
Abstract
N6-methyladenosine (m6A) is methylation that occurs in the N6-position of adenosine, which is the most prevalent internal modification on eukaryotic mRNA. Accumulating evidence suggests that m6A modulates gene expression, thereby regulating cellular processes ranging from cell self-renewal, differentiation, invasion and apoptosis. M6A is installed by m6A methyltransferases, removed by m6A demethylases and recognized by reader proteins, which regulate of RNA metabolism including translation, splicing, export, degradation and microRNA processing. Alteration of m6A levels participates in cancer pathogenesis and development via regulating expression of tumor-related genes like BRD4, MYC, SOCS2 and EGFR. In this review, we elaborate on recent advances in research of m6A enzymes. We also highlight the underlying mechanism of m6A in cancer pathogenesis and progression. Finally, we review corresponding potential targets in cancer therapy.
Similar content being viewed by others
Background
Post-transcriptional modification has emerged as an important regulator of a variety of physiological processes and disease progression, attracting accumulating attention in bioscience research. Among numerous RNA modifications, N6-methyladenosine (m6A) is the most abundant mRNA modification. An average of 1000 nucleotides are found to contain 1–2 m6A residues [1, 2]. Mainly occurring in the RRACH sequence (where R = A or G, H = A, C, or U) [3, 4], m6A is enriched near the stop codon, 3′ untranslated region (UTR) and long internal exon [5, 6]. M6A can also be found in RNA of bacteria and viruses [7, 8].
M6A can be installed by the methyltransferase complex (MTC) and removed by demethylases [9, 10]. M6A alters target gene expression, thus influencing the corresponding cell processes and physiological function. In molecular mechanism, m6A participates in almost all steps of RNA metabolism including mRNA translation, degradation, splicing, export and folding [11, 12]. Roles of m6A in various cancers have been reported recently [87,88,]. In this review, we focus on the up-to-date progress in m6A enzymes. We describe functions of m6A in tumorigenesis and cancer progression. Finally, we discuss future research directions of m6A.
Regulators of m6A
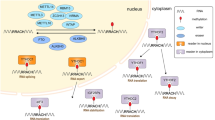
Regulators of m6A can be divided into 3 types: writers, erasers and readers. M6A is catalyzed by the methyltransferase complex (MTC), also called “writers”. Demethylase, also termed as “eraser”, removes m6A. RNA reader protein recognizes m6A, binds the RNA and implements corresponding functions (Table 1, Fig. 1). Crosslink among writers, erasers and readers, is involved in cancer pathogenesis and progression [35, 36].

Mechanism of m6A. The m6A methylation is catalyzed by the writer complex including METTL3, METTL14, WTAP, VIRMA, RBM15, and ZC3H13. The m6A modification is removed by demethylase FTO or ALKBH5. Reader proteins recognize m6A and determine target RNA fate.
Writers
M6A is installed co-transcriptionally through the methyltransferase complex (MTC) that consists of METTL3 catalytic subunit and other accessory subunits including METTL14, WTAP, VIRMA, RBM15, and ZC3H13 [37]. METTL14 forms a stable complex with METTL3 and plays a key role in substrate recognition [13, 14, 38]. Wilms Tumor 1 associated protein (WTAP) ensures the localization of the METTL3-METTL14 heterodimer to the nuclear speckle and promotes catalytic activity [16, 19]. RNA binding motif 15 (RBM15) binds the m6A complex and recruits it to special RNA site [17, 39]. ZC3H13 enhances m6A through bridging WTAP to the mRNA-binding factor Nito [20, 40]. VIRMA directs m6A in 3′ UTR and near stop codon by recruiting the MTC to modulate region-selective methylation [18]. In addition, METTL16 is a newly discovered writer that catalyzes m6A modification in U6-snRNA and participates in pre-RNA splicing [15].
METTL3
In glioblastoma (GBM), METTL3 exerts an oncogenic effect through modulating nonsense-mediated mRNA decay of splicing factors and alternative splicing isoform switches. Loss of METTL3 results in higher level of BCL-XS isoform and NCOR2α isoform and inhibition of GSC growth and self-renewal [41]. In gastric cancer, up-regulated METTL3 promotes stability of ZMYM1, thus enhancing EMT process in vitro and metastasis in vivo [42]. Moreover, METTL3 can also participate in regulation of target mRNA in a post-modification way, therefore partially acting as a reader [43, 44].
METTL3 modulates hematopoietic stem cells (HSC) self-renewal through enhancing expressions of self-renewal-related genes such as Nr4a2, p21, Bmi-1 and Prdm16 [45]. Depletion of METTL3 leads to a significant suppression of HSC reconstitution potential [46].
In human non-small cell lung carcinoma, METTL3, which undergoes sumoylation, has a reduced ability to catalyze m6A, resulting in enhanced tumorigenesis [47,48,49].
R-loops are three-stranded nucleic acid structures. M6A enhances co-transcriptional R-loops, impairing readthrough activity of Pol II for efficient termination [50].
METTL3 participates in neurogenesis and development through installing m6A in histone methyltransferase Ezh2 and enhancing expression of it [51].
METTL3 suppresses autophagic flux and enhances apoptosis in hypoxia/reoxygenation -treated cardiomyocytes by installing m6A in TFEB and inhibiting its expression, an essential regulator of lysosomal biogenesis and autophagy genes [52].
METTL3 facilitates the stability of viral RNA-dependent RNA polymerase 3D and boosts type 71 enterovirus replication by inducing sumoylation and ubiquitination of the polymerase [53].
METTL14
METTL14 is markedly elevated in Epstein–Barr virus (EBV) latently infected cells, promoting cell proliferation. Mechanistically, METTL14 contributes to oncogenesis through inducing m6A on the essential EBV latent antigen EBNA3C and thereby enhancing expression and stability of it. Interestingly, EBNA3C also increases expression and stability of METTL14 [54].
METTL14 participates in erythropoiesis by promoting the translation of related genes including those coding for SETD histone methyltransferases, ribosomal components, and polyA RNA binding proteins [55].
Histone H3 trimethylation at H3K36me3 (Lys36) mediates m6A deposition, which can be recognized and bound by METTL14. METTL14 promotes the binding of the MTC to adjacent RNA polymerase II, therefore transferring the MTC to actively transcribed nascent RNAs [56].
METTL14 mediates the self-renewal capabilities of HSCs through promoting the expression of self-renewal-related genes such as Bmi-1 and Prdm16 [45].
METTL14 regulates post-implantation embryonic development via promoting the conversion from naive state to primed state of the epiblast. Loss of METTL14 impairs the priming and further differentiation competence of embryonic stem cells [57]. Moreover, the role of METTL14 in regulation of cancer stem cell has been revealed [58].
METTL16
METTL16 binds a subset of mRNAs and methylates long noncoding RNA (lncRNA) and U6 small nuclear RNA (U6 snRNA) [59, 60]. The UACAGAGAA sequence is required for METTL16-mediated-methylation and the N-terminal module of METTL16 is essential for RNA binding [61, 62].
METTL16 regulates S-adenosylmethionine (SAM) homeostasis. SAM is an important methyl-group donor in DNA and histone methylation. MAT2A encodes the SAM synthetase. Under loss-of-SAM conditions, METTL16 induces the splicing of a retained intron, thus promoting expression of MAT2A and level of SAM [63].
METTL16 participates in catalyzing m6A in A43 of the U6 small nuclear RNA [15].
Erasers
Employing ferrous iron as cofactor and α-ketoglutarate as cosubstrate, eraser removes m6A, thereby functioning as demethylase [64].
FTO
FTO not only controls mRNA splicing by inhibiting SRSF2 binding at splice sites [65], but also regulates adipogenesis by increasing the relative expression of the pro-adipogenic RUNX1T1-S isoform [66]. FTO blocks YTHDF2-mediated mRNA degradation by reducing m6A levels of cyclin A2 and cyclin-dependent kinase 2, thereby promoting fat cell cycle progression and adipogenesis [67].
FTO regulates leukemogenesis by modulating proliferation, differentiation and apoptosis of acute myeloid leukemia (AML) cells [68].
