
Abstract
Regulation of the PI-3 kinase (PI3K)/Akt signalling pathway is essential for maintaining the integrity of fundamental cellular processes, cell growth, survival, death and metabolism, and dysregulation of this pathway is implicated in the development and progression of cancers. Receptor tyrosine kinases (RTKs) are major upstream regulators of PI3K/Akt signalling. The phosphatase and tensin homologue (PTEN), a well characterised tumour suppressor, is a prime antagonist of PI3K and therefore a negative regulator of this pathway. Loss or inactivation of PTEN, which occurs in many tumour types, leads to overactivation of RTK/PI3K/Akt signalling driving tumourigenesis. Cellular PTEN levels are tightly regulated by a number of transcriptional, post-transcriptional and post-translational regulatory mechanisms. Of particular interest, transcription of the PTEN pseudogene, PTENP1, produces sense and antisense transcripts that exhibit post-transcriptional and transcriptional modulation of PTEN expression respectively. These additional levels of regulatory complexity governing PTEN expression add to the overall intricacies of the regulation of RTK/PI-3 K/Akt signalling. This review will discuss the regulation of oncogenic PI3K signalling by PTEN (the regulator) with a focus on the modulatory effects of the sense and antisense transcripts of PTENP1 on PTEN expression, and will further explore the potential for new therapeutic opportunities in cancer treatment.
Similar content being viewed by others

Background
The phosphatase and tensin homologue (PTEN) is essential for normal cell maintenance and is well characterised as a key tumour suppressor [1]. PTEN is pivotal in the regulation of the receptor tyrosine kinase (RTK) PI-3 kinase (PI3K)/Akt signalling pathway and, as such, even small changes in PTEN expression have been shown to have major consequences for normal cellular function [2,3,4,5]. The PTEN protein translocates between the nucleus and the cytoplasm enabling PTEN-specific compartmentalised functions [6, 7]. At the molecular level, PTEN expression and cellular abundance is tightly regulated at the transcriptional, post-translational and post-transcriptional levels. In recent years, there has been much interest in the PTEN pseudogene (PTENP1) as a novel negative and positive modulator of PTEN expression.
The PI3K/Akt pathway is activated subsequent to RTK activation. Hyperactivation of PI3K/Akt signalling has been reported in many types of human cancers, thus targeting the regulators in this pathway has attractive therapeutic potential. As such, a large number RTKs and PI3K candidates are under development and a few are now being used successfully in cancer patient treatments. Nevertheless, adverse side effects and therapeutic resistance to RTK/PI3K inhibition remains problematic.
This review provides an overview of PTEN as a major regulator of RTK//PI3K/Akt activation and, in turn, discusses the regulation of PTEN by well characterised mechanisms, and more recently, by a novel mechanism involving regulation of PTEN by its pseudogene (PTENP1).
The clinical importance of PTEN inactivation in cancer and other diseases and the therapeutic potential of PTEN and PTENP1 modulation of the RTK/PI3K/Akt is discussed.
PTEN sequence and structure
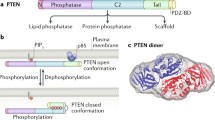
The PTEN gene is encoded in 9 exons and has a 1212 nucleotide (nt) open reading frame. The gene encodes a polypeptide of 403 amino acids with a relative molecular mass of 47 kDa [8,9,10,11,12]. The PTEN protein consists of two major domains, the N-terminal phosphatase catalytic domain (residues 7–185) and a C-terminal domain (residues 186–351) [13,14,15] (Fig. 1). These two domains together form a minimal catalytic unit and comprise almost the entire protein, excluding only a very short N-terminal tail. The N-terminal phosphatase domain of PTEN contains a consensus PI (4,5) P2-binding motif. The C-terminal domain of PTEN contains the lipid binding C2 domain which confers affinity for phospholipid membranes in vitro. The C2 domain is believed to be required for the correct positioning of PTEN at the plasma membrane, the site of the lipid substrates of PTEN [13, 16,17,18]. The C-terminal tail of PTEN, consisting of the last 50 amino acids, also contains several phosphorylation sites that are critical for protein stability. Protein stability is dependent on the phosphorylation of S380, T382, and T383. Mutations within these sites reduce both the protein half-life and PTEN phosphatase activity [19]. Phosphorylation-defective mutants of PTEN have decreased protein stability and dephosphorylated PTEN is degraded by proteasome-mediated mechanisms [20, 21].

