
Abstract
Autophagy, as a type II programmed cell death, plays crucial roles with autophagy-related (ATG) proteins in cancer. Up to now, the dual role of autophagy both in cancer progression and inhibition remains controversial, in which the numerous ATG proteins and their core complexes including ULK1/2 kinase core complex, autophagy-specific class III PI3K complex, ATG9A trafficking system, ATG12 and LC3 ubiquitin-like conjugation systems, give multiple activities of autophagy pathway and are involved in autophagy initiation, nucleation, elongation, maturation, fusion and degradation. Autophagy plays a dynamic tumor-suppressive or tumor-promoting role in different contexts and stages of cancer development. In the early tumorigenesis, autophagy, as a survival pathway and quality-control mechanism, prevents tumor initiation and suppresses cancer progression. Once the tumors progress to late stage and are established and subjected to the environmental stresses, autophagy, as a dynamic degradation and recycling system, contributes to the survival and growth of the established tumors and promotes aggressiveness of the cancers by facilitating metastasis. This indicates that regulation of autophagy can be used as effective interventional strategies for cancer therapy.
Similar content being viewed by others

Introduction
Fifty years ago, Christian de Duve, a Belgian scientist, firstly coined the term autophagy at the Ciba Foundation symposium on lysosomes in 1963 [1, 2], for which he shared the Nobel Prize in Physiology or Medicine in 1974 with Albert Claude and George E. Palade. There are three morphologically and mechanistically distinct types of autophagy in cells: macroautophagy, microautophagy and chaperone mediated autophagy [3], and usually macroautophagy is referred to as autophagy [4].
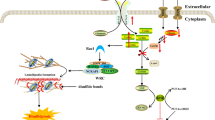
Autophagy is an intracellular evolutionarily conserved catabolic degradation process in which cytoplasmic macromolecules, aggregated proteins, damaged organelles or pathogen are delivered to lysosomes, and digested by lysosomal hydrolases to generate nucleotides, amino acids, fatty acids, sugars, and ATP, and ultimately recycled into the cytosol [5,6,7,8,9,10,11,12,13] (Fig. 1). This cellular self-digestion mediated by lysosome sustains, on the one hand, cell metabolism and survival during starvation and stress, and eliminates, on the other hand, damaged proteins and organelles to maintain protein and organelle quality and quantity [14, 15].

Schematic overview of autophagy. a Initiation, activation of ULK1 complex and multiple ATG proteins are engaged and localized to PAS. b Nucleation, ATG proteins and lipids are recruited to form phagophore; Elongation, cytoplasm and organelles are wrapped and engulfed during elongation of the phagophore; Maturation, completion and transport of the autophagosome. c Fusion, docking and fusion between autophagosome and lysosome. d Degradation, degradation of the cargos inside the autolysosome. e The ULK1 kinase core complex including ULK1, ATG13, FIP200, and ATG101. f The class III PI3K complex I including Beclin1, VPS34, VPS15, and ATG14L. g The ATG9A/ATG2-WIPI1/2 trafficking system including ATG9A, ATG2, and WIPI1/2. h The ATG12-conjugation system including ATG12, ATG7, ATG10, ATG5, and ATG16L. i The LC3-conjugation system including ProLC3, ATG4, LC3-I, ATG7, ATG3, and LC3-II (LC3-I/PE)
Although autophagy was found over 50 years ago, only within decade lots of studies elucidated the functions and roles of this ubiquitous process. Recent studies have indicated that autophagy plays a greater variety of pathophysiological roles in many disease processes, including cancer, neurodegeneration, autoimmune diseases, aging, cell death, heart disease and infection, and aids cell to clear damaged proteins, organelles, pathogens or aggregates, and has been proposed as a cell death mechanism, programmed cell death type II [16,17,18,19,20,21], whereas apoptosis is distinctively programmed cell death type I [22,23,24]. The potential ability of autophagy to modulate cell death makes it a therapeutic target in cancer [25, 26].
With its basic role in the turnover of proteins and organelles, autophagy has multiple physiological and pathophysiological functions. During tumorigenesis, autophagy plays an important role. In this review, the molecular basis of autophagy and its roles in cancer are summarized.
Molecular basis of autophagy
Only a small amount of autophagy in cells is involved in maintaining homeostasis in physiological condition. When cells are stimulated by intracellular and extracellular factors e.g. starvation, hypoxia [27], some small molecular compounds [28], oxidation, and pathogen invasion [3, 29], a large number of autophagy is induced by the transduction of cellular signaling pathways, and many important autophagy-related proteins and their complex involved in the autophagic process [30].
Process of autophagy
Physiologically, autophagy is an evolutionarily conserved, self-degradative, normal physiological process in cells, which is composed of several closely related steps including induction of autophagy, assembly and formation of autophagosome, autophagosome docking and fusion with lysosomal membranes, and degradation and recirculation of intra-autophagosomal contents in autophagolyosome [17, 31] (Fig. 1a-d).
Induction of autophagy
Induction of autophagy can be triggered by several intracellular and extracellular stimulus, e.g. nutrient starvation including depletion of total amino acids and serum starvation that strongly induces a high level of autophagy [27], oxidative stress that induces autophagy in order to recycle damaged organelles (e.g. mitochondria) and eliminate proteins aggregates [32], and inhibitors of TOR such as rapamycin and CCI-779 [17]. Under nutrient-rich condition, the active mTORC1 kinase hyperphosphorylates ATG13 and blocks the interaction of ATG13 with ULK1and FIP200. When cells are induced by those intracellular and extracellular stimulating factors, the ATG13 anchors ULK1 to a pre-autophagosomal structure (PAS), and then the almost all autophagy-related (Atg) proteins gather hierarchically onto the PAS (Fig. 1a), which is reported to be a crucial site of the cytoplasm to vacuole targeting (Cvt) and autophagosome formation [2, 33, 34].
As a dock structure for recruitment of ATG proteins, PAS plays a critical role during induction of autophagy [34, 35] Under autophagy-inducing conditions, the functional unit ULK1/Atg1 (including ULK1, ATG13, FIP200, and ATG101) acts as autophagy initiation complex, in which the ATG13 is a crucial protein for the PAS localization of ULK1 (Atg1 in yeast) and the interaction of FIP200 with ULK1, while the FIP200 (Atg11 and Atg17 in yeast) functions as a scaffold for downstream ATG protein assembly at the PAS. Once the ATG13 and ULK1 target to the PAS, all of these multiple ATG proteins are initially engaged and localized to the PAS, that is the initiation of autophagy [2, 33, 34] (Fig. 1a). Subsequently, the other functional units, including ULK1 complex, PI3K complex, ATG9A system, ATG12-conjugation system, and LC3-conjugation system, are targeted to the PAS in a hierarchical manner and involved in assembly and formation of autophagosome [12, 36,37,38,39].
Assembly and formation of autophagosome
Final formation of mature autophagosome includes nucleation of the multiple Atg proteins at PAS, elongation of the isolation membrane, and maturation of autophagosome, and four functional units are involved in these processes (Fig. 1b). The multiple Atg proteins gathering onto the PAS lead to the formation of a phagophore (or an isolation membrane) [40, 41]. The PAS is a potential nucleating site for forming the isolation membrane and recruits multiple Atg proteins. This nucleation process is initiated by the ULK1/Atg1 complex [42]. In response to nutrient starvation, the ULK1/Atg1 protein forms a complex with Atg13, FIP200/Atg17, Atg29, and Atg31, and this complex further associates with itself to generate the PAS scaffold complex, then the PI3K complex is gathered to the PAS and involved in forming phagophore through ATG14L interacting and binding to the ATG13 at PAS; and the ATG9A positive membrane vesicles associating with ATG2-WIPI complex (Atg2-Atg18 complex in yeast) are tethered to the PAS via interacting with the FIP200 (Atg17 and Atg11 in yeast). The multiple Atg proteins coordinate to generate the isolation membrane [42]. Once the first small ATG9A positive vesicles are fused at the PAS to form a phagophore, the bowl-shaped membrane is elongated continuously, and wraps and engulfs portions of cytoplasm and organelles. Finally, the isolation membrane, mediated by two ubiquitin-like ATG conjugation pathways, Atg12-Atg5 and Atg8/LC3 conjugation systems, forms a closed bilayer membrane structure, mature autophagosome with an inner and outer membrane [43] (Fig. 1b).
