-38, the active metabolite of Irinotecan (CPT-11), can overcome hypoxia-induced chemoresistance [217] and inhibit the radiation-induced up-regulation of HIF-1α [218].
Additional data suggested that melatonin may also be a potent anti-tumor agent, inhibiting hypoxia-mediated tumor survival, angiogenesis, invasion, and migration [219]. Melatonin can block tumor angiogenesis by reducing HIF-1α protein expression in tumors [220]. Specifically, melatonin can inhibit the sphingosine kinase 1 (SPHK1) signaling pathway and impairs ROS generation in hypoxic cancer cells [221]. Under hypoxia, SPHK1 is activated by ROS to promote the accumulation of HIF-1α and initiate its transcriptional activity [221, 222]. Digoxin is also able to inhibit HIF-1α protein translation and HIF-2α mRNA expression, leading to anti- tumor effects [223].
Conclusion and future perspective
Hypoxia is a critical factor that affects the communication between tumor cells and TMAs. Hypoxia influences the crosstalk between TAMs and tumor cells via vast multifunctional exosomes, cytokines, growth factors, cellular debris, oncometabolites, and a variety of ligands and receptors on the cell surface. Hypoxia-induced interaction between tumor cells and TAMs promotes tumor proliferation, migration, invasion, angiogenesis, drug resistance, EMT, and cancer stem cell self-renewal. In addition, hypoxia also promotes macrophage phagocytosis, which inhibits tumor cell proliferation. Therefore, hypoxia is a double-edged sword and a non-negligible factor in anti-tumor treatment, which needs further research to evaluate the evidences.
As Semenza stated in 2017, "It is ironic that hypoxic cancer cells… would be the last to capture the attention of oncologists." [224]. The majority of studies are still performed under normoxia, frequently ignoring the importance of hypoxia. As a result, more emphasis should be placed on hypoxic exploration. Oxygen level and duration have an impact on the stabilization of HIF-1α and HIF-2α [225], which in turn activates different signal pathways. Thus, more attention need to be paid on the detailed hypoxia level [61]. Plenty of studies have proved that HIF pathway is the core mechanism of cell hypoxic adaptation. However, the observation of oxygen sensing by KDMs suggested that additional oxygen sensors independent of HIF may be presented. The mechanisms of tumor hypoxic adaptation are more complex than currently envisaged. In clinical studies, HIF inhibitors are currently divided into three categories: small molecule drugs, nucleic acid drugs and drug repurposing. Applying strategies to suppress the negative effects of hypoxia appears to be useful to overcome malignant tumors, especially an already-approved HIF-2α inhibitor that has shown promising therapeutic activity, which has greatly enhanced our confidence in developing HIF inhibitors. In the future, we anticipate HIF pathway inhibitors being a cornerstone of cancer treatment.
Availability of data and materials
Not applicable.
Abbreviations
- TAMs:
-
Tumor-associated macrophages
- TME:
-
Tumor microenvironment
- EMT:
-
Epithelial mesenchymal transition
- HbCO:
-
Carboxyhemoglobin
- IH:
-
Intermittent hypoxia
- H-R cycles:
-
Cycles of hypoxia and reoxygenation
- HRE:
-
Hypoxia response element
- HIF-1:
-
Hypoxia-inducible factor 1
- ARNT:
-
Aryl hydrocarbon receptor nuclear translocator
- PHDs:
-
Prolyl hydroxylases
- FIH:
-
Factor inhibiting HIF
- VHL:
-
Von Hippel-Lindau
- KDMs:
-
Histone lysine demethylases
- PIM:
-
Proviral insertion site in Moloney murine leukaemia virus
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
Long noncoding RNAs
- circRNAs:
-
Circular RNA
- IL:
-
Interleukin
- ncRNAs:
-
Non-coding RNAs
- TERF2IP:
-
Telomeric repeat binding factor 2 interacting protein
- SOCS:
-
Suppressor of cytokine signaling
- IGF1R:
-
Insulin-like growth factor 1 receptor
- INSR:
-
Insulin receptor
- IRS:
-
Insulin receptor substrate
- OXPHOS:
-
Oxidative phosphorylation
- CDK8:
-
Cyclin-dependent kinase 8
- CREB3:
-
CAMP responsive element binding protein 3
- PTEN:
-
Phosphatase and tensin homolog deleted on chromosome ten
- PIP3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- ceRNA:
-
Competitive endogenous RNA
- TLRs:
-
Toll-like receptors
- HMMR-AS1:
-
Hyaluronan-mediated motility receptor antisense RNA 1
- ARID3A:
-
A-T rich interacting domain 3a
- OSA:
-
Obstructive sleep apnea
- OSM:
-
Oncostatin M
- mTORC2:
-
MTOR signaling complex 2
- CCL8:
-
C–C chemokine ligand 8
- Zeb1:
-
Zinc finger E-box binding homeobox 1
- MCP-1:
-
Monocyte chemoattractant protein-1
- VEGF:
-
Vascular endothelial growth factor
- MYDGF:
-
Myeloid-derived growth factor
- Gal‐3:
-
Galectin-3
- ROS:
-
Reactive oxygen species
- HMGB1:
-
High-mobility group box 1
- HGF:
-
Hepatocyte growth factor
- CD47:
-
Cluster of differentiation 47
- SIRPα:
-
Signal-regulatory protein α
- Nrp-1:
-
Neuropilin-1
- TRIF:
-
TIR domain-containing adapter-inducing interferon-β
- TCA:
-
Tricarboxylic acid
- HK2:
-
Hexokinase2
- PFKP:
-
Phosphofructokinase, platelet
- PKM2:
-
Pyruvate kinase M2
- SUCNR1:
-
Succinate receptor
- AR:
-
Androgen receptor
- LOXL2:
-
Lysyl oxidase-like 2
- GM-CSF:
-
Granulocyte–macrophage-colony-stimulating factors
- siRNAs:
-
Small interfering RNAs
- ASOs:
-
Antisense oligonucleotides
- LNA:
-
Locked nucleic acid
- RNAi:
-
RNA interference
- dsRNAs:
-
Double-stranded RNAs
- CPT:
-
Camptothecin
- CSCs:
-
Cancer stem cells
- NDC:
-
Nanoparticle-drug conjugate
- SPHK1:
-
Sphingosine kinase 1
- S1P:
-
Sphingosine 1-phosphate
References
Rickard AG, Palmer GM, Dewhirst MW. Clinical and Pre-clinical Methods for Quantifying Tumor Hypoxia. Adv Exp Med Biol. 2019;1136:19–41.
Codony VL, Tavassoli M. Hypoxia-induced therapy resistance: Available hypoxia-targeting strategies and current advances in head and neck cancer. Transl Oncol. 2021;14(3):101017.