PTEN protein structure and sites of post-translational modification. PTEN is composed of 403 amino acids and is characterised by five functional domains: a phosphatidylinositol-4,5-bisphosphate (PIP2)-binding domain (PBD), a phosphatase domain containing the catalytic core, a C2 domain with putative ubiquitination sites, two PEST (proline, glutamic acid, serine, threonine) domains for degradation, and a PDZ interaction motif for protein-protein interactions. Post-translational regulation of PTEN occurs by ubiquitination (Ub) of Lys residues within the PBD and C2 domain, by oxidation, SUMOylation within the C2 domain, and acetylation on protein tyrosine phosphatase (PTPase) and PDZ-binding sites. Furthermore, PTEN is regulated by phosphorylation of specific serine and threonine residues within the C2 domain and C-tail terminal of PTEN (Modified from [14, 15])
PTEN cellular function and regulation of PTEN nuclear-cytoplasmic transport
Subcellular localisation of PTEN is crucial for its normal cellular function and its role as a tumour suppressor. PTEN translocates between the cytoplasm and nucleus of the cell and is known to have specific functions in both cellular compartments [6]. In the cytoplasm, PTEN interacts with its cytoplasmic targets to regulate cell proliferation, cell cycle progression, apoptosis, cell adhesion, migration and invasion. In the nucleus, PTEN plays a role in maintaining chromosomal stability and in DNA double strand break repair [6, 22], hence maintaining genome integrity. The mechanism(s) by which PTEN can translocate between the nucleus and cytoplasm of cells has not been completely characterised as PTEN does not appear to contain a traditional or consensus nuclear localisation signal (NLS), although putative NLS-like sequences have been identified [7].
The tumour suppressive role of cytoplasmic PTEN is through antagonism of PI3K/Akt signalling and the role of nuclear PTEN is to maintain chromosomal integrity and centromere stability. Mislocalisation of PTEN between the nucleus and the cytoplasm may lead to malignant growth, thus, the subcellular localisation of PTEN is closely regulated and several regulatory mechanisms have been identified. PTEN lacks a typical NLS, and monoubiquitination, active transport and passive diffusion has been identified as transport mechanisms for PTEN [23]. Monoubiquitination, catalysed by the ubiquitin-protein ligase, developmental downregulated-4-1 (NEDD4–1), enhances PTEN transport to the nucleus [24]. Nuclear pores are large enough to allow proteins of less than 60 kDa to pass through [25], thus making PTEN a perfect candidate for passage through the nuclear pore by diffusion. Ran (Ras-related nuclear protein) GTPase is able to actively transport PTEN into the nucleus [26]. A cytoplasmic localisation signal has been identified in the N-terminal domain of PTEN, spanning residues 19–25. Mutations in these residues (except residue 22) appear to increase nuclear localisation of PTEN, however the mechanism is not known [27]. Furthermore, mutations occurring at PTEN phosphorylation sites also appear to alter its nuclear-cytoplasmic localisation [26]. The stage of the cell cycle can also modulate the subcellular localisation of PTEN and the nuclear-cytoplasmic partitioning of PTEN can differentially regulate cell cycle progression and apoptosis [28]. The cell cycle dependent PTEN localisation can be regulated by Ca2+ mediated interaction with the major vault protein (MVP) [29]. Bipartite nuclear localisation sequences in PTEN are required for MVP mediated nuclear import and four such bipartite NLS have been identified and are responsible for MVP interaction [28].
