
Abstract
The human genome encodes 538 protein kinases that transfer a γ-phosphate group from ATP to serine, threonine, or tyrosine residues. Many of these kinases are associated with human cancer initiation and progression. The recent development of small-molecule kinase inhibitors for the treatment of diverse types of cancer has proven successful in clinical therapy. Significantly, protein kinases are the second most targeted group of drug targets, after the G-protein-coupled receptors. Since the development of the first protein kinase inhibitor, in the early 1980s, 37 kinase inhibitors have received FDA approval for treatment of malignancies such as breast and lung cancer. Furthermore, about 150 kinase-targeted drugs are in clinical phase trials, and many kinase-specific inhibitors are in the preclinical stage of drug development. Nevertheless, many factors confound the clinical efficacy of these molecules. Specific tumor genetics, tumor microenvironment, drug resistance, and pharmacogenomics determine how useful a compound will be in the treatment of a given cancer. This review provides an overview of kinase-targeted drug discovery and development in relation to oncology and highlights the challenges and future potential for kinase-targeted cancer therapies.
Similar content being viewed by others

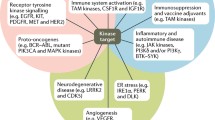

Background
Kinases are enzymes that transfer a phosphate group to a protein while phosphatases remove a phosphate group from protein. Together, these two enzymatic processes modulate numerous activities of proteins in a cell, often in response to an external stimulus [1]. Approximately 538 known kinases are encoded in the human genome, and these kinases maintain cellular function by turning protein function on, while corresponding phosphatases reverse this action [2, 3]. These counter mechanisms greatly improve the plasticity of epigenome by regulating protein activity in virtually every imaginable way. Biochemically, protein kinases catalyze the following reaction [3]:
Recent advances in our understanding of the fundamental molecular mechanisms underlying cancer cell signaling have elucidated a crucial role for kinases in the carcinogenesis and metastases of various types of cancer [4]. Since most protein kinases promote cell proliferation, survival and migration, when constitutively overexpressed, or active, they are also associated with oncogenesis [5]. Genome-wide studies of kinase mutations have revealed genetically inherited variants of specific kinases are causally associated with cancer initiation, promotion, progression as well as recurrence [4, 6]. Over the last three decades, multiple human malignancies have been identified to be associated with modulation and dysfunction of protein and lipid kinases and deactivated phosphatases on account of chromosomal reshuffling and genetic mutations [7,8,9]. Apart from the oncological issues, dysregulation of kinases has been demonstrated in many human disorders including immune, neurological and infectious diseases [10,11,12,13]. However, there is probably no greater clinical niche for kinases as the key targets for developing drugs than in cancer therapy. Kinome, the complete set of protein kinases encoded in its genome has become an attractive target for the treatment of numerous types of cancer. Single and multiple kinase inhibitors, both synthetic and natural molecules, are now targeted therapeutic strategies for treatment of human malignancies. The ROCK kinase inhibitor fasudil for treating cerebral vasospasms was the first approved small molecule for clinical use [14]. Kinase inhibitors now account for a quarter of all current drug discovery research and development efforts. Key oncogenic kinase drug targets include the PIK3CA, BRAF, and epidermal growth factor receptor (EGFR), which activates significant tumor cell signaling pathways and is related to the mutations and/or deletions in phosphatase and tensin homolog (PTEN), a phosphatase that negatively regulates PI3K [6, 7, 15]. Approximately 538 kinases are encoded in the human genome. Apart from this wide range of kinase-based drug targets, inhibition of distinct kinase signaling pathways can be less cytotoxic to non-cancerous cells, thus presenting the selective killing of tumor cells with considerably lower toxic manifestations [16, 17]. Interestingly, specific-kinase inhibitors, currently in clinical treatments, e.g., imatinib and dasatinib, produce more favorable outcome compared to conventional cytotoxic therapy [18, 19]. These kinase inhibitors have achieved a significant increase in patient survival rate in myeloid leukemia (CML) and gastrointestinal stromal tumors (GIST), thus translating basic molecular research into effective patient treatment. Due to improved clinical efficacy, U.S. Food and Drug Administration (FDA) has approved many small-molecule kinase inhibitors for clinical use (Fig. 1). These kinase inhibitors include target kinome members such as EGFR, ERBB2, VEGFRs, Kit, PDGFRs, ABL, SRC and mTOR, all providing improved clinical outcome and patient health status [4, 20]. The majority of these inhibitors target the ATP-binding site [21, 22], while a few of the non-ATP competitive kinase inhibitors target novel allosteric sites [23]. Consequently, the inhibition of kinase activity in treated patients prompts multiple anti-proliferative mechanisms, which leads to clinical remission of cancer.

Chemical structures of representative kinase inhibitors used for treatment of various human cancers
The current procedure for developing robust and selective kinase inhibitors has swiftly evolved from synthesizing analogs of staurosporine to sophisticated structure-based design methodologies, facilitated by molecular docking, crystallography and nuclear magnetic resonance spectroscopy [24, 25]. Since 2001, more than 10,000 patent applications for kinase inhibitors have been filed in the United States alone. In addition to the small-molecule kinase inhibitors, kinase-targeted antibodies have also demonstrated efficacy in various cancers, for example, cetuximab in colorectal and head and neck cancer, and trastuzumab in breast cancer [26, 27]. Trastuzumab and cetuximab bind to the extracellular domain of HER2 and EGFR respectively, and block the binding of the natural ligand, thus avoiding conformational rearrangement essential to the activation of the kinase and its downstream kinase-signaling pathways. Currently, FDA has approved 35 drugs (31 for cancer therapy) including orally effective direct protein kinase inhibitors that target a limited number of enzymes (Table 1). However, despite these encouraging results, the problems with drug resistance, toxicity, and compromised efficacy present critical challenges in both clinical and experimental oncology [3]. Furthermore, problems in the synthesis of novel kinase inhibitors have plagued drug development through an inadequate understanding of the selectivity of the kinase inhibitors [16, 28]. A key challenge in the clinical assessment is to identify the most efficient combination of kinase targets and then develop treatment combinations for targeted cancer. These issues have prompted research initiatives that may override various limitation of kinase inhibition, particularly evading the treatment-related drug resistance. In this current review, the authors examined the status, novel methodologies of drug design and validation of the prospective kinase inhibitors for clinical usage.
Role of kinases in cancer
Targeting the kinases harboring oncogenic transformational capacity and metastasis has led to a notable change in the clinical management of cancer (Fig. 2). Hundreds of kinases play overlapping and intricate roles in cell transformation, tumor initiation, survival and proliferation. Diving kinases while justifying their coinciding functionalities is difficult. However, in order to understand and discuss their oncogenic undertakings, they can be vaguely categorized based on their hallmark roles in cancer. The first group is the kinases that play a fundamental role in the primary oncogenic transformation and thus present themselves as prospective drug targets. Cytoplasmic tyrosine kinases are critical conveyers of extracellular signals, and mutations in these kinases have been reported to occur in various oncogenic conditions. This category includes the PI3K family of dual specific protein/lipid kinases, which are the most frequently mutated kinases implicated in 30–50% of human cancers [29]. PI3KCA, perhaps the most notable member of PI3K family is associated with the pathology of colorectal cancer [30], breast cancer [31], ovarian cancer [32], endometrial carcinoma [33], and hepatocellular carcinoma [34]. The PI3KCA kinase catalyzes the production of PIP3, a phospholipid which activates downstream signaling components such as protein kinase AKT and promotes tumor cell growth and survival [35]. Similarly, active form of the protein kinase Akt/PKB contributes to oncogenic transformation of cells [36]. Likewise, V599E and V600E mutations in BRAF kinase are associated with various carcinomas while BRAF somatic missense mutations occur in 66% of malignant melanomas [37]. The oncogenic mutations in JAK2 kinase such as single point mutation (Val617Phe) and JAK2 exon 12 mutations are implicated in both myeloproliferative disorders and myelodysplastic syndromes [38, 39]. Similarly, genetic alterations in other kinases such as ALK, IGF-1R, c-Kit, FGFR1–4, c-Met, c-Ret, c-SRC, regulate fundamental molecular mechanisms for tumor cell growth and development [9, 40]. Apart from tumor initiation, kinases are also vital for tumor cell survival and proliferation and may be present as downstream members of oncogenic kinase pathways. This category of kinases includes EGFR, a receptor tyrosine kinase, which has been shown to prevent autophagic cell death by maintaining intracellular glucose levels through interaction and stabilization of the sodium/glucose cotransporter 1 (SGLT1) [41]. Oncogenic alterations in EGFR make up approximately 45% of mutations in the tyrosine kinase domain [42, 43]. This leads to the loss of the inhibitory regulatory domains for dimerization resulting in hyper-proliferation of cancer cells via G1/S cell cycle progression [44, 45]. Other crucial members of the kinase family are aurora kinases (Aurora A-C). Aurora kinases are strategic kinases involved in defective spindle pole organization, and their pathophysiology correlates strongly with their oncogenic functions [46]. Aurora-A is an oncogenic kinase, and its amplification is documented in 10–25% of ovarian cancers [47]. Interestingly, Aurora A gene was originally named BTAK (breast tumor activated kinase) because its mRNA is overexpressed in breast cancer and is involved in the oncogenic transformation of breast cells [48]. Aurora A phosphorylates p53 at Ser215 and inhibits p53-DNA binding, disrupting cell cycle check activities [49]. It is also related to the activation of NF-κB, which boosts cancer cell survival by evading apoptosis [50]. Similar to Aurora-A, Aurora B and C are overexpressed in tumor cells and help cell survival, metastasis, and avoidance of apoptosis [51,52,53]. Other examples of tumor cell survival kinases include MEK1 [54], MEK2 [54], mTOR [55], and S6 kinase [56] which are all downstream members of MAPK, PI3K–Akt and EGFR pathway, respectively. In recent years, the mechanistic basis for developing kinase inhibitors from the second class of kinases has improved significantly. Types of serine/threonine kinases include MAP kinases (activated by protein phosphatases), ERK and stress-activated JNK and p38. Currently, there are about 30 Aurora kinase inhibitors in different stages of pre-clinical and clinical development [57]. The third category of kinases implicated in oncogenesis includes kinases overexpressed in tumors and surrounding tissues of cancers, which are important for the maintenance of tumors in the host. These include mutations in neurotrophic growth factor receptor which are involved in pilocytic astrocytoma, the most common childhood brain tumor [58]. Other examples include VEGFRs, fibroblast growth factor receptor (FGFR) kinases, protein kinase CK2 and TrkB [9, 16]. Overall, oncogenic kinases underlie and define multiple features of cancer including rapid proliferation, survival, growth, and metastasis, and have promoted the development of a plethora of kinase inhibitors. The fourth category of kinases, RTK with 58 known members and 20 subfamilies, were discovered more than a quarter of a century ago [40]. These kinases have a similar molecular architecture, and their mutations and aberrant activation are associated with carcinogenesis and angiogenesis. Four principal mechanisms are involved in abnormal RTK activation in human cancers; these include autocrine activation, chromosomal translocations, RTK overexpression, and gain-of-function mutations. RTKs are activated by growth factor binding by inducing receptor dimerization or in some cases subset of RTKs forms oligomers even in the absence of activating ligand [59, 60]. Principal members of RTK include 20 members including EGFR and others [61]. EGFR represent RTKs family as the well-studied kinase, implicated in several human cancers including lung cancer [62], glioblastoma [63], breast cancer [64], cervical carcinoma [65] and related mutations [66]. Several small-molecule inhibitors and monoclonal antibodies have been approved by FDA against RTKs for cancer therapy. The key drugs include Imatinib (against PDGFR, KIT, Abl, Arg), Sorafenib (against Raf, VEGFR, PDGFR, Flt3, KIT) and Lapatinib (against EGFR, ErbB2).