Alharbi M, Lai A, Sharma S, Kalita-de Croft P, Godbole N, Campos A, et al. Extracellular Vesicle Transmission of Chemoresistance to ovarian cancer cells is associated with hypoxia-induced expression of glycolytic pathway proteins, and prediction of epithelial ovarian cancer disease recurrence. Cancers (Basel). 2021;13(14):3388.
Li X, Wu Y, Zhang R, Bai W, Ye T, Wang S. Oxygen-Based Nanocarriers to Modulate Tumor Hypoxia for Ameliorated Anti-Tumor Therapy: Fabrications, Properties, and Future Directions. Front Mol Biosci. 2021;8:683519.
Jing X, Yang F, Shao C, Wei K, Xie M, Shen H, et al. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol Cancer. 2019;18(1):157.
Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer. 2016;2(12):758–70.
Kumar B, Adebayo AK, Prasad M, Capitano ML, Wang R, Bhat-Nakshatri P, et al. Tumor collection/processing under physioxia uncovers highly relevant signaling networks and drug sensitivity. Sci Adv. 2022;8(2):eabh3375.
Tang M, Bolderson E, O’Byrne KJ, Richard DJ. Tumor Hypoxia Drives Genomic Instability. Front Cell Dev Biol. 2021;9:626229.
Rakesh R, PriyaDharshini LC, Sakthivel KM, Rasmi RR. Role and regulation of autophagy in cancer. Biochim Biophys Acta Mol Basis Dis. 2022;1868(7):166400.
Liu S, Cao X, Wang D, Zhu H. Iron metabolism: State of the art in hypoxic cancer cell biology. Arch Biochem Biophys. 2022;723:109199.
Hao X, Ren Y, Feng M, Wang Q, Wang Y. Metabolic reprogramming due to hypoxia in pancreatic cancer: Implications for tumor formation, immunity, and more. Biomed Pharmacother. 2021;141:111798.
Davis L, Recktenwald M, Hutt E, Fuller S, Briggs M, Goel A, et al. Targeting HIF-2α in the Tumor Microenvironment: Redefining the Role of HIF-2α for Solid Cancer Therapy. Cancers (Basel). 2022;14(5):1259.
Schegoleva AA, Khozyainova AA, Gerashchenko TS, Zhuikova LD, Denisov EV. Metastasis prevention: targeting causes and roots. Clin Exp Metastasis. 2022;39(4):505–19.
Kaushik N, Kim S, Suh Y, Lee SJ. Proinvasive extracellular matrix remodeling for tumor progression. Arch Pharm Res. 2019;42(1):40–7.
Buyuk B, Jin S, Ye K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell Mol Bioeng. 2022;15(1):1–13.
Xiang L, Semenza GL. Hypoxia-inducible factors promote breast cancer stem cell specification and maintenance in response to hypoxia or cytotoxic chemotherapy. Adv Cancer Res. 2019;141:175–212.
Mortezaee K, Majidpoor J. The impact of hypoxia on immune state in cancer. Life Sci. 2021;286:120057.
Ikeda S, Tagawa H. Impact of hypoxia on the pathogenesis and therapy resistance in multiple myeloma. Cancer Sci. 2021;112(10):3995–4004.
He G, Peng X, Wei S, Yang S, Li X, Huang M, et al. Exosomes in the hypoxic TME: from release, uptake and biofunctions to clinical applications. Mol Cancer. 2022;21(1):19.
Chang WH, Lai AG. The hypoxic tumour microenvironment: A safe haven for immunosuppressive cells and a therapeutic barrier to overcome. Cancer Lett. 2020;487:34–44.
Terceiro LEL, Edechi CA, Ikeogu NM, Nickel BE, Hombach-Klonisch S, Sharif T, et al. The Breast Tumor Microenvironment: A Key Player in Metastatic Spread. Cancers (Basel). 2021;13(19):4798.
Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. Int J Mol Sci. 2021;22(13):6995.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20.
Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–88.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Front Immunol. 2020;11:583084.
He D, Wang D, Lu P, Yang N, Xue Z, Zhu X, et al. Single-cell RNA sequencing reveals heterogeneous tumor and immune cell populations in early-stage lung adenocarcinomas harboring EGFR mutations. Oncogene. 2021;40(2):355–68.
Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: implications on therapeutic strategies. Exp Hematol Oncol. 2021;10(1):60.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19(6):369–82.
Zheng X, Weigert A, Reu S, Guenther S, Mansouri S, Bassaly B, et al. Spatial Density and Distribution of Tumor-Associated Macrophages Predict Survival in Non-Small Cell Lung Carcinoma. Cancer Res. 2020;80(20):4414–25.
Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y, et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74(1):24–30.
Díaz-Bulnes P, Saiz ML, López-Larrea C, Rodríguez RM. Crosstalk Between Hypoxia and ER Stress Response: A Key Regulator of Macrophage Polarization. Front Immunol. 2019;10:2951.
Han Y, Wang X, Xia K, Su T. A novel defined hypoxia-related gene signature to predict the prognosis of oral squamous cell carcinoma. Ann Transl Med. 2021;9(20):1565.
Yang M, McKay D, Pollard JW, Lewis CE. Diverse Functions of Macrophages in Different Tumor Microenvironments. Cancer Res. 2018;78(19):5492–503.
Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126(10):3672–9.
He Z, Zhang S. Tumor-Associated Macrophages and Their Functional Transformation in the Hypoxic Tumor Microenvironment. Front Immunol. 2021;12:741305.
Nazon C, Pierrevelcin M, Willaume T, Lhermitte B, Weingertner N, Marco AD, et al. Together Intra-Tumor Hypoxia and Macrophagic Immunity Are Driven Worst Outcome in Pediatric High-Grade Osteosarcomas. Cancers (Basel). 2022;14(6):1482.
Wang Y, Yu J, Luo Z, Shi Q, Liu G, Wu F, et al. Engineering Endogenous Tumor-Associated Macrophage-Targeted Biomimetic Nano-RBC to Reprogram Tumor Immunosuppressive Microenvironment for Enhanced Chemo-Immunotherapy. Adv Mater. 2021;33(39):e2103497.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51.
Wei J, Chen Z, Hu M, He Z, Jiang D, Long J, et al. Characterizing Intercellular Communication of Pan-Cancer Reveals SPP1+ Tumor-Associated Macrophage Expanded in Hypoxia and Promoting Cancer Malignancy Through Single-Cell RNA-Seq Data. Front Cell Dev Biol. 2021;9:749210.