Regulation of PTEN abundance and activity
Since PTEN is involved in, and plays a central role in many cellular processes, the level of PTEN is tightly regulated by a number of cellular mechanisms which act at the transcriptional, post-transcriptional and post-translational levels and, as mentioned, small decreases in PTEN abundance or activity, may lead to tumourigenesis [2,3,4,5]. These regulatory mechanisms maintain the activity and abundance of PTEN at the required level under normal physiological conditions [30]. There are a number of well-established and documented regulatory mechanisms acting to modulate PTEN gene expression and protein abundance, stability and activity. However, more recently, PTEN regulation by the processed pseudogene of PTEN (PTENP1) is gaining much interest as an added level of complexity to the stringent regulation of PTEN expression.
In this section, we provide an overview of the well documented mechanisms of PTEN regulation, discuss the more recently defined mechanisms of PTEN regulation by small non-coding RNAs, microRNAs (miRNAs) and the exciting emerging field of pseudogene long non-coding RNAs (lncRNA). Importantly, we describe how the web of interactions between PTEN, PTEN-targeting miRNAs and the sense and antisense transcripts of the PTEN pseudogene, PTENP1, regulate RTK-dependent PI3K/Akt signalling [31,32,33].
Transcriptional regulation of PTEN
A number of transcription factors bind directly to the PTEN promoter to either activate or repress PTEN transcription. Such factors include the early growth response transcriptional factor 1 (EGR1), peroxisome proliferator-activated receptor gamma (PPARγ) [34, 35], activating transcription factor 2 (ATF2) [36] and the tumour suppressor, p53 [37]. p53 and PTEN share regulatory interactors and regulate each other in a feedback loop mechanism [38]. p53 upregulates PTEN transcription by binding to the functional p53 binding element upstream of the PTEN promoter [39]. PTEN is transcriptionally repressed by the zinc finger-like proteins SNAIL and SLUG, which are transcription factors competing with p53 for the PTEN promoter binding region [40]. Other transcription factors such as the polycomb group protein, CBF-1 and c-Jun, nuclear factor kappa-B and the antisense transcript of the PTEN pseudogene (PTENP1(AS)), also bind to the PTEN promoter and negatively regulate PTEN transcription [2, 15, 41, 42].
Post-transcriptional regulation of PTEN by miRNA
Recent advances in genomic technology have revolutionised the way we view cellular regulation, providing a greater appreciation and understanding of the complexity of non-coding genes and non-coding gene function(s). Once regarded as junk DNA, these non-coding genes have been shown to be critical in gene regulation and to play important roles in disease development and control. PTEN is regulated at the post-transcriptional level by miRNAs which are comprised of small ncRNAs approximately 14–24 nt in length [31]. These ncRNAs bind to their target messenger RNA (mRNA) at seed regions, known as miRNA recognition elements [43, 44], which are located within the 3’untranslated region (UTR) of the specific target mRNAs [45, 46]. Recent studies have revealed miRNA binding sites are also present in the coding regions, the 5’UTR region and even the promoter region of target mRNAs [46,47,48]. miRNA function is dependent on binding affinity with the target mRNA, therefore, binding of miRNAs can either lead to degradation of target through perfect complementary binding or inhibition of translation through imperfect binding [49, 50]. PTEN is known to be post-transcriptionally regulated by miRNAs binding within its 3’UTR, which results in blockage of translation, and a consequent decrease in PTEN abundance [51]. miRNAs commonly known to bind to, and repress PTEN include miR-17, miR-19, miR-21, miR-26, and miR-214 [32, 52, 53]. MiRNAs have been shown to possess functional roles in cancer development and progression [54], and a variety of oncogenic miRNAs (oncomirs) have recently been shown to bind specifically to PTEN transcripts, blocking PTEN translation, and to be cancer-type dependent. Overexpressing PTEN-specific miRNAs has the potential to enhance cancer progression, and specific PTEN-targeting oncomirs have been linked to hepatocellular carcinomas, prostate cancer, clear-cell renal carcinoma, breast cancer and endometrial cancer (Table 1). In 2010, a processed pseudogene of PTEN (PTENP1) was found to be transcribed to produce a transcript with high sequence similarity with the PTEN transcript. Further, this pseudogene transcript was ascribed a novel function by acting as a ‘decoy’ for miRNA binding of PTEN-targeting miRNAs, as discussed in more detail below [32].