Autophagosome fusion with lysosomal membranes
Autophagosome docking and fusion with lysosomal membranes require the mature autophagosomes which will be transported to the perinuclear region for the autophagosome-lysosome fusion [44]. Autophagosomes can be formed randomly throughout the cytoplasm, whereas lysosomes are predominantly found in the perinuclear region. Therefore, once mature autophagosomes have been generated, they need to be delivered to the perinuclear region [45]. As long as autophagosomes arrive at the perinuclear region, they dock and fuse with lysosome immediately, and then form autophagolyosome (Fig. 1c).
Degradation and recirculation of autophagosomal contents
When autophagosome fuses with lysosomes to form autophagolyosome, many enzymes in lysosomes, e.g. lysosomal hydrolases, can degrade the inner membrane of the autophagosome and the cytoplasm-derived macromolecules, e.g. proteins and organelles, in the autophagosome into amino acids or peptides for reuse by cells (Fig. 1d).
Autophagy-related proteins
Although autophagic structures by electron microscopy examination were firstly reported by Christian de Duve under 60 years ago, the molecular mechanism of autophagy regulation remained mostly unknown until discovery of yeast Atg genes in the 1990s, which greatly promoted the mechanistic understanding of autophagy and clarified the fact that autophagy plays important roles in various biological processes [46,47,48,49]. Functionally, multiple autophagy-related proteins regulate and control various stages of the autophagy formation, including initiation of autophagy, nucleation of the multiple Atg proteins at PAS, elongation of the isolation membrane, and maturation of autophagosome, trafficking of mature autophagosomes, autophagosome docking and fusion with lysosomal membranes, and degradation of intra-autophagosomal contents in autophagolyosome by a hierarchical manner [17, 31].
So far, more than 40 genes encoding Atg proteins have been identified in yeast [49], and most of the genes (e.g. Atg1-Atg10, Atg12-Atg14, Atg16-Atg18) are conserved between yeast and mammalian, which indicates that autophagy is an evolutionarily conserved process [50]. Klionsky et al. (2003) collectively named the genes encoding these proteins as ATG (AuTophaGy), which is used to represent the autophagy gene and its encoding protein [50] (Table 1; Fig. 1).
Regulation and signaling of autophagy
In mammal cells, the starvation-induced autophagy is regulated by about 20 core ATG proteins, which can be classified into several functional units: (1) the ULK kinase core complex including ULK1/2, ATG13, RB1CC1/FIP200, and ATG101, (2) the autophagy-specific class III phosphatidylinositol 3-kinase (PI3K) complex including VPS34, VPS15, Beclin1, and ATG14L, (3) the ATG9A trafficking system including ATG9A, WIPI1/2, and ATG2A, (4) the ATG12 ubiquitin-like conjugation system including ATG12, ATG7, ATG10, ATG5, and ATG16L1, and (5) the LC3 ubiquitin-like conjugation system including LC3A/B/C, ATG7, ATG3, and ATG4A/B/C/D. These ATG proteins are recruited hierarchically proximal to the vacuole and organize the pre-autophagosomal structure (PAS) that is essential for autophagosome formation [12, 36,37,38,39] (Fig. 1e-i; Table 2).
ULK/Atg1 kinase core complex
During autophagy, autophagosome biogenesis commences at the PAS. In yeast, the Atg1 kinase core complex, consisting of the subunits Atg1, Atg13, Atg17, Atg29, and Atg31, is thought to play an essential and crucial role in the initiation of autophagy at the PAS, and has similar function to the ULK kinase core complex in mammal cells [92]. The ULK/Atg1 complex is mainly involved in receiving signals of cellular stimulation, recruiting ATG/Atg proteins to the PAS, organizing the vesicle cluster to form the phagophore, and governing elongation of the phagophore and formation of autophagosome [54, 93].
In human cells, the ULK1/2 is thought to serve similar and conserved functions as the yeast Atg1 [54]. In yeast, the core subunits of the Atg1 kinase complex are Atg1 and Atg13 [94]. When the cell is stimulated by starvation or other external stress, the target of rapamycin kinase complex (TOR) is inactivated, and then the Atg13 is dephosphorylated and binds greatly to Atg1 to form an activated Atg1-Atg13 dimer [95, 96]. The Atg17, Atg29, and Atg31 can assembly and form a trimeric complex Atg17-Atg31-Atg29 at the PAS [2, 97], and then serve as a preexisting scaffold for the recruitment of Atg1-Atg13 upon activation [98]. In mammal, the ULK1/2, a homologous protein of the kinase Atg1, forms a ULK1/2 kinase complex with ATG13 (homologous to Atg13 in yeast), FIP200 (homologous to Atg17 in yeast) and ATG101 (no homolog in yeast) [99] (Fig. 1e; Table 2).
Atg17 (FIP200) is the earliest protein to arrive at the PAS and adapts a highly elongated crescent shape [37, 93, 100]. The Atg17 is required specifically and associates physically with Atg1-Atg13 (ULK1/2-ATG13) complex, and the interaction between Atg17 and Atg1 is mediated by Atg13, indicating that Atge13 directly binds to both Atg1 and Atg17 and the Atg17-Atg13 complex formation plays an important role in normal autophagosome formation via binding to and activating the Atg1 kinase [33, 93, 96, 101, 102].
The class III PI3K complex I
In mammalian cells, the class III PI3K complex has two distinct types: complex I (PI3KC3-CI) and complex II (PI3KC3-CII). The both complexes share three core subunits: VPS34 (Vps34 in yeast), VPS15 (Vps15 in yeast), and Beclin1 (Atg6/Vps30 in yeast), and each complex contains a unique component: ATG14L/Barkor (Atg14 in yeast) of the autophagy-related complex I and Vps38 of the Vps-related complex II, which determines the localization of its own complex in the cell (Fig. 1f). The ATG14L (Atg14) can associate with and anchor the PI3KC3-CI to the PAS [103,104,105,106] and the Vps38 can localize the PI3KC3-CII to vacuolar and endosomal membranes [103, 107].
In the autophagy-specific PI3KC3-CI, VPS34, a catalytic PI(3) kinase, catalyzes phosphatidylinositol (PI) phosphorylation to form phosphatidylinositol 3-phosphate (PtdIns(3) P or PI3P). The PtdIns(3) P on autophagic membranes is essential for the elongation and completion of autophagosomes for it can bind and recruit the membrane-bound protein ATG18 to the bilayer membrane [108, 109].
In yeast, Atg6 mediates interaction with Atg14 that is crucial for localizing the PI3KC3-CI to PAS [110, 111]. The sole Class III PI3K, Vps34, is associated with the protein kinase Vps15, which functions as a Vps34 regulatory subunit [112]. Recently, it is reported that the interaction of Vps15-Vps34 with Atg14-Atg6 is mediated by Atg38, which was shown to play a crucial role in the complex integrity [113]. In mammalian cells, Beclin1 is a central regulator, which interacts with a multitude of proteins including ATG14L, UVRAG, Rubicon, and Bcl-2, etc. [114,115,116,117]. The Beclin1 has three functional domains including a N-terminal Bcl-2 homology 3 (BH3) domain, interacting with the Bcl-2 family protein Bcl-XL [118,119,120], a central coiled-coil domain (CCD), mediating interaction of Beclin1 with ATG14L and UVRAG [121], and a C-terminal evolutionarily conserved domain (ECD), mediating the interaction of Beclin1 with VPS34 and activation of VPS34 kinase activity to regulate the size and number of autophagosomes [110, 111, 116, 122] (Fig. 1f; Table 2).
The ATG9A/Atg9 trafficking system
After the ULK/Atg1 complex is formed, the next step is recruitment of ATG9A/Atg9-containing cytoplasmic vesicles (ATG9A/Atg9 vesicles), which is a crucial step of autophagosome formation and plays an essential role in the nucleation step of autophagosome formation in eukaryotes (from yeast to mammals) [73, 123, 124]. The ATG9A/Atg9 can be phosphorylated by ULK/Atg1, and then the phosphorylated ATG9A/Atg9 is required for the recruitment of LC3/Atg8 and WIPI1/2/Atg18 to the site of autophagosome formation and the expansion and elongation of phagophore [125].