Korbecki J, Kojder K, Barczak K, Simińska D, Gutowska I, Chlubek D, et al. Hypoxia Alters the Expression of CC Chemokines and CC Chemokine Receptors in a Tumor-A Literature Review. Int J Mol Sci. 2020;21(16):5647.
Jiang H, Zhao H, Zhang M, He Y, Li X, Xu Y, et al. Hypoxia Induced Changes of Exosome Cargo and Subsequent Biological Effects. Front Immunol. 2022;13:824188.
Campillo N, Falcones B, Otero J, Colina R, Gozal D, Navajas D, et al. Differential Oxygenation in Tumor Microenvironment Modulates Macrophage and Cancer Cell Crosstalk: Novel Experimental Setting and Proof of Concept. Front Oncol. 2019;9:43.
Dehne N, Mora J, Namgaladze D, Weigert A, Brüne B. Cancer cell and macrophage cross-talk in the tumor microenvironment. Curr Opin Pharmacol. 2017;35:12–9.
Qi L, Chen J, Yang Y, Hu W. Hypoxia Correlates With Poor Survival and M2 Macrophage Infiltration in Colorectal Cancer. Front Oncol. 2020;10:566430.
Singleton DC, Macann A, Wilson WR. Therapeutic targeting of the hypoxic tumour microenvironment. Nat Rev Clin Oncol. 2021;18(12):751–72.
Baradaran A, Asadzadeh Z, Hemmat N, Baghbanzadeh A, Shadbad MA, Khosravi N, et al. The cross-talk between tumor-associated macrophages and tumor endothelium: Recent advances in macrophage-based cancer immunotherapy. Biomed Pharmacother. 2022;146:112588.
Batoon L, McCauley LK. Cross Talk Between Macrophages and Cancer Cells in the Bone Metastatic Environment. Front Endocrinol (Lausanne). 2021;12:763846.
Sung PS. Crosstalk between tumor-associated macrophages and neighboring cells in hepatocellular carcinoma. Clin Mol Hepatol. 2021;28(3):333–50.
Ge Z, Ding S. The Crosstalk Between Tumor-Associated Macrophages (TAMs) and Tumor Cells and the Corresponding Targeted Therapy. Front Oncol. 2020;10:590941.
Horsman MR, Vaupel P. Pathophysiological Basis for the Formation of the Tumor Microenvironment. Front Oncol. 2016;6:66.
Gottwald J, Han K, Milosevic M, Yeung I, Jaffray DA. Impact of PET scanner non-linearity on the estimation of hypoxic fraction in cervical cancer patients. Phys Med. 2022;93:1–7.
Bayer C, Shi K, Astner ST, Maftei CA, Vaupel P. Acute versus chronic hypoxia: why a simplified classification is simply not enough. Int J Radiat Oncol Biol Phys. 2011;80(4):965–8.
Vaupel P, Mayer A. Hypoxia in tumors: pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv Exp Med Biol. 2014;812:19–24.
Michiels C, Tellier C, Feron O. Cycling hypoxia: a key feature of the tumor microenvironment. Biochim Biophys Acta. 2016;1866(1):76–86.
Vaupel P, Mayer A. Tumor hypoxia: causative mechanisms, microregional heterogeneities, and the role of tissue-based hypoxia markers. Adv Exp Med Biol. 2016;923:77–86.
Saxena K, Jolly MK. Acute vs. chronic vs. cyclic hypoxia: their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules. 2019;9(8):339.
Luo W, Wang Y. Hypoxia mediates tumor malignancy and therapy resistance. Adv Exp Med Biol. 2019;1136:1–18.
Young K, Lawlor RT, Ragulan C, Patil Y, Mafficini A, Bersani S, et al. Immune landscape, evolution, hypoxia-mediated viral mimicry pathways and therapeutic potential in molecular subtypes of pancreatic neuroendocrine tumours. Gut. 2021;70(10):1904–13.
Xiong Z, Liu H, He C, Li X. Hypoxia contributes to poor prognosis in primary IDH-wt GBM by inducing tumor cells mes-like transformation trend and inhibiting immune cells activity. Front Oncol. 2021;11:782043.
Godet I, Doctorman S, Wu F, Gilkes DM. Detection of hypoxia in cancer models: significance, challenges, and advances. Cells. 2022;11(4):686.
Hompland T, Fjeldbo CS, Lyng H. Tumor Hypoxia as a Barrier in Cancer Therapy: Why Levels Matter. Cancers (Basel). 2021;13(3):499.
Huang Y, Fan J, Li Y, Fu S, Chen Y, Wu J. Imaging of Tumor Hypoxia With Radionuclide-Labeled Tracers for PET. Front Oncol. 2021;11:731503.
Mo T, Brandal SHB, Köhn-Luque A, Engebraaten O, Kristensen VN, Fleischer T, et al. Quantification of tumor hypoxia through unsupervised modelling of consumption and supply hypoxia MR imaging in breast Cancer. Cancers (Basel). 2022;14(5):1326.
Bader SB, Dewhirst MW, Hammond EM. Cyclic Hypoxia: An Update on Its Characteristics, Methods to Measure It and Biological Implications in Cancer. Cancers (Basel). 2020;13(1):23.
Chakraborty AA, Laukka T, Myllykoski M, Ringel AE, Booker MA, Tolstorukov MY, et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science. 2019;363(6432):1217–22.
Gallipoli P, Huntly BJP. Histone modifiers are oxygen sensors. Science. 2019;363(6432):1148–9.
Janssens LK, Stove CP. Sensing an Oxygen Sensor: Development and Application of Activity-Based Assays Directly Monitoring HIF Heterodimerization. Anal Chem. 2021;93(43):14462–70.
Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272(36):22642–7.
Xiong Q, Liu B, Ding M, Zhou J, Yang C, Chen Y. Hypoxia and cancer related pathology. Cancer Lett. 2020;486:1–7.
Taylor CT, Scholz CC. The effect of HIF on metabolism and immunity. Nat Rev Nephrol. 2022;18(9):573–87.
Domene C, Jorgensen C, Schofield CJ. Mechanism of molecular oxygen diffusion in a hypoxia-sensing prolyl hydroxylase using multiscale simulation. J Am Chem Soc. 2020;142(5):2253–63.
Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–65.
Tarade D, Lee JE, Ohh M. Evolution of metazoan oxygen-sensing involved a conserved divergence of VHL affinity for HIF1α and HIF2α. Nat Commun. 2019;10(1):3293.
Qian H, Zou Y, Tang Y, Gong Y, Qian Z, Wei G, et al. Proline hydroxylation at different sites in hypoxia-inducible factor 1α modulates its interactions with the von Hippel-Lindau tumor suppressor protein. Phys Chem Chem Phys. 2018;20(27):18756–65.