Pseudogenes and post-transcriptional regulation of PTEN by its pseudogene, PTENP1
The post-transcriptional regulation of PTEN by PTENP1 is a novel mechanism and sets a paradigm for regulation of cognate genes by their pseudogenes. This regulatory mechanism may provide new targets for cancer therapy or novel designs for cancer therapeutics.
Pseudogenes
Knowledge of pseudogenes has existed for many years but their importance as post-transcriptional regulators of gene expression has only been recognised in recent years [55]. Since their initial identification, pseudogenes have been described in a wide range of species from bacteria [56], insects [57], plants [58] and animals [53]. Approximately 50% of transcribed pseudogenes in multicellular organisms exhibit evolutionarily conserved sequences across species, strongly suggesting a functional role for pseudogenes in humans and other organisms [59]. Pseudogenes are generally labelled as non-functional relatives of active genes that, over time, have lost their protein-coding ability, but share high sequence similarity with their cognate protein-coding genes. Despite the high sequence similarity, pseudogenes often contain nucleotide changes which prevent their translation to functional proteins. In the genome, pseudogenes are classified as either unitary pseudogenes, non-processed pseudogenes or processed pseudogenes. Unitary pseudogenes are those originating from native functional genes but which have lost their function due to mutations. Non-processed pseudogenes are a consequence of gene duplication while processed pseudogenes exist as an outcome of retrotransposition of mRNA transcripts [60, 61] (Fig. 2). Pseudogenes have generally been labelled as “junk” DNA as they are non-protein-coding sequences and their function, until recently, has been a mystery. Almost exact copies of their cognate genes, pseudogenes often harbour premature stop codons, deletions/insertions and frameshift mutations that cause their translation to non-functional proteins [62]. Because pseudogenes have lost the ability to produce full-length proteins, for many years, the assumption has been that they are non-functional, redundant, and evolutionary gene failures [63]. Whilst it has also been hypothesised for some time that antisense pseudogenes may bind to the sense parent gene transcript to regulate gene expression [64]. Although many pseudogenes are not transcribed due to inactive promoters, or their integration into silent regions of the genome, important roles have recently been highlighted through the discovery that some pseudogenes have the potential to regulate their protein-coding counterparts [32, 33, 61, 65]. Importantly, pseudogenes have recently been identified as modulators of disease processes, especially cancer [54, 66].

Pseudogene types shown to occur in the human genome. a Unitary pseudogenes are once functional gene sequences that have lost gene function due to the accumulation of mutations over time. b Non-processed pseudogenes are the result of direct duplication of existing genes, after which the duplicated version becomes inactivated due to the accumulation of mutations in sequences essential for gene expression. c Processed pseudogenes are the result of retrotransposition events. In this case, the mature mRNA transcript of a gene is reverse transcribed into a cDNA copy, which is then integrated into the genome of the organism. The site of integration of pseudogenes is random (Adapted from [61])
Pseudogenes are categorised as members of the LncRNA family, however some pseudogene transcripts have been shown to be processed into short interfering RNAs thereby regulating the coding genes through RNAi signalling [67]. As non-coding RNAs, pseudogenes offer an attractive control mechanism for gene regulation.