In melanoma, FTO impairs IFNγ-induced killing in melanoma cells in vitro via up-regulating PD-1, CXCR4, and SOX10 through suppressing YTHDF2-mediated-degradation and inhibits response to anti-PD-1 blockade immunotherapy [69].
ALKBH5
In head and neck squamous cell carcinomas, ALKBH5 is up-regulated and promotes cell viability [70].
In epithelial ovarian cancer (EOC), up-regulated ALKBH5 impairs the autophagy and facilitates cell proliferation and invasion [71].
ALKBH5-directed m6A demethylation is involved in splicing and the production of longer 3′ -UTR mRNAs [72, 73].
Readers
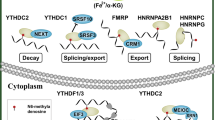
“Readers” recognize and bind m6A sites, leading to different destinies of target RNA [74, 75] (Fig. 2). In YTH domain family, the YT521-B homology (YTH) domain functions as the module for recognizing m6A [5, 76, 77]. YTHDF1 enhances mRNA translation and protein synthesis through interacting with initiation factors [30]. YTHDF2 induces degradation of the transcripts by selectively binding m6A-modified mRNA and recruiting them to mRNA decay sites [32]. YTHDF3 respectively enhances RNA translation via interacting with YTHDF1 and promotes RNA degradation by associating with YTHDF2 [33, 34]. YTHDC1 contributes to RNA splicing and export [27, 28]. YTHDC2 increases the translation efficiency of target RNA but reduces the abundance of them [29]. Recently, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) including IGF2BP1–3, are identified to bind m6A and function as readers. IGF2BPs promote RNA expression by enhancing RNA stability [78,79,80]. Eukaryotic initiation factor 3 (EIF3) facilitates cap-independent translation [23]. HNRNP (heterogeneous nuclear ribo nucleo protein) A2B1 mediates the alternative splicing of target RNAs and enhances primary miRNA processing by interacting with the microRNA microprocessor complex protein DGCR8 [24], while HNRNPC participates in pre-mRNA processing [25, 81,82,83,84].

Functions of readers on RNA. Readers participate in a variety of steps in RNA metabolism including translation, splicing, export, degradation and so on.
YTHDF1
Tumor neoantigens are involved in anti-tumor immune response and immunotherapy. YTHDF1 promotes lysosomal proteases-directed-degradation of neoantigens in dendritic cells by recognizing their m6A modification and enhancing their translation, thereby suppressing dendritic cells presenting tumor neoantigens to T cells and promoting tumor cells to evade immunosurveillance [85].
YTHDF2
YTHDF2 recruits the CCR4–NOT deadenylase complex by a direct association between the N-terminal region of YTHDF2 and the SH domain of the CNOT1 subunit, initiating deadenylation and decay of m6A-containing mRNAs [31]. Moreover, YTHDF2 mediates m6A methylated RNA endoribonucleolytic cleavage through forming a complex with HRSP12 and RNase P/MRP [86].
YTHDF2 interacts with miRNA [87]. In prostate cancer, YTHDF2 expression can be suppressed by miR-493-3p, therefore impairing cell proliferation and migration [88].
YTHDF2 affects the homeostasis of HSC. YTHDF2 deficiency prevents degradation of mRNAs of both survival-related genes and Wnt target genes during hematopoietic stresses, which synergistically enhances the regenerative capacity of HSCs [89].
M6A inhibits circular RNA (circRNA) immunity through functioning as a marker for “self”. Unmodified circRNA forms a complex with activated pattern recognization receptor RIG-I and K63-polyubiquitin, resulting in MAVS filamentation, IRF3 dimerization, and interferon production. YTHDF2 suppresses circRNA immunity by binding m6A-modificated circRNA, leading to the “self circRNA” being ignored and failure of RIG-I activation [90].
YTHDF2 recognizes and binds the nuclear receptor peroxisome proliferator-activator a (PPaRa), leading to decay of PPaRa [91].
YTHDF3
LncRNA GAS5 enhances YAP phosphorylation to increase ubiquitination and degradation of it, thereby impairing YAP signaling and inhibiting colorectal cancer (CRC) cell proliferation and metastasis. YTHDF3 recognizes m6A modified GAS5 and induces decay of it [92].
CircRNAs regulate numerous biological processes [93]. M6A participates in efficient initiation of protein translation from circRNAs in cells, requiring initiation factor eIF4G2 and YTHDF3 [94].
In seminoma, YTHDF3 and VIRMA are significantly overexpressed and there is a positive correlation between their expression [95].
YTHDC1
M6A level is higher in RNA X-inactive specific transcript (XIST). YTHDC1 recognizes m6A in XIST and regulates XIST function [17].
M6A of MAT2A mediated by METTL16 is read by YTHDC1. Inhibition of YTHDC1 and METTL16 damages SAM-responsive modulation of MAT2A [96].
YTHDC2
YTHDC2 is an RNA-induced ATPase with a 3′ -to-5′ RNA helicase activity [97]. Germ cells down-regulated YTHDC2 enter meiosis but process prematurely to abnormal metaphase and apoptosis [98, 99].
IGF2BPs
IGF2BPs enhance expression of target mRNA by promoting the stability of target mRNA through binding to mRNA stabilizers like ELAVL1 and MATR3 under normal conditions and by promoting the storage of target mRNA through translocating to stress granules under stress condition [26].
IGF2BP1 promotes tumor cell malignant phenotype by preferentially associating upstream of miRNA binding sites in the 3′ UTR of target mRNAs and then antagonizing miRNA-impaired gene expression [100]. Serum response factor (SRF) controls expression of oncogenes like FOXK1 and PDLIM7. IGF2BP1 can inhibit the microRNA-mediated degradation of SRF expression and thereby acting as a cancer promoter [78, 101]. In metastatic melanomas, expression of sequestosome 1/SQSTM1/p62 (p62) is higher, associated with poor patient prognosis. Mechanistically, p62 enhances stability of FERMT2 and other pro-metastatic factors through interacting with IGF2BP1 [102]. In HCC, TCAM1P-004 interacts with IGF2BP1, thereby suppressing IGF2 translation and enhancing DDIT3 expression, resulting in inhibition of cell proliferation and promotion of cell apoptosis [103].
In colorectal carcinoma, IGF2BP2 recognizes m6A in the coding sequence (CDS) regions of target gene SOX2 and prevents it degradation, contributing to colorectal cancer pathogenesis and progression [80].
IGF2BP3 shows oncogenic features and is markedly up-regulated in a variety of cancer types, associated with poor patient survival. IGF2BP3 functions as a potential oncogene across multiple cancer types [104]. In pancreatic cancer, the DNA methylation level of IGF2BP3 is significantly reduced and the expression IGF2BP3 is higher, associated with patient overall survival [105]. IGF2BP3 participates in the fetal–adult hematopoietic switch through interacting with RNA-binding protein Lin28b. In B-cell progenitors, Lin28b and IGF2BP3 promote stability of mRNAs such as B-cell regulators Pax5 and Arid3a [79].
EIF3
EIF3 is essential for specialized translation initiation through interacting with the 5′ cap, contributing to assembly of translation initiation complexes on eIF3-specialized mRNA [106].
EIF3 is highly expressed in colorectal cancer and contributes to mTOR signaling. EIF3 knockdown promotes autophagy [107].
In head and neck squamous cell carcinoma, DDX3 promotes the association of the cap-binding complex with mRNAs and enhances recruitment of EIF3, thereby contributing to the expression of metastatic-related gene expression [108].
Roles of m6A in cancer
Effect of m6A on cancer is reflected in the change of these tumor-related genes. Systematic study of the crosslink between substrate genes, m6A modification and post-modification regulation will contribute to reveal the mechanism of m6A in cancer comprehensively (Table 3) [116, 117]. Recently, many studies choose one or two of m6A regulators to explore its aberrant expression and underlying mechanism in cancer (Table 2). Here, we detail the role of m6A in cancer pathogenesis and progression (Fig. 3).