Mammalian ATG9A, the yeast Atg9 homolog, is the sole multi-spanning transmembrane protein within the core machinery of autophagosome formation and has 6 highly conserved transmembrane helices and 2 cytosolic NH2- and COOH-terminal domains that are involved in interactions with other ATG components in both yeast and mammals [126,127,128].
The ATG9A/Atg9 self-interacts and self-associates within membranes into a higher-order assembly [129]. Recent studies indicated that the majority of Atg9 in the yeast are incorporated on small cytoplasmic vesicles with diameters of 30–60 nm, namely Atg9 vesicles [123, 130]. It is estimated that 3 Atg9 vesicles contain approximately 30 molecules of Atg9 each assemble at the PAS [123, 131], and subsequently the Atg9 is integrated into the outer autophagosomal membrane. Once the autophagosomes fuse with vacuole, the Atg9 are recycled as new Atg9 vesicles [102, 123, 132]. The level of Atg9 expressed in cells correlates with the frequency of autophagosome formation and the number of autophagosome [133]. The Atg9 vesicles are originated and transported from the Golgi apparatus [73, 102, 123, 127]. In normal physiological conditions, Atg9 localizes to and cycles between the trans-Golgi network (TGN) and early and late post-Golgi endosomes [134, 135]. A recent study suggests that autophagosome formation occurs where ATG9 vesicles coalesce with the ER [136].
In yeast, Atg9 recycling from PAS is regulated by the Atg2-Atg18 complex [37, 102]. The Atg18 and Atg2 are peripheral membrane proteins. The Atg18 as a downstream effector of class III PI (3) K localizes to PAS via binding to PtdIns(3) P [137]. Recent reports showed that the Atg18-Atg2 complex may play an important role in transporting the membrane structures during autophagosome formation through binding to Atg9 and form an Atg9·Atg2-Atg18 complex on the surface of the PAS and further regulate cycling of Atg9 [102, 138, 139].
In mammals, the WIPI (WD-repeat protein interacting with phosphoinositides) proteins, including WIPI1, WIPI2, WIPI3, and WIPI4, have a similar function to the Atg18 [88, 140, 141]. The WIPI1/2-ATG2 complex is involved in forming ATG9A·WIPI1/2-ATG2 trafficking system, mediating and regulating cycling of ATG9A, and promoting formation of LC3-positive autophagosomes in autophagy [140]. The WIPI1/2-ATG2 (Atg18-Atg2 in yeast) complex localizes to the expanding edge of the isolation membrane and plays a key role in the elongation and/or closure of the isolation membrane [43, 142] (Fig. 1g; Table 2).
The ATG12/Atg12-conjugation system
During autophagosome formation, two ubiquitin-like conjugation systems are required including the ATG12/Atg12 and LC3/Atg8 conjugation systems, and as many as eight ATG proteins are involved in both conjugation systems, which contribute to and are tightly associated with expansion of autophagosomal membrane.
In yeast, the Atg12, a ubiquitin-like protein, is covalently linked to its substrate Atg5 and forms an irreversible Atg12~Atg5 conjugate [38]. The Atg12-conjugation system is similar to the E1-E2-E3 activation and ligase present in the ubiquitination pathway, in which Atg12 is activated by Atg7, an E1-like enzyme [143], and then is transferred to Atg10, an E2-like enzyme [144], and is finally conjugated to its substrate protein Atg5 [38]. The Atg12~Atg5 conjugation has no typical E3 enzyme. The Atg5 of the Atg12~Atg5 conjugate further interacts with a small coiled-coil protein, Atg16, to form a ~ 350-kDa Atg12~Atg5-Atg16 complex [145, 146].
In mammals, the ATG12, activating by the E1 enzyme ATG7, is conjugated to ATG5 via the E2 enzyme Atg10 and then the ATG12-ATG5 conjugate can be stabilized by ATG16L proteins and further form ATG12-ATG5- ATG16L complex of approximately 800 kDa, which is important for the formation of the LC3 conjugation system [58, 62, 147] (Fig. 1h; Table 2).
The LC3/Atg8 -conjugation system
The LC3/Atg8-conjugation system is located downstream of the ATG12/Atg12 system in the context of Atg protein organization.
In yeast, the Atg8, another ubiquitin-like protein in yeast, is covalently linked to phosphatidylethanolamine (PE) after its C-terminal Arg117 residue is removed by a cysteine protease, Atg4, to expose to Gly116 [148]. The Atg8 is activated by the E1-like enzyme Atg7 [143], and then transferred to the E2-like enzyme Atg3 [149], and eventually the Atg3 conjugates Atg8 with the PE through an amide bond [39]. The Atg8-PE conjugate can be cleaved by Atg4 to release free Atg8, indicating that the Atg8-PE is reversible [148]. Most of Atg8 exist in the unconjugated form under normal conditions, but when autophagy is induced by starvation, most of Atg8 are activated, transferred, and converted to the PE-conjugated form [150].
In mammalian cells, there are several homologues of yeast Atg8 including LC3, GATE16, GABARAP and ATG8L. The LC3 has been best investigated and characterized as an autophagosome marker in mammalian cells [151,152,153,154,155], which forms an Atg8-like conjugation system, called the LC3-conjugation system (Fig. 1i; Table 2).
LC3, microtubule-associated protein light chain 3, is a soluble protein with a molecular mass of approximately 17 kDa. The LC3 is firstly synthesized as a precursor protein (proLC3) [156], then the C-terminal peptide of the proLC3 precursor is cleaved by mammalian ATG4B homologues to form LC3-I with an exposed C-terminal glycine [151, 157, 158]. Catalyzed by mammalian ATG7 and ATG3 homologues, cytosolic LC3-I is then activated by the E1 enzyme ATG7 and transferred to the E2 enzyme ATG3, and finally is modified to a membrane-bound protein, LC3-II, by conjugating to the amino group of the lipid phosphatidylethanolamine (PE) (LC3-I/PE). Then, the Atg12-Atg5-Atg16 complex acts as an E3 enzyme for the conjugation reaction of LC3-II (LC3-I/PE) [154, 155, 157] (Fig. 1i), which corresponds to the Atg8-PE-conjugated form in yeast [151, 159]. The ATG4B has been reported that it is the sole enzyme to efficiently cleave LC3 precursors and LC3-I/PE among four human homologues of yeast Atg4 (Atg4A-D) [151]. Thus, the both ubiquitin-like systems are intimately involved in formation of PAS, assembly and formation of autophagosome, and subsequent biogenesis of autophagy.
Autophagy in cancer
Physiologically, autophagy, by eliminating damaged proteins and organelles during stress and aging, plays critical roles in regulating organismal development, cooperating with the adaptive immune system, sustaining energy homeostasis and maintaining protein and organelle quality control [11, 160,161,162,163,164].
In diseases, such as neurodegenerative diseases [165, 166], infectious diseases [11, 167, 168], and metabolic diseases [14], dysfunctional autophagy leads to the accumulation of abnormal and damaged proteins and organelles and formation of intracellular aggregates, and then prevents the ability of autophagy to battle and eliminate infectious pathogens [11, 161, 167, 169].
In cancer, autophagy can play neutral, tumor-suppressive, or tumor-promoting roles in different contexts and stages of cancer development [25, 170,171,172,173], which is determined by nutrient availability, microenvironment stress, pathogenic conditions, and the presence of an immune system.
Dual role of autophagy in cancer
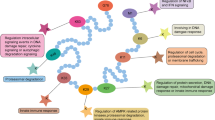
In cancer development, autophagy plays a dual role depending on type, stage or genetic context of the cancers [174,175,176,177,178,179]. On the one hand, via its protein and organelle quality control function, autophagy can maintain genome stability, prevent chronic tissue damage, cell injury, and inflammation, and inhibit accumulation of oncogenic p62 protein aggregates, and then prevent tumor initiation, proliferation, invasion, and metastasis, thereby function as a tumor suppressive mechanism, especially in the early stage of tumorigenesis [180,181,182]. Autophagy is important for the quality control of the cells such as removing damaged mitochondria, and its defective proteins (e.g., heterozygous knockdown Beclin1 and Atg7 in mice) promote the malignant transformation and spontaneous tumors [183,184,185]; on the other hand, once the tumors progress to late stage, autophagy can function as a cellular protective, survival, and defense mechanism, maintain functional mitochondria, reduce DNA damage, and enhance the survival and resistance of the cancer cells against stress (e.g., nutrient deprivation, hypoxia, DNA damage metabolic stress, and chemotherapy), and then sustain tumor metabolism, growth, and survival and then mediate tumor promotion and development, finally promotes tumorigenesis and causes resistance to therapeutic agents [180, 182, 186]. It is reported that autophagy can contribute to the aggressiveness of the cancers by facilitating metastasis [187,188,189] (Fig. 2). The effect of autophagy on cancers is dependent on multiple factors including tumor microenvironment, cancer type and stage, and genetic background.