The PTENP1 pseudogene is evolutionarily conserved over many species, although the phylogenetic evolutionary history is complex (reviewed in [53]). During evolution, formation of the PTEN/PTENP1 gene families occurred through multiple gene duplication events. The human PTENP1 or ψPTEN is a processed pseudogene of PTEN located on chromosome 9p13.3. This pseudogene possesses extensive sequence identity to PTEN with only 18 nucleotide mismatches within the coding region [60, 68]. Sequence similarity between the 3′ untranslated regions (UTR) of PTEN and PTENP1 can be considered as occurring in two regions, the 5′ region with 95% sequence identity and the 3′ region with < 50% sequence identity [32]. Expression of PTENP1 leads to the production of three transcripts, two of which are antisense to PTEN (PTENP1 sense and antisense transcripts). One antisense transcript acts through binding chromatin remodelling complexes which alter H3K27me3 prevalence at the PTEN parental gene promoter [69]. The other antisense transcript is needed to stabilise the PTENP1 sense transcript, which lacks a poly-A tail.
The PTENP1 sense transcript acts as a ‘sponge’ to mop up PTEN-targeting microRNAs
Many pseudogenes, while not being able to produce a functional protein are transcribed and act at the RNA level to regulate their coding counterparts, in part, by acting as decoys for microRNA binding [67]. Some pseudogenes display a tissue-specific pattern of action, and in their role as microRNA decoys, have the potential capacity to regulate oncogenes and tumour suppressor genes with tissue specificity (reviewed in [70]). Expression of the PTENP1 sense transcript is positively correlated with PTEN cellular abundance, consistent with a mechanism whereby the sense pseudogene transcript acts as a ‘sponge’ or ‘decoy’ for microRNAs that would otherwise bind the PTEN transcript and deactivate it (Fig. 3) [32]. Most interestingly, the 3’UTR sequences of PTEN and PTENP1 share common microRNA binding sites. PTENP1 was one of the first pseudogenes reported to be transcribed as a lncRNA and reported to function as ‘sponge’, or ‘decoy’, for miRNA binding to liberate PTEN from miRNA repression, hence restoring PTEN function [32]. Through binding of PTEN-targeting miRNAs, PTENP1 sense ultimately reduces the cellular concentration of these specific miRNAs. The PTEN/PTENP1 regulatory cycle is supported by experiments in which knockdown of PTENP1 results in decreased PTEN mRNA and protein levels [33].

Regulation of PTEN by the sense and antisense transcripts of its processed pseudogene PTENP1: regulating the regulator of PI3K signalling. PTENP1 is transcribed into a sense and 2 antisense transcripts (a and b). In the cytoplasm, the sense transcript (PTENP1(S)) acts as competing endogenous RNA, competing with PTEN for the binding of PTEN-targeting miRNAs and thus freeing PTEN from miRNA-mediated repression and increasing PTEN cellular abundance. Of the 2 antisense PTENP1 transcripts, PTENP1(AS)α and PTENP1(AS)β produced, PTENP1(AS)α acts in the nucleus to negatively regulate PTEN transcription by recruiting chromatin-repressor proteins, the Enhancer of Zeste Homolog 2 and DNA methyltransferase 3a (EZH2) and DNA methyltransferase 3a (DNMT3a) to the PTEN promoter. Conversely, also in the cytoplasm, PTENP1(AS)β acts to stabilise the PTENP1(S) transcript through RNA-RNA interactions, as this the sense transcript lacks a poly(A) tail, and hence reinforces the miRNA ‘sponging’ activity of PTENP1(S) (modified from [42])
Given that PTEN is a tumour suppressor gene, the PTENP1 pseudogene has been described as a tumour suppressor lncRNA pseudogene. Through its binding of PTEN-targeting miRNAs (Table 1), PTENP1 protects PTEN from miRNA binding and inhibition of PTEN translation [32]. Thus, PTENP1 acts as a repressor (molecular sponge) of the repressors (miRNAs) of PTEN function, and, in turn, regulates the regulator (PTEN) downstream of the RTK-dependent PI3K/Akt signalling pathway. These counteracting mechanisms illustrate the importance and complexity of the PTENP1 pseudogene as a lncRNA-mediator or regulator of PTEN expression and function.