The role of m6A in cancers. The effect of m6A on cancer is reflected in the regulation of cancer-related gene expression. The m6A modification promotes cancer pathogenesis and progression through enhancing oncogene expression and inhibiting tumor suppressor gene expression. The m6A modification hampers cancer pathogenesis and progression through inhibiting oncogene expression and enhancing tumor suppressor gene expression.
m6A functions as a tumor promoter
m6A promotes expression of oncogenes
In bladder cancer, METTL3 and oncogene CDCP1 are up-regulated, correlating with bladder cancer progression status. METTL3 elevated m6A level of CDCP1, thus promoting its translation modulated by YTHDF1 [109]. AF4/FMR2 family member 4 (AFF4) is a direct upstream regulator of MYC and can enhance MYC expression. METTL3 promotes expressions of MYC and AFF4 [129]. Inhibition of METTL3 significantly hampered bladder tumor cell proliferation, migration, invasion, and survival in vitro and impairs cell proliferation in vivo [109, 129]. METTL3 promotes maturation of pri-miR221/222 [130].
In lung adenocarcinoma, the expression of METTL3 is elevated. METTL3 enhances translation of oncogene BRD4 through forming a mRNA loop with EIF3, thus facilitating translation of the oncogene by promoting ribosome recycling, indicating that METTL3 can not only catalyze m6A but also participate in the post-methylation regulation of target mRNA therefore functioning as a reader [43]. METTL3 can also promote translation of oncogenes such as EGFR and TAZ by associating with EIF3 [44]. METTL3 depletion leads to increased cell apoptosis and decreased cell growth and invasion, impairing tumorigenicity in mouse xenografts [43, 44].
In AML, METTL3 is elevated, facilitating translation of oncogenes like SP1 through relieving ribosome stalling. SP1 regulates expression of oncogene c-MYC [119]. METTL3 enhances translation of oncogenes such as c-MYC, BCL2 and PTEN by installing m6A [118]. METTL14 is highly expressed in AML, which promotes AML development and maintenance and self-renewal of leukemia stem/initiation cells via enhancing translation and inhibiting decay of MYB and MYC [58].
In ovarian cancer, IGF2BP1 is elevated and associated with a poor prognosis. SRF contributes to tumor cell proliferation and metastasis, and transcription of oncogenes like FOXK1 and PDLIM7 [131, 132]. IGF2BP1 enhances expression of SRF through inhibiting the miRNA-mediated degradation of it in an m6A-dependent way, resulting in increased expression of SRF, FOXK1 and PDLIM7 [78, 100]. METTL3 stimulates AXL mRNA translation and epithelial-mesenchymal transition, thereby promoting growth and invasion of ovarian tumors [126, 133].
In liver cancer, levels of METTL3 and YTHDF1 are higher, related with worse overall survival. M6A in CDS of Snail enhances Snail expression through YTHDF1-mediated translation, a key transcription factor of EMT. Low level of m6A due to loss of METTL3 hampers EMT of cancer cells [115, 134].
In breast cancer, METTL3 is elevated. HBXIP (hepatitis B X-interacting protein) enhances the malignant phenotypes of breast tumor. METTL3 promotes expression of HBXIP. Interestingly, HBXIP also facilitates METTL3 expression by inhibiting miRNA let-7 g, which reduces METTL3 expression through targeting its 3′ UTR [122].
In pancreatic tumors and in smokers, expression of miR-25-3p is elevated, associated with worse prognosis. Cigarette smoke condensate promotes m6A, thus contributing to maturation of oncogene miR-25-3p in pancreatic tumor. MiR-25-3p inhibits PH domain leucine-rich repeat protein phosphatase 2 (PHLPP2), leading to the activation of oncogenic AKT-p70S6K signaling [135].
In CRC, YTHDF1 is highly expressed. Knockdown of YTHDF1 suppresses the CRC cell tumorigenicity and colonosphere formation ability through impairing Wnt/β-catenin pathway activity [136]. Moreover, c-MYC enhances YTHDF1 expression [137]. METTL3 promotes expression of SRY (sex determining region Y)-box 2 (SOX2) through IGF2BP2-directed suppression of RNA degradation [80]. In CRC cells, overexpression of RP11 due to m6A-directed-nuclear accumulation promotes cell dissemination and induces EMT [138].
In nasopharyngeal carcinoma (NPC), the m6A level of oncogenic lncRNA FAM225A is elevated, resulting in enhanced stability and higher expression of FAM225A. FAM225A exerts a cancer-promoting role through functioning as a ceRNA and sponging miR-590-3p and miR-1275, causing higher expression of ITGB3 and activation of the FAK/PI3K/Akt pathway [139]. M6A regulates NPC cell proliferation [140, 141].
In cutaneous squamous cell carcinoma (cSCC), up-regulated METTL3 promotes △Np63 expression, thereby enhancing cSCC cell proliferation and tumor growth [142].
m6A inhibits expression of tumor suppressor genes
In human hepatocellular carcinoma (HCC), highly expressed METTL3 restrains expression of SOCS2 via YTHDF2-mediated degradation [143, 144]. Depletion of METTL3 results in a decrease in cell proliferation, colony formation, and migration in vitro and inhibits tumor cell proliferation and metastasis in vivo [113]. The expression of VIRMA is also higher in HCC tissue, leading to increased cell proliferation and invasion by enhancing m6A modification and inhibiting expression of ID2 mRNA [145]. WTAP is also elevated, resulting in increased cell proliferation and colony formation through facilitating m6A in ETS1 and causing epigenetic silencing of ETS1 [146].
In clear cell renal cell carcinoma (ccRCC), higher m6A level due to less FTO inhibits expression of PGC-1α through reducing its stability. Higher level of m6A and lower expression of PGC-1α results in enhanced tumor growth [128].
In pancreatic cancer, the expression of ALKBH5 is reduced, resulting in elevated m6A level and reduced expression of tumor suppressor gene KCNK15-AS1, leading to enhanced migration and invasion of pancreatic tumor cells [127].
m6A functions as a tumor suppressor
m6A inhibits expression of oncogenes
In endometrial tumors, the m6A level is lower which is caused by reduced METTL3 expression or METTL14 mutation. M6A methylation inhibits activation of the AKT pathway through YTHDF2-mediated degradation of the positive AKT regulator mTORC2. Attenuated m6A methylation leads to enhanced cell proliferation, colony formation, migration and invasion [110].
In HCC, m6A acts as a tumor suppressor through YTHDF2-directed degradation of EGFR by binding the m6A site in the 3′ UTR of this mRNA. YTHDF2 inhibits ERK/MAPK signaling cascades by destabilizing the EGFR mRNA [112].
In breast cancer, hypoxia facilitates expression of ALKBH5, resulting in reduction of NANOG mRNA m6A level and enhancement of mRNA stabilization. Elevated NANOG promotes breast cancer stem cells (BCSCs) enrichment [120].
In lung squamous cell carcinoma (LUSC), the expression of FTO is elevated. FTO facilitates oncogene MZF1 expression through inhibiting m6A methylation and increasing mRNA stabilization [22, 147].
In glioblastoma, elevated ALKBH5 reduces m6A methylation and facilitates expression of oncogene FOXM1, leading to enhancement of glioblastoma stem-like cells (GSCs) self-renewal and tumorigenicity. Furthermore, lncRNA plays a dynamic role in transcriptional and translational regulation of cancer [148]. Nuclear lncRNA FOXM1-AS promotes the association between ALKBH5 with FOXM1 nascent RNA, leading to enhanced FOXM1 expression and GSC pathogenesis [21].
m6A promotes expression of tumor suppressor genes
In endometrial tumors, m6A methylation inhibits activation of the AKT pathway through YTHDF1-mediated translation of the negative AKT regulator PHLPP2. Attenuated m6A methylation leads to enhanced cell proliferation, colony formation, migration and invasion [110].