Categorization of different kinases implicated in human cancer. CTK: cytoplasmic tyrosine kinase, S/T Kinase: serine/threonine kinase, LK: lipid kinase, RTK: receptor tyrosine kinase. SK1: Sphingosine kinase 1, PI3K: phosphoinositide 3-kinase, PKCi: Protein kinase Ci, mTOR: mammalian target of rapamycin, CDKs: cyclin-dependent kinases, ATM: Ataxia telangiectasia mutated, Akt: protein kinase B, S6K: ribosomal protein S6 kinase, STK11/LKB1: Serine/threonine kinase 11 or liver kinase B1, PLKs: Polo-like kinases, b-Raf: B-Raf proto-oncogene, Aur A & B: Aurora Kinase A & B, c-SRC: Proto-oncogene tyrosine-protein kinase Src, c-YES: c-Yes proto-oncogene (pp62c-Yes), Abl: Abelson murine leukemia viral oncogene homolog 1, JAK-2: Janus kinase 2, RON: Recepteur d’Origine Nantais, FGFRs: Fibroblast growth factor receptors, c-Met: c-MET proto-oncogene, c-Ret: c-RET proto-oncogene, IGF-IR: Insulin-like growth factor 1 receptor, EGFR: Epidermal growth factor receptor, PDGFR-α: Platelet-derived growth factor receptor α, c-Kit: proto-oncogene c-Kit or Mast/stem cell growth factor receptor, Flt3,Flt-4: Fms-like tyrosine kinase 3, 4, PDGFR-β: Platelet-derived growth factor receptor β, ALK: Anaplastic lymphoma kinase, HER-2: human epidermal growth factor receptor-2
Kinase discovery and development timeline
The development of kinase inhibitors for the treatment of human cancers started in mid 1970s (Fig. 3). In 1978, the first oncogene was found to be a protein kinase [67]. This discovery was supported by a successive finding in 1981 when tumor-promoting phorbol esters was shown to exhibit hyperactivation of protein kinase C (PKC) [68]. In the coming years, naphthalene sulphonamides, the first protein kinase inhibitors were synthesized and served as a basic design for developing further molecules [69]. During this time, staurosporine, an antifungal drug was shown to be a nanomolar inhibitor of PKC [70]. This drug was later used as a parent compound to produce various analogs as potential inhibitors of PKC. In the 1991 the 3-D structure of protein kinase A (PKA) was elucidated, and it became apparent that the residues that were involved in binding ATP were conserved from kinase to kinase [71, 72]. This discovery perpetuated a myth that it was “impossible” to develop protein-kinase inhibitors with the requisite potency and specificity. However, with the discovery of cellular targets for cyclosporin and subsequent development of HA1077, an inhibitor of several protein kinases, the field of kinase inhibitors rapidly progressed [14, 73, 74]. Finally, the breakthrough occurred in 2001 when imatinib, a phenyl-amino-pyrimidine derivative targeting the inactive conformation of the ABL1 kinase, was approved for the treatment of CML (Fig. 4). Starting with a 2-phenylaminopyrimidine derivative, chemists added a 3′ pyridyl group, benzamide, a flag methyl instead of N-methylpiperazine to synthesize a drug candidate called CGP57148B (later changed to imatinib) [75]. Clinical targeting of BCR-ABL gene, formed by the fusion of ABL gene from chromosome 9 to the BCR gene on chromosome 22, also called the Philadelphia chromosome, improved the clinical management of leukemia patients [76, 77]. Owing to its’ broad-spectrum nature imatinib has since then been approved for various other oncology indications. Following the FDA approval of imatinib, different strategies have been used for the development of single and multi-target kinase inhibitors for cancer treatment [78]. More active drugs, such as nilotinib, with a selectivity profile similar to imatinib, were approved for imatinib-resistant CML [79, 80]. Later on, the indolinone-derivative sunitinib with a broad spectrum activity targeting VEGFR, PDGFR, FGFR, KIT, and FLT3, was approved for the treatment of renal cell carcinoma, as well as second-line therapy in the imatinib-resistant gastrointestinal stromal tumor (GIST) [81]. Sorafenib was later approved for the treatment of renal cell and hepatocellular carcinoma due to its ability to bind to the inactive conformation of the VEGFR kinase [82]. Similarly, in the year 2009, pazopanib, a 2-amino pyrimidine targeting VEGFR, PDGFR, and KIT was approved for the treatment of advanced renal cell carcinoma [83]. A quick surge in clinical approval of kinase inhibitors started following the approval of everolimus (mTOR inhibitor) in 2009 for the treatment of metastatic renal cell carcinoma (mRCC), astrocytoma and breast tumors [84,85,86]. In the year 2011, four kinase inhibitors, vemurafenib, vandetanib, ruxolitinib, and crizotinib were approved for the treatment of melanoma, thyroid cancer, myelofibrosis and ALK-positive non-small cell lung cancer [87,88,89,90]. Successively, in 2012 and 2013 ten new kinase inhibitors were approved by FDA for the treatment of various malignancies. Since the initial development of imatinib, 28 kinase inhibitors have been approved by FDA with Brigatinib and Osimertinib being the latest approvals [91, 92]. Apart from the approved kinase inhibitors, there is more than three thousand ongoing Phase I-III clinical trials for hundreds of new kinase inhibitors. It is therefore beyond the scope of this mini-review to discuss all the protein kinase inhibitors that are in clinical Phase I–III.

Timeline of key events in the development of protein-kinase inhibitors for the treatment of cancer

Interruption of the BCR-Abl pathway can be achieved by Gleevec (imatinib mesylate)
Types of kinase inhibitors
Kinase inhibitors are very efficacious for the treatment of cancer especially targeting specific mutations that chiefly drive tumorigenesis. They are categorized according to their capacity to catalyze the transfer of the terminal phosphate of ATP to the substrates that usually contain a serine, threonine or tyrosine residue (Table 2). Many reviewers have categorized types of kinase inhibitors according to their mechanism of action. Initially, small molecule protein kinase inhibitors were divided into three classes, termed as types I, II, and III kinase inhibitors [93]. Dar and Sakot defined the type I kinase inhibitor as “a small molecule that binds to the active conformation of a kinase in the ATP pocket,” the type II inhibitor as “a small molecule that binds to an inactive (usually Asp-Phe-Gly (DFG)-OUT) confirmation of a kinase,” and the type III inhibitor as “a non-ATP competitive inhibitor” or allosteric inhibitor [93, 94]. Later on, Zuccotto et al. introduced a new class of kinase inhibitors, i.e. type I½ inhibitors, which bind to the protein kinases with the DFG-Asp in and C-helix out conformation [95]. Later, Gavrin and Saiah further divided the allosteric effectors into two subclasses (III and IV) where the type III inhibitors bind within the cleft between the small and large lobes adjacent to the ATP binding pocket and type IV inhibitors bind outside of the cleft and the phosphor-acceptor region [96]. Afterwards, bivalent molecules that span two regions of the protein kinase domain were labeled as type V inhibitors [97]. Finally, small molecules that form covalent adducts with the target enzyme were recently termed as covalent inhibitors [94]. The classification described herein uses these parameters with added subdivisions and criteria, labeling them as types I, II, allosteric, and substrate directed and covalent inhibitors.
Type I kinase inhibitors
Type I kinase inhibitors represent ATP-competitors that mimic the purine ring of the adenine moiety of ATP. Functionally, they interact with the conformational phosphorylated active catalytic site of the kinases. These kinase inhibitors bind to the active conformational site and alter the structural conformation otherwise favorable to phosphotransfer [98, 99]. Type I inhibitors usually contain a heterocyclic ring system that occupies the purine binding site, where it serves as a scaffold for side chains that occupy adjacent hydrophobic regions [100]. These hydrophilic regions of the enzyme occupied by the ribose moiety of ATP may be used to exploit the solubility of the drugs or other active compounds [98]. To date, many Type I kinase inhibitors for the treatment of cancer have been approved by the FDA viz. bosutinib, crizotinib, dasatinib, erlotinib, gefitinib, lapatinib, pazopanib, ruxolitinib, sunitinib, and vemurafenib. Apart from the large-scale clinical success, Type I kinase inhibitors also come with adverse side-effects. Type I inhibitors display an inclination for low kinase selectivity as the targeted ATP pocket is conserved through the kinome; therefore, increasing the potential for off-target side effects. This little selectivity for targeted kinases may result in cardiotoxicity and possible deterioration in cardiac function [101, 102].
Type II kinase inhibitors
Type II kinase inhibitors act by targeting the inactive conformation of kinases and interact with the catalytic site of the unphosphorylated inactive conformation of kinases [103]. Type II kinase inhibitors exploit new interactions inside the lipophilic pocket derived from the change of confirmation of the phenylalanine residue of the “Asp-Phe-Gly (DFG)” N-terminal loop conformation of kinases [16, 103]. These inhibitors interact reversibly with the target kinase which leads to the formation of single or multiple hydrogen bonds with the protein in the ‘hinge region’ and also causes extra interactions in the open DFG-out conformation [98, 103]. These lipophilic interactions have a high degree of selectivity towards unwanted kinases affecting an increase in the safety profile of Type II kinase inhibitors. Type II inhibitors also display a high conservation of distinctive H-bond pattern between the inhibitor and the glutamic and aspartic acids of the kinase [98, 104]. Due to the exclusivity of inactive protein kinase conformations, it was theorized than type II kinase inhibitors would be more selective. However, there is considerable overlap of selectivity between type I and type II inhibitors. The discovery of Type II kinase inhibitors such as imatinib and sorafenib was serendipitous, and it wasn’t until much later that their mode of action was discovered. The role of imatinib in the consequent development of small molecule protein kinase inhibitors cannot be overstated. All Type II inhibitors share a similar pharmacophore and hydrogen bonds that interact with DFG-out kinase conformational structure as revealed by the discovery of the Type II kinase inhibitor co-crystal structure [105]. Since canonical ATP-binding sites of activated kinases, the target sites of Type I inhibitors, do not share these features, this pocket is conserved to a lesser extent across the kinome, and hence promises better prospects for the rational design of selective inhibitors [100, 103]. Overall, Type II kinase inhibitors display high selectivity towards kinase inhibition as compared to Type I kinase inhibitors along with the profound impact on cellular activity.
Type III or allosteric inhibitors
The third class of kinase inhibitors bind outside the catalytic domain/ATP-binding site and modulates kinase activity in an allosteric manner. Some authors have divided the allosteric inhibitors into two subtypes where type A inhibitors bind to an allosteric site next to the adenine-binding pocket whereas the type B inhibitors bind elsewhere [97]. Overall, Allosteric or Type III inhibitors exhibit the highest degree of target kinase selectivity as they exploit binding sites and physiological mechanisms that are exclusive to a particular kinase [106]. With respect to ATP, these drugs are steady-state noncompetitive or uncompetitive inhibitors because ATP cannot prevent their interaction with the target kinase. One of the earliest allosteric inhibitors was CI-1040, an orally active, highly specific, small-molecule inhibitor of the MEK1/MEK2 pathway [107]. A recent chemical proteomics study confirms the allosteric activity of type III inhibitors as they showed a higher selectivity, but also stated that these are special cases as most of them are designated MEK1/2 inhibitors that bind to a particular cavity adjacent to the ATP-binding site [108]. Another allosteric kinase inhibitor GnF2 binds to the myristate binding site of BCR–ABL1 [109]. GnF2 also displays sound IL-3 reversible anti-proliferative and apoptotic effect on two mutants identified as E255V and Y253H [109]. Likewise, TAK-733 binds to the MEK1-ATP complex in the gate area and the back cleft adjacent to the ATP-binding pocket; however, it cannot bind to the adenine pocket owing to its occupation by ATP [110]. Other examples include RO0281675 and analogs thereof [111, 112]. Overall, targeting kinases using allosteric inhibitors is thought to be a crucial approach for overcoming hurdles in kinase inhibitor research, such as limited selectivity, off-target side effects, and drug resistance. In future, more active and target specific allosteric inhibitors will be discovered as larger stress is placed on cell-based assays in which kinases are explored in their native cellular context.
Substrate-directed inhibitors
These are also called Type IV kinase inhibitors and undergo a reversible interaction outside the ATP pocket, located in the kinase substrate-binding site. These inhibitors don’t compete with ATP and offer a higher degree of selectivity against targeted kinases [113]. Substrate-directed inhibitors include ATP-noncompetitive inhibitors such as ON012380 which are targeted against Philadelphia chromosome-positive leukemias [114]. More importantly, ON012380 was found to override imatinib resistance at physiologically relevant concentrations of < 10 nM [115].
Type V or covalent inhibitors
The covalent kinase inhibitors form an irreversible covalent bond with the kinase active site and target a catalytic nucleophile cysteine within the active site of the enzyme [116, 117]. The chemical rationale for developing Type V inhibitors is based on exposed cysteine side chain in the ATP site which can be targeted for covalent reaction with a drug candidate with an electrophilic Michael acceptor in the right position [118, 119]. This type of kinase inhibition takes place via trapping of a solvent-exposed cysteine residue either by SN2 displacement of a leaving group or by reacting with a Michael acceptor incorporated within the kinase inhibitor [113, 120, 121]. Covalent inhibitors target respective kinase by formation of a rapidly reversible collision complex followed by an irreversible enzyme-inhibitor complex [122]. Afatinib (targets EGFR (ErbB1), ErbB2, and ErbB4) and ibrutinib are currently FDA-approved drugs that form a covalent bond with their target kinase. Afatinib, unlike the first-generation EGFR-TKIs such as gefitinib and erlotinib, is a mutant-selective EGFR inhibitor with low toxicity profile despite its irreversible mechanism [123]. Similar to Afatinib, ibrutinib also targets mutant-EGFR kinase with a distinct binding conformation [124]. Both of these kinase inhibitors initiate Michael reaction with the addition of a nucleophile (the -SH of cysteine) to an α, β unsaturated carbonyl compound [125]. C481 within hinge region of the Bruton tyrosine-protein kinase is hypothesized to form a covalent link with ibrutinib [126]. A recently approved kinase inhibitor, neratinib (HKI-272), inhibits Herceptin-2 (HER-2), and prevents recurrence in patients with early-stage HER2-positive breast cancer [127]. Overexpression of HER-2 is seen in 25–30% of breast cancer patients and predicts a poor outcome in patients with primary disease. Likewise, CL-387785, a covalent inhibitor, overcomes resistance caused by T790 M mutation of the epidermal growth factor receptor (EGFR) [128]. These kinase inhibitors also display an extended dissociation half-life which minimizes off-target side effects [118]. Other advantages include prolonged pharmacodynamics, suitability for rational design, high potency, and ability to validate pharmacological specificity through mutation of the reactive cysteine residue [119]. The approved covalent kinase inhibitors (Ibrutinib, Afatinib, and Neratinib) have shown that small molecules containing weak reactive electrophiles can be mutant specific in action with low toxicity. These kinase inhibitors have initiated resurgence of interest in covalent inhibitors, and feature an acrylamide functionality to specifically target the cysteine side chains of kinases. Example include a recent study showing nine irreversible EGFR and two BTK inhibitors with higher kinase inhibitory selectivity than reversible compounds [108]. The Type V or covalent kinase inhibitors have substantial potential for exploration as 200 different kinases have a cysteine chain located near the ATP pocket.