Post-translational regulation of PTEN
A number of post-translational mechanisms regulate PTEN activity and stability (half-life) and these include phosphorylation, oxidation, acetylation, ubiquitination and SUMOylation (Fig. 4).

Regulation of PTEN, a major regulator of the PI3K/AKT signalling pathway. Growth factors bind receptor tyrosine kinases (RTKs) on the extracellular cell membrane, which leads to the recruitment and binding of PI3K (directly or through adaptor proteins) to its cytoplasmic domain through its regulatory subunit (P85). Activated PI3K phosphorylates of PI(4,5)P2 to PI(3,4,5)P3, which occurs through its catalytic subunit (P110). The serine/threonine kinases Akt and PDK1 are recruited to the membrane after binding to the pleckstrin homology (PH) domain of PI(3,4,5)P3. PDK1 and mTORC2 phosphorylate and activate Akt, which phosphorylates a number of downstream protein targets with the overall effect of enhancing cell proliferation, metabolism and survival whilst inhibiting apoptosis. PTEN is a major negative regulator of PI3K/Akt signalling through its phosphoinositide phosphatase activity which acts to directly antagonise PI3K activity by dephosphorylating PI(3,4,5)P3 to PI(4,5)P2. PTEN abundance and activity is highly regulated through various complementary mechanisms working at the transcriptional, post-transcriptional and post-translational levels (modified from [14])
Phosphorylation
PTEN catalytic activity may be modulated by phosphorylation of specific sites in the C2 and C-tail domains. Phosphorylation of particular serine and threonine residues (Ser380, Thr382, Thr383 and Ser385) in the C-terminal tail of PTEN, catalysed by the action of casein kinase 2 and glycogen synthase kinase 3ß [71], results in decreased phosphatase activity. The decreased phosphatase activity is associated with greater protein stability, or protein half-life, as a consequence of the generation of a closed-conformation through interaction of the C-terminal tail with the C2 domain [20]. Dephosphorylation results in a catalytically active open-conformation, but with decreased PTEN stability and cellular half-life [1, 2, 72].
Oxidation
PTEN oxidation by H2O2 facilitates disulphide bond formation between the catalytic Cys124 and Cys71 residues, resulting in a conformational change which alters the PTEN substrate binding site and leads to loss of PTEN phosphatase activity. PTEN oxidation is reversible through the action of thiol compounds, such as thioredoxin [73], and through PTEN interacting with peroxiredoxin-1 to prevent disulphide bond formation [74].
Acetylation
In response to growth factor stimulation, lysine acetyltransferase 2B (KAT2B), also known as PCAF, acetylates PTEN on lysine residues 125 and 128, which are located within the catalytic site of PTEN, and this results in the inactivation of PTEN phosphatase activity and the stimulation of PI3K signalling [75]. Another PTEN acetylation site is located in the PTEN PDZ-binding domain at Lys 402, which is driven by the CREB-binding protein. Acetylation of Lys 402 results in the negative regulation of PTEN activity [76]. Acetylation of PTEN may be reversed by the action of sirtuin 1, which restores the phosphatase activity of PTEN [77, 78].
Ubiquitination
Ubiquitination is a post-translational regulatory mechanism influencing the degradation [24] and compartmentalisation of PTEN [79]. The C-terminal tail and C2 domains of PTEN interact with each other to form a loop, which contains a major ubiquitination site (Lys 289). PTEN can be ubiquitinated by neural precursor cell expressing NEDD4–1 [79]. Polyubiquitination of PTEN results in decreased protein stability leading to PTEN degradation by proteasome mediated decay mechanisms [24], whereas monoubiquitination of PTEN on Lys13 and Lys289 promotes the nuclear transport of PTEN [24, 80].