In breast cancer, the expression of FTO is higher. FTO promoted breast cancer cell proliferation, colony formation and metastasis by reducing BNIP3 methylation and promoting BNIP3 degradation [121]. Premature polyadenylation (pPA) of tumor suppressor genes frequently truncates these genes, leading to inhibition of their functions. These genes such as MAGI3 undergo pPA at the intron downstream of its long internal exon, which is the m6A site. In breast cancer cells with enhanced MAGI3 pPA, m6A modification in this site is obviously diminished [149].
In HCC, Jin-zhao Ma and colleagues have found that METTL14 positively manipulates primary microRNA126 processing via an m6A-dependent manner, which impairs the metastatic potential of HCC. Overexpression of METTL14 inhibits the migration and invasiveness of HepG2 cells in vitro and restrains cell proliferation and metastasis in vivo. METTL14 not only enhances the recognition and binding of microprocessor protein DGCR8 to pri-mi126, but also promotes the subsequent processing to mature miRNA [125].
The dual role of m6A in cancer
Accumulating evidence shows that m6A plays a dual role in cancer [150, 151]. On the one hand, m6A regulates expression of oncogenes or tumor suppressor genes, thus affecting cancer progression. On the other hand, the m6A level and m6A enzymes expression and activity can be modulated, thereby influencingz the role of m6A in cancer.
m6A affects cancer progression by modulating target gene
Based on current researches, how m6A influences cancer progression via regulating target genes depends on three elements: (1). Whether the target gene is an oncogene or a tumor suppressor gene (Fig. 4); (2). The aberrant m6A level in cancer (which is decided by the change of expression or activity of writer or eraser); (3). The post-modification regulation on target mRNA (which is decided by readers, both the reader’ s corresponding role to RNA and the change of reader expression and function contribute to this element). The final effect of readers on target mRNA can be divided into two types: positive-reader-role and negative-reader-role. The former is to promote the expression of RNA, while the latter is to inhibit the expression of RNA. More detailed, the positive-reader-role is shown in: 1). Reader promotes target gene translation, splicing, export or stabilization and so on, while the expression of reader is up-regulated or unchanged; 2). Reader promotes the degradation of target mRNA, while the expression of reader is down-regulated. The negative-reader-role is the opposite. For instance, in colorectal carcinoma, the expression of METTL3 is elevated, resulting in higher m6A level in oncogene SOX2. IGF2BP2 is up-regulated and enhances target gene stability, thereby exert a positive-reader-role. Therefore, SOX2 expression is facilitated and m6A plays a cancer-promoting role [80]. In endometrial cancer, the m6A level is lower, leading to inhibited YTHDF1-mediated-tranlation of tumor suppressor gene PHLPP2 and impaired YTHDF2-directed-degradation to oncogene mTORC2, therefore suggesting the cancer-suppressing role of m6A [110].

The total effect of m6A on cancer. The total effect of m6A on cancer depends roughly on three elements: (1). Whether the target RNA works as a tumor promoter or a tumor suppressor; (2). The change of m6A level in tumor cells; (3). The post-methylation regulation on target mRNA.
Factors affecting m6A modification and m6A enzymes in cancer
In HCC, miR-145 up-regulates m6A level by targeting 3′ UTR of YTHDF2, leading to suppression of cell proliferation [152].
In non-small-cell lung carcinoma (NSCLC), miR-33a decreases expression of METTL3, thereby attenuating expression of target genes EGFR, TAZ and DNMT3A and suppressing NSCLC cell proliferation [153].
In lung cancer, sumoylation reduces activity of METTL3 catalyzing m6A, resulting in enhanced tumorigenesis [49].
In gastric cancer, up-regulated METTL3 enhances m6A methylation in HDGF and promotes the IGF2BP3-directed-stability of it, therefore facilitating cell proliferation and metastasis in vitro and in vivo. P300-mediated H3K27 acetylation activation in METTL3 promoter promotes transcription of it [111].
In ccRCC, methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is overexpressed. MTHFD2 enhances m6A modification of HIF-2α and promotes translation of it, thereby resulting in malignant phenotypes [154].
In HCC, high level of nicotinamide N-methyltransferase (NNMT) contributes to vascular invasion and distant metastasis. NNMT inhibits m6A methylation, therefore enhancing CD44v3 formation. NNMT-modulated CD44 m6A methylation improves the RNA stability by impairing ubiquitin-mediated degradation [155].
Therapeutic strategy based on m6A
Research aimed at uncovering the mechanism has shown that m6A serves as a regulator in cancer [156, 157].
Meclofenamic acid (MA) is one of selective FTO inhibitors by competing with FTO binding sites. As the ethyl ester derivative of MA, MA2 suppresses GSC growth and self-renewal in vitro and inhibits tumor growth in vivo [158, 159].
As another inhibitor of FTO, FB23–2 impairs proliferation and enhances differentiation of AML cells [68].
As a metabolite by mutant IDH1/2 enzymes, R-2-hydroxyglutarate (R-2HG) elevates m6A level and enhances the YTHDF2-mediated-degradation of MYC, ASB2, and RARA [160].
In CRC, carbonic anhydrase IV suppresses Wnt signaling pathway through targeting the WTAP–WT1–TBL1 axis [161].
In epithelial ovarian cancer, elevated m6A level contributes to resistance to Poly (ADP-ribose) polymerase inhibitors (PARPi) through up-regulating Wnt signaling pathway by enhancing stability of FZD10. Restraining Wnt signaling pathway in combination with PARPi therefore may be a potential therapeutic strategy for EOC [162].
In glioblastoma, up-regulated METTL3 promotes GSCs maintenance and glioblastoma progression. Loss of METTL3 results in increased GSCs sensitivity to γ-irradiation [124].
In leukemia, rapidly acquired tolerance to tyrosine kinase inhibitors (TKIs) destroys effective therapy, which proves to be driven by overexpression of FTO. The FTO-mediated demethylation promotes stability of proliferation-related genes [114] Table 3.
In cervical squamous cell carcinoma (CSCC), FTO enhances the chemo-radiotherapy resistance by up-regulating β-catenin via reducing m6A level [123].
Conclusion
Epigenetics has become a hot topic in scientific research nowadays [37, 163]. The m6A methylation participates in regulation of cancer malignant phenotype by controlling the expression of cancer-related genes [164,165,166]. Abnormal level of m6A methylation contributes to tumor pathogenesis and progression. Although m6A has been the focus of many studies in recent years [167, 168], our knowledge about it is far from complete. How expression and activity of readers are regulated in cancer still remains a mistery. The mechanism that readers coordinate their functions since they all bind to m6A but with different functions needs being further elucidated. Little is known about whether there is RNA sequence specificity for readers.
Although m6A enzyme inhibitors have shown tumor-regulating roles in a variety of cancers, more efficacy drugs and novel therapeutic strategies related to m6A are expected to be explored.
Availability of data and materials
Not applicable.
Abbreviations
- AFF4:
-
AF4/FMR2 family member 4
- ALKBH5:
-
Alkb homologue 5
- AML:
-
Acute myeloid leukemia
- BCSC:
-
Breast cancer stem cell
- ccRCC:
-
Clear cell renal cell carcinoma
- CEBPZ:
-
CCAAT-box binding factor
- CircRNA:
-
Circular RNA
- CSCC:
-
Cervical squamous cell carcinoma
- ECM:
-
Extracellular matrix
- EIF3:
-
Eukaryotic initiation factor 3
- EOC:
-
Epithelial ovarian cancer
- FTO:
-
Fat mass and obesity-associated protein
- GSC:
-
Glioblastoma stem-like cell
- HCC:
-
Human hepatocellular carcinoma
- HNRNP:
-
Heterogeneous nuclear ribo nucleo protein
- HSC:
-
Hematopoietic stem cells
- IGF2BPs:
-
Insulin-like growth factor 2 mRNA-binding proteins
- LUSC:
-
Lung squamous cell carcinoma
- M6A:
-
N6-methyladenosine
- METTL3:
-
Methyltransferase-like 3
- MTC:
-
Methyltransferase complex
- NPC:
-
Nasopharyngeal carcinoma
- PARP:
-
Poly (ADP-ribose) polymerase inhibitors
- PHLPP2:
-
PH domain leucine-rich repeat protein phosphatase 2
- pPA:
-
Premature polyadenylation
- PPaRa:
-
Peroxisome proliferator-activator a
- RBM15:
-
RNA binding motif 15
- SAM:
-
S-adenosylmethionine
- SOX2:
-
SRY (sex determining region Y)-box 2
- SUMO:
-
Small ubiquitin-like modification
- TKI:
-
Tyrosine kinase inhibitors
- U6 snRNA:
-
U6 small nuclear RNA
- UTR:
-
Untranslated region
- WTAP:
-
Wilms Tumor 1 associated protein
- XIST:
-
X-inactive specific transcript
- YTHDF:
-
YT521-B homology domain-containing protein family
References
Krug RM, Morgan MA, Shatkin AJ. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J Virol. 1976;20:45–53.