Biochemical mechanism
Biochemically, kinase inhibitors are classified according to the activation state of the protein kinase target including the nature of DFG-Asp (active in, inactive out), the C-helix (active in, inactive out), and the regulatory spine (active linear, inactive distorted). Apart from type III or allosteric inhibitors, all the FDA-approved kinase inhibitors form hydrogen bonds with one or more hinge residues. Overall, most kinase inhibitors form: (i) hydrophobic contacts with catalytic spine residues; (ii) contact with the RS3 R-spine residue within the C-helix; (iii) interaction with the gatekeeper residue; and (iv) residues that occur just before the DFG-D of the activation segment [94, 129]. The following section briefly discusses the biochemical mechanism of action of FDA-approved kinase inhibitors.
Frequent mutations in various protein kinases present specific kinase inhibition as a therapeutically relevant approach in oncology. Kinase inhibitors have evolved to target many different regulatory and inhibitory mechanisms. There are various mechanisms by which kinase inhibitors bind to their target kinases broadly classified into kinase inhibitors that bind either covalently or non-covalently to or around the ATP binding site. Primarily, kinases bind with ATP in a cleft between the N- and C-terminal lobes of the kinase domain. In this domain, the adenine group of ATP is bound by two hydrophobic surfaces and interact via hydrogen bonds to the connector of two lobes, called the “hinge region” [130,131,132]. The cleft of ATP contains various elements such as the flexible activation loop (A-loop), along with closed conformations which are responsible for the catalytic activity of the kinase [133, 134]. The active or inactive state of the protein kinase is determined by the position of the A-loop, including the DFG motif at its N-terminal, which has various conformations [28, 98, 134, 135]. The only component of kinases that does not vary between the active and inactive states is the catalytic loop. The active state of the protein kinase when the Asp in the DFG motif coordinates one magnesium ion, which prepares the phosphates of ATP for the transfer of the phosphoryl group. The Phe in the DFG motif packs under the helix-C positioning both helix-C and A-loop for catalysis [98, 133, 136]. Protein kinases return to their inactive conformation once kinase transfers the phosphoryl group from ATP to tyrosine, serine or threonine of the substrate protein. This process also involves the returning of the A-loop to the closed position by the change of A-loop from the DFG-in to the DFG-out conformation [98, 137, 138]. However, ribose binding and the phosphate binding site of ATP usually remains unexplored by the majority of kinase inhibitors [134, 139]. Based on the biochemical mechanisms of action, kinase inhibitors are categorized as covalent and non-covalent kinase inhibitors. The non-covalent kinase inhibitors are classified into those who either bind or do not bind to the hinge region of the kinase [140]. The DFG-in or Type I kinase inhibitors bind to hinge region and represent the vast majority of non-covalent kinase inhibitors [98]. In these kinase inhibitors, the Asp in the DFG motif coordinates the phosphates of ATP, and the Phe in the DFG motif stabilizes the position of helix-C and the A-loop for catalysis [20]. However, the ATP-binding pocket is highly preserved among members of the kinase family, and it is hard to find highly selective Type I kinase inhibitors. Moreover, the pre-clinical to clinical translation of Type I kinase inhibitors is hindered as they compete with high levels of intracellular ATP leading to a discrepancy between biochemical and cellular analysis. Contrary to the Type I inhibitors, Type II inhibitors bind to the DFG-out confirmation of kinases. These inhibitors induce a conformational shift in the target enzyme such that the target kinase is no longer able to function. Type II inhibitors use an additional hydrophobic pocket adjacent to the ATP site exposed by the movement of A-loop from DFG-in to DFG-out conformation [141]. This gives the Type II inhibitors higher selectivity as they recognize novel regions of the active cleft outside the highly conserved ATP-binding site. Like Type II kinase inhibitors, the allosteric inhibitors or Type III inhibitors also display high selectivity as they explore binding sites and regulatory mechanisms that are unique to a particular kinase. They contain a heterocyclic system that forms one or two hydrogen bonds with the kinase hinge residue. Like Type II inhibitors, they also induce the DFG-out confirmation and move phenylalanine side chain to a new position [98, 99]. Examples include compounds such as CI-1040, which inhibit MEK kinase by occupying a pocket adjacent to the ATP-binding site [107]. Interestingly, exploration of allosteric kinase inhibitors also helps to recognize unique kinase activation targets, which could be explored for therapeutic intervention in other diseases states. Recently, there has been an increased interest in the development of irreversible (covalent) kinase inhibitors that form covalent bonds with cysteine or other nucleophilic residues in the ATP-binding pocket. These inhibitors have typically been developed by incorporation of an electrophilic moiety into an inhibitor that already possesses submicromolar binding affinity to the target of interest. The covalent kinase inhibitors bind to a cysteine residue in or around the active site, thus preventing the binding of ATP to the protein kinase [117, 127]. These kinase inhibitors undergo the “Michael reaction”, which is a reaction that triggers the addition of a nucleophile, such as a cysteine, to an α, β unsaturated carbonyl functionality. Nucleophile additions cause the formation of adducts at the electrophilic β-position and inactivate kinases by irreversibly blocking the binding of ATP to kinase [142]. These kinase inhibitors are highly selective as they overcome endogenous ATP competition and target a specific cysteine at the corresponding position in a kinase. Various covalent kinase inhibitors target kinases such as BTK [143], Fes [144], VEGF-R2 [145], and RSK2 [146] through their ability to bind to a cysteine residue.
Recent clinical developments
Traditional cancer therapies follow palliative as well as off-targeted approaches in oncology. In contrast, kinase inhibitors symbolize a class of targeted cancer therapeutic agents with limited nonspecific toxicities. So far, 28 inhibitors with activity targeted to one or multiple kinases have been approved for clinical use. With over 500 members, the kinase family has received a high degree of attention from academic researchers as well as pharmaceutical industries [147]. After the clearance of possible hindrances, owing to the high degree of active site similarities and possible off-target activity, kinase inhibitors have gained scientific limelight [21, 24, 78, 148, 149]. In a 13-year summary of targeted therapies including kinase inhibitors, the clinical success rate of kinase inhibitors was superior to other cancer therapies [150, 151]. Nevertheless, this clinical success does come with exceptions; attempts to control cytotoxicity during treatment, particularly with sunitinib and EGFR/VEGF-system targeting drugs have yielded disappointing results [152,153,154,155]. Overall, during the last 5 years, Aurora kinases [156], casein kinase II [157], cyclin-dependent kinases [158], focal adhesion kinase [159], protein kinase B [160], phosphatidylinositol 4,5-bisphosphate 3-kinase delta and gamma [161], polo-like kinase I [162], tyrosine-protein kinase SYK [163], high affinity nerve growth factor receptor family [164] and Wee1-like protein kinase [165] have been targeted in Phase I clinical trials. Although recent developments have shown Aurora kinases as major new targets in kinase inhibitor development [166, 167]. After initial hurdles, two compounds palbociclib and ribociclib have passed the phase III clinical trials and are in clinical use [168].
Recent kinase developments include precision therapy based on tumor genomic data. The ability to perform genetic studies of tumors and follow-up treatment decisions based on the identification of tumorigenesis drivers has resulted in significant benefits for patients in need of effective systemic therapy. The detailed information regarding all the clinical trials is out of the scope of this mini-review; however, a few important developments are highlighted. A small number of small molecule tyrosine kinase inhibitors have recently received FDA approval for treatment of non-small cell lung cancer (NSCLC) with EGFR mutations or ALK translocations [169]. Afatinib, a second-generation, non-competitive kinase inhibitor targeting all members of the ErbB family of receptors (also known as Her-2/neu) was approved in 2013 as frontline therapy for NSCLC patients with EGFR-deletion 19 and L858R mutations [170]. Despite several challenges that need to be overcome, reviewed in [171, 172], precision medicine has yielded important dividends for patients with advanced cancers [173]. In order to counter currently undruggable targets and acquired resistance, immunotherapy has gained widespread recognition in recent years [174]. Additionally, kinase targeted antibody therapy for hematological malignancies, and solid tumors have become established over the past 20 years. Key examples of antibody constructs targeting kinases include Trastuzumab and T-DM1 (targeting ERBB2/HER2) in breast and bladder cancer [175, 176], Bevacizumab (targeting VEGF) in ovarian, metastatic colon cancer and glioblastoma [177], Cetuximab, Panitumumab and necitumumab (targeting EGFR) in colorectal cancer and NSCLC [178]. Other experimental candidates include scFv, affibody and minibody (ERBB2/HER2 and FGFR1) [179,180,181,182], Protein–Fc (VEGFR1 and VEGFR2) [183] and Intact IgG (EGFR, ERBB2, and VEGF) in breast and lung cancer studies. Also, there is an increased development of PI3K and mTOR inhibiting compounds. Dual PI3K/mTOR inhibitors in advanced clinical trials include NVP-BEZ235 (glioblastomas) [184], XL765 (breast cancer) [185], GDC0980 (mRCC) [186], PF04691502 (breast cancer) [187], GSK2126458 (colorectal, breast, non-small cell lung, and pancreatic cancers) [188], Quinacrine (various leukemias) [189, 190] and PKI587 (advanced solid malignancies) [191]. Also, buparlisib and idelalisib, both PI3K inhibitors, have entered phase III clinical trials [192, 193]. In line with PI3K/mTOR inhibitors, various kinase inhibitors have entered into clinical trials for gastrointestinal cancers [194], thyroid carcinoma [195], breast cancer [196], and endocrine tumors [197]. Many previously approved kinase inhibitors are being tested in clinical trials against BRAF and cyclin-dependent kinases 4/6 mutations [20, 99]. BRAF somatic mutation, particularly BRAF V600E/K, drive tumorigenesis through constitutive activation of the downstream MAPK pathway [198]. Multiple drugs including vemurafenib, dabrafenib, PLX3603, ARQ736, CEP-32496, BMS-908662, BGB283, encorafenib in combination with other chemotherapies are being targeted for BRAF-mutated cancers [199]. It is now suggested that dabrafenib, a selective BRAF inhibitor may target other kinases indicating polypharmacology (that is, drugs that act on more than one target) [108]. A paper published by Klaeger and colleagues explains the potential of 243 clinically evaluated kinase drugs [108]. Although multiple new kinases have been targeted during the last 5 years, a large share of the cancer kinome is still untargeted. Furthermore, use of these targeted therapies is not without limitations. Reservations on the use of kinase inhibitors include the development of resistance and the lack of tumor response in the general population and these constraints still need to be resolved.
Natural bioactives as kinase inhibitors
Overexpression of kinases is observed in multiple carcinomas. In recent years, there has been a major paradigm shift in discovery and screening of natural compounds as potential kinase inhibitors. Emerging data has revealed numerous mechanisms by which natural compounds mitigate kinase mutations. Classically, many of the biological actions of small molecule compounds, especially polyphenols, have been credited with their antioxidant properties, either through their reducing capacities or their possible influence on intracellular redox states. These small molecule bioactives can directly bind receptor tyrosine kinases and alter their phosphorylation state to regulate multiple cell signaling pathways (Fig. 5). Elevated levels of the EGFR and HER-2 have been identified as common components of multiple cancer types and appear to promote solid tumor growth [200, 201]. EGFR inhibition is exhibited by multiple polyphenols including resveratrol [202], quercetin [203], curcumin [204], and green tea extracts [205]. HER-2 overexpression in tumor cells is also attenuated by these bioactives [206,207,208]. Fibroblast growth factors are involved in a variety of cellular processes, such as tumor cell proliferation, drug resistance, and angiogenesis [209]. Oncogenic alterations of RTK kinases including FGFR1, FGFR3, and FGFR4 are inhibited by natural compounds [210,211,212]. Similarly, curcumin and chrysin block expression of receptor d’origine nantais (RON) in tumor cells [213, 214]. The product of the human SRC gene, c-Src, is found to be over-expressed and highly activated in a wide variety of human cancers [215]. It is also accompanied by elevated levels of Abl [216] and JAK-2 kinases [217]. Interestingly, the overexpression and translocation of oncogenic cytoplasmic tyrosine kinases such as c-SRC [218], Abl [219], c-Met [220] and JAK-2 [221, 222] are tempered by natural compounds. Serine/threonine kinases, within the kinase family, play vital roles regarding their involvement in human cancers. Akt, a crucial kinase modulates diverse cellular processes involved in the regulation of cell survival, cell cycle progression and cellular growth [223]. Up to date, more than 50 proteins have been identified as the phosphorylation substrates of Akt. Resveratrol modulates expression of Akt in breast [224], uterine [225], prostate [226, 227], skin [228] and glioma cells [229]. It targets the kinases at ATP-binding site competitively and reversibly [230, 231].