Lee P, Chandel NS, Simon MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol. 2020;21(5):268–83.
Diao X, Ye F, Zhang M, Ren X, Tian X, Lu J, et al. Identification of oleoylethanolamide as an endogenous ligand for HIF-3α. Nat Commun. 2022;13(1):2529.
Casillas AL, Chauhan SS, Toth RK, Sainz AG, Clements AN, Jensen CC, et al. Direct phosphorylation and stabilization of HIF-1α by PIM1 kinase drives angiogenesis in solid tumors. Oncogene. 2021;40(32):5142–52.
Zhao D, Zheng S, Wang X, Liu H, Zhao K, Li L, et al. iASPP is essential for HIF-1α stabilization to promote angiogenesis and glycolysis via attenuating VHL-mediated protein degradation. Oncogene. 2022;41(13):1944–58.
Najafi M, Goradel NH, Farhood B, Salehi E, Solhjoo S, Toolee H, et al. Tumor microenvironment: Interactions and therapy. J Cell Physiol. 2019;234(5):5700–21.
Li J, Xu P, Wu D, Guan M, Weng X, Lu Y, et al. Hypoxic stress suppresses lung tumor-secreted exosomal miR101 to activate macrophages and induce inflammation. Cell Death Dis. 2021;12(8):776.
Qian M, Wang S, Guo X, Wang J, Zhang Z, Qiu W, et al. Hypoxic glioma-derived exosomes deliver microRNA-1246 to induce M2 macrophage polarization by targeting TERF2IP via the STAT3 and NF-κB pathways. Oncogene. 2020;39(2):428–42.
Lee JY, Ryu D, Lim SW, Ryu KJ, Choi ME, Yoon SE, et al. Exosomal miR-1305 in the oncogenic activity of hypoxic multiple myeloma cells: a biomarker for predicting prognosis. J Cancer. 2021;12(10):2825–34.
Chen X, Zhou J, Li X, Wang X, Lin Y, Wang X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018;435:80–91.
Xiao L, He Y, Peng F, Yang J, Yuan C. Endometrial Cancer Cells Promote M2-Like Macrophage Polarization by Delivering Exosomal miRNA-21 under Hypoxia Condition. J Immunol Res. 2020;2020:9731049.
Zhu X, Shen H, Yin X, Yang M, Wei H, Chen Q, et al. Macrophages derived exosomes deliver miR-223 to epithelial ovarian cancer cells to elicit a chemoresistant phenotype. J Exp Clin Cancer Res. 2019;38(1):81.
Wang X, Luo G, Zhang K, Cao J, Huang C, Jiang T, et al. Hypoxic Tumor-Derived Exosomal miR-301a Mediates M2 Macrophage Polarization via PTEN/PI3Kγ to Promote Pancreatic Cancer Metastasis. Cancer Res. 2018;78(16):4586–98.
Xu J, Zhang J, Zhang Z, Gao Z, Qi Y, Qiu W, et al. Hypoxic glioma-derived exosomes promote M2-like macrophage polarization by enhancing autophagy induction. Cell Death Dis. 2021;12(4):373.
Chen X, Ying X, Wang X, Wu X, Zhu Q, Wang X. Exosomes derived from hypoxic epithelial ovarian cancer deliver microRNA-940 to induce macrophage M2 polarization. Oncol Rep. 2017;38(1):522–8.
Lu Q, Wang X, Zhu J, Fei X, Chen H, Li C. Hypoxic Tumor-Derived Exosomal Circ0048117 Facilitates M2 Macrophage Polarization Acting as miR-140 Sponge in Esophageal Squamous Cell Carcinoma. Onco Targets Ther. 2020;13:11883–97.
Wang X, Zhou Y, Dong K, Zhang H, Gong J, Wang S. Exosomal lncRNA HMMR-AS1 mediates macrophage polarization through miR-147a/ARID3A axis under hypoxia and affects the progression of hepatocellular carcinoma. Environ Toxicol. 2022;37(6):1357–72.
Park JE, Dutta B, Tse SW, Gupta N, Tan CF, Low JK, et al. Hypoxia-induced tumor exosomes promote M2-like macrophage polarization of infiltrating myeloid cells and microRNA-mediated metabolic shift. Oncogene. 2019;38(26):5158–73.
Liu Y, Lu M, Chen J, Li S, Deng Y, Yang S, et al. Extracellular vesicles derived from lung cancer cells exposed to intermittent hypoxia upregulate programmed death ligand 1 expression in macrophages. Sleep Breath. 2021;26(2):893–906.
Shrivastava R, Asif M, Singh V, Dubey P, Ahmad Malik S, Lone MU, et al. M2 polarization of macrophages by Oncostatin M in hypoxic tumor microenvironment is mediated by mTORC2 and promotes tumor growth and metastasis. Cytokine. 2019;118:130–43.
Piao H, Fu L, Wang Y, Liu Y, Wang Y, Meng X, et al. A positive feedback loop between gastric cancer cells and tumor-associated macrophage induces malignancy progression. J Exp Clin Cancer Res. 2022;41(1):174.
Chen XJ, Deng YR, Wang ZC, Wei WF, Zhou CF, Zhang YM, et al. Hypoxia-induced ZEB1 promotes cervical cancer progression via CCL8-dependent tumour-associated macrophage recruitment. Cell Death Dis. 2019;10(7):508.
Yu X, Li Z, Zhang Y, Xu M, Che Y, Tian X, et al. β-elemene inhibits radiation and hypoxia-induced macrophages infiltration via Prx-1/NF-κB/HIF-1α signaling pathway. Onco Targets Ther. 2019;12:4203–11.
Yin X, Han S, Song C, Zou H, Wei Z, Xu W, et al. Metformin enhances gefitinib efficacy by interfering with interactions between tumor-associated macrophages and head and neck squamous cell carcinoma cells. Cell Oncol (Dordr). 2019;42(4):459–75.
Ma F, Zhang B, Ji S, Hu H, Kong Y, Hua Y, et al. Hypoxic Macrophage-Derived VEGF Promotes Proliferation and Invasion of Gastric Cancer Cells. Dig Dis Sci. 2019;64(11):3154–63.
Casazza A, Laoui D, Wenes M, Rizzolio S, Bassani N, Mambretti M, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709.
Wang X, Mao J, Zhou T, Chen X, Tu H, Ma J, et al. Hypoxia-induced myeloid derived growth factor promotes hepatocellular carcinoma progression through remodeling tumor microenvironment. Theranostics. 2021;11(1):209–21.
Wang L, Li YS, Yu LG, Zhang XK, Zhao L, Gong FL, et al. Galectin-3 expression and secretion by tumor-associated macrophages in hypoxia promotes breast cancer progression. Biochem Pharmacol. 2020;178:114113.