SUMOylation
The attachment of small ubiquitin-related modifiers (SUMO) to proteins is also a post-translational regulatory mechanism of PTEN [81]. SUMOylation of PTEN on Lys266 facilitates the recruitment of PTEN to the plasma membrane [82], whereas, SUMOylation of PTEN on Lys254 partakes in controlling the nuclear localisation of PTEN [83]. SUMO proteins are related to ubiquitin [81]. SUMOylation of the Lys289 residue, a major monoubiquitination site for PTEN, results in PTEN nuclear localisation [24, 80]. Conversely, SUMOylation at Lys289 results in the recruitment of PTEN to the plasma membrane, implicating a possible competitive action for the modification of Lys293 [15, 81].
The RTK/PI3K/Akt Signalling pathway and regulation by PTEN
RTKs are a family of transmembrane proteins with inherent phosphotyrosine kinase activity which remain inactive in the plasma membrane until activated by ligand. The RTK family consists of a number of sub-families, including the epidermal growth factor receptors (EGFRs), platelet derived growth factor receptors (PDGFRs), fibroblast growth factor receptors (FGFRs), vascular endothelial growth factor receptors (VEGFRs), insulin growth factor receptors (IGFRs), and hepatocyte growth factor receptors (HGFRs) [84,85,86]. Activation of the RTKs by their cognate growth factors, cytokines, hormones or other extracellular signalling molecules, triggers the activation of the PI3K signalling pathway. Tight regulation of cell proliferation by RTKs and their ligands is critical in cancer prevention [87].
The Class IA PI3Ks are heterodimeric enzymes consisting of a p110α /β /δ catalytic subunit and a p85 regulatory subunit and are directly activated by RTKs such as the insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-IR) [88, 89]. Class IB PI3K heterodimers consist of a p110γ catalytic subunit and a p101 regulatory subunit and are activated downstream of G-protein-coupled receptors (GPCRs). Class IA and IB PI3Ks are activated upon extracellular stimulation of RTKs or GPCRs, and, once activated, phosphorylate the D3-position of the inositol ring of phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) to generate phosphatidylinositol 3,4,5-triphosphate (PtdIns(3,4,5)P3) at the plasma membrane. Both PtdIns(3,4,5)P3 and PtdIns(3,4)P2 facilitate the recruitment of pleckstrin homology-domain containing proteins, such as the serine/threonine kinase Akt [3,4,5], to the plasma membrane. Upon phosphoinositide binding, Akt is phosphorylated at Threonine-308 (Thr308) by phosphoinositide-dependent kinase 1 (PDK1) and at Serine-473 (Ser473) by the mammalian target of rapamycin complex 2 (mTORC2), leading to activation of its kinase activity and the subsequent phosphorylation of a number of target protein [90, 91].
PTEN is a well characterised negative regulator of PI3K-dependent Akt signalling. As a phosphoinositide phosphatase, PTEN acts as a direct antagonist of PI3K action through dephosphorylation of PtdIns(3,4,5)P3 at the D3-position of the inositol ring to form PtdIns(4,5)P2 [92,93,94]. Loss of PTEN, which occurs in many tumours, drives PI3K/Akt hyperactivation. The phosphoprotein phosphatase activity of PTEN has been linked to cancer signalling through dephosphorylation of protein targets such as focal adhesion kinase (FAK), insulin receptor substrate 1, c-SRC or PTEN itself [12, 95,96,97]. However, it is well established that, of the two activities of PTEN, it is the phosphoinositide phosphatase activity that plays the major tumour suppressor role [11, 98]. Of most importance, Akt hyperactivation resulting from the loss of PTEN lipid phosphatase function is the foremost oncogenic driving force in PTEN-deficient cancers. The protein phosphatase activity of PTEN is thought to be most important in the regulation of cell adhesion, cell migration, tumour metastasis and angiogenesis [99, 100]. Due to its importance in maintaining normal physiological functions in the cell, tight regulation of PTEN abundance and activity is essential for balancing cellular homeostasis (i.e. balancing cell proliferation and cell death).