Dual role of autophagy in tumorigenesis. Tumorigenesis begins with an oncogene mutation in the epithelial cell that makes the cell more likely to divide. The genetically altered or abnormal cells and its descendants grow and divide uncontrolled and rapidly at Hyperplasia stage. At Dysplasia stage, the overgrowing cells change their original form and behavior, have increased growth potential, and consist of more immature cells than mature. In situ cancer, the cells grow rapidly, but do not go into the process of maturation, have lost their tissue identity, and grow without regulation. In the malignant tumor (invasive cancer), the overgrowing cells invade neighboring areas and blood circulation systems from the primary tumor site by rupturing basal membrane. Metastases occur when cancer cells reach to the distant parts through lymphatic system and blood circulation. Autophagy plays dual roles during tumorigenesis including tumor-suppressing role during the early stage and cancer promoting role during the late stage
Autophagy suppresses tumorigenesis
Autophagy, the lysosome-mediated cellular self-digestion, acts as a cellular quality-control mechanism to sustain cell metabolism and its protein and organelle quality control during starvation, eliminates damaged proteins and organelles that accumulate during stress, and suppress chronic tissue damage, then prevent tumor initiation, especially in the early stage of tumorigenesis [11, 180]. Several indirect evidences indicate that autophagy acts as a tumor suppressor (Fig. 2).
Defective autophagy contributes to tumorigenesis
Through the identification of Beclin1, an essential autophagy gene, autophagy is first linked to human cancer. The Beclin1, as a haploid-insufficient tumor suppressor, is mono-allelically deleted in human hepatocellular carcinoma (HCC), breast, ovarian, and prostate cancers [114, 115, 190, 191] and in mice tumor prone [192]. It is reported that the expression of Beclin1 in cancer tissues was down-regulated in 44 patients with hepatocellular carcinoma, and it was concluded that autophagy might inhibit tumorigenesis [190]. The spontaneous frequency of malignancies is higher in the Beclin1+/− mouse model [192, 193], indicating that autophagy is a tumor-suppression mechanism [11, 25, 190, 194].
A number of studies on the ATG genes relevance to human cancers showed that other ATG genes are also oncogenically associated, including ATG2B, ATG5, ATG9B, ATG12 and ATG16L1. The frameshift mutations with mononucleotide repeats have been found in ATG2B, ATG5, ATG9B and ATG12 genes in gastric cancer and colorectal cancer, which may be involved in cancer development by deregulating the autophagy process [195]. The homozygote deletion of ATG5 predisposed to liver tumors with high penetrance mouse model [196]; the somatic point mutations of ATG5 are also identified in 135 patient samples of gastric cancer, colorectal cancer, and hepatocellular carcinoma [197]. The compartment-specific expression of ATG16L1 in epithelial cancer cells inhibited tumor growth [198].
Taken together, whether the expression of the intact autophagy genes is downregulated in cancers or the spontaneous frequency of cancer malignancies is increased due to autophagy-related gene deficiency, indicating that the intact autophagy functions as a cancer suppression mechanism by limiting genome damage and mutation and constraining tumor initiation.
Autophagy inhibits necrosis and inflammation
Autophagy is a central regulator of the inflammasome, and the chronic inflammation is a common future of early cancer development [199,200,201,202]. The oncogene activation can cause neoplasia and inflammation, and the inflammatory conditions can increase cancer risk. The autophagy-deficient tumors display an increased level of necrosis and inflammation, indicating that the intact autophagy can inhibit neoplasia, inflammation and cancer [203, 204]. Defective autophagy lead to tissue damage, necrosis, chronic inflammation, and genetic instability, which can increase the incidence of cancer by altering the tumor microenvironment, elevating oxidative stress and creating cancer-causing mutations [204, 205]. In autophagy-defective cells and tissues, the failure to eliminate damaged proteins and organelles leads cellular dysfunction and death, and then stimulates an inflammation condition, and creates ultimately a cancer-prone environment [206].
Among mammal ATG proteins, Beclin-1, ATG5, ATG7, ATG12, ATG16L1 and LC3B are the most studied with respect to inflammation [207], and defects in autophagy are linked to many inflammatory diseases [208, 209] and cancer [210].
Accumulation of p62/SQSTM1 promotes tumorigenesis
The p62, also called sequestosome 1 (SQSTM1) in humans, a multifunctional adaptor protein, is a selective substrate of autophagy. In intact autophagy, the p62/SQSTM1 possesses a short LC3 interaction region (LIR) that facilitates direct interaction with LC3 and causes p62 to be specifically degraded by autophagy, while defective autophagy is a mechanism for p62 upregulation commonly observed in human tumors, so the level of p62 has been used as a marker for inhibition of autophagy or defects in autophagic degradation [211,212,213].
The aberrant accumulation of p62 has been detected in the cases of gastrointestinal cancer [214], prostate cancer [215, 216], hepatocellular carcinoma [217,218,219], breast cancer [220, 221], lung adenocarcinoma [222], suggesting that p62 accumulation correlates with cancer progression and autophagy suppresses tumorigenesis by limiting p62 accumulation [213, 223, 224].
Autophagy mediates cancer promotion
Once the tumors progress to late stage, autophagy can promote the survival and growth of the established tumors by removing toxic oxygen radicals or damaged proteins, maintaining mitochondrial function, sustaining metabolism and survival in stress, and preventing diversion of tumor progression to benign oncocytomas [180,181,182]. Many investigations have shown that autophagy is the major contributor for cancer cells substantially survival [225,226,227]. It is reported that autophagy can contribute to the aggressiveness of the cancers by facilitating metastasis [187,188,189]. Moreover, autophagy as a cellular defense mechanism may reduce the effect of treatments of most chemotherapeutic agents (Fig. 2).
Autophagy prevents cancer cell damage
Autophagy is robustly activated in cancer cells under a multitude of stress conditions, including starvation, growth factor deprivation, hypoxia, damaging stimuli and proteasome inhibition, so elevated levels of autophagy have been observed in many tumor types, e.g. the essential autophagy gene Beclin1 was upregulated in colorectal cancer, gastric cancer, liver cancer, breast cancer, and cervical cancer [228,229,230,231], suggesting that the enhancement of autophagy can promote tumorigenesis and overexpression of the Beclin1 plays a crucial role in tumor formation.
Autophagy functioning as a cancer promotion mechanism is mainly based on its role involved in removing damaged mitochondria, inhibiting DNA damage, maintaining genome stability, limiting inflammation, and finally preventing cancer cell damage under the conditions of stress [14, 213, 232]. Normal mitochondrial function, e.g. mitochondrial respiration, is required for tumorigenesis [233], t the accumulation of morphologically abnormal mitochondria and mitochondrial dysfunction have been found in the autophagy-defective tumors [196, 234,235,236], indicating that intact autophagy can remove damaged mitochondria and contribute to tumorigenesis. The activation of the DNA damage response, gene amplification, DNA copy number variations and an elevated mutation rate has been found in the autophagy-deficient cancer cells [237]. Autophagy prevents genome damage and promotes tumor cell survival in a model of mammary cancer [194]. Autophagy is induced in hypoxic tumor regions and is required for tumor cell survival and for limiting inflammation [226]. (preventing cancer cell damage). All of these evidences indicate that the survival function of autophagy can be commandeered by tumors to prevent cell damage and promote tumorigenesis under conditions of metabolic stress.
Autophagy promotes cancer metastasis
During cancer progression, metastasis is an extremely complex process that indicates a more advanced stage and a poorer prognosis and accounts for most cancer-related deaths [238]. The metastasis of primary tumor can be divided into a series of stages including invasion of tumor cells from the primary tumor site, intravasation and survival in blood circulation systems, dissemination of the malignant cancer cells through the circulation systems to reach a capillary bed and adhere to the vessel walls, extravasation of the cancer cells at a distant site, and finally colonization of disseminated tumor cells at their destination organs [239,240,241,242].