Structures of key natural bioactives which pharmacologically modulate kinases
Furthermore, myricetin has been reported to target Akt to inhibit cell transformation and proliferation by directly binding to the ATP-binding site of Akt [232]. Similar effects are also exhibited by curcumin [233], quercetin [234, 235], green tea molecules [236], anthocyanins [237] and other polyphenols [238,239,240]. Hyperactivity of cyclin-dependent kinases (CDKs) is one of the key mechanisms underlying tumorigenesis [241]. The overexpression of CDKs is inhibited by various small molecule compounds [242,243,244,245,246]. Likewise, hyperactivity of mTOR pathway is also downregulated by natural compounds [229, 247,248,249]. The mTOR pathway is a critical effector in cell-signaling pathways and is commonly deregulated in human cancers. Furthermore, small molecule compounds also inhibit the activity of polo-like and Aurora kinases [207, 210, 250, 251]. B-Raf kinases, key kinases intimately involved in cancer cell proliferation [252], are also inhibited by natural plant compounds such as curcumin, luteolin, quercetin and ursolic acid [253, 254]. Finally, the overexpression of oncogenic lipid kinases such as PI3K and SK1 is also mitigated by small molecule bioactives. More than 30% of various solid tumor types were recently found to contain mutations in PI3K [255]. Well explored bioactive molecules such as resveratrol [228], curcumin [256], quercetin [235] and green tea polyphenols [257] inhibit PI3K pathway. Similar to the parent compounds, metabolites of bioactives also inhibit PI3K pathway [258]. Sphingosine kinase 1 (SphK1) is also an important component of carcinogenesis as it converts the proapoptotic lipids ceramide and sphingosine into the anti-apoptotic lipid sphingosine-1-phosphate [259]. Inhibition of SphK1 is exhibited by few chelating bioactives [260,261,262]. Oncogenic kinases are vital proteins that couple extracellular signals with intracellular signaling pathways, which contribute to all stages of cancer development. Accumulated data reveals that plant compounds, particularly polyphenols, exert anti-cancer effects through acting on protein kinase signaling pathways. Many natural bioactives bind directly to oncogenic protein kinases and then alter their phosphorylation state, thus mitigating cell signaling pathways in carcinogenesis processes.
Challenges and limitations
Despite numerous advances, scientists are still trying to understand pathophysiology and application of kinase inhibitors for therapeutic benefit in clinical oncology. Kinase inhibition triggers a strong discerning pressure for cells to acquire resistance to chemotherapy through kinase mutations [263]. Thus, the treatment and pathology of cancer are further complicated by the plethora of such mutations that occur in different kinases [264]. There are two types of chemotherapy resistance: de novo resistance, which refers to the failure of an agent to produce any detectable response after initial treatment and acquired resistance. Multiple mechanisms including the targeted kinase, the structure of inhibitor, and the underlying genetic features of the tumor contribute to treatment failure and both types of resistance. Acquired resistance refers to the progression of a tumor that initially responds to treatment and subsequently becomes resistant to treatment despite continual administration of the inhibitor. Interestingly, most of the kinase resistant cases fall into the acquired resistant category. Treatment resistance associated with kinase inhibitors is induced by changes in the kinase gatekeeper residue as hydrophobic interactions on this site are crucial for the binding affinity of the inhibitor [265, 266]. Since a small gatekeeper residue allows an inhibitor to access the “gated” hydrophobic regions of the binding pocket, changes in this region hinder activity of kinase inhibitors. The gatekeeper residue has no interaction with ATP but is usually in contact with Type I and Type II kinase inhibitors and sterically impedes inhibitor binding [98]. These mutations mainly lead to in the substitution of one amino acid for another in the protein made by a gene, thus conferring resistance to cell cycle termination and chemo drugs. A classic example is induction of imatinib resistance due to gatekeeper mutations in Thr 315 (coded by ACT) in BCR-ABL kinase [254]. Other examples of such gatekeeper mutations include T790 (EGFR) [267], G697R (FLT3) [268], BCR–ABL1 (T315I) [269], PDGFRα (T674I) [270] and KIT (T670I) [271] oncogenic mutations. In the case of the EGFR kinase, the T790 M mutation induces resistance to quinazoline inhibitors by increasing affinity for the natural substrate ATP [272]. It is one of the most common mutations in which methionine substitutes for threonine at amino acid position 790, conferring a growth advantage to cancer cells alongside drug-resistant variant of the targeted kinase [273]. Similarly, 20% of cases of acquired TKI resistance involve amplification of the MET gene [274]. These events thereby provide signalling redundancy and eliminate consequences of clinical kinase inactivation. Furthermore, the lipid modifying PI3K together with the Ras-Raf-MAPK also undergoes several resistance-inducing mutations [275]. Interestingly, these mutations cause a minute or no change in kinase activity but confer inhibitor resistance to kinase inhibitors [276]. An example includes gatekeeper mutation T790 M in EGFR which causes gefitinib and erlotinib resistance via hyper affinity for ATP [277, 278]. Overcoming gatekeeper-mutation induced drug-resistance in the clinic is extremely difficult and requires structural fine-tuning of the drug candidates. To surmount resistance to inhibitors gefitinib and erlotinib, kinase inhibitors that bind covalently to the ATP-binding site of EGFR are been developed [117, 279]. Such next-generation EGFR inhibitors selectively target the inhibitor-sensitizing mutations and display an improved safety profile by sparing wild-type EGFR activity in normal cells. A recent study using chemical proteomics analyzed 243 clinically evaluated kinase drugs and showed that some kinase inhibitors are highly selective, especially KIs targeting mutant EGFR [108]. Likewise, G-loop mutations in ABL, p38α, FGFR1, CK2α1, JNK3, AURORA-A, ROCK1 and CDK5 kinases prompt oncogenic or drug-sensitizing mutations [280]. Another clinical challenge associated with kinase inhibitors is variation in clinical results from combinations of kinase inhibitors. Examples of clinical failure include combined gefitinib and trastuzumab treatment in breast cancer, erlotinib and bevacizumab in renal cell carcinoma, and cetuximab and bevacizumab in colorectal cancer. Conversely, combinations of lapatinib and pertuzumab with trastuzumab in breast cancer, and combination of bevacizumab and erlotinib in NSCLC have exhibited clinical success. Further, in some cases, the combinations of kinase targeting agents reduced patient survival compared with the treatment using single drug [281]. However, these discrepancies are proposed due to misinterpretation of the preclinical data, rather than a failure of the preclinical model itself [282, 283]. Additionally, these preclinical studies of drug combinations are probably biased towards validating well-characterized targets thereby limiting their ability to prioritize novel targets. Further, many kinase inhibitors are associated with toxicities and off-target effects such as cardiotoxicity, hypertension, hypothyroidism, skin reactions and proteinuria [284, 285]. Looking specifically, inhibition of EGFR is associated with dermatological problems, VEGFR inhibition with cardiotoxicity, HER2 and ALK inhibition with gastric irregularities and dermatological problems, and BCR-ABL inhibition causes cytopenia, in addition to cardiotoxicity and cardiac complications [286, 287]. Another challenge is in translating RNAi therapy into drugs, particularly in kinase inhibition. The majority of drug targets cannot be battered by shRNA (or gene knockout) as most shRNAs cannot be replicated by drugs since most proteins cannot be translated to therapy [288]. Thus, clinical resistance to kinase inhibitors remains the major limitation to kinase-based -therapies. Resistance to chemotherapy has also been well recognized as a significant challenge in oncology, a problem also confronted by kinase inhibitors. Beyond the stated illustrative examples, numerous other pathways outside the scope of this review can influence the clinical activity of kinase inhibitors.
Numerous follow-up strategies are being employed to overcome the challenge of kinase inhibitor resistance. A first approach is to develop inhibitors that can tolerate diverse amino acids at the gatekeeper position [289, 290]. A second approach is to target the kinase with inhibitors that bind at alternative binding sites [115, 291]. A third approach involves targeting other pathways that may be required for kinase transformation [292]. These approaches have been demonstrated to work in cell line studies, and strategies are being developed for their clinical use. However, it is also vital to consider the possibility that multiple different resistance mechanisms might develop concurrently in patients, thereby challenging clinical ability to overcome acquired resistance to kinase inhibitors.
Future developments
Even though only a small fraction of the kinome is currently being targeted, kinase inhibitor drug discovery has progressed dramatically in the past decade. Clinical evaluation of kinase inhibitors has shown that therapeutic responses vary widely in individual patients and across patient populations, and seem to depend on many diverse factors. Many new candidate molecules have entered clinical trials, and much more are still at the preclinical stage. Most of the current kinase inhibitor discoveries have developed through rational drug design rather than through random screening and analysis of structure-activity relationships. An important strategy required for future development is to understand the basis of unexpected toxicities related to kinase inhibitors. Improvement in the documentation of toxicities of kinase inhibitor would provide a valuable database for understanding whether there are particular kinases of which inhibition should be avoided or specific substructures that result in problematic metabolites. This strategy will help to develop kinases with better selectivity benefitting the vast patient population. Also, there is a critical need for better ways to monitor target kinase inhibition in humans using minimally invasive techniques. This may include monitoring of cancer biomarkers that may serve as benchmarks for the clinical development of kinase inhibitors. The development of such technologies will help to discover and eradicate tumors using targeted kinase inhibition with minimal toxicities. There is also an urgent need for developing more non-ATP-competitive kinase inhibitors as the current collection of kinase inhibitors is limited to ABL, IKK, AKT, CHK1, MEK, SRC, IGF1R inhibitors [99, 293,294,295,296]. Furthermore, there is need to develop sophisticated modeling of chemotherapy resistance in response to kinase inhibitors. This will help to overcome kinase resistance and allow for the systematic application of combinations of kinase inhibitors. Furthermore, novel pre-clinical models are required to identify the best cocktails of kinase inhibitors combined with natural bioactives. Advanced high-throughput cell-based screening using well-defined phosphorylation readouts should be established. However, it may prove challenging to screen and develop natural kinase inhibitors using the cellular readout only. It is also important to understand that kinase inhibitors are not only important for the treatment of cancer, but also help us better understand the physiological roles of kinases. In the field of oncology, kinase inhibitors are proving to be well tolerated compared with conventional cytotoxic chemotherapeutic treatments. The future of kinase-targeted therapeutics in cancer appears promising, and implementation of these strategies will help to achieve therapeutic advances and overcome treatment hindrances.
Conclusions
By transferring the γ-phosphate from the ATP-cofactor onto diverse substrates, kinases regulate key cellular functions. As many human diseases result from mutations and overexpression of kinases, this enzyme class symbolizes an important targeted strategy for drug development. Kinases also play indispensable roles in signaling pathways that regulate tumor cell functions. Deregulation of kinases leads to a variety of pathophysiological changes triggering cancer cell proliferation and metastases. Hyperactivation of kinases also increases anti-apoptotic effects. Currently, about one-third of all protein targets under research in the pharmaceutical industry are kinase-based. Kinase inhibitors represent targeted therapy resultant of the understanding of molecular genetics and molecular signaling pathways. Most of the FDA-approved kinase inhibitors target ATP binding site of kinase enzymes and display therapeutic indications against tumorigenesis. This class of therapeutics represents a transformation from conventional chemotherapy to targeted cancer treatment. Kinase inhibitors have overcome a major drawback of traditional cancer treatment as it effectively discriminates between normal non-malignant cells and rapidly proliferating cancer cells. This leads to fewer off-target effects and low toxicities in the cancer patient population. Kinase inhibitors are also often useful in combination with cytotoxic chemotherapy or radiation therapy. A vital challenge for clinical use of kinase inhibitors in the prevention of drug-resistant cancer stem cells. This phenomenon occurs due to cellular pressure to compensate for the loss of function of an important kinase. Pharmacogenomic factors including gene polymorphisms also contribute to primary kinase drug-resistance. Due to the clinical importance of kinase inhibitors, multiple strategies are required to overcome resistance mechanisms and develop more effective targeted therapies. A key approach is to allosterically induce and stabilize inactive kinase conformations. In the future, scientific advances may eventually allow scientists to combine mutagenesis screens through next generation sequencing and proteomic techniques with the computational modeling of compound interactions with all possible mutant variants of a targeted kinase. This will lead to the development of well-tolerated kinase inhibitors compared to traditional chemotherapeutic treatments. Overall, kinase inhibitors represent a new and promising approach to cancer therapy, one that is already providing beneficial clinical effects.
Abbreviations
- ABL:
-
Abelson murine leukemia viral oncogene
- Abl:
-
Abelson murine leukemia viral oncogene homolog 1
- Akt:
-
Protein kinase B
- ALK:
-
Anaplastic lymphoma kinase
- ATM:
-
Ataxia telangiectasia mutated
- Aur A & B:
-
Aurora kinase A & B, B-Raf
- BRAF:
-
Proto-oncogene
- BTK:
-
Bruton agammaglobulinemia tyrosine kinase
- CDK:
-
Cyclin-dependent kinase
- CHK1:
-
Checkpoint kinase 1
- c-Kit:
-
Proto-oncogene c-Kit or Mast/stem cell growth factor receptor
- c-Met:
-
c-MET proto-oncogene
- c-Ret:
-
c-RET proto-oncogene
- c-SRC:
-
Proto-oncogene tyrosine-protein kinase
- CTK:
-
Cytoplasmic tyrosine kinase
- c-YES:
-
c-Yes proto-oncogene (pp62c-Yes)
- EGFR:
-
Epidermal growth factor receptor
- ERBB2:
-
V-Erb-B2 avian erythroblastic leukemia viral oncogene homolog
- Fes:
-
Feline sarcoma oncogene
- FGFRs:
-
Fibroblast growth factor receptors
- Flt3, Flt-4:
-
Fms-like tyrosine kinase 3, 4
- HER-2:
-
Human epidermal growth factor receptor-2
- IGR-R:
-
Insulin-like growth factor 1 receptor
- IKK:
-
IκB kinase
- JAK2:
-
Janus kinase 2
- KIT:
-
V-Kit hardy-zuckerman 4 feline sarcoma viral oncogene homolog
- LK:
-
Lipid kinase
- MAPK:
-
Mitogen-activated protein kinases
- MEK:
-
MEK kinase gene
- mRCC:
-
Metastatic renal cell carcinoma
- mTOR:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PDGFRs:
-
Platelet-derived growth factor receptors
- PDGFR-α:
-
Platelet-derived growth factor receptor α
- PDGFR-β:
-
Platelet-derived growth factor receptor β
- PI3K:
-
Phosphatidylinositol-3-kinase
- PI3KCA:
-
Phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha
- PIP3:
-
Phosphatidylinositol-3, 4, 5-triphosphate,
- PKCi:
-
Protein kinase Ci
- PLKs:
-
Polo-like kinases
- PTEN:
-
Phosphatase and tensin homolog
- RNAi:
-
RNA interference
- ROCK1:
-
Rho-associated, coiled-coil-containing protein kinase 1
- RON:
-
Recepteur d’Origine Nantais
- RSK2:
-
Ribosomal protein kinase 2
- RTK:
-
Receptor tyrosine kinase
- S/T Kinase:
-
Serine/threonine kinase
- S6K:
-
Ribosomal protein S6 kinase
- SGLT1:
-
Sodium/glucose cotransporter 1
- shRNA:
-
A small hairpin RNA
- SK1:
-
Sphingosine kinase 1
- SRC:
-
Proto-oncogene tyrosine-protein kinase c
- STK11/LKB1:
-
Serine/threonine kinase 11 or liver kinase B1
- Trkb:
-
Tropomyosin-related kinase B
- VEGFR-2:
-
Vascular endothelial growth factor receptor 2
- VEGFRs:
-
Vascular endothelial growth factor receptors
References
Coussens L, Parker PJ, Rhee L, Yang-Feng TL, Chen E, Waterfield MD, Francke U, Ullrich A. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science. 1986;233:859–67.
Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34.
Fabbro D, Cowan-Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR review 14. Br J Pharmacol. 2015;172:2675–700.
Köstler WJ, Zielinski CC. Targeting Receptor Tyrosine Kinases in Cancer. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease. New York: Spring; 2015. p. 225–78.
Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011;30:3477–88.
Kittler H, Tschand P. Driver mutations in the mitogen-activated protein kinase pathway: the seeds of good and evil. Br J Dermat. 2018;178:26-27.
Bardelli A, Parsons DW, Silliman N, Ptak J, Szabo S, Saha S, Markowitz S, Willson JK, Parmigiani G, Kinzler KW. Mutational analysis of the tyrosine kinome in colorectal cancers. Science. 2003;300:949.
Bartram CR, de Klein A, Hagemeijer A, van Agthoven T, van Kessel AG, Bootsma D, Grosveld G, Ferguson-Smith MA, Davies T, Stone M: Translocation of c-abl oncogene correlates with the presence of a Philadelphia chromosome in chronic myelocytic leukaemia. 1983.
Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–83.
Sato S, Sanjo H, Takeda K, Ninomiya-Tsuji J, Yamamoto M, Kawai T, Matsumoto K, Takeuchi O, Akira S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol. 2005;6:1087–95.
Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–98.
Chong ZZ, Shang YC, Wang S, Maiese K. A critical kinase cascade in neurological disorders: PI3K, Akt and mTOR. Future Neurol. 2012;7:733–48.
Tabit CE, Shenouda SM, Holbrook M, Fetterman JL, Kiani S, Frame AA, Kluge MA, Held A, Dohadwala MM, Gokce N. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95.
Shibuya M, Suzuki Y, Sugita K, Saito I, Sasaki T, Takakura K, Nagata I, Kikuchi H, Takemae T, Hidaka H. Effect of AT877 on cerebral vasospasm after aneurysmal subarachnoid hemorrhage: results of a prospective placebo-controlled double-blind trial. J Neurosurg. 1992;76:571–7.
Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127.
Davies S, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105.
Druker BJ, Guilhot F, O'Brien SG, Gathmann I, Kantarjian H, Gattermann N, Deininger MW, Silver RT, Goldman JM, Stone RM. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–17.
Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399–401.
Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino) thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–61.
Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–36.
Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–32.
Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009;37:359–65.
Kirkland LO, McInnes C. Non-ATP competitive protein kinase inhibitors as anti-tumor therapeutics. Biochem Pharmacol. 2009;77:1561–71.
Tamaoki T, Nomoto H, Takahashi I, Kato Y, Morimoto M, Tomita F. Staurosporine, a potent inhibitor of phospholipidCa++ dependent protein kinase. Biochem Biophys Res Commun. 1986;135:397–402.
Shukla S, Chufan E, Singh S, Skoumbourdis A, Kapoor K, Boxer M, Duveau D, Thomas C, Talele T, Ambudkar S. Elucidation of the structural basis of interaction of the BCR-ABL kinase inhibitor, nilotinib (Tasigna) with the human ABC drug transporter P-glycoprotein. Leukemia. 2014;
Jonker DJ, O'Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au H-J, Berry SR, Krahn M, Price T, Simes RJ. Cetuximab for the treatment of colorectal cancer. N Engl J Med. 2007;357:2040–8.
Hudis CA. Trastuzumab—mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51.
Noble ME, Endicott JA, Johnson LN. Protein kinase inhibitors: insights into drug design from structure. Science. 2004;303:1800–5.
Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554.
Ogino S, Lochhead P, Giovannucci E, Meyerhardt J, Fuchs C, Chan A. Discovery of colorectal cancer PIK3CA mutation as potential predictive biomarker: power and promise of molecular pathological epidemiology. Oncogene. 2014;33:2949–55.
Vora SR, Juric D, Kim N, Mino-Kenudson M, Huynh T, Costa C, Lockerman EL, Pollack SF, Liu M, Li X. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26:136–49.
Levine DA, Bogomolniy F, Yee CJ, Lash A, Barakat RR, Borgen PI, Boyd J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin Cancer Res. 2005;11:2875–8.
Oda K, Stokoe D, Taketani Y, McCormick F. High frequency of coexistent mutations of PIK3CA and PTEN genes in endometrial carcinoma. Cancer Res. 2005;65:10669–73.
Lee JW, Soung YH, Kim SY, Lee HW, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. PIK3CA gene is frequently mutated in breast carcinomas and hepatocellular carcinomas. Oncogene. 2005;24:1477–80.
Czech MP. PIP2 and PIP3: complex roles at the cell surface. Cell. 2000;100:603–6.
Mirza AM, Kohn AD, Roth RA, McMahon M. Oncogenic transformation of cells by a conditionally active form of the protein kinase Akt/PKB. Cell Growth Differ Publ Am Assoc Cancer Res. 2000;11:279–92.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, Vassiliou GS, Bench AJ, Boyd EM, Curtin N. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61.
Steensma DP, Dewald GW, Lasho TL, Powell HL, McClure RF, Levine RL, Gilliland DG, Tefferi A. The JAK2 V617F activating tyrosine kinase mutation is an infrequent event in both “atypical” myeloproliferative disorders and myelodysplastic syndromes. Blood. 2005;106:1207–9.
Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–65.
Weihua Z, Tsan R, Huang W-C, Wu Q, Chiu C-H, Fidler IJ, Hung M-C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell. 2008;13:385–93.
Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500.
Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339–46.
Chen S-J, Luan J, Zhang H-S, Ruan C-P, Xu X-Y, Li Q-Q, Wang N-H. EGFR-mediated G1/S transition contributes to the multidrug resistance in breast cancer cells. Mol Biol Rep. 2012;39:5465–71.
Sahin Ö, Fröhlich H, Löbke C, Korf U, Burmester S, Majety M, Mattern J, Schupp I, Chaouiya C, Thieffry D. Modeling ERBB receptor-regulated G1/S transition to find novel targets for de novo trastuzumab resistance. BMC Syst Biol. 2009;3:1.
Marumoto T, Zhang D, Saya H. Aurora-A—a guardian of poles. Nat Rev Cancer. 2005;5:42–50.
Gritsko TM, Coppola D, Paciga JE, Yang L, Sun M, Shelley SA, Fiorica JV, Nicosia SV, Cheng JQ. Activation and overexpression of centrosome kinase BTAK/aurora-a in human ovarian cancer. Clin Cancer Res. 2003;9:1420–6.
Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–200.
Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, Cheng JQ. Aurora-a abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–82.
Sun C, Chan F, Briassouli P, Linardopoulos S. Aurora kinase inhibition downregulates NF-κB and sensitises tumour cells to chemotherapeutic agents. Biochem Biophys Res Commun. 2007;352:220–5.
Qi G, Ogawa I, Kudo Y, Miyauchi M, Siriwardena B, Shimamoto F, Tatsuka M, Takata T. Aurora-B expression and its correlation with cell proliferation and metastasis in oral cancer. Virchows Arch. 2007;450:297–302.
Chieffi P, Cozzolino L, Kisslinger A, Libertini S, Staibano S, Mansueto G, De Rosa G, Villacci A, Vitale M, Linardopoulos S. Aurora B expression directly correlates with prostate cancer malignancy and influence prostate cell proliferation. Prostate. 2006;66:326–33.
Khan J, Ezan F, Crémet J-Y, Fautrel A, Gilot D, Lambert M, Benaud C, Troadec M-B, Prigent C. Overexpression of active aurora-C kinase results in cell transformation and tumour formation. PLoS One. 2011;6:e26512.
Keith WM, Kenichi N, Sara W. Recent advances of MEK inhibitors and their clinical progress. Curr Top Med Chem. 2007;7:1364–78.
Faivre S, Kroemer G, Raymond E. Current development of mTOR inhibitors as anticancer agents. Nat Rev Drug Discov. 2006;5:671–88.
Hu Y, Fang X, Dunham SM, Prada C, Stachowiak EK, Stachowiak MK. 90-kDa ribosomal S6 kinase is a direct target for the nuclear fibroblast growth factor receptor 1 (FGFR1) ROLE IN FGFR1 SIGNALING. J Biol Chem. 2004;279:29325–35.
Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M, Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Investig New Drugs. 2012;30:2411–32.
Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz H-J, Zichner T, Lambert SR, Ryzhova M, Quang DAK. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–32.
Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–12.
Ward CW, Lawrence MC, Streltsov VA, Adams TE, McKern NM. The insulin and EGF receptor structures: new insights into ligand-induced receptor activation. Trends Biochem Sci. 2007;32:129–37.
Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25.
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81.
Libermann TA, Nusbaum HR, Razon N, Kris R, Lax I, Soreq H, Whittle N, Waterfield MD, Ullrich A, Schlessinger J: Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. 1985.
Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244:707.
Eswarakumar V, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005;16:139–49.
Brewer MR, Choi SH, Alvarado D, Moravcevic K, Pozzi A, Lemmon MA, Carpenter G. The juxtamembrane region of the EGF receptor functions as an activation domain. Mol Cell. 2009;34:641–51.
Collett MS, Erikson R. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc Natl Acad Sci. 1978;75:2021–4.
Castagna M, Takai Y, Kaibuchi K, Sano K, Kikkawa U, Nishizuka Y. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257:7847–51.
Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide-dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–41.
Ward NE, O'Brian CA. Kinetic analysis of protein kinase C inhibition by staurosporine: evidence that inhibition entails inhibitor binding at a conserved region of the catalytic domain but not competition with substrates. Mol Pharmacol. 1992;41:387-92.
Knighton DR, Zheng J, Ten Eyck LF, Ashford VA, Xuong N-H, Taylor SS, Sowadski JM. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate-dependent protein kinase. Science. 1991;253:407–14.
Zheng J, Knighton DR, Taylor SS, Xuong NH, Sowadski JM, Eyck LF. Crystal structures of the myristylated catalytic subunit of cAMP-dependent protein kinase reveal open and closed conformations. Protein Sci. 1993;2:1559-73.
Asano T, Ikegaki I, Satoh SI, Seto M, Sasaki Y. A Protein Kinase Inhibitor, Fasudil (AT-877): A Novel Approach to Signal Transduction Therapy. Cardiovasc Drug Rev. 1998;16:76–87.
Liu J, Farmer JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin a and FKBP-FK506 complexes. Cell. 1991;66:807–15.
Hopkin MD, Baxendale IR, Ley SV. A flow-based synthesis of Imatinib: the API of Gleevec. Chem Commun. 2010;46:2450–2.
Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood. 1975;45:321–34.
Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, Wang C, Davies SM, Gaynon PS, Trigg M. Improved early event-free survival with imatinib in Philadelphia chromosome–positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol. 2009;27:5175–81.
Capdeville R, Buchdunger E, Zimmermann J, Matter A. Glivec (STI571, imatinib), a rationally developed, targeted anticancer drug. Nat Rev Drug Discov. 2002;1:493–502.
Steinberg M. Dasatinib: a tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia and philadelphia chromosome—positive acute lymphoblastic leukemia. Clin Ther. 2007;29:2289–308.
Kantarjian H, Giles F, Wunderle L, Bhalla K, O'Brien S, Wassmann B, Tanaka C, Manley P, Rae P, Mietlowski W. Nilotinib in imatinib-resistant CML and Philadelphia chromosome–positive ALL. N Engl J Med. 2006;354:2542–51.
Motzer RJ, Hoosen S, Bello CL, Christensen JG: Sunitinib malate for the treatment of solid tumours: a review of current clinical data. 2006.
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc J-F, de Oliveira AC, Santoro A, Raoul J-L, Forner A. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–90.
Sternberg C, Szczylik C, Lee E, Salman P, Mardiak J, Davis I, Pandite L, Chen M, McCann L, Hawkins R. A randomized, double-blind phase III study of pazopanib in treatment-naive and cytokine-pretreated patients with advanced renal cell carcinoma (RCC). In: ASCO annual meeting proceedings; 2009. p. 5021.
Pal SK, Azad AA, Bhatia S, Drabkin HA, Costello B, Sarantopoulos J, Kanesvaran R, Lauer R, Starodub AN, Hauke RJ: A phase I/II trial of BNC105P with everolimus in metastatic renal cell carcinoma (mRCC. Clin Cancer Res 2015:clincanres. 3370.2014.