Beemon K, Keith J. Localization of N6-methyladenosine in the Rous sarcoma virus genome. J Mol Biol. 1977;113:165–79.
Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. Rna. 1997;3:1233–47.
Wei CM, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16:1672–6.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–46.
Deng X, Chen K, Luo GZ, Weng X, Ji Q, Zhou T, He C. Widespread occurrence of N6-methyladenosine in bacterial mRNA. Nucleic Acids Res. 2015;43:6557–67.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proc Natl Acad Sci U S A. 1974;71:3971–5.
Dai D, Wang H, Zhu L, Jin H, Wang X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 2018;9:124.
Batista PJ. The RNA Modification N (6)-methyladenosine and Its Implications in Human Disease. Genomics Proteomics Bioinform. 2017;15:154–63.
Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucleic Acids Res. 2019.
Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, Pan T. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45:6051–63.
Wang P, Doxtader KA, Nam Y. Structural Basis for Cooperative Function of Mettl3 and Mettl14 Methyltransferases. Mol Cell. 2016;63:306–17.
Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. Corrigendum: Structural basis of N (6)-adenosine methylation by the METTL3-METTL14 complex. Nature. 2017;542:260.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C, Sloan KE, Bohnsack MT. Human METTL16 is a N (6)-methyladenosine (m (6) A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 2017;18:2004–14.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–96.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M. Jaffrey SR: m (6) A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, Cheng T, Gao M, Shu X, Ma H, et al. VIRMA mediates preferential m (6) A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4:10.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, Masiello I, Hares T, Villasenor R, Hess D, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m (6) A machinery component Wtap/Fl (2) d. Genes Dev. 2018;32:415–29.
Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, Chen Y, Sulman EP, Xie K, Bogler O, et al. m (6) A Demethylase ALKBH5 Maintains Tumorigenicity of Glioblastoma Stem-like Cells by Sustaining FOXM1 Expression and Cell Proliferation Program. Cancer Cell. 2017;31:591–606.e596.
Liu J, Ren D, Du Z, Wang H, Zhang H. Jin Y: m (6) A demethylase FTO facilitates tumor progression in lung squamous cell carcinoma by regulating MZF1 expression. Biochem Biophys Res Commun. 2018;502:456–64.
Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m (6) A Promotes Cap-Independent Translation. Cell. 2015;163:999–1010.
Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 Is a Mediator of m (6) A-Dependent Nuclear RNA Processing Events. Cell. 2015;162:1299–308.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N (6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–4.
Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, Zhao BS, Mesquita A, Liu C, Yuan CL, et al. Recognition of RNA N (6)-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. 2018;20:285–95.
Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, Sha J, Huang X, Guerrero L, Xie P, et al. YTHDC1 mediates nuclear export of N (6)-methyladenosine methylated mRNAs. Elife. 2017;6.
Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, Gregory BD, Schultz RM, Wang PJ. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 2018;14:e1007412.
Hsu PJ, Zhu Y, Ma H, Guo Y, Shi X, Liu Y, Qi M, Lu Z, Shi H, Wang J, et al. Ythdc2 is an N (6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27:1115–27.
Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. N (6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–99.
Du H, Zhao Y, He J, Zhang Y, Xi H, Liu M, Ma J, Wu L. YTHDF2 destabilizes m (6) A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C. YTHDF3 facilitates translation and decay of N (6)-methyladenosine-modified RNA. Cell Res. 2017;27:315–28.
Li A, Chen YS, Ping XL, Yang X, Xiao W, Yang Y, Sun HY, Zhu Q, Baidya P, Wang X, et al: Cytoplasmic m (6) A reader YTHDF3 promotes mRNA translation. In Cell Res. Volume 27. England2017: 444–447.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N (6)-methyladenosine modification in cancers: current status and perspectives. Cell Res. 2018;28:507–17.
Panneerdoss S, Eedunuri VK, Yadav P, Timilsina S, Rajamanickam S, Viswanadhapalli S, Abdelfattah N, Onyeagucha BC, Cui X, Lai Z, et al: Cross-talk among writers, readers, and erasers of m (6) A regulates cancer growth and progression. Sci Adv 2018, 4:eaar8263.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA Modifications in Gene Expression Regulation. Cell. 2017;169:1187–200.
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–5.
Zhang S. Mechanism of N (6)-methyladenosine modification and its emerging role in cancer. Pharmacol Ther. 2018;189:173–83.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, Jiao F, Liu H, Yang P, Tan L, et al: Zc3h13 Regulates Nuclear RNA m (6) A Methylation and Mouse Embryonic Stem Cell Self-Renewal. Mol Cell 2018, 69:1028–1038.e1026.
Li F, Yi Y, Miao Y, Long W, Long T, Chen S, Cheng W, Zou C, Zheng Y, Wu X, et al. N6-methyladenosine Modulates Nonsense-mediated mRNA Decay in Human Glioblastoma. Cancer Res. 2019.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, Zhao G. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. Mrna circularization by mettl3-eif3h enhances translation and promotes oncogenesis. Nature. 2018;561:556–60.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m (6) A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol Cell. 2016;62:335–45.
Yao QJ, Sang L, Lin M, Yin X, Dong W, Gong Y, Zhou BO: Mettl3-Mettl14 methyltransferase complex regulates the quiescence of adult hematopoietic stem cells. Cell Res. 28. England2018: 952–954.
Lee H, Bao S, Qian Y, Geula S, Leslie J, Zhang C, Hanna JH, Ding L. Stage-specific requirement for Mettl3-dependent m (6) A mRNA methylation during haematopoietic stem cell differentiation. Nat Cell Biol. 2019;21:700–9.
Hay RT. SUMO: a history of modification. Mol Cell. 2005;18:1–12.
Gill G. SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev. 2004;18:2046–59.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, Li L, Chen R, Wang Y, Deng R, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic Acids Res. 2018;46:5195–208.
Yang X, Liu QL, Xu W, Zhang YC, Yang Y, Ju LF, Chen J, Chen YS, Li K. Ren J, et al: m (6) A promotes R-loop formation to facilitate transcription termination. Cell Res. 2019.
Chen J, Zhang YC, Huang C, Shen H, Sun B, Cheng X, Zhang YJ, Yang YG, Shu Q, Yang Y. Li X: m (6) A Regulates Neurogenesis and Neuronal Development by Modulating Histone Methyltransferase Ezh2. Genomics Proteomics Bioinformatics. 2019.
Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, Jin J, Ding X, Wu S, Huang H, et al. METTL3 and ALKBH5 oppositely regulate m (6) A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy. 2019:1–19.
Hao H, Hao S, Chen H, Chen Z, Zhang Y, Wang J, Wang H, Zhang B, Qiu J, Deng F, Guan W. N6-methyladenosine modification and METTL3 modulate enterovirus 71 replication. Nucleic Acids Res. 2019;47:362–74.
Lang F, Singh RK, Pei Y, Zhang S, Sun K, Robertson ES. EBV epitranscriptome reprogramming by METTL14 is critical for viral-associated tumorigenesis. PLoS Pathog. 2019;15:e1007796.
Kuppers DA, Arora S, Lim Y, Lim AR, Carter LM, Corrin PD, Plaisier CL, Basom R, Delrow JJ, Wang S, et al. N (6)-methyladenosine mRNA marking promotes selective translation of regulons required for human erythropoiesis. Nat Commun. 2019;10:4596.