Jiang J, Wang GZ, Wang Y, Huang HZ, Li WT, Qu XD. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp Cell Res. 2018;367(1):81–8.
Susen RM, Bauer R, Olesch C, Fuhrmann DC, Fink AF, Dehne N, et al. Macrophage HIF-2α regulates tumor-suppressive Spint1 in the tumor microenvironment. Mol Carcinog. 2019;58(11):2127–38.
Martins F, Oliveira R, Cavadas B, Pinto F, Cardoso AP, Castro F, et al. Hypoxia and Macrophages Act in Concert Towards a Beneficial Outcome in Colon Cancer. Cancers (Basel). 2020;12(4):818.
Chen XJ, Wu S, Yan RM, Fan LS, Yu L, Zhang YM, et al. The role of the hypoxia-Nrp-1 axis in the activation of M2-like tumor-associated macrophages in the tumor microenvironment of cervical cancer. Mol Carcinog. 2019;58(3):388–97.
Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67(5):1872–89.
Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559–63.
Geeraerts X, Fernández-Garcia J, Hartmann FJ, de Goede KE, Martens L, Elkrim Y, et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 2021;37(13):110171.
Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-Derived Succinate Promotes Macrophage Polarization and Cancer Metastasis via Succinate Receptor. Mol Cell. 2020;77(2):213-27.e5.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977.
Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750.
Cocozza F, Grisard E, Martin-Jaular L, Mathieu M. Théry C. SnapShot: Extracellular Vesicles. Cell. 2020;182(1):262-.e1.
Li B, Cao Y, Sun M, Feng H. Expression, regulation, and function of exosome-derived miRNAs in cancer progression and therapy. Faseb J. 2021;35(10):e21916.
Qian M, Chen Z, Guo X, Wang S, Zhang Z, Qiu W, et al. Exosomes derived from hypoxic glioma deliver miR-1246 and miR-10b-5p to normoxic glioma cells to promote migration and invasion. Lab Invest. 2021;101(5):612–24.
Deregowska A, Wnuk M. RAP1/TERF2IP-A Multifunctional Player in Cancer Development. Cancers (Basel). 2021;13(23):5970.
Kashfi K, Kannikal J, Nath N. Macrophage Reprogramming and Cancer Therapeutics: Role of iNOS-Derived NO. Cells. 2021;10(11):3194.
Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. 2020;877:173090.
Kono Y, Kawakami S, Higuchi Y, Yamashita F, Hashida M. In vitro evaluation of inhibitory effect of nuclear factor-kappaB activity by small interfering RNA on pro-tumor characteristics of M2-like macrophages. Biol Pharm Bull. 2014;37(1):137–44.
Wang CZ, Deng F, Li H, Wang DD, Zhang W, Ding L, et al. MiR-101: a potential therapeutic target of cancers. Am J Transl Res. 2018;10(11):3310–21.
Zhang K, Dong C, Chen M, Yang T, Wang X, Gao Y, et al. Extracellular vesicle-mediated delivery of miR-101 inhibits lung metastasis in osteosarcoma. Theranostics. 2020;10(1):411–25.
Ježek J, Smethurst DGJ, Stieg DC, Kiss ZAC, Hanley SE, Ganesan V, et al. Cyclin C: The Story of a Non-Cycling Cyclin. Biology (Basel). 2019;8(1):3.
Sehgal PB. Interleukin-6 at the Host-Tumor Interface: STAT3 in Biomolecular Condensates in Cancer Cells. Cells. 2022;11(7):1164.
Udagawa K, Niki Y, Kikuchi T, Fukuhara Y, Takeda Y, Miyamoto T, et al. Overexpression of Interleukin-1α Suppresses Liver Metastasis of Lymphoma: Implications for Antitumor Effects of CD8+ T-cells. J Histochem Cytochem. 2021;69(4):245–55.
Lin D, Mei Y, Lei L, Binte Hanafi Z, Jin Z, Liu Y, et al. Immune suppressive function of IL-1α release in the tumor microenvironment regulated by calpain 1. Oncoimmunology. 2022;11(1):2088467.
Chiu JW, Binte Hanafi Z, Chew LCY, Mei Y, Liu H. IL-1α Processing, Signaling and Its Role in Cancer Progression. Cells. 2021;10(1):92.
Xue H, Yuan G, Guo X, Liu Q, Zhang J, Gao X, et al. A novel tumor-promoting mechanism of IL6 and the therapeutic efficacy of tocilizumab: Hypoxia-induced IL6 is a potent autophagy initiator in glioblastoma via the p-STAT3-MIR155-3p-CREBRF pathway. Autophagy. 2016;12(7):1129–52.
Zhang H, McCarty N. Tampering with cancer chemoresistance by targeting the TGM2-IL6-autophagy regulatory network. Autophagy. 2017;13(3):627–8.
Wen ZF, Liu H, Gao R, Zhou M, Ma J, Zhang Y, et al. Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J Immunother Cancer. 2018;6(1):151.
Chen L, Christian DA, Kochanowsky JA, Phan AT, Clark JT, Wang S, et al. The Toxoplasma gondii virulence factor ROP16 acts in cis and trans, and suppresses T cell responses. J Exp Med. 2020;217(3):e20181757.
Wang Q, Wang J, Xiang H, Ding P, Wu T, Ji G. The biochemical and clinical implications of phosphatase and tensin homolog deleted on chromosome ten in different cancers. Am J Cancer Res. 2021;11(12):5833–55.
Lee YR, Chen M, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor: new modes and prospects. Nat Rev Mol Cell Biol. 2018;19(9):547–62.
Ye D, Gong M, Deng Y, Fang S, Cao Y, Xiang Y, et al. Roles and clinical application of exosomal circRNAs in the diagnosis and treatment of malignant tumors. J Transl Med. 2022;20(1):161.
Patra MC, Shah M, Choi S. Toll-like receptor-induced cytokines as immunotherapeutic targets in cancers and autoimmune diseases. Semin Cancer Biol. 2020;64:61–82.
Kashani B, Zandi Z, Pourbagheri-Sigaroodi A, Bashash D, Ghaffari SH. The role of toll-like receptor 4 (TLR4) in cancer progression: A possible therapeutic target? J Cell Physiol. 2021;236(6):4121–37.
Qian S, Han X, Sha X, Tian F, Huang H, Jiang P, et al. Aqueous extract of cimicifuga dahurica reprogramming macrophage polarization by activating TLR4-NF-κB signaling pathway. J Inflamm Res. 2022;15:1027–46.