Massarweh S, Romond E, Black EP, Van Meter E, Shelton B, Kadamyan-Melkumian V, Stevens M, Elledge R. Erratum to: a phase II study of combined fulvestrant and everolimus in patients with metastatic estrogen receptor (ER)-positive breast cancer after aromatase inhibitor (AI) failure. Breast Cancer Res Treat. 2015:1–1.
Abdel-Rahman O, Fouad M. Everolimus-based combination for the treatment of advanced gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs): biological rationale and critical review of published data. Tumor Biol. 2015:1–12.
Scholtens A, Foppen MG, Blank C, van Thienen J, van Tinteren H, Haanen J. Vemurafenib for BRAF V600 mutated advanced melanoma: results of treatment beyond progression. Eur J Cancer. 2015;
Chougnet CN, Borget I, Leboulleux S, de la Fouchardiere C, Bonichon F, Criniere L, Niccoli P, Bardet S, Schneegans O, Zanetta S. Vandetanib for the treatment of advanced medullary thyroid cancer outside a clinical trial: results from a French cohort. Thyroid. 2015;
Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT: Three-year efficacy, overall survival, and safety of ruxolitinib therapy in patients with myelofibrosis from the COMFORT-I study. Haematologica 2015:haematol. 2014.115840.
Costa DB, Shaw AT, Ou S-HI, Solomon BJ, Riely GJ, Ahn M-J, Zhou C, Shreeve SM, Selaru P, Polli A: Clinical experience with Crizotinib in patients with advanced ALK-rearranged non–small-cell lung cancer and brain metastases. J Clin Oncol 2015:JCO. 2014.2059. 0539.
Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, Michellys P-Y, Awad MM, Yanagitani N, Kim S. The ALK inhibitor ceritinib overcomes crizotinib resistance in non–small cell lung cancer. Cancer Discovery. 2014;4:662–73.
Shaw AT, Kim D-W, Mehra R, Tan DS, Felip E, Chow LQ, Camidge DR, Vansteenkiste J, Sharma S, De Pas T. Ceritinib in ALK-rearranged non–small-cell lung cancer. N Engl J Med. 2014;370:1189–97.
Dar AC, Shokat KM. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu Rev Biochem. 2011;80:769–95.
Roskoski R. Classification of small molecule protein kinase inhibitors based upon the structures of their drug-enzyme complexes. Pharmacol Res. 2016;103:26–48.
Zuccotto F, Ardini E, Casale E, Angiolini M. Through the “gatekeeper door”: exploiting the active kinase conformation. J Med Chem. 2009;53:2681–94.
Gavrin LK, Saiah E. Approaches to discover non-ATP site kinase inhibitors. Med Chem Comm. 2013;4:41–51.
Lamba V, Ghosh I. New directions in targeting protein kinases: focusing upon true allosteric and bivalent inhibitors. Curr Pharm Des. 2012;18:2936–45.
Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol. 2006;2:358–64.
Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39.
Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–51.
Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov. 2011;10:111–26.
Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol. 2010;244:190–5.
Kufareva I, Abagyan R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J Med Chem. 2008;51:7921–32.
Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33.
Berndt N, Karim RM, Schönbrunn E. Advances of small molecule targeting of kinases. Curr Opin Chem Biol. 2017;39:126–32.
Eglen R, Reisine T. Drug discovery and the human kinome: recent trends. Pharmacol Ther. 2011;130:144–56.
Allen LF, Sebolt-Leopold J, Meyer MB. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). In Seminars in oncology. Amsterdam: Elsevier; 2003. p. 105–16.
Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig P-A, Reinecke M, Ruprecht B, Petzoldt S, Meng C. The target landscape of clinical kinase drugs. Science. 2017;358:eaan4368.
Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P. Allosteric inhibitors of Bcr-abl–dependent cell proliferation. Nat Chem Biol. 2006;2:95–102.
Dong Q, Dougan DR, Gong X, Halkowycz P, Jin B, Kanouni T, O’Connell SM, Scorah N, Shi L, Wallace MB. Discovery of TAK-733, a potent and selective MEK allosteric site inhibitor for the treatment of cancer. Bioorg Med Chem Lett. 2011;21:1315–9.
Grimsby J, Sarabu R, Corbett WL, Haynes N-E, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–3.
Guertin KR, Grimsby J. Small molecule glucokinase activators as glucose lowering agents: a new paradigm for diabetes therapy. Curr Med Chem. 2006;13:1839–43.
Blanc J, Geney R, Menet C. Type II kinase inhibitors: an opportunity in cancer for rational design. Anti-Cancer Agents Med Chem (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 2013;13:731–47.
Maekawa T, Ashihara E, Kimura S. The Bcr-Abl tyrosine kinase inhibitor imatinib and promising new agents against Philadelphia chromosome-positive leukemias. Int J Clin Oncol. 2007;12:327–40.
Gumireddy K, Baker SJ, Cosenza SC, John P, Kang AD, Robell KA, Reddy MR, Reddy EP. A non-ATP-competitive inhibitor of BCR-ABL overrides imatinib resistance. Proc Natl Acad Sci U S A. 2005;102:1992–7.
Cohen MS, Zhang C, Shokat KM, Taunton J. Structural bioinformatics-based design of selective, irreversible kinase inhibitors. Science. 2005;308:1318–21.
Kwak EL, Sordella R, Bell DW, Godin-Heymann N, Okimoto RA, Brannigan BW, Harris PL, Driscoll DR, Fidias P, Lynch TJ. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci U S A. 2005;102:7665–70.
Leproult E, Barluenga S, Moras D, Wurtz J-M, Winssinger N. Cysteine mapping in conformationally distinct kinase nucleotide binding sites: application to the design of selective covalent inhibitors. J Med Chem. 2011;54:1347–55.
Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–59.
Singh J, Petter RC, Baillie TA, Whitty A. The resurgence of covalent drugs. Nat Rev Drug Discov. 2011;10:307–17.
Barf T, Kaptein A. Irreversible protein kinase inhibitors: balancing the benefits and risks. J Med Chem. 2012;55:6243–62.
Zhao Z, Liu Q, Bliven S, Xie L, Bourne PE. Determining cysteines available for covalent inhibition across the human kinome. J Med Chem. 2017;60:2879–89.
Liao B-C, Lin C-C, Yang JC-H. Second and third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr Opin Oncol. 2015;27:94–101.
Wang A, Yan X-E, Wu H, Wang W, Hu C, Chen C, Zhao Z, Zhao P, Li X, Wang L. Ibrutinib targets mutant-EGFR kinase with a distinct binding conformation. Oncotarget. 2016;7:69760.
Schwartz PA, Murray BW. Protein kinase biochemistry and drug discovery. Bioorg Chem. 2011;39:192–210.
Roskoski R. Ibrutinib inhibition of Bruton protein-tyrosine kinase (BTK) in the treatment of B cell neoplasms. Pharmacol Res. 2016;113:395–408.
Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004;64:3958–65.
Kobayashi S, Ji H, Yuza Y, Meyerson M, Wong K-K, Tenen DG, Halmos B. An alternative inhibitor overcomes resistance caused by a mutation of the epidermal growth factor receptor. Cancer Res. 2005;65:7096–101.
Moebitz H. The ABC of protein kinase conformations. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2015;1854:1555–66.
Cohen P. Protein kinases—the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–15.
Vieth M, Sutherland JJ, Robertson DH, Campbell RM. Kinomics: characterizing the therapeutically validated kinase space. Drug Discov Today. 2005;10:839–46.
Chen H, Ma J, Li W, Eliseenkova AV, Xu C, Neubert TA, Miller WT, Mohammadi M. A molecular brake in the kinase hinge region regulates the activity of receptor tyrosine kinases. Mol Cell. 2007;27:717–30.
Cowan-Jacob SW, Ramage P, Stark W, Fendrich G, Jahnke W. Structural biology of protein tyrosine kinases. In: Protein Tyrosine Kinases. New York: Springer; 2006. p. 187–230.
Nolen B, Taylor S, Ghosh G. Regulation of protein kinases: controlling activity through activation segment conformation. Mol Cell. 2004;15:661–75.
Johnson LN, Noble ME, Owen DJ. Active and inactive protein kinases: structural basis for regulation. Cell. 1996;85:149–58.
Scapin G. Protein kinase inhibition: different approaches to selective inhibitor design. Curr Drug Targets. 2006;7:1443–54.
Angell RM, Angell TD, Bamborough P, Bamford MJ, Chung C-W, Cockerill SG, Flack SS, Jones KL, Laine DI, Longstaff T. Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg Med Chem Lett. 2008;18:4433–7.
Han S, Mistry A, Chang JS, Cunningham D, Griffor M, Bonnette PC, Wang H, Chrunyk BA, Aspnes GE, Walker DP. Structural characterization of proline-rich tyrosine kinase 2 (PYK2) reveals a unique (DFG-out) conformation and enables inhibitor design. J Biol Chem. 2009;284:13193–201.
Traxler P, Bold G, Buchdunger E, Caravatti G, Furet P, Manley P, O'Reilly T, Wood J, Zimmermann J. Tyrosine kinase inhibitors: from rational design to clinical trials. Med Res Rev. 2001;21:499–512.
Fabbro D, Ruetz S, Buchdunger E, Cowan-Jacob SW, Fendrich G, Liebetanz J, Mestan J, O'Reilly T, Traxler P, Chaudhuri B. Protein kinases as targets for anticancer agents: from inhibitors to useful drugs. Pharmacol Ther. 2002;93:79–98.
Garuti L, Roberti M, Bottegoni G. Non-ATP competitive protein kinase inhibitors. Curr Med Chem. 2010;17:2804–21.
Garuti L, Roberti M, Bottegoni G. Irreversible protein kinase inhibitors. Curr Med Chem. 2011;18:2981–94.
Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2:58–61.
Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G. Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell. 2008;134:793–803.
Wissner A, Fraser HL, Ingalls CL, Dushin RG, Floyd MB, Cheung K, Nittoli T, Ravi MR, Tan X, Loganzo F. Dual irreversible kinase inhibitors: quinazoline-based inhibitors incorporating two independent reactive centers with each targeting different cysteine residues in the kinase domains of EGFR and VEGFR-2. Bioorg Med Chem. 2007;15:3635–48.
Cohen MS, Hadjivassiliou H, Taunton J. A clickable inhibitor reveals context-dependent autoactivation of p90 RSK. Nat Chem Biol. 2007;3:156–60.
Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Amsterdam: Seminars in oncology Elsevier; 2006. p. 407–20.
García-Echeverría C, Traxler P, Evans DB. ATP site-directed competitive and irreversible inhibitors of protein kinases. Med Res Rev. 2000;20:28–57.
Cervantes F, Vannucchi AM, Kiladjian J-J, Al-Ali HK, Sirulnik A, Stalbovskaya V, McQuitty M, Hunter DS, Levy RS, Passamonti F. Three-year efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for myelofibrosis. Blood. 2013;122:4047–53.
Walker I, Newell H. Do molecularly targeted agents in oncology have reduced attrition rates? Nat Rev Drug Discov. 2009;8:15–6.
Toniatti C, Jones P, Graham H, Pagliara B, Draetta G. Oncology drug discovery: planning a turnaround. Cancer Discovery. 2014;4:397–404.
Yr L, Zhu W, Jl Z, Jq H, Zhao YZ, Zhang W, Han BH, Yao YH, Jiang LY, Li SQ. The evaluation of efficacy and safety of sunitinib on EGFR-TKI pretreated advanced non-small cell lung cancer patients in China. Clin Respir J. 2014;8:206–12.
Takeda M, Nakagawa K. Toxicity profile of epidermal growth factor receptor tyrosine kinase inhibitors in patients with epidermal growth factor receptor gene mutation-positive lung cancer. Mole Clin Oncol. 2017;6:3–6.
Fakih M, Vincent M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr Oncol. 2010;17:S18.
Hayman SR, Leung N, Grande JP, Garovic VD. VEGF inhibition, hypertension, and renal toxicity. Curr Oncol Rep. 2012;14:285–94.
Hilton JF, Shapiro GI. Aurora kinase inhibition as an anticancer strategy. J Clin Oncol. 2014;32:57–9.
Ahmed K, Unger G, Kren BT, Trembley JH. Targeting CK2 for cancer therapy using a nanomedicine approach. In Protein kinase CK2 cellular function in normal and disease states. New York: Springer; 2015. p. 299–315.
Gallorini M, Cataldi A, di Giacomo V. Cyclin-dependent kinase modulators and cancer therapy. BioDrugs. 2012;26:377–91.
Cance WG, Kurenova E, Marlowe T, Golubovskaya V. Disrupting the scaffold to improve focal adhesion kinase–targeted cancer therapeutics. Sci Signal. 2013;6:pe10.
Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. In: Phosphoinositide 3-kinase in health and disease. New York: Springer; 2011. p. 31–56.
Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6:88.
WeiSZ L, Efferth T. Polo-like kinase 1 as target for cancer therapy. Exp Hematol Oncol. 2012;1:38.
Krisenko MO, Geahlen RL. Calling in SYK: SYK's dual role as a tumor promoter and tumor suppressor in cancer. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2015;1853:254–63.
Anagnostopoulou V, Pediaditakis I, Alkahtani S, Alarifi SA, Schmidt E-M, Lang F, Gravanis A, Charalampopoulos I, Stournaras C. Differential effects of dehydroepiandrosterone and testosterone in prostate and colon cancer cell apoptosis: the role of nerve growth factor (NGF) receptors. Endocrinology. 2013;154:2446–56.
Sharma A, Madhunapantula SV, Gowda R, Berg A, Neves RI, Robertson GP. Identification of aurora kinase B and Wee1-like protein kinase as downstream targets of V600E B-RAF in melanoma. Am J Pathol. 2013;182:1151–62.