Huang H, Weng H, Zhou K, Wu T, Zhao BS, Sun M, Chen Z, Deng X, Xiao G, Auer F, et al. Histone H3 trimethylation at lysine 36 guides m (6) A RNA modification co-transcriptionally. Nature. 2019;567:414–9.
Meng TG, Lu X, Guo L, Hou GM, Ma XS, Li QN, Huang L, Fan LH, Zhao ZH, Ou XH, et al. Mettl14 is required for mouse postimplantation development by facilitating epiblast maturation. FASEB J. 2019;33:1179–87.
Weng H, Huang H, Wu H, Qin X, Zhao BS, Dong L, Shi H, Skibbe J, Shen C, Hu C, et al: METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via mRNA m (6) A Modification. Cell Stem Cell 2018, 22:191–205.e199.
Fitzsimmons CM, Batista PJ. It's complicated… m (6) A-dependent regulation of gene expression in cancer. Biochim Biophys Acta Gene Regul Mech. 1862;2019:382–93.
Brown JA, Kinzig CG, DeGregorio SJ, Steitz JA. Methyltransferase-like protein 16 binds the 3′-terminal triple helix of MALAT1 long noncoding RNA. Proc Natl Acad Sci U S A. 2016;113:14013–8.
Doxtader KA, Wang P, Scarborough AM, Seo D, Conrad NK, Nam Y: Structural Basis for Regulation of METTL16, an S-Adenosylmethionine Homeostasis Factor. Mol Cell 2018, 71:1001–1011.e1004.
Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, McCarthy AA, Pillai RS: Methylation of Structured RNA by the m (6) A Writer METTL16 Is Essential for Mouse Embryonic Development. Mol Cell 2018, 71:986–1000.e1011.
Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, Tu BP, Conrad NK: The U6 snRNA m (6) A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169:824–835.e814.
Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM: The AlkB Family of Fe (II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond*. In J Biol Chem. Volume 2902015: 20734–20742.
Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, Hao YJ, Ping XL, Chen YS, Wang WJ, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014;24:1403–19.
Merkestein M, Laber S, McMurray F, Andrew D, Sachse G, Sanderson J, Li M, Usher S, Sellayah D, Ashcroft FM, Cox RD. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun. 2015;6:6792.
Wu R, Liu Y, Yao Y, Zhao Y, Bi Z, Jiang Q, Liu Q, Cai M, Wang F, Wang Y, Wang X. FTO regulates adipogenesis by controlling cell cycle progression via m (6) A-YTHDF2 dependent mechanism. Biochim Biophys Acta Mol Cell Biol Lipids. 1863;2018:1323–30.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, Ni T, Zhang ZS, Zhang T, Li C, et al: Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35:677–691.e610.
Yang S, Wei J, Cui YH, Park G, Shah P, Deng Y, Aplin AE, Lu Z, Hwang S, He C. He YY: m (6) A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat Commun. 2019;10:2782.
Pilzys T, Marcinkowski M, Kukwa W, Garbicz D, Dylewska M, Ferenc K, Mieczkowski A, Kukwa A, Migacz E, Wolosz D, et al. ALKBH overexpression in head and neck cancer: potential target for novel anticancer therapy. Sci Rep. 2019;9:13249.
Zhu H, Gan X, Jiang X, Diao S, Wu H, Hu J. ALKBH5 in BCL-2hibited autophagy of epithelial ovarian cancer through miR-7 and BCL-2. J Exp Clin Cancer Res. 2019;38:163.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, Zheng H, Klungland A, Yan W. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115:E325–e333.
Chen XY, Zhang J, Zhu JS. The role of m (6) A RNA methylation in human cancer. Mol Cancer. 2019;18:103.
Casella G, Tsitsipatis D, Abdelmohsen K, Gorospe M: mRNA methylation in cell senescence. Wiley Interdiscip Rev RNA 2019:e1547.
Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. m6A-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20.
Xu C, Wang X, Liu K, Roundtree IA, Tempel W, Li Y, Lu Z, He C, Min J. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–9.
Muller S, Glass M, Singh AK, Haase J, Bley N, Fuchs T, Lederer M, Dahl A, Huang H, Chen J, et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic Acids Res. 2019;47:375–90.
Wang S, Chim B, Su Y, Khil P, Wong M, Wang X, Foroushani A, Smith PT, Liu X, Li R, et al. Enhancement of LIN28B-induced hematopoietic reprogramming by IGF2BP3. Genes Dev. 2019;33:1048–68.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, Chen ZH, Zeng ZL, Wang F, Zheng J, et al. METTL3 facilitates tumor progression via an m (6) A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18:112.
Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–15.
McCloskey A, Taniguchi I, Shinmyozu K. Ohno M: hnRNP C tetramer measures RNA length to classify RNA polymerase II transcripts for export. Science. 2012;335:1643–6.
Cienikova Z, Damberger FF, Hall J, Allain FH, Maris C. Structural and mechanistic insights into poly (uridine) tract recognition by the hnRNP C RNA recognition motif. J Am Chem Soc. 2014;136:14536–44.
Zarnack K, Konig J, Tajnik M, Martincorena I, Eustermann S, Stevant I, Reyes A, Anders S, Luscombe NM, Ule J. Direct competition between hnRNP C and U2AF65 protects the transcriptome from the exonization of Alu elements. Cell. 2013;152:453–66.
Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, Huang X, Wang J, Dougherty U, Bissonnette MB, et al: Author Correction: Anti-tumour immunity controlled through mRNA m (6) A methylation and YTHDF1 in dendritic cells. In Nature. Volume 568. England2019: E3.
Park OH, Ha H, Lee Y, Boo SH, Kwon DH, Song HK, Kim YK: Endoribonucleolytic Cleavage of m (6) A-Containing RNAs by RNase P/MRP Complex. Mol Cell 2019, 74:494–507.e498.
Xie B, Deng Z, Pan Y, Fu C, Fan S, Tao Y, Zhou J, Xiao D. Post-transcriptional regulation DPC4 gene by miR-190 in colorectal cancer cells. J Cancer Res Ther. 2018;14:838–43.
Li J, Meng S, Xu M, Wang S, He L, Xu X, Wang X, Xie L. Downregulation of N (6)-methyladenosine binding YTHDF2 protein mediated by miR-493-3p suppresses prostate cancer by elevating N (6)-methyladenosine levels. Oncotarget. 2018;9:3752–64.
Wang H, Zuo H, Liu J, Wen F, Gao Y, Zhu X, Liu B, Xiao F, Wang W, Huang G, et al: Loss of YTHDF2-mediated m (6) A-dependent mRNA clearance facilitates hematopoietic stem cell regeneration. In Cell Res. Volume 28. England2018: 1035–1038.
Chen YG, Chen R, Ahmad S, Verma R, Kasturi SP, Amaya L, Broughton JP, Kim J, Cadena C, Pulendran B, et al: N6-Methyladenosine Modification Controls Circular RNA Immunity. Mol Cell 2019, 76:96–109.e109.
Zhong X, Yu J, Frazier K, Weng X, Li Y, Cham CM, Dolan K, Zhu X, Hubert N, Tao Y, et al: Circadian Clock Regulation of Hepatic Lipid Metabolism by Modulation of m (6) A mRNA Methylation. Cell Rep 2018, 25:1816–1828.e1814.
Ni W, Yao S, Zhou Y, Liu Y, Huang P, Zhou A, Liu J, Che L, Li J. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m (6) A reader YTHDF3. Mol Cancer. 2019;18:143.
Zhong Y, Du Y, Yang X, Mo Y, Fan C, Xiong F, Ren D, Ye X, Li C, Wang Y, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer. 2018;17:79.
Yang Y, Fan X, Mao M, Song X, Wu P, Zhang Y, Jin Y, Chen LL, Wang Y, Wong CC, et al. Extensive translation of circular RNAs driven by N (6)-methyladenosine. Cell Res. 2017;27:626–41.