Autophagy plays a complex and stage-specific role and promotes multiple steps during cancer metastasis [243]. During the early stage of metastasis, the autophagy may act as a suppressor of metastasis by preventing tumor necrosis and restricting inflammatory cell infiltration [243]; on the other hand, in the advanced stages of metastasis, the autophagy may act as a promoter of metastasis by promoting dissemination of the malignant cancer cells in the circulation [244], enhancing colonization of detached metastatic cell in the destination organs [245], and inducing metastatic cells to enter dormancy and survive in the new environment [246].
Autophagy is upregulated during cancer metastasis. Once the metastatic cancer cells successfully establish distant colonies in their destination organs, autophagy begins to play a critical role and the autophagic flux is induced to respond various environmental stress including hypoxia, nutrient deprivation and detachment from the ECM [187, 240, 247, 248]. Using the autophagy marker, LC3B, various studies have identified an association between increased autophagy and metastasis in several types of cancer including breast cancer metastasis [249, 250], melanoma metastases [251], hepatocellular carcinoma [189], and glioblastoma [252]. These evidences indicate that autophagy promotes cancer metastasis and enhances the aggressiveness of cancer cells [253].
Autophagy inhibit cancer therapy
Cancer cells have common characteristics including increased metabolic demands, high level cellular proliferation, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activated invasion and metastasis, and enhanced cellular stress, which require autophagy to be activated to maintain energy, enhance stress tolerance, limit damage, and prevent death in these cells.
Autophagy plays a cytoprotective or pro-survival role in cancer cells and can be induced by most cancer treatments including radiation therapy [254,255,256], chemotherapy [257, 258], histone deacetylase inhibitors in colon cancer cells [259], arsenic trioxide (As2O3) in malignant glioma cells [260, 261], Temozolomide (TMZ) in malignant glioma cells [262], γ-irradiation in breast cancer, prostate cancer, colon cancer and malignant glioma [263,264,265], resveratrol in ovarian cancer [266], TNFα in breast cancer cells [267], IFNγ in Hepatocellular carcinoma (HCC) [268], imatinib lung carcinoma cell [269], rapamycin in malignant glioma cells [270], and tamoxifen in breast cancer and Glioblastoma [271, 272], and the autophagy, in turn, functions as a cellular defense and protection mechanism to prevent cancer cell death upon treatment, enable a state of dormancy in residual cancer cells post treatment, contribute to cancer recurrence and metastasis, and inhibit cancer therapy and tumor cell killing [246, 273].
Given the pro-survival role, the inhibition of the autophagy has been shown to enhance and increase the efficacy of anticancer therapy, implying that autophagy inhibition is a potential valuable approach in combination with other anticancer therapeutic approaches to enhance cancer treatment [181, 182].
Conclusions and perspectives
Autophagy, as a cell survival pathway, plays an important role in cancer, and can help to prevent bioenergetic failure by metabolic stress and maintain protein and organelle quality and quantity, and contributes to all aspects of tumorigenesis, including tumor initiation, progression and development, and maintenance of the malignant state. Cancer cells divide relentlessly, and they are also metabolically stressed. As cancer cells grow, spread, and form solid tumors or flood the blood with abnormal cells, they always face an acute problem increasing hypoxia and nutrient deprivation, which may promote their death and prevent their growth, progression, and development, and autophagy is essential for surviving these stresses and maintaining tumorigenesis. Autophagy also plays key roles in controlling the tumor microenvironment, in suppressing tumor during the early stage and promoting cancer during the late stage, and in the therapeutic response.
Autophagy has a dual role both in progression and inhibition of cancer. Hitherto many data support a dynamic role of autophagy in cancer, both as a tumor suppressor early in progression and as a cancer promotor later in tumor maintenance and therapeutic resistance. In the early tumorigenesis, autophagy, as a survival pathway and quality-control mechanism, contributes to normal cell physiology metabolism and provides biological materials and energy in response to stress, and as a dynamic degradation and quality-control mechanism, eliminates damaged proteins and organelles and prevents tumor initiation. Once the tumors progress to late stage and are established and subjected to the environmental stresses including limited angiogenesis, nutrient deprivation, and hypoxia, autophagy, as a dynamic degradation and recycling system, contributes to the survival and growth of the established tumors and promotes aggressiveness of the cancers by facilitating metastasis.
Regulation of autophagy can be used as effective interventional strategies for cancer prevention and therapy by preventing cancer development, limiting tumor progression, and increasing the efficiency of cancer treatment. On the one hand, autophagy, as one type of programmed cell death, is ubiquitous in various cancer, functions as a tumor suppressor pathway, facilitates the degradation of oncogenic molecules, and finally prevents development of cancers. So defective or inadequate levels of autophagy can lead to cancer. Investigations showed that all chemotherapeutic agents and radiotherapies induce cancer metabolic stress and concomitant inhibition of autophagy, indicating that the autophagy regulation represents a significant direction in the development of anticancer therapies. On the other hand, autophagy, the type II programmed cell death, is involved in several signaling pathways during tumorigenesis via coordinating with apoptosis, the type I programmed cell death. Under stress conditions such as hypoxic or low-nutrition environments autophagy facilitates the survival of tumor cells, and at same time, apoptosis prevents the survival of cancer cells, indicating that autophagy and apoptosis, as two catabolic pathways, are essential for organismal homeostasis and tumor microenvironment. Investigations have now shown that autophagy and apoptosis are interconnected and coordinated by several molecular nodes of crosstalk, such as interaction of Beclin1 with Bcl-2, UVRAG with Bif-1, and ATG12 with the Mcl-1, etc.
So far, some standard cancer treatments have saved, or at least prolonged, many lives. However, the most severe clinical issue is the frequent tumors progression and cancer recurrence after treatment, mainly due to therapeutic resistance. It can be sure that autophagy can facilitate the tumor cells survival and deal with anticancer therapy. Therefore, in the near future, standard cancer treatment combining with regulation of autophagy activity, promoting or preventing by autophagy inducers or inhibitors based on tumorigenesis and cancer stages, can be considered as a potential anticancer therapy. However, further investigations should be done to understand and clarify how autophagy contributes to the development and treatment of cancer, how the autophagy pathway can be targeted and regulated, and how the activity of autophagy pathway can be monitored and quantified during cancer prevention and therapy.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- As2O3 :
-
Arsenic Trioxide
- ATG:
-
autophagy-related proteins, such as ATG1, ATG4, ATG5 ATG7 etc.
- BH3:
-
Bcl-2 homology 3 domain
- CCD:
-
Coiled-coil domain
- Cvt:
-
Cytoplasm to vacuole targeting
- ECD:
-
Evolutionarily conserved domain
- ECM:
-
Extracellular matrix
- ER:
-
Endoplasmic reticulum
- FIP200:
-
FAK-family interacting protein of 200 kDa
- GABARAP:
-
γ-aminobutyric-acid-type-A-receptor-associated protein
- GATE16:
-
Golgi-associated ATPase enhancer of 16 kDa
- HCC:
-
Hepatocellular carcinoma
- LC3:
-
Microtubule-associated protein 1A/1B-light chain 3
- LC3-I:
-
The cytosolic form of LC3
- LC3-II:
-
The conjugate form of LC3-I with phosphatidylethanolamine (PE) (LC3-I/PE)
- LIR:
-
LC3 interaction region
- p62/SQSTM1:
-
a ubiquitin-binding protein p62, sequestosome 1
- PAS:
-
Pre-autophagosomal structure
- PI:
-
Phosphatidylinositol
- PI3KC3-CI and PI3KC3-CII:
-
Class III phosphatidylinositol 3-kinase complex I and II
- ProLC3:
-
The precursor protein form of LC3
- PtdIns(3)P:
-
Phosphatidylinositol 3-phosphate (PI3P)
- RB1CC1:
-
RB1-inducible coiled-coil protein 1
- TGN:
-
Trans-Golgi network
- TMZ:
-
Temozolomide
- TOR:
-
Target of rapamycin
- ULK1/2:
-
Unc51-like kinase 1 and 2
- UVRAG:
-
UV radiation resistance-associated gene
- VPS15:
-
Vacuolar protein sorting 15
- VPS34:
-
Vacuolar protein sorting 34
- WIPI1/2:
-
WD-repeat protein interacting with phosphoinositides proteins 1 and 2
References
De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92.