Fedorov O, Müller S, Knapp S. The (un) targeted cancer kinome. Nat Chem Biol. 2010;6:166–9.
Katayama H, Brinkley WR, Sen S. The aurora kinases: role in cell transformation and tumorigenesis. Cancer Metastasis Rev. 2003;22:451–64.
Knapp S, Sundström M. Recently targeted kinases and their inhibitors—the path to clinical trials. Curr Opin Pharmacol. 2014;17:58–63.
Morgensztern D, Campo MJ, Dahlberg SE, Doebele RC, Garon E, Gerber DE, Goldberg SB, Hammerman PS, Heist RS, Hensing T. Molecularly targeted therapies in non–small-cell lung cancer annual update 2014. J Thorac Oncol. 2015;10:S1–S63.
Sequist LV, Yang JC-H, Yamamoto N, O'Byrne K, Hirsh V, Mok T, Geater SL, Orlov S, Tsai C-M, Boyer M. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34.
Gainor JF, Shaw AT. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J Clin Oncol. 2013;31:3987–96.
Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81.
Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–5.
Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–51.
von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, Suter T, Arahmani A, Rouchet N, Clark E. Adjuvant Pertuzumab and Trastuzumab in early HER2-positive breast cancer. N Engl J Med. 2017;
Hayashi T, Seiler R, Oo HZ, Jäger W, Moskalev I, Awrey S, Dejima T, Todenhöfer T, Li N, Fazli L. Targeting HER2 with T-DM1, an antibody cytotoxic drug conjugate, is effective in HER2 over expressing bladder cancer. J Urol. 2015;194:1120–31.
Ferrara N, Hillan KJ, Gerber H-P, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391.
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45.
Sokolowska-Wedzina A, Chodaczek G, Chudzian J, Borek A, Zakrzewska M, Otlewski J: High-affinity internalizing human scFv-fc antibody for targeting FGFR1-overexpressing lung cancer. Mol Cancer Res 2017:molcanres. 0136.2016.
Mohammadi M, Nejatollahi F, Ghasemi Y, Faraji SN. Anti-metastatic and anti-invasion effects of a specific anti-MUC18 scFv antibody on breast cancer cells. Appl Biochem Biotechnol. 2017;181:379–90.
Sandberg D, Tolmachev V, Velikyan I, Olofsson H, Wennborg A, Feldwisch J, Carlsson J, Lindman H, Sörensen J. Intra-image referencing for simplified assessment of HER2-expression in breast cancer metastases using the Affibody molecule ABY-025 with PET and SPECT. Eur J Nucl Med Mol Imaging. 2017:1–10.
Hu S-z, Shively L, Raubitschek A, Sherman M, Williams LE, Wong JY, Shively JE, Wu AM. Minibody: a novel engineered anti-carcinoembryonic antigen antibody fragment (single-chain Fv-C H 3) which exhibits rapid, high-level targeting of xenografts. Cancer Res. 1996;56:3055–61.
Jafari R, M Zolbanin N, Rafatpanah H, Majidi J, Kazemi T. Fc-fusion proteins in therapy: An updated view. Curr Med Chem. 2017;24:1228–37.
Mukherjee B, Tomimatsu N, Amancherla K, Camacho CV, Pichamoorthy N, Burma S. The dual PI3K/mTOR inhibitor NVP-BEZ235 is a potent inhibitor of ATM-and DNA-PKCs-mediated DNA damage responses. Neoplasia. 2012;14:34–IN38.
Yu P, Laird AD, Du X, Wu J, Won K-A, Yamaguchi K, Hsu PP, Qian F, Jaeger CT, Zhang W. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014;13:1078–91.
Powles T, Oudard S, Escudier BJ, Brown JE, Hawkins RE, Castellano DE, Ravaud A, Staehler MD, Rini BI, Lin W: A randomized phase II study of GDC-0980 versus everolimus in metastatic renal cell carcinoma (mRCC) patients (pts) after VEGF-targeted therapy (VEGF-TT). In ASCO annual meeting proceedings 2014: 4525.
Dowsett M, Koehler M, Millham R, Borzillo G, A'Hern R, Pierce K, Barton J, Giorgetti C. Phase Ii Randomized Study Of Pre-Operative Pf-04691502 Plus Letrozole Compared With Letrozole (L) In Patients With Estrogen Receptor-Positive, Her2-Negative Early Breast Cancer (BC). In: Annals Of Oncology. Oxford Univ Press Great Clarendon St, Oxford; 2012: 44–44.
Bedard PL, Grilley-Olson JE, Cornfeld M, Cartee L, Warwick S, Razak AA, Stayner L-A, Wu Y, Greenwood R, Viana-Gilmartin V. Abstract CT205: a phase I dose-escalation study of trametinib (T) in combination with continuous or intermittent GSK2126458 (GSK458) in patients (pts) with advanced solid tumors. Cancer Res. 2014;74:CT205.
Haïk S, Marcon G, Mallet A, Tettamanti M, Welaratne A, Giaccone G, Azimi S, Pietrini V, Fabreguettes J-R, Imperiale D. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13:150–8.
Eriksson A, Österroos A, Hassan S, Gullbo J, Rickardson L, Jarvius M, Nygren P, Fryknäs M, Höglund M, Larsson R. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015;5:e307.
Shapiro GI, Bell-McGuinn KM, Molina JR, Bendell J, Spicer J, Kwak EL, Pandya SS, Millham R, Borzillo G, Pierce KJ. First-in-human study of PF-05212384 (PKI-587), a small-molecule, intravenous, dual inhibitor of PI3K and mTOR in patients with advanced cancer. Clin Cancer Res. 2015.
Bauer TM, Patel MR, Infante JR. Targeting PI3 kinase in cancer. Pharmacol Ther. 2015;146:53-60.
Eradat HA, Coutre SE, Barrientos JC, Rai KR, Farber CM, Hillmen P, Sharman JP, Ghia P, Coiffier B, Walewski JA. A phase III, randomized, double-blind, placebo-controlled study evaluating the efficacy and safety of idelalisib (GS-1101) in combination with bendamustine and rituximab for previously treated chronic lymphocytic leukemia (CLL). In: ASCO annual meeting proceedings; 2013. p. TPS7133.
Mikhail S, Albanese C, Pishvaian MJ. Cyclin-dependent kinase inhibitors and the treatment of gastrointestinal cancers. Am J Pathol. 2015;85:1185-97.
Marotta V, Sciammarella C, Vitale M, Colao A, Faggiano A. The evolving field of kinase inhibitors in thyroid cancer. Crit Rev Oncol Hematol. 2015;93:60–73.
Mohamed A, Krajewski K, Cakar B, Ma CX. Targeted therapy for breast cancer. Am J Pathol. 2013;183:1096–112.
Marotta V, Franzese MD, Del Prete M, Chiofalo MG, Ramundo V, Esposito R, Marciello F, Pezzullo L, Carratù A, Vitale M. Targeted therapy with kinase inhibitors in aggressive endocrine tumors. Expert Opin Pharmacother. 2013;14:1187–203.
Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–703.
Yadav V, Chen SH, Yue YG, Buchanan S, Beckmann RP, Peng SB. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacol Ther. 2015;149:139-49.
Nicholson R, Gee J, Harper M. EGFR and cancer prognosis. Eur J Cancer. 2001;37:9–15.
Ménard S, Pupa SM, Campiglio M, Tagliabue E. Biologic and therapeutic role of HER2 in cancer. Oncogene. 2003;22:6570–8.
Jeong KJ, Cho KH, Panupinthu N, Kim H, Kang J, Park CG, Mills GB, Lee HY. EGFR mediates LPA-induced proteolytic enzyme expression and ovarian cancer invasion: inhibition by resveratrol. Mol Oncol. 2013;7:121–9.
Huang C-Y, Chan C-Y, Chou I-T, Lien C-H, Hung H-C, Lee M-F. Quercetin induces growth arrest through activation of FOXO1 transcription factor in EGFR-overexpressing oral cancer cells. J Nutr Biochem. 2013;24:1596–603.
Sun X-D, Liu X-E, Huang D-S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol Med Rep. 2012;6:1267–70.
Masuda M, Wakasaki T, Toh S, Shimizu M, Adachi S. Chemoprevention of head and neck cancer by green tea extract: EGCG—the role of EGFR signaling and “lipid raft”. J Oncol. 2011;2011.
Thomas T, Venkiteswaran S. Polyphenolic phytochemical curcumin suppresses HER-2 protein phosphorylation and gene expression in breast cancer cells. Cancer Res. 2012;72:5445.
Boly R, Gras T, Lamkami T, Guissou P, Serteyn D, Kiss R, Dubois J. Quercetin inhibits a large panel of kinases implicated in cancer cell biology. Int J Oncol. 2011;38:833–42.
Khan A, Aljarbou AN, Aldebasi YH, Faisal SM, Khan MA. Resveratrol suppresses the proliferation of breast cancer cells by inhibiting fatty acid synthase signaling pathway. Cancer Epidemiol. 2014;38:765–72.
Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34:280–300.
Fu S, Kurzrock R. Development of curcumin as an epigenetic agent. Cancer. 2010;116:4670–6.
Chadalapaka G, Jutooru I, Safe S. Celastrol decreases specificity proteins (Sp) and fibroblast growth factor receptor-3 (FGFR3) in bladder cancer cells. Carcinogenesis. 2012;33:886–94.
Sakurai R, Villarreal P, Husain S, Liu J, Sakurai T, Tou E, Torday JS, Rehan VK. Curcumin protects the developing lung against long-term hyperoxic injury. Am J Phys Lung Cell Mol Phys. 2013;305:L301–11.
Narasimhan M, Ammanamanchi S. Curcumin blocks RON tyrosine kinase–mediated invasion of breast carcinoma cells. Cancer Res. 2008;68:5185–92.
Xia Y, Lian S, Khoi PN, Yoon HJ, Han JY, Chay KO, Kim KK, Jung YD. Chrysin inhibits cell invasion by inhibition of Recepteur d'origine Nantais via suppressing early growth response-1 and NF-κB transcription factor activities in gastric cancer cells. Int J Oncol.
Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–42.
Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer. 2013;13:559–71.
Dutta P, Li WX. Role of the JAK-STAT Signalling pathway in cancer. eLS. https://doi.org/10.1002/9780470015902.a0025214
Byun S, Lee KW, Jung SK, Lee EJ, Hwang MK, Lim SH, Bode AM, Lee HJ, Dong Z. Luteolin inhibits protein kinase Cε and c-Src activities and UVB-induced skin cancer. Cancer Res. 2010;70:2415–23.
Wolanin K, Magalska A, Mosieniak G, Klinger R, Mckenna S, Vejda S, Sikora E, Piwocka K. Curcumin affects components of the chromosomal passenger complex and induces mitotic catastrophe in apoptosis-resistant Bcr-Abl-expressing cells. Mol Cancer Res. 2006;4:457–69.
Milligan SA, Burke P, Coleman DT, Bigelow RL, Steffan JJ, Carroll JL, Williams BJ, Cardelli JA. The green tea polyphenol EGCG potentiates the Antiproliferative activity of c-met and epidermal growth factor receptor inhibitors in non–small cell lung cancer cells. Clin Cancer Res. 2009;15:4885–94.
Rajasingh J, Raikwar HP, Muthian G, Johnson C, Bright JJ. Curcumin induces growth-arrest and apoptosis in association with the inhibition of constitutively active JAK–STAT pathway in T cell leukemia. Biochem Biophys Res Commun. 2006;340:359–68.
Wang G, Wang J, Chen X, Du S, Li D, Pei Z, Lan H, Wu L. The JAK2/STAT3 and mitochondrial pathways are essential for quercetin nanoliposome-induced C6 glioma cell death. Cell Death Dis. 2013;4:e746.
Vara JÁF, Casado E, de Castro J, Cejas P, Belda-Iniesta C, González-Barón M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204.
Li Y, Liu J, Liu X, Xing K, Wang Y, Li F, Yao L. Resveratrol-induced cell inhibition of growth and apoptosis in MCF7 human breast cancer cells are associated with modulation of phosphorylated Akt and caspase-9. Appl Biochem Biotechnol. 2006;135:181–92.
Sexton É, Van Themsche C, Leblanc K, Parent S, Lemoine P, Asselin E. Resveratrol interferes with AKT activity and triggers apoptosis in human uterine cancer cells. Mol Cancer. 2006;5:45.
Aziz MH, Nihal M, Fu VX, Jarrard DF, Ahmad N. Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3′-kinase/Akt pathway and Bcl-2 family proteins. Mol Cancer Ther. 2006;5:1335–41.
Sheth S, Jajoo S, Kaur T, Mukherjea D, Sheehan K, Rybak LP, Ramkumar V. Resveratrol reduces prostate cancer growth and metastasis by inhibiting the Akt/MicroRNA-21 pathway. PLoS One. 2012;7:e51655.
Roy P, Kalra N, Prasad S, George J, Shukla Y. Chemopreventive potential of resveratrol in mouse skin tumors through regulation of mitochondrial and PI3K/AKT signaling pathways. Pharm Res. 2009;26:211–7.
Jiang H, Shang X, Wu H, Gautam SC, Al-Holou S, Li C, Kuo J, Zhang L, Chopp M. Resveratrol downregulates PI3K/Akt/mTOR signaling pathways in human U251 glioma cells. J Exp Ther Oncol. 2009;8:25.
Fröjdö S, Cozzone D, Vidal H, Pirola L. Resveratrol is a class IA phosphoinositide 3-kinase inhibitor. Biochem J. 2007;406:511–8.