Lobo J, Costa AL, Cantante M, Guimaraes R, Lopes P, Antunes L, Braga I, Oliveira J, Pelizzola M, Henrique R. Jeronimo C: m (6) A RNA modification and its writer/reader VIRMA/YTHDF3 in testicular germ cell tumors: a role in seminoma phenotype maintenance. J Transl Med. 2019;17:79.
Shima H, Matsumoto M, Ishigami Y, Ebina M, Muto A, Sato Y, Kumagai S, Ochiai K, Suzuki T, Igarashi K. S-Adenosylmethionine Synthesis Is Regulated by Selective N (6)-Adenosine Methylation and mRNA Degradation Involving METTL16 and YTHDC1. Cell Rep. 2017;21:3354–63.
Wojtas MN, Pandey RR, Mendel M, Homolka D, Sachidanandam R, Pillai RS: Regulation of m (6) A Transcripts by the 3′-->5′ RNA Helicase YTHDC2 Is Essential for a Successful Meiotic Program in the Mammalian Germline. Mol Cell 2017, 68:374–387.e312.
Jain D, Puno MR, Meydan C, Lailler N, Mason CE, Lima CD, Anderson KV. Keeney S: ketu mutant mice uncover an essential meiotic function for the ancient RNA helicase YTHDC2. Elife. 2018;7.
Bailey AS, Batista PJ, Gold RS, Chen YG, de Rooij DG, Chang HY, Fuller MT. The conserved RNA helicase YTHDC2 regulates the transition from proliferation to differentiation in the germline. Elife. 2017;6.
Muller S, Bley N, Glass M, Busch B, Rousseau V, Misiak D, Fuchs T, Lederer M, Huttelmaier S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic Acids Res. 2018;46:6285–303.
Busch B, Bley N, Muller S, Glass M, Misiak D, Lederer M, Vetter M, Strauss HG, Thomssen C, Huttelmaier S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic Acids Res. 2016;44:3845–64.
Karras P, Riveiro-Falkenbach E, Canon E, Tejedo C, Calvo TG, Martinez-Herranz R, Alonso-Curbelo D, Cifdaloz M, Perez-Guijarro E, Gomez-Lopez G, et al. p62/SQSTM1 Fuels Melanoma Progression by Opposing mRNA Decay of a Selective Set of Pro-metastatic Factors. Cancer Cell. 2019;35:46–63.e10.
Xu F, Li CH, Wong CH, Chen GG, Lai PBS, Shao S, Chan SL, Chen Y. Genome-Wide Screening and Functional Analysis Identifies Tumor Suppressor Long Noncoding RNAs Epigenetically Silenced in Hepatocellular Carcinoma. Cancer Res. 2019;79:1305–17.
Li Y, Xiao J, Bai J, Tian Y, Qu Y, Chen X, Wang Q, Li X, Zhang Y, Xu J: Molecular characterization and clinical relevance of m (6) A regulators across 33 cancer types. In Mol Cancer. Volume 18. England2019: 137.
Chen H, Kong Y, Yao Q, Zhang X, Fu Y, Li J, Liu C, Wang Z: Three hypomethylated genes were associated with poor overall survival in pancreatic cancer patients. Aging (Albany NY) 2019, 11:885–897.
Lee AS, Kranzusch PJ, Doudna JA, Cate JH. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature. 2016;536:96–9.
Yang C, Zhang Y, Du W, Cheng H, Li C. Eukaryotic translation initiation factor 3 subunit G promotes human colorectal cancer. Am J Transl Res. 2019;11:612–23.
Chen HH, Yu HI, Yang MH, Tarn WY. DDX3 Activates CBC-eIF3-Mediated Translation of uORF-Containing Oncogenic mRNAs to Promote Metastasis in HNSCC. Cancer Res. 2018;78:4512–23.
Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, Zhou Z, Yuan Z, Su J, Wu B, et al. Dynamic m (6) A mRNA methylation reveals the role of METTL3-m (6) A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38:4755–72.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, Tienda SM, Chryplewicz A, Zhu AC. Yang Y, et al: m (6) A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20:1074–83.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, Jiang Z, Zhang Y, Xu G, Zhang J, et al. METTL3-mediated m (6) A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2019.
Zhong L, Liao D, Zhang M, Zeng C, Li X, Zhang R, Ma H, Kang T. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett. 2019;442:252–61.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, Tsang LH, Ho DW, Chiu DK, Lee JM, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–70.
Yan F, Al-Kali A, Zhang Z, Liu J, Pang J, Zhao N, He C, Litzow MR, Liu S. A dynamic N (6)-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018;28:1062–76.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, Luo G, Tauler J, Du J, Lin S, et al. RNA m (6) A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nat Commun. 2019;10:2065.
Luo J, Liu H, Luan S, He C, Li Z. Aberrant Regulation of mRNA m (6) A Modification in Cancer Development. Int J Mol Sci. 2018;19.
Chang G, Leu JS, Ma L, Xie K, Huang S. Methylation of RNA N (6)-methyladenosine in modulation of cytokine responses and tumorigenesis. Cytokine. 2018.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, Chou T, Chow A, Saletore Y, MacKay M, et al. The N (6)-methyladenosine (m (6) A)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23:1369–76.
Barbieri I, Tzelepis K, Pandolfini L, Shi J, Millan-Zambrano G, Robson SC, Aspris D, Migliori V, Bannister AJ, Han N, et al. Promoter-bound METTL3 maintains myeloid leukaemia by m (6) A-dependent translation control. Nature. 2017;552:126–31.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, He X, Semenza GL. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m (6) A-demethylation of NANOG mRNA. Proc Natl Acad Sci U S A. 2016;113:E2047–56.
Niu Y, Lin Z, Wan A, Chen H, Liang H, Sun L, Wang Y, Li X, Xiong XF, Wei B, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer. 2019;18:46.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, Liu Y, Zhang X, Zhang W, Ye L. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, Zhe H. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, Somasundaram K. Essential role of METTL3-mediated m (6) A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Ma JZ, Yang F, Zhou CC, Liu F, Yuan JH, Wang F, Wang TT, Xu QG, Zhou WP, Sun SH. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N (6) -methyladenosine-dependent primary MicroRNA processing. Hepatology. 2017;65:529–43.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, Duan P. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151:356–65.
He Y, Hu H, Wang Y, Yuan H, Lu Z, Wu P, Liu D, Tian L, Yin J, Jiang K, Miao Y. ALKBH5 Inhibits Pancreatic Cancer Motility by Decreasing Long Non-Coding RNA KCNK15-AS1 Methylation. Cell Physiol Biochem. 2018;48:838–46.
Zhuang C, Luo X, Huang X, Yao L, Li J, Li Y, Xiong T, Ye J, Zhang F, Gui Y. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1alpha signalling axis. J Cell Mol Med. 2019;23:2163–73.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, Liang Y, Zhu F, Zhang Y, Zhang X, et al. The m (6) A methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38:3667–80.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, Yuan WB, Lu JC, Zhou ZJ, Lu Q, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18:110.
Ro S. Multi-phenotypic Role of Serum Response Factor in the Gastrointestinal System. J Neurogastroenterol Motil. 2016;22:193–200.
Medjkane S, Perez-Sanchez C, Gaggioli C, Sahai E, Treisman R. Myocardin-related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol. 2009;11:257–68.
Antony J, Tan TZ, Kelly Z, Low J, Choolani M, Recchi C, Gabra H, Thiery JP, Huang RY: The GAS6-AXL signaling network is a mesenchymal (Mes) molecular subtype-specific therapeutic target for ovarian cancer. Sci Signal 2016, 9:ra97.
Zhou Y, Yin Z, Hou B, Yu M, Chen R, Jin H, Jian Z. Expression profiles and prognostic significance of RNA N6-methyladenosine-related genes in patients with hepatocellular carcinoma: evidence from independent datasets. Cancer Manag Res. 2019;11:3921–31.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, Li S, Tan L, Mai D, Li G, et al. Excessive miR-25-3p maturation via N (6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10:1858.
Bai Y, Yang C, Wu R, Huang L, Song S, Li W, Yan P, Lin C, Li D, Zhang Y. YTHDF1 Regulates Tumorigenicity and Cancer Stem Cell-Like Activity in Human Colorectal Carcinoma. Front Oncol. 2019;9:332.