Fan CS, Chen LL, Hsu TA, Chen CC, Chua KV, Li CP, et al. Endothelial-mesenchymal transition harnesses HSP90α-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol Oncol. 2019;12(1):138.
Fan CS, Chen CC, Chen LL, Chua KV, Hung HC, Hsu JT, et al. Extracellular HSP90α Induces MyD88-IRAK complex-associated ikkα/β-nf-κb/irf3 and jak2/tyk2-stat-3 signaling in macrophages for tumor-promoting m2-polarization. Cells. 2022;11(2):229.
Yao RW, Wang Y, Chen LL. Cellular functions of long noncoding RNAs. Nat Cell Biol. 2019;21(5):542–51.
Liu W, Ma J, Cheng Y, Zhang H, Luo W, Zhang H. HMMR antisense RNA 1, a novel long noncoding RNA, regulates the progression of basal-like breast cancer cells. Breast Cancer (Dove Med Press). 2016;8:223–9.
Gottlieb CE, Mills AM, Cross JV, Ring KL. Tumor-associated macrophage expression of PD-L1 in implants of high grade serous ovarian carcinoma: A comparison of matched primary and metastatic tumors. Gynecol Oncol. 2017;144(3):607–12.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
Adekoya TO, Richardson RM. Cytokines and Chemokines as Mediators of Prostate Cancer Metastasis. Int J Mol Sci. 2020;21(12):4449.
Mughees M, Kaushal JB, Sharma G, Wajid S, Batra SK, Siddiqui JA. Chemokines and cytokines: axis and allies in prostate cancer pathogenesis. Semin Cancer Biol. 2022;16:S1044–579X(22)00041–4.
Li E, Yang X, Du Y, Wang G, Chan DW, Wu D, et al. CXCL8 Associated Dendritic Cell Activation Marker Expression and Recruitment as Indicators of Favorable Outcomes in Colorectal Cancer. Front Immunol. 2021;12: 667177.
Cheng L, Zhou MY, Gu YJ, Chen L, Wang Y. ZEB1: New advances in fibrosis and cancer. Mol Cell Biochem. 2021;476(4):1643–50.
Singh S, Anshita D, Ravichandiran V. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. 2021;101(Pt B):107598.
Karpuz T, Araz M, Korkmaz L, Kılınc I, Findik S, Karaagaç M, et al. The Prognostic Value of Serum Semaphorin3A and VEGF Levels in Patients with Metastatic Colorectal Cancer. J Gastrointest Cancer. 2020;51(2):491–7.
Maione F, Molla F, Meda C, Latini R, Zentilin L, Giacca M, et al. Semaphorin 3A is an endogenous angiogenesis inhibitor that blocks tumor growth and normalizes tumor vasculature in transgenic mouse models. J Clin Invest. 2009;119(11):3356–72.
Dumond A, Pagès G. Neuropilins, as Relevant Oncology Target: Their Role in the Tumoral Microenvironment. Front Cell Dev Biol. 2020;8:662.
Houseright RA, Miskolci V, Mulvaney O, Bortnov V, Mosher DF, Rindy J, et al. Myeloid-derived growth factor regulates neutrophil motility in interstitial tissue damage. J Cell Biol. 2021;220(8):e202103054.
Xu H, Liu H, Liu C, Shangguan X, Cheng X, Zhang R, et al. Molecular characterization and antibacterial ability of galectin-3 and galectin-9 in Onychostoma macrolepis. Dev Comp Immunol. 2022;128:104333.
Kataoka Y, Igarashi T, Ohshio Y, Fujita T, Hanaoka J. Predictive importance of galectin-3 for recurrence of non-small cell lung cancer. Gen Thorac Cardiovasc Surg. 2019;67(8):704–11.
Wang C, Zhou X, Ma L, Zhuang Y, Wei Y, Zhang L, et al. Galectin-3 may serve as a marker for poor prognosis in colorectal cancer: A meta-analysis. Pathol Res Pract. 2019;215(10):152612.
Liu Y, Xie L, Wang D, Li D, Xu G, Wang L, et al. Galectin-3 and β-catenin are associated with a poor prognosis in serous epithelial ovarian cancer. Cancer Manag Res. 2018;10:3963–71.
Gu X, Meng H, Wang J, Wang R, Cao M, Liu S, et al. Hypoxia contributes to galectin-3 expression in renal carcinoma cells. Eur J Pharmacol. 2021;890:173637.
Zhang H, Liu P, Zhang Y, Han L, Hu Z, Cai Z, et al. Inhibition of galectin-3 augments the antitumor efficacy of PD-L1 blockade in non-small-cell lung cancer. FEBS Open Bio. 2021;11(3):911–20.
Owusu BY, Galemmo R, Janetka J, Klampfer L. Hepatocyte Growth Factor, a Key Tumor-Promoting Factor in the Tumor Microenvironment. Cancers (Basel). 2017;9(4):35.
Jia X, Yan B, Tian X, Liu Q, Jin J, Shi J, et al. CD47/SIRPα pathway mediates cancer immune escape and immunotherapy. Int J Biol Sci. 2021;17(13):3281–7.
Karsch-Bluman A, Benny O. Necrosis in the Tumor Microenvironment and Its Role in Cancer Recurrence. Adv Exp Med Biol. 2020;1225:89–98.
Sørensen MD, Kristensen BW. Tumour-associated CD204(+) microglia/macrophages accumulate in perivascular and perinecrotic niches and correlate with an interleukin-6-enriched inflammatory profile in glioblastoma. Neuropathol Appl Neurobiol. 2022;48(2):e12772.
Raymond MH, Davidson AJ, Shen Y, Tudor DR, Lucas CD, Morioka S, et al. Live cell tracking of macrophage efferocytosis during Drosophila embryo development in vivo. Science. 2022;375(6585):1182–7.
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8.
Kiss M, Vande Walle L, Saavedra PHV, Lebegge E, Van Damme H, Murgaski A, et al. IL1β Promotes Immune Suppression in the Tumor Microenvironment Independent of the Inflammasome and Gasdermin D. Cancer Immunol Res. 2021;9(3):309–23.
Hagemann T, Lawrence T, McNeish I, Charles KA, Kulbe H, Thompson RG, et al. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J Exp Med. 2008;205(6):1261–8.
Hoover AA, Hufnagel DH, Harris W, Bullock K, Glass EB, Liu E, et al. Increased canonical NF-kappaB signaling specifically in macrophages is sufficient to limit tumor progression in syngeneic murine models of ovarian cancer. BMC Cancer. 2020;20(1):970.
Meier-Soelch J, Mayr-Buro C, Juli J, Leib L, Linne U, Dreute J, et al. Monitoring the Levels of Cellular NF-κB Activation States. Cancers (Basel). 2021;13(21):5351.