Kawamata T, et al. Organization of the pre-autophagosomal structure responsible for autophagosome formation. Mol Biol Cell. 2008;19(5):2039–50.
Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–9.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12.
Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463–77.
Cuervo AM. Autophagy: in sickness and in health. Trends Cell Biol. 2004;14(2):70–7.
Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306(5698):990–5.
Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8(11):931–7.
Levine B. Cell biology: autophagy and cancer. Nature. 2007;446(7137):745–7.
Goswami SK, Das DK. Autophagy in the myocardium: dying for survival? Exp Clin Cardiol. 2006;11(3):183–8.
Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–32.
Rogov V, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53(2):167–78.
Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330(6009):1344–8.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41.
Mizushima N. The pleiotropic role of autophagy: from protein metabolism to bactericide. Cell Death Differ. 2005;12(Suppl 2):1535–41.
Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–73.
Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–55.
Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12(9):814–22.
Mizushima N, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–75.
Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–9.
Kondo Y, Kondo S. Autophagy and cancer therapy. Autophagy. 2006;2(2):85–90.
Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–10.
Yu L, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci U S A. 2006;103(13):4952–7.
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–7.
White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15(17):5308–16.
Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–80.
Tsvetkov AS, et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107(39):16982–7.
Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–76.
Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194(2):341–61.
Hansen TE, Johansen T. Following autophagy step by step. BMC Biol. 2011;9:39.
Ureshino RP, et al. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid Redox Signal. 2014;21(1):123–37.
Yamamoto H, et al. The intrinsically disordered protein Atg13 mediates Supramolecular assembly of autophagy initiation complexes. Dev Cell. 2016;38(1):86–99.
Suzuki K, et al. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20(21):5971–81.
Kotani T, et al. The Atg2-Atg18 complex tethers pre-autophagosomal membranes to the endoplasmic reticulum for autophagosome formation. Proc Natl Acad Sci U S A. 2018;115(41):10363–8.
Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14(12):759–74.
Suzuki K, et al. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12(2):209–18.
Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395(6700):395–8.
Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408(6811):488–92.
Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12(9):831–5.
Militello RD, Colombo MI. A membrane is born: origin of the autophagosomal compartment. Curr Mol Med. 2011;11(3):197–203.
Cheong H, et al. The Atg1 kinase complex is involved in the regulation of protein recruitment to initiate sequestering vesicle formation for nonspecific autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2008;19(2):668–81.
Suzuki K, et al. Fine mapping of autophagy-related proteins during autophagosome formation in Saccharomyces cerevisiae. J Cell Sci. 2013;126(Pt 11):2534–44.
Wijdeven RH, et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun. 2016;7:11808.
Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct Funct. 2008;33(1):109–22.
Matsuura A, et al. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene. 1997;192(2):245–50.
Clark SL Jr. Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol. 1957;3(3):349–62.
Novikoff AB. The proximal tubule cell in experimental hydronephrosis. J Biophys Biochem Cytol. 1959;6(1):136–8.
Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23.
Klionsky DJ, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5(4):539–45.
Kamada Y, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010;30(4):1049–58.
Cheong H, et al. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci U S A. 2011;108(27):11121–6.
Hara T, Mizushima N. Role of ULK-FIP200 complex in mammalian autophagy: FIP200, a counterpart of yeast Atg17? Autophagy. 2009;5(1):85–7.
Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr Opin Cell Biol. 2010;22(2):132–9.
Tang Z, et al. Atg2A/B deficiency switches cytoprotective autophagy to non-canonical caspase-8 activation and apoptosis. Cell Death Differ. 2017;24(12):2127–38.
Velikkakath AK, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23(5):896–909.
Besteiro S, et al. Autophagy protein Atg3 is essential for maintaining mitochondrial integrity and for normal intracellular development of toxoplasma gondii tachyzoites. PLoS Pathog. 2011;7(12):e1002416.
Metlagel Z, et al. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proc Natl Acad Sci U S A. 2013;110(47):18844–9.
Radoshevich L, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. 2010;142(4):590–600.
Li M, et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286(9):7327–38.
Lang T, et al. Aut2p and Aut7p, two novel microtubule-associated proteins are essential for delivery of autophagic vesicles to the vacuole. EMBO J. 1998;17(13):3597–607.
Otomo C, et al. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol. 2013;20(1):59–66.
Matsushita M, et al. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem. 2007;282(9):6763–72.
Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17(10):839–49.
Kang R, et al. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011;18(4):571–80.
Yuan W, Stromhaug PE, Dunn WA Jr. Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol Biol Cell. 1999;10(5):1353–66.
Hong SB, et al. Insights into noncanonical E1 enzyme activation from the structure of autophagic E1 Atg7 with Atg8. Nat Struct Mol Biol. 2011;18(12):1323–30.
Noda NN, et al. Structural basis of Atg8 activation by a homodimeric E1, Atg7. Mol Cell. 2011;44(3):462–75.
He H, et al. Post-translational modifications of three members of the human MAP 1LC3 family and detection of a novel type of modification for MAP 1LC3B. J Biol Chem. 2003;278(31):29278–87.
Rogov VV, et al. Structural and functional analysis of the GABARAP interaction motif (GIM). EMBO Rep. 2017;18(8):1382–96.
Weidberg H, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29(11):1792–802.
Reggiori F, et al. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012;2012:219625.
Mari M, et al. An Atg9-containing compartment that functions in the early steps of autophagosome biogenesis. J Cell Biol. 2010;190(6):1005–22.
Zhao Q, et al. Dual roles of two isoforms of autophagy-related gene ATG10 in HCV-subgenomic replicon mediated autophagy flux and innate immunity. Sci Rep. 2017;7(1):11250.
Hong SB, et al. Structure of the autophagic E2 enzyme Atg10. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 10):1409–17.
Yamaguchi M, et al. Structural insights into Atg10-mediated formation of the autophagy-essential Atg12-Atg5 conjugate. Structure. 2012;20(7):1244–54.
Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. 2013;9(3):424–5.
Alers S, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7(12):1423–33.
Ganley IG, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–305.
Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–91.
Kim HJ, et al. Beclin-1-interacting autophagy protein Atg14L targets the SNARE-associated protein Snapin to coordinate endocytic trafficking. J Cell Sci. 2012;125(Pt 20):4740–50.
Ma B, et al. Dapper1 promotes autophagy by enhancing the Beclin1-Vps34-Atg14L complex formation. Cell Res. 2014;24(8):912–24.
Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–96.
Fujita N, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008;19(5):2092–100.
Hwang S, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012;11(4):397–409.
Romanov J, et al. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J. 2012;31(22):4304–17.
Hara T, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510.
Proikas-Cezanne T, et al. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207–17.
Graef M. Membrane tethering by the autophagy ATG2A-WIPI4 complex. Proc Natl Acad Sci U S A. 2018;115(42):10540–1.
Hosokawa N, et al. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5(7):973–9.
Suzuki H, et al. Structure of the Atg101-Atg13 complex reveals essential roles of Atg101 in autophagy initiation. Nat Struct Mol Biol. 2015;22(7):572–80.
Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22(1):R29–34.
Ragusa MJ, Stanley RE, Hurley JH. Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell. 2012;151(7):1501–12.
Stjepanovic G, et al. Assembly and dynamics of the autophagy-initiating Atg1 complex. Proc Natl Acad Sci U S A. 2014;111(35):12793–8.
Wong PM, et al. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124–37.
Kamada Y, et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150(6):1507–13.
Kabeya Y, et al. Characterization of the Atg17-Atg29-Atg31 complex specifically required for starvation-induced autophagy in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2009;389(4):612–5.
Yeh YY, et al. The identification and analysis of phosphorylation sites on the Atg1 protein kinase. Autophagy. 2011;7(7):716–26.
Alers S, et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32(1):2–11.
Chew LH, et al. Structural characterization of the Saccharomyces cerevisiae autophagy regulatory complex Atg17-Atg31-Atg29. Autophagy. 2013;9(10):1467–74.
Kabeya Y, et al. Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell. 2005;16(5):2544–53.
Reggiori F, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004;6(1):79–90.
Kihara A, et al. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–30.
Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes--Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17(4):1527–39.
Obara K, Ohsumi Y. Dynamics and function of PtdIns(3) P in autophagy. Autophagy. 2008;4(7):952–4.
Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol. 2011;2011:713435.