Nishizawa Y, Konno M, Asai A, Koseki J, Kawamoto K, Miyoshi N, Takahashi H, Nishida N, Haraguchi N, Sakai D, et al. Oncogene c-Myc promotes epitranscriptome m (6) A reader YTHDF1 expression in colorectal cancer. Oncotarget. 2018;9:7476–86.
Wu Y, Yang X, Chen Z, Tian L, Jiang G, Chen F, Li J, An P, Lu L. Luo N, et al: m (6) A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18:87.
Zheng ZQ, Li ZX, Zhou GQ, Lin L, Zhang LL, Lv JW, Huang XD, Liu RQ, Chen F, He XJ, et al. Long Noncoding RNA FAM225A Promotes Nasopharyngeal Carcinoma Tumorigenesis and Metastasis by Acting as ceRNA to Sponge miR-590-3p/miR-1275 and Upregulate ITGB3. Cancer Res. 2019;79:4612–26.
Lai W, Jia J, Yan B, Jiang Y, Shi Y, Chen L, Mao C, Liu X, Tang H, Gao M, et al. Baicalin hydrate inhibits cancer progression in nasopharyngeal carcinoma by affecting genome instability and splicing. Oncotarget. 2018;9:901–14.
Wang H, Zhou Y, Oyang L, Han Y, Xia L, Lin J, Tang Y, Su M, Tan S, Tian Y, et al. LPLUNC1 stabilises PHB1 by counteracting TRIM21-mediated ubiquitination to inhibit NF-kappaB activity in nasopharyngeal carcinoma. Oncogene. 2019;38:5062–75.
Zhou R, Gao Y, Lv D, Wang C, Wang D, Li Q. METTL3 mediated m (6) A modification plays an oncogenic role in cutaneous squamous cell carcinoma by regulating DeltaNp63. Biochem Biophys Res Commun. 2019;515:310–7.
Shi LP, Liang M, Li FF, Li T, Lai DH, Xie QL, Yin YF, Liu YF. MiR-492 exerts tumor-promoting function in prostate cancer through repressing SOCS2 expression. Eur Rev Med Pharmacol Sci. 2019;23:992–1001.
Miyamoto A, Masuhiro Y, Seki T, Hanazawa S, Shiba H. A designed cell-penetrating human SOCS2 protein suppresses GH-dependent cancer cell proliferation. Biosci Biotechnol Biochem. 2019;83:300–8.
Cheng X, Li M, Rao X, Zhang W, Li X, Wang L, Huang G. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 2019;12:3421–8.
Chen Y, Peng C, Chen J, Chen D, Yang B, He B, Hu W, Zhang Y, Liu H, Dai L, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18:127.
Cayir A, Barrow TM, Guo L, Byun HM. Exposure to environmental toxicants reduces global N6-methyladenosine RNA methylation and alters expression of RNA methylation modulator genes. Environ Res. 2019;175:228–34.
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, Shi Y, Chen L, Xiao D, Yu F, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019.
Ni TK, Elman JS, Jin DX, Gupta PB, Kuperwasser C. Premature polyadenylation of MAGI3 is associated with diminished N (6)-methyladenosine in its large internal exon. Sci Rep. 2018;8:1415.
Wang S, Chai P, Jia R. Novel insights on m (6) A RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. 2018;17:101.
He L, Li J, Wang X, Ying Y, Xie H, Yan H, Zheng X, Xie L. The dual role of N6-methyladenosine modification of RNAs is involved in human cancers. J Cell Mol Med. 2018;22:4630–9.
Yang Z, Li J, Feng G, Gao S, Wang Y, Zhang S, Liu Y, Ye L, Li Y, Zhang X. MicroRNA-145 Modulates N (6)-Methyladenosine Levels by Targeting the 3′-Untranslated mRNA Region of the N (6)-Methyladenosine Binding YTH Domain Family 2 Protein. J Biol Chem. 2017;292:3614–23.
Du M, Zhang Y, Mao Y, Mou J, Zhao J, Xue Q, Wang D, Huang J, Gao S, Gao Y. MiR-33a suppresses proliferation of NSCLC cells via targeting METTL3 mRNA. Biochem Biophys Res Commun. 2017;482:582–9.
Green NH, Galvan DL, Badal SS, Chang BH, LeBleu VS, Long J, Jonasch E, Danesh FR. MTHFD2 links RNA methylation to metabolic reprogramming in renal cell carcinoma. Oncogene. 2019;38:6211–25.
Li J, You S, Zhang S, Hu Q, Wang F, Chi X, Zhao W, Xie C, Zhang C, Yu Y, et al. Elevated N-methyltransferase expression induced by hepatic stellate cells contributes to the metastasis of hepatocellular carcinoma via regulation of the CD44v3 isoform. Mol Oncol. 2019;13:1993–2009.
Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol. 2019;20:608–24.
Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The Critical Role of RNA m (6) A Methylation in Cancer. Cancer Res. 2019;79:1285–92.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, Gan J, Jiang H, Jia GF, Luo C, Yang CG. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, Lu Z, Huang Y, Yang CG. Riggs AD, et al: m (6) A RNA Methylation Regulates the Self-Renewal and Tumorigenesis of Glioblastoma Stem Cells. Cell Rep. 2017;18:2622–34.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al: R-2HG Exhibits Anti-tumor Activity by Targeting FTO/m (6) A/MYC/CEBPA Signaling. Cell 2018, 172:90–105.e123.
Zhang J, Tsoi H, Li X, Wang H, Gao J, Wang K, Go MY, Ng SC, Chan FK, Sung JJ, Yu J. Carbonic anhydrase IV inhibits colon cancer development by inhibiting the Wnt signalling pathway through targeting the WTAP-WT1-TBL1 axis. Gut. 2016;65:1482–93.
Fukumoto T, Zhu H, Nacarelli T, Karakashev S, Fatkhutdinov N, Wu S, Liu P, Kossenkov AV, Showe LC, Jean S, et al. N (6)-Methylation of Adenosine of FZD10 mRNA Contributes to PARP Inhibitor Resistance. Cancer Res. 2019;79:2812–20.
Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science. 2018;361:1346–9.
Chen B, Li Y, Song R, Xue C, Xu F. Functions of RNA N6-methyladenosine modification in cancer progression. Mol Biol Rep. 2019;46:1383–91.
Bi Z, Liu Y, Zhao Y, Yao Y, Wu R, Liu Q, Wang Y, Wang X. A dynamic reversible RNA N (6) -methyladenosine modification: current status and perspectives. J Cell Physiol. 2019;234:7948–56.
Pinello N, Sun S, Wong JJL: Aberrant expression of enzymes regulating m6A mRNA methylation: implication in cancer. In Cancer Biol Med. Volume 152018: 323–334.
Shi H, Wei J, He C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol Cell. 2019;74:640–50.
Liu J, Harada BT, He C. Regulation of Gene Expression by N (6)-methyladenosine in Cancer. Trends Cell Biol. 2019;29:487–99.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National Natural Science Foundation of China (No. 81572900). The Fundamental Research Funds for the Central Universities of Central South University (No. 1053320171187 and No. 2018zzts232). The Undergraduate Training Programs for Innovation and Entrepreneurship (UTPIE, XCX20190537 (L. He)).
Author information
Authors and Affiliations
Contributions
Liuer He finished the manuscript and abstract; Huiyu Li consulted relevant literatures and completed English revision; Anqi Wu and Yulong Peng completed the figures and tables; Gang Yin provided constructive feedback and guidance; Guang Shu completed critical revisions and proofread the manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have agreed on the contents of the manuscript.
Competing interests
The authors declare that they do not have any conflicts of interest related to this study. This manuscript has been read and approved by all the authors and has not been submitted to or is not under consider for publication elsewhere.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
He, L., Li, H., Wu, A. et al. Functions of N6-methyladenosine and its role in cancer. Mol Cancer 18, 176 (2019). https://doi.org/10.1186/s12943-019-1109-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1109-9