Park D, Jeong H, Lee MN, Koh A, Kwon O, Yang YR, Noh J, Suh P-G, Park H, Ryu SH. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci Rep. 2016;6:21772.
Kumamoto T, Fujii M, Hou D-X. Akt is a direct target for myricetin to inhibit cell transformation. Mol Cell Biochem. 2009;332:33–41.
Weir NM, Selvendiran K, Kutala VK, Tong L, Vishwanath S, Rajaram M, Tridandapani S, Anant S, Kuppusamy P. Curcumin induces G2/M arrest and apoptosis in cisplatin-resistant human ovarian cancer cells by modulating Akt and p38 MAPK. Cancer Biol Therapy. 2007;6:178–84.
Granado-Serrano AB, Martín MA, Bravo L, Goya L, Ramos S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J Nutr. 2006;136:2715–21.
Gulati N, Laudet B, Zohrabian VM, Murali R, Jhanwar-Uniyal M. The antiproliferative effect of quercetin in cancer cells is mediated via inhibition of the PI3K-Akt/PKB pathway. Anticancer Res. 2006;26:1177–81.
Tang FY, Nguyen N, Meydani M. Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. Int J Cancer. 2003;106:871–8.
Shin DY, Lee WS, Lu JN, Kang MH, Ryu CH, Kim GY, Kang HS, Shin SC, Choi YH. Induction of apoptosis in human colon cancer HCT-116 cells by anthocyanins through suppression of Akt and activation of p38-MAPK. Int J Oncol. 2009;35:1499–504.
Kavitha K, Kowshik J, Kishore TKK, Baba AB, Nagini S. Astaxanthin inhibits NF-κB and Wnt/β-catenin signaling pathways via inactivation of Erk/MAPK and PI3K/Akt to induce intrinsic apoptosis in a hamster model of oral cancer. Biochimica et Biophysica Acta (BBA)-General Subjects. 2013;1830:4433–44.
Lin Y, Shi R, Wang X, Shen H-M. Luteolin, a flavonoid with potentials for cancer prevention and therapy. Curr Cancer Drug Targets. 2008;8:634.
Kaur P, Shukla S, Gupta S. Plant flavonoid apigenin inactivates Akt to trigger apoptosis in human prostate cancer: an in vitro and in vivo study. Carcinogenesis. 2008;29:2210–7.
Sharma PS, Sharma R, Tyagi R. Inhibitors of cyclin dependent kinases: useful targets for cancer treatment. Curr Cancer Drug Targets. 2008;8:53–75.
Wolter F, Akoglu B, Clausnitzer A, Stein J. Downregulation of the cyclin D1/Cdk4 complex occurs during resveratrol-induced cell cycle arrest in colon cancer cell lines. J Nutr. 2001;131:2197–203.
Shan B-E, Wang M-X, Li R-Q. Quercetin inhibit human SW480 colon cancer growth in association with inhibition of cyclin D1 and survivin expression through Wnt/β-catenin signaling pathway. Cancer Investig. 2009;27:604–12.
Mukhopadhyay A, Banerjee S, Stafford LJ, Xia C, Liu M, Aggarwal BB. Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene. 2002;21:8852–61.
Aggarwal BB, Banerjee S, Bharadwaj U, Sung B, Shishodia S, Sethi G. Curcumin induces the degradation of cyclin E expression through ubiquitin-dependent pathway and up-regulates cyclin-dependent kinase inhibitors p21 and p27 in multiple human tumor cell lines. Biochem Pharmacol. 2007;73:1024–32.
Thangapazham RL, Singh AK, Sharma A, Warren J, Gaddipati JP, Maheshwari RK. Green tea polyphenols and its constituent epigallocatechin gallate inhibits proliferation of human breast cancer cells in vitro and in vivo. Cancer Lett. 2007;245:232–41.
Van Aller GS, Carson JD, Tang W, Peng H, Zhao L, Copeland RA, Tummino PJ, Luo L. Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res Commun. 2011;406:194–9.
Li M, Zhang Z, Hill DL, Wang H, Zhang R. Curcumin, a dietary component, has anticancer, chemosensitization, and radiosensitization effects by down-regulating the MDM2 oncogene through the PI3K/mTOR/ETS2 pathway. Cancer Res. 2007;67:1988–96.
Adhami VM, Syed DN, Khan N, Mukhtar H. Dietary flavonoid fisetin: a novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem Pharmacol. 2012;84:1277–81.
Seo YJ, Kim BS, Chun SY, Park YK, Kang KS, Kwon TG. Apoptotic effects of genistein, biochanin-a and apigenin on LNCaP and PC-3 cells by p21 through transcriptional inhibition of polo-like kinase-1. J Korean Med Sci. 2011;26:1489–94.
Xie F, Lang Q, Zhou M, Zhang H, Zhang Z, Zhang Y, Wan B, Huang Q, Yu L. The dietary flavonoid luteolin inhibits aurora B kinase activity and blocks proliferation of cancer cells. Eur J Pharm Sci. 2012;46:388–96.
Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–9.
Hasima N, Aggarwal BB. Cancer-linked targets modulated by curcumin. Int J Biochem Mol Biol. 2012;3:328.
Xavier CP, Lima CF, Preto A, Seruca R, Fernandes-Ferreira M, Pereira-Wilson C. Luteolin, quercetin and ursolic acid are potent inhibitors of proliferation and inducers of apoptosis in both KRAS and BRAF mutated human colorectal cancer cells. Cancer Lett. 2009;281:162–70.
Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82.
Chen L-X, He Y-J, Zhao S-Z, Wu J-G, Wang J-T, Zhu L-M, Lin T-T, Sun B-C, Li X-R. Inhibition of tumor growth and vasculogenic mimicry by curcumin through down-regulation of the EphA2/PI3K/MMP pathway in a murine choroidal melanoma model. Cancer Biol Therapy. 2011;11:229–35.
Khan N, Afaq F, Saleem M, Ahmad N, Mukhtar H. Targeting multiple signaling pathways by green tea polyphenol (−)-epigallocatechin-3-gallate. Cancer Res. 2006;66:2500–5.
Wu JC, Lai CS, Badmaev V, Nagabhushanam K, Ho CT, Pan MH. Tetrahydrocurcumin, a major metabolite of curcumin, induced autophagic cell death through coordinative modulation of PI3K/Akt-mTOR and MAPK signaling pathways in human leukemia HL-60 cells. Mol Nutr Food Res. 2011;55:1646–54.
Heffernan-Stroud LA, Obeid LM. Sphingosine kinase 1 in cancer. Adv Cancer Res. 2012;201.
Lim KG, Gray AI, Pyne S, Pyne NJ. Resveratrol dimers are novel sphingosine kinase 1 inhibitors and affect sphingosine kinase 1 expression and cancer cell growth and survival. Br J Pharmacol. 2012;166:1605–16.
Kim Y, Kim W-J, Cha E-J. Quercetin-induced growth inhibition in human bladder cancer cells is associated with an increase in Ca2+−activated K+ channels. Korean J Physiol Pharmacol. 2011;15:279–83.
Brizuela L, Dayon A, Doumerc N, Ader I, Golzio M, Izard J-C, Hara Y, Malavaud B, Cuvillier O. The sphingosine kinase-1 survival pathway is a molecular target for the tumor-suppressive tea and wine polyphenols in prostate cancer. FASEB J. 2010;24:3882–94.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9.
Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401–9.
Chell V, Balmanno K, Little A, Wilson M, Andrews S, Blockley L, Hampson M, Gavine P, Cook S. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene. 2013;32:3059–70.
Foguel D, Cordeiro Y, Ventura S, Graña-Montes AA, Cortines J. The importance of a gatekeeper residue on. 2014.
Chen R, An T, Zhao J, Duan J, Wang Z, Zhuo M, Wang S, Wu M, Li Z, Yang X. Non-invasive, Dynamic and Quantitative Analysis of T790M mutation in matched plasma DNA of pre-and post-EGFR-TKIs treatment for advanced NSCLC. 第 13 届全国肺癌学术大会论文汇编. 2013.
Pauwels D, Sweron B, Cools J. The N676D and G697R mutations in the kinase domain of FLT3 confer resistance to the inhibitor AC220. Haematologica. 2012;97:1773–4.
Gibbons DL, Pricl S, Kantarjian H, Cortes J, Quintás-Cardama A. The rise and fall of gatekeeper mutations? The BCR-ABL1 T315I paradigm. Cancer. 2012;118:293–9.
Metzgeroth G, Erben P, Martin H, Mousset S, Teichmann M, Walz C, Klippstein T, Hochhaus A, Cross N, Hofmann W. Limited clinical activity of nilotinib and sorafenib in FIP1L1-PDGFRA positive chronic eosinophilic leukemia with imatinib-resistant T674I mutation. Leukemia. 2012;26:162–4.
Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, Eisenberg BL, von Mehren M, Fletcher CD, Sandau K. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74.
Yun C-H, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong K-K, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci. 2008;105:2070–5.
Suda K, Onozato R, Yatabe Y, Mitsudomi T. EGFR T790M mutation: a double role in lung cancer cell survival? J Thorac Oncol. 2009;4:1–4.
Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale C-M, Zhao X, Christensen J. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–43.
Janku F, Wheler JJ, Westin SN, Moulder SL, Naing A, Tsimberidou AM, Fu S, Falchook GS, Hong DS, Garrido-Laguna I. PI3K/AKT/mTOR inhibitors in patients with breast and gynecologic malignancies harboring PIK3CA mutations. J Clin Oncol. 2012;30:777–82.
Blencke S, Zech B, Engkvist O, Greff Z, Őrfi L, Horváth Z, Kéri G, Ullrich A, Daub H. Characterization of a conserved structural determinant controlling protein kinase sensitivity to selective inhibitors. Chem Biol. 2004;11:691–701.
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73.
Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92.
Heymach JV, Nilsson M, Blumenschein G, Papadimitrakopoulou V, Herbst R. Epidermal growth factor receptor inhibitors in development for the treatment of non–small cell lung cancer. Clin Cancer Res. 2006;12:4441s–5s.
Patel RY, Doerksen RJ. Protein kinase− inhibitor database: structural variability of and inhibitor interactions with the protein kinase P-loop. J Proteome Res. 2010;9:4433–42.
Knight ZA, Lin H, Shokat KM. Targeting the cancer kinome through polypharmacology. Nat Rev Cancer. 2010;10:130.
Warburton C, Dragowska WH, Gelmon K, Chia S, Yan H, Masin D, Denyssevych T, Wallis AE, Bally MB. Treatment of HER-2/neu overexpressing breast cancer xenograft models with trastuzumab (Herceptin) and gefitinib (ZD1839): drug combination effects on tumor growth, HER-2/neu and epidermal growth factor receptor expression, and viable hypoxic cell fraction. Clin Cancer Res. 2004;10:2512–24.
Normanno N, Campiglio M, Perrone F, De Luca A, Menard S. Is the gefitinib plus trastuzumab combination feasible in breast cancer patients? Ann Oncol. 2005;16:1709.
Shah DR, Shah RR, Morganroth J. Tyrosine kinase inhibitors: their on-target toxicities as potential indicators of efficacy. Drug Saf. 2013;36:413–26.
Orphanos GS, Ioannidis GN, Ardavanis AG. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009;48:964–70.
Mayor S. Targeting cardiovascular complications. Lancet Oncol. 2006;7:282.
Moslehi JJ, Deininger M. Tyrosine kinase inhibitor–associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33:4210–8.
Bernards R, Brummelkamp TR, Beijersbergen RL. shRNA libraries and their use in cancer genetics. Nat Methods. 2006;3:701–6.
Modugno M, Casale E, Soncini C, Rosettani P, Colombo R, Lupi R, Rusconi L, Fancelli D, Carpinelli P, Cameron AD. Crystal structure of the T315I Abl mutant in complex with the aurora kinases inhibitor PHA-739358. Cancer Res. 2007;67:7987–90.
Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, Huntly B, Fabbro D, Fendrich G, Hall-Meyers E. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7:129–41.
Gumireddy K, Reddy MR, Cosenza SC, Nathan RB, Baker SJ, Papathi N, Jiang J, Holland J, Reddy EP. ON01910, a non-ATP-competitive small molecule inhibitor of Plk1, is a potent anticancer agent. Cancer Cell. 2005;7:275–86.
Copland M, Pellicano F, Richmond L, Allan EK, Hamilton A, Lee FY, Weinmann R, Holyoake TL. BMS-214662 potently induces apoptosis of chronic myeloid leukemia stem and progenitor cells and synergizes with tyrosine kinase inhibitors. Blood. 2008;111:2843–53.
Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–4.
Barnett S, Defeo-Jones D, Fu S, Hancock P, Haskell K, Jones R, Kahana J, Kral A, Leander K, Lee L. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385:399–408.
Converso A, Hartingh T, Garbaccio RM, Tasber E, Rickert K, Fraley ME, Yan Y, Kreatsoulas C, Stirdivant S, Drakas B. Development of thioquinazolinones, allosteric Chk1 kinase inhibitors. Bioorg Med Chem Lett. 2009;19:1240–4.
Fabbro D, Manley PW, Jahnke W, Liebetanz J, Szyttenholm A, Fendrich G, Strauss A, Zhang J, Gray NS, Adrian F. Inhibitors of the Abl kinase directed at either the ATP-or myristate-binding site. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics. 2010;1804:454–62.
Author information
Authors and Affiliations
Contributions
All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bhullar, K.S., Lagarón, N.O., McGowan, E.M. et al. Kinase-targeted cancer therapies: progress, challenges and future directions. Mol Cancer 17, 48 (2018). https://doi.org/10.1186/s12943-018-0804-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0804-2