Hagemann T, Biswas SK, Lawrence T, Sica A, Lewis CE. Regulation of macrophage function in tumors: the multifaceted role of NF-kappaB. Blood. 2009;113(14):3139–46.
Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L, et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66(23):11432–40.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Rackov G, Hernández-Jiménez E, Shokri R, Carmona-Rodríguez L, Mañes S, Álvarez-Mon M, et al. p21 mediates macrophage reprogramming through regulation of p50–p50 NF-κB and IFN-β. J Clin Invest. 2016;126(8):3089–103.
Yu Z, Li Y, Li Y, Zhang J, Li M, Ji L, et al. Bufalin stimulates antitumor immune response by driving tumor-infiltrating macrophage toward M1 phenotype in hepatocellular carcinoma. J Immunother Cancer. 2022;10(5):e004297.
Kes MMG, Van den Bossche J, Griffioen AW, Huijbers EJM. Oncometabolites lactate and succinate drive pro-angiogenic macrophage response in tumors. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188427.
Niu D, Wu Y, Lei Z, Zhang M, Xie Z, Tang S. Lactic acid, a driver of tumor-stroma interactions. Int Immunopharmacol. 2022;106:108597.
Jiang H, Wei H, Wang H, Wang Z, Li J, Ou Y, et al. Zeb1-induced metabolic reprogramming of glycolysis is essential for macrophage polarization in breast cancer. Cell Death Dis. 2022;13(3):206.
Rawat D, Chhonker SK, Naik RA, Mehrotra A, Trigun SK, Koiri RK. Lactate as a signaling molecule: Journey from dead end product of glycolysis to tumor survival. Front Biosci (Landmark Ed). 2019;24(2):366–81.
Shan T, Chen S, Chen X, Wu T, Yang Y, Li S, et al. M2-TAM subsets altered by lactic acid promote T-cell apoptosis through the PD-L1/PD-1 pathway. Oncol Rep. 2020;44(5):1885–94.
Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, Xavier JB. Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci U S A. 2017;114(11):2934–9.
Zhao Y, Zhao B, Wang X, Guan G, Xin Y, Sun YD, et al. Macrophage transcriptome modification induced by hypoxia and lactate. Exp Ther Med. 2019;18(6):4811–9.
Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167(2):457-70.e13.
Chong AS, Anderson PC. Molecular Dynamics Simulations of the Hypoxia-Inducible Factor PAS-B Domain Confirm That Internally Bound Water Molecules Function To Stabilize the Protein Core for Ligand Binding. Biochemistry. 2020;59(4):450–9.
Scheuermann TH, Li Q, Ma HW, Key J, Zhang L, Chen R, et al. Allosteric inhibition of hypoxia inducible factor-2 with small molecules. Nat Chem Biol. 2013;9(4):271–6.
Visweswaran V, Pavithran K. Belzutifan: a narrative drug review. Curr Drug Res Rev. 2022;14(2):88–95.
Courtney KD, Infante JR, Lam ET, Figlin RA, Rini BI, Brugarolas J, et al. Phase I dose-escalation trial of pt2385, a first-in-class hypoxia-inducible factor-2α antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J Clin Oncol. 2018;36(9):867–74.
Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, et al. A Small-Molecule Antagonist of HIF2α Is Efficacious in Preclinical Models of Renal Cell Carcinoma. Cancer Res. 2016;76(18):5491–500.
Deeks ED. Belzutifan: First Approval. Drugs. 2021;81(16):1921–7.
Zhou J, Gong K. Belzutifan: a novel therapy for von Hippel-Lindau disease. Nat Rev Nephrol. 2022;18(4):205–6.
Caruso C. FDA OK’s HIF2α Inhibitor Belzutifan. Cancer Discov. 2021;11(10):2360–1.
Hasanov E, Jonasch E. MK-6482 as a potential treatment for von Hippel-Lindau disease-associated clear cell renal cell carcinoma. Expert Opin Investig Drugs. 2021;30(5):495–504.
Gläsker S, Vergauwen E, Koch CA, Kutikov A, Vortmeyer AO. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. Onco Targets Ther. 2020;13:5669–90.
Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N Engl J Med. 2021;385(22):2036–46.
Kamihara J, Hamilton KV, Pollard JA, Clinton CM, Madden JA, Lin J, et al. Belzutifan, a Potent HIF2α Inhibitor, in the Pacak-Zhuang Syndrome. N Engl J Med. 2021;385(22):2059–65.
Wehn PM, Rizzi JP, Dixon DD, Grina JA, Schlachter ST, Wang B, et al. Design and Activity of Specific Hypoxia-Inducible Factor-2α (HIF-2α) Inhibitors for the Treatment of Clear Cell Renal Cell Carcinoma: Discovery of Clinical Candidate ( S)-3-((2,2-Difluoro-1-hydroxy-7-(methylsulfonyl)-2,3-dihydro-1 H-inden-4-yl)oxy)-5-fluorobenzonitrile (PT2385). J Med Chem. 2018;61(21):9691–721.
Courtney KD, Ma Y, Diaz de Leon A, Christie A, Xie Z, Woolford L, et al. HIF-2 Complex Dissociation, Target Inhibition, and Acquired Resistance with PT2385, a First-in-Class HIF-2 Inhibitor, in Patients with Clear Cell Renal Cell Carcinoma. Clin Cancer Res. 2020;26(4):793–803.
Cho H, Kaelin WG. Targeting HIF2 in Clear Cell Renal Cell Carcinoma. Cold Spring Harb Symp Quant Biol. 2016;81:113–21.
Xu J, Zheng L, Chen J, Sun Y, Lin H, Jin RA, et al. Increasing AR by HIF-2α inhibitor (PT-2385) overcomes the side-effects of sorafenib by suppressing hepatocellular carcinoma invasion via alteration of pSTAT3, pAKT and pERK signals. Cell Death Dis. 2017;8(10):e3095.
Lee K, Kim HM. A novel approach to cancer therapy using PX-478 as a HIF-1α inhibitor. Arch Pharm Res. 2011;34(10):1583–5.
Palayoor ST, Mitchell JB, Cerna D, Degraff W, John-Aryankalayil M, Coleman CN. PX-478, an inhibitor of hypoxia-inducible factor-1alpha, enhances radiosensitivity of prostate carcinoma cells. Int J Cancer. 2008;123(10):2430–7.
Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther. 2008;7(1):90–100.
Luo F, Lu FT, Cao JX, Ma WJ, Xia ZF, Zhan JH, et al. HIF-1α inhibition promotes the efficacy of immune checkpoint blockade in the treatment of non-small cell lung cancer. Cancer Lett. 2022;531:39–56.