Burda P, et al. Retromer function in endosome-to-Golgi retrograde transport is regulated by the yeast Vps34 PtdIns 3-kinase. J Cell Sci. 2002;115(Pt 20):3889–900.
Nagy P, et al. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in drosophila. FEBS Lett. 2014;588(3):408–13.
Noda T, et al. Regulation of membrane biogenesis in autophagy via PI3P dynamics. Semin Cell Dev Biol. 2010;21(7):671–6.
Noda NN, et al. Structure of the novel C-terminal domain of vacuolar protein sorting 30/autophagy-related protein 6 and its specific role in autophagy. J Biol Chem. 2012;287(20):16256–66.
Fogel AI, et al. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol. 2013;33(18):3675–88.
Backer JM. The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410(1):1–17.
Araki Y, et al. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203(2):299–313.
Aita VM, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59(1):59–65.
Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–6.
Furuya N, et al. The evolutionarily conserved domain of Beclin 1 is required for Vps34 binding, autophagy and tumor suppressor function. Autophagy. 2005;1(1):46–52.
Itakura E, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–72.
Liang XH, et al. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72(11):8586–96.
Feng W, et al. Molecular basis of Bcl-xL's target recognition versatility revealed by the structure of Bcl-xL in complex with the BH3 domain of Beclin-1. J Mol Biol. 2007;372(1):223–35.
Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282(17):13123–32.
Li X, et al. Imperfect interface of Beclin1 coiled-coil domain regulates homodimer and heterodimer formation with Atg14L and UVRAG. Nat Commun. 2012;3:662.
Huang W, et al. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res. 2012;22(3):473–89.
Yamamoto H, et al. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol. 2012;198(2):219–33.
Orsi A, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23(10):1860–73.
Papinski D, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014;53(3):471–83.
Noda T, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol. 2000;148(3):465–80.
Young AR, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006;119(Pt 18):3888–900.
Yamada T, et al. Endothelial nitric-oxide synthase antisense (NOS3AS) gene encodes an autophagy-related protein (APG9-like2) highly expressed in trophoblast. J Biol Chem. 2005;280(18):18283–90.
He C, et al. Self-interaction is critical for Atg9 transport and function at the phagophore assembly site during autophagy. Mol Biol Cell. 2008;19(12):5506–16.
Wang J, et al. Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc Natl Acad Sci U S A. 2013;110(24):9800–5.
Feng Y, et al. Phosphorylation of Atg9 regulates movement to the phagophore assembly site and the rate of autophagosome formation. Autophagy. 2016;12(4):648–58.
Reggiori F, et al. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy. 2005;1(2):101–9.
Jin M, et al. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr Biol. 2014;24(12):1314–22.
Jia S, et al. Mammalian Atg9 contributes to the post-Golgi transport of lysosomal hydrolases by interacting with adaptor protein-1. FEBS Lett. 2017;591(24):4027–38.
Shirahama-Noda K, et al. TRAPPIII is responsible for vesicular transport from early endosomes to Golgi, facilitating Atg9 cycling in autophagy. J Cell Sci. 2013;126(Pt 21):4963–73.
Karanasios E, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat Commun. 2016;7:12420.
Obara K, et al. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem. 2008;283(35):23972–80.
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–31.
Sun LL, et al. Global analysis of fission yeast mating genes reveals new autophagy factors. PLoS Genet. 2013;9(8):e1003715.
Polson HE, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–22.
Muller AJ, Proikas-Cezanne T. Function of human WIPI proteins in autophagosomal rejuvenation of endomembranes? FEBS Lett. 2015;589(14):1546–51.
Suzuki H, et al. Structural biology of the core autophagy machinery. Curr Opin Struct Biol. 2017;43:10–7.
Tanida I, et al. Apg7p/Cvt2p: a novel protein-activating enzyme essential for autophagy. Mol Biol Cell. 1999;10(5):1367–79.
Shintani T, et al. Apg10p, a novel protein-conjugating enzyme essential for autophagy in yeast. EMBO J. 1999;18(19):5234–41.
Kuma A, et al. Formation of the similar to 350-kDa Apg12-Apg5 center dot Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem. 2002;277(21):18619–25.
Mizushima N, Noda T, Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J. 1999;18(14):3888–96.
Mizushima N, et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci. 2003;116(Pt 9):1679–88.
Kirisako T, et al. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J Cell Biol. 2000;151(2):263–76.
Yamada Y, et al. The crystal structure of Atg3, an autophagy-related ubiquitin carrier protein (E2) enzyme that mediates Atg8 lipidation. J Biol Chem. 2007;282(11):8036–43.
Huang WP, et al. The itinerary of a vesicle component, Aut7p/Cvt5p, terminates in the yeast vacuole via the autophagy/Cvt pathways. J Biol Chem. 2000;275(8):5845–51.
Kabeya Y, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117(Pt 13):2805–12.
Tanida I, et al. GATE-16 and GABARAP are authentic modifiers mediated by Apg7 and Apg3. Biochem Biophys Res Commun. 2003;300(3):637–44.
Tanida I, et al. Atg8L/Apg8L is the fourth mammalian modifier of mammalian Atg8 conjugation mediated by human Atg4B, Atg7 and Atg3. FEBS J. 2006;273(11):2553–62.
Tanida I, et al. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002;277(16):13739–44.
Tanida I, et al. The human homolog of Saccharomyces cerevisiae Apg7p is a protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. J Biol Chem. 2001;276(3):1701–6.
Yang Z, et al. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J Biol Chem. 2015;290(44):26549–61.
Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8.
Tanida I, Ueno T, Kominami E. Human light chain 3/MAP 1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279(46):47704–10.
Tanida I, et al. Lysosomal turnover of GABARAP-phospholipid conjugate is activated during differentiation of C2C12 cells to myotubes without inactivation of the mTor kinase-signaling pathway. Autophagy. 2006;2(4):264–71.
Mizushima N. Physiological functions of autophagy. Curr Top Microbiol Immunol. 2009;335:71–84.
Ravikumar B, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90(4):1383–435.
Uchiyama Y, et al. Autophagy-physiology and pathophysiology. Histochem Cell Biol. 2008;129(4):407–20.
Wang L, Ye X, Zhao T. The physiological roles of autophagy in the mammalian life cycle. Biol Rev Camb Philos Soc. 2019;94(2):503-16.
Kuma A, Mizushima N. Physiological role of autophagy as an intracellular recycling system: with an emphasis on nutrient metabolism. Semin Cell Dev Biol. 2010;21(7):683–90.
Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–97.
Nah J, Yuan J, Jung YK. Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells. 2015;38(5):381–9.
Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24(1):69–79.
Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol. 2018;16(6):341-54.
Sridhar S, et al. Autophagy and disease: always two sides to a problem. J Pathol. 2012;226(2):255–73.
White E. The role for autophagy in cancer. J Clin Invest. 2015;125(1):42–6.
Levine B. Unraveling the role of autophagy in cancer. Autophagy. 2006;2(2):65–6.
Yang ZJ, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–41.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17(9):528-42.
Eskelinen EL. The dual role of autophagy in cancer. Curr Opin Pharmacol. 2011;11(4):294–300.
Singh SS, et al. Dual role of autophagy in hallmarks of cancer. Oncogene. 2018;37(9):1142–58.
Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32(7):955–63.
Jin S, et al. Autophagy regulation and its dual role in blood cancers: a novel target for therapeutic development (review). Oncol Rep. 2018;39(6):2473–81.
Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat Commun. 2014;5:3056.
Cristofani R, et al. Dual role of autophagy on docetaxel-sensitivity in prostate cancer cells. Cell Death Dis. 2018;9(9):889.
Barnard RA, et al. Autophagy inhibition delays early but not late-stage metastatic disease. J Pharmacol Exp Ther. 2016;358(2):282–93.
Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell. 2013;155(6):1216–9.
White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12(6):401–10.
Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7(3):297–300.
Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42.
Anding AL, Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. 2017;41(1):10–22.
Wu WK, et al. The autophagic paradox in cancer therapy. Oncogene. 2012;31(8):939–53.
Fung C, et al. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19(3):797–806.
Macintosh RL, et al. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle. 2012;11(10):2022–9.
Peng YF, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9(12):2056–68.
Ding ZB, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68(22):9167–75.
Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy. 2007;3(1):28–31.
Yue Z, et al. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A. 2003;100(25):15077–82.
Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112(12):1809–20.
Karantza-Wadsworth V, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21(13):1621–35.
Kang MR, et al. Frameshift mutations of autophagy-related genes ATG2B, ATG5, ATG9B and ATG12 in gastric and colorectal cancers with microsatellite instability. J Pathol. 2009;217(5):702–6.
Takamura A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25(8):795–800.
An CH, et al. Mutational and expressional analyses of ATG5, an autophagy-related gene, in gastrointestinal cancers. Pathol Res Pract. 2011;207(7):433–7.
Capparelli C, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11(12):2285–302.
Rodgers MA, et al. Regulation where autophagy intersects the inflammasome. Antioxid Redox Signal. 2014;20(3):495–506.
Shi CS, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13(3):255–63.
Harris J, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem. 2011;286(11):9587–97.
Saitoh T, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456(7219):264–8.
Kwong C, Gilman-Sachs A, Beaman K. Tumor-associated a2 vacuolar ATPase acts as a key mediator of cancer-related inflammation by inducing pro-tumorigenic properties in monocytes. J Immunol. 2011;186(3):1781–9.
White E, et al. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22(2):212–7.
Mantovani A, et al. Cancer-related inflammation. Nature. 2008;454(7203):436–44.
Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40.
Virgin HW, Levine B. Autophagy genes in immunity. Nat Immunol. 2009;10(5):461–70.
Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13(10):722–37.
Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–35.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420(6917):860–7.
Bjorkoy G, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–14.
Pankiv S, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–45.
Mathew R, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–75.
Su Y, et al. The diversity expression of p62 in digestive system cancers. Clin Immunol. 2005;116(2):118–23.
Kitamura H, et al. Cytosolic overexpression of p62 sequestosome 1 in neoplastic prostate tissue. Histopathology. 2006;48(2):157–61.
Valencia T, et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell. 2014;26(1):121–35.
Stumptner C, et al. Analysis of intracytoplasmic hyaline bodies in a hepatocellular carcinoma. Demonstration of p62 as major constituent. Am J Pathol. 1999;154(6):1701–10.
Saito T, et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun. 2016;7:12030.
Umemura A, et al. p62, Upregulated during Preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell. 2016;29(6):935–48.
Thompson HG, et al. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22(15):2322–33.
Li SS, et al. p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis. 2017;38(11):1092–103.
Inoue D, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103(4):760–6.
Huang J, et al. Adipocyte p62/SQSTM1 suppresses tumorigenesis through opposite regulations of metabolism in adipose tissue and tumor. Cancer Cell. 2018;33(4):770–84 e6.
Parkhitko A, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108(30):12455–60.
Jin S, et al. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007;120(Pt 3):379–83.
Degenhardt K, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64.
Lum JJ, et al. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120(2):237–48.
Ahn CH, et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS. 2007;115(12):1344–9.
Tang H, et al. Hepatitis B virus X protein sensitizes cells to starvation-induced autophagy via up-regulation of beclin 1 expression. Hepatology. 2009;49(1):60–71.
Karantza-Wadsworth V, White E. Role of autophagy in breast cancer. Autophagy. 2007;3(6):610–3.
Sun Y, et al. Over-expression of the Beclin1 gene upregulates chemosensitivity to anti-cancer drugs by enhancing therapy-induced apoptosis in cervix squamous carcinoma CaSki cells. Cancer Lett. 2010;294(2):204–10.
Robert T, et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature. 2011;471(7336):74–9.
Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A. 2010;107(19):8788–93.
Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25(5):460–70.
Wei H, et al. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25(14):1510–27.
Yang S, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25(7):717–29.
Mathew R, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21(11):1367–81.
Maishman T, et al. Local recurrence and breast oncological surgery in Young women with breast Cancer: the POSH observational cohort study. Ann Surg. 2017;266(1):165–72.
Alsarraj J, Hunter KW. Bromodomain-containing protein 4: a dynamic regulator of breast Cancer metastasis through modulation of the extracellular matrix. Int J Breast Cancer. 2012;2012:670632.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64.
Klein CA. Cancer. The metastasis cascade. Science. 2008;321(5897):1785–7.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168(4):670–91.
Kenific CM, Thorburn A, Debnath J. Autophagy and metastasis: another double-edged sword. Curr Opin Cell Biol. 2010;22(2):241–5.
Sosa MS, Bragado P, Aguirre-Ghiso JA. Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer. 2014;14(9):611–22.
Peng YF, et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PLoS One. 2013;8(9):e74407.
Lu Z, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118(12):3917–29.
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–92.
Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–93.
Lazova R, et al. Punctate LC3B expression is a common feature of solid tumors and associated with proliferation, metastasis, and poor outcome. Clin Cancer Res. 2012;18(2):370–9.
Zhao H, et al. High expression of LC3B is associated with progression and poor outcome in triple-negative breast cancer. Med Oncol. 2013;30(1):475.
Lazova R, Klump V, Pawelek J. Autophagy in cutaneous malignant melanoma. J Cutan Pathol. 2010;37(2):256–68.
Galavotti S, et al. The autophagy-associated factors DRAM1 and p62 regulate cell migration and invasion in glioblastoma stem cells. Oncogene. 2013;32(6):699–712.
Zheng HY, et al. Autophagy enhances the aggressiveness of human colorectal cancer cells and their ability to adapt to apoptotic stimulus. Cancer Biol Med. 2012;9(2):105–10.
Tam SY, Wu VW, Law HK. Influence of autophagy on the efficacy of radiotherapy. Radiat Oncol. 2017;12(1):57.
Classen F, et al. Autophagy induced by ionizing radiation promotes cell death over survival in human colorectal cancer cells. Exp Cell Res. 2019;374(1):29–37.
Zois CE, Koukourakis MI. Radiation-induced autophagy in normal and cancer cells: towards novel cytoprotection and radio-sensitization policies? Autophagy. 2009;5(4):442–50.
Garbar C, et al. Chemotherapy treatment induces an increase of autophagy in the luminal breast cancer cell MCF7, but not in the triple-negative MDA-MB231. Sci Rep. 2017;7(1):7201.
Sui X, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:e838.
Zhang J, et al. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy. 2015;11(4):629–42.
Kanzawa T, et al. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63(9):2103–8.
Kanzawa T, et al. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene. 2005;24(6):980–91.
Kanzawa T, et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11(4):448–57.
Ito H, et al. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int J Oncol. 2005;26(5):1401–10.
Paglin S, et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61(2):439–44.
Yao KC, et al. Molecular response of human glioblastoma multiforme cells to ionizing radiation: cell cycle arrest, modulation of the expression of cyclin-dependent kinase inhibitors, and autophagy. J Neurosurg. 2003;98(2):378–84.
Opipari AW Jr, et al. Resveratrol-induced autophagocytosis in ovarian cancer cells. Cancer Res. 2004;64(2):696–703.
Sivaprasad U, Basu A. Inhibition of ERK attenuates autophagy and potentiates tumour necrosis factor-alpha-induced cell death in MCF-7 cells. J Cell Mol Med. 2008;12(4):1265–71.
Li P, et al. Interferon-gamma induces autophagy with growth inhibition and cell death in human hepatocellular carcinoma (HCC) cells through interferon-regulatory factor-1 (IRF-1). Cancer Lett. 2012;314(2):213–22.
Ertmer A, et al. The anticancer drug imatinib induces cellular autophagy. Leukemia. 2007;21(5):936–42.
Takeuchi H, et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65(8):3336–46.
Graham CD, et al. Tamoxifen induces cytotoxic autophagy in Glioblastoma. J Neuropathol Exp Neurol. 2016;75(10):946–54.
Scarlatti F, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279(18):18384–91.
Kondo Y, et al. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5(9):726–34.
Acknowledgements
Not applicable.
Funding
The study was funded by the grants from the National Nature Science Foundation of China (81770952, 81873681), the Nature Science Foundation of Henan Province (162300410296), and the “23456” project of Henan Provincial People’s Hospital (ZC201600020).
Author information
Authors and Affiliations
Contributions
BM and SH carried out the design of this review. BM drafted the manuscript and prepared the Figs. BM and XL made substantial contributions to the conception. BM, XL, and SH collected the related references and revised the manuscript. BM and SH revised and improved the language. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, X., He, S. & Ma, B. Autophagy and autophagy-related proteins in cancer. Mol Cancer 19, 12 (2020). https://doi.org/10.1186/s12943-020-1138-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-1138-4