Wang H, Jia R, Zhao T, Li X, Lang M, Lan C, et al. HIF-1α mediates tumor-nerve interactions through the up-regulation of GM-CSF in pancreatic ductal adenocarcinoma. Cancer Lett. 2019;453:10–20.
Korbecki J, Simińska D, Gąssowska-Dobrowolska M, Listos J, Gutowska I, Chlubek D, et al. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int J Mol Sci. 2021;22(19):10701.
Chen WL, Wang CC, Lin YJ, Wu CP, Hsieh CH. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J Transl Med. 2015;13:389.
Santana-Viera L, Ibba ML, Rotoli D, Catuogno S, Esposito CL. Emerging Therapeutic RNAs for the Targeting of Cancer Associated Fibroblasts. Cancers (Basel). 2020;12(6):1365.
Wu J, Contratto M, Shanbhogue KP, Manji GA, O’Neil BH, Noonan A, et al. Evaluation of a locked nucleic acid form of antisense oligo targeting HIF-1α in advanced hepatocellular carcinoma. World J Clin Oncol. 2019;10(3):149–60.
Gupta A, Andresen JL, Manan RS, Langer R. Nucleic acid delivery for therapeutic applications. Adv Drug Deliv Rev. 2021;178:113834.
Sharma R, Dong Y, Hu Y, Ma VP, Salaita K. Gene Regulation Using Nanodiscs Modified with HIF-1-α Antisense Oligonucleotides. Bioconjug Chem. 2022;33(2):279–93.
Smith CIE, Zain R. Therapeutic Oligonucleotides: State of the Art. Annu Rev Pharmacol Toxicol. 2019;59:605–30.
Greenberger LM, Horak ID, Filpula D, Sapra P, Westergaard M, Frydenlund HF, et al. A RNA antagonist of hypoxia-inducible factor-1alpha, EZN-2968, inhibits tumor cell growth. Mol Cancer Ther. 2008;7(11):3598–608.
Kara G, Calin GA, Ozpolat B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv Drug Deliv Rev. 2022;182:114113.
Wong SC, Cheng W, Hamilton H, Nicholas AL, Wakefield DH, Almeida A, et al. HIF2α-Targeted RNAi Therapeutic Inhibits Clear Cell Renal Cell Carcinoma. Mol Cancer Ther. 2018;17(1):140–9.
Brugarolas J, Beckermann K, Rini BI, Vogelzang NJ, Tat Lam E, Hamilton JC. Initial results from the phase 1 study of ARO-HIF2 to silence HIF2-alpha in patients with advanced ccRCC (AROHIF21001). J Clin Oncol. 2022;40:4.
Hua Y, Dai X, Xu Y, Xing G, Liu H, Lu T, et al. Drug repositioning: Progress and challenges in drug discovery for various diseases. Eur J Med Chem. 2022;234:114239.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32.
Conley SJ, Baker TL, Burnett JP, Theisen RL, Lazarus D, Peters CG, et al. CRLX101, an investigational camptothecin-containing nanoparticle-drug conjugate, targets cancer stem cells and impedes resistance to antiangiogenic therapy in mouse models of breast cancer. Breast Cancer Res Treat. 2015;150(3):559–67.
Bertozzi D, Marinello J, Manzo SG, Fornari F, Gramantieri L, Capranico G. The natural inhibitor of DNA topoisomerase I, camptothecin, modulates HIF-1α activity by changing miR expression patterns in human cancer cells. Mol Cancer Ther. 2014;13(1):239–48.
Schmidt KT, Chau CH, Strope JD, Huitema ADR, Sissung TM, Price DK, et al. Antitumor Activity of NLG207 (Formerly CRLX101) in Combination with Enzalutamide in Preclinical Prostate Cancer Models. Mol Cancer Ther. 2021;20(5):915–24.
Murono K, Tsuno NH, Kawai K, Sasaki K, Hongo K, Kaneko M, et al. SN-38 overcomes chemoresistance of colorectal cancer cells induced by hypoxia, through HIF1alpha. Anticancer Res. 2012;32(3):865–72.
Okuno T, Kawai K, Hata K, Murono K, Emoto S, Kaneko M, et al. SN-38 Acts as a Radiosensitizer for Colorectal Cancer by Inhibiting the Radiation-induced Up-regulation of HIF-1α. Anticancer Res. 2018;38(6):3323–31.
Bastani S, Akbarzadeh M, Rastgar Rezaei Y, Farzane A, Nouri M, Mollapour Sisakht M, et al. Melatonin as a Therapeutic Agent for the Inhibition of Hypoxia-Induced Tumor Progression: A Description of Possible Mechanisms Involved. Int J Mol Sci. 2021;22(19):10874.
Kim KJ, Choi JS, Kang I, Kim KW, Jeong CH, Jeong JW. Melatonin suppresses tumor progression by reducing angiogenesis stimulated by HIF-1 in a mouse tumor model. J Pineal Res. 2013;54(3):264–70.
Cho SY, Lee HJ, Jeong SJ, Lee HJ, Kim HS, Chen CY, et al. Sphingosine kinase 1 pathway is involved in melatonin-induced HIF-1α inactivation in hypoxic PC-3 prostate cancer cells. J Pineal Res. 2011;51(1):87–93.
Cuvillier O, Ader I, Bouquerel P, Brizuela L, Gstalder C, Malavaud B. Hypoxia, therapeutic resistance, and sphingosine 1-phosphate. Adv Cancer Res. 2013;117:117–41.
Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A. 2008;105(50):19579–86.
Semenza GL. Hypoxia-inducible factors: coupling glucose metabolism and redox regulation with induction of the breast cancer stem cell phenotype. Embo j. 2017;36(3):252–9.
Podkalicka P, Stępniewski J, Mucha O, Kachamakova-Trojanowska N, Dulak J, Łoboda A. Hypoxia as a Driving Force of Pluripotent Stem Cell Reprogramming and Differentiation to Endothelial Cells. Biomolecules. 2020;10(12):1614.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from National Natural Science Foundation of China (No. 82073281, 81573462, 82073884, U20A20413), Shenyang S&T Projects [19–109–4–09, 20–204–4–22].
Author information
Authors and Affiliations
Contributions
LZ and RB conceived of the presented idea. RB and YL wrote the manuscript. MW and LZ provided the guidance throughout the revision of this manuscript. LJ and YY revised the paper. All authors contributed to and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Bai, R., Li, Y., Jian, L. et al. The hypoxia-driven crosstalk between tumor and tumor-associated macrophages: mechanisms and clinical treatment strategies. Mol Cancer 21, 177 (2022). https://doi.org/10.1186/s12943-022-01645-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01645-2