Clinical importance of PTEN mutations and PTEN deletions in cancer and other diseases
Germline mutations of PTEN have been linked to three autosomal dominant inherited cancer syndromes with overlapping features: Cowden Syndrome (CS), Bannayan Riley Ruvalcaba syndrome (BRRS), and Proteus syndrome (PS), all characterised by increased susceptibility to cancer [101]. These syndromes are notable for the presence of hamartomas, benign tumours in which differentiation is normal, but cells are highly disorganised. In these seemingly unrelated syndromes, PTEN germline mutations account for 80% of CS, 60% of BRRS, 20% of PS patients. A detailed comparative list of these PTEN mutations (CS, BRRS and PS), including their gene position, any associated amino acid changes and disease associations is provided in Table 3 in reference [102]. The features of CS include hamartomatous overgrowth of tissues and a predisposition to developing tumours of the breast, thyroid, endometrium and, in some instances, colon cancer [102]. An additional feature of CS is an increase in insulin sensitivity, which has been linked with PTEN haploinsufficiency-associated enhancement of PI3K/Akt signalling [103]. The majority of CS patients have macrocephaly and some patients also have autism spectrum disorders related to germline mutations of PTEN [104,105,106,107,108]. Over 80 different germline PTEN mutations have been identified, with specific mutations, including the R130X, Y178X nonsense and H93R, D252G, F241S missense mutations shown to be associated with the autism and macrocephaly characteristics and leading to the proposal that PTEN sequencing may allow genetic phenotyping and subsequent diagnosis of a subset of autistic patients [99].
BRRS is a rare hereditary autosomal dominant syndrome identified by developmental delays, megencephaly, speckled penis and lipomatosis [109]. There is some overlap in the germline mutations between CS and BRRS, however each syndrome has distinct PTEN germline mutations and, overall, distinct CS-associated mutations are located mainly in the 5′ exon-encoded region whereas BRRS distinct mutations occur mainly in the 3′-encoded C2 domain region [102].
The aetiology of PS is mostly considered as a germline mosaic mutation with features such as lipomas, overgrowth and benign neoplasms (hamartomas) [109]. At least three unique PS-associated PTEN mutations have been identified, W111R, C211X, M35 T and PS-like has a common mutation linked with both CS and BRRS [102].
Germline PTEN mutations associated with the hamartoma syndromes, as described above, are associated with patient predisposition to cancer. However, most cancers are associated with somatic alterations of PTEN being described in over 50% of all tumours of various types. In fact, PTEN is one of the most common targets for mutations in human sporadic cancers, with a mutational frequency rivalling that of p53 [1, 8, 9, 110, 111]. PTEN has been shown to be lost or inactivated by multiple mechanisms in a wide spectrum of human cancer types (Table 2). The spectrum of cancer-associated somatic mutations encompasses insertions, deletions, point mutations and epigenetic changes. Interestingly, in glioblastomas, loss of heterozygosity at the PTEN locus occurs in 60–80% of tumours and somatic mutations in 20–40% of such tumours [112]. Interestingly, haploinsufficiency or inactivation of a single PTEN allele has been shown to be sufficient for cancer development [3]. For example, key hereditary PTEN cancer-associated germline mutations and common somatic mutations with increased cancer risk have been identified in colorectal cancers [111, 113], breast cancers [114, 115], prostate cancers [116] and gliomas [117]. In tumours, PTEN is inactivated by various mechanisms, including not only mutations, but also deletions, transcriptional silencing through promoter hypermethylation, subcellular mislocalisation, and alterations of cellular stability and protein half-life as well as multiple mutations (reviewed in: [1, 110]. Loss of PTEN is commonly observed in glioblastoma, thyroid, breast, endometrial, ovarian, prostate, colorectal cancers, and melanoma [8, 9, 110, 111].
As cellular PTEN concentration strongly influences cancer development, and subsequent cancer severity [5], maintenance and control of cellular PTEN levels is critical for preventing oncogenesis. For example, loss of PTEN is associated with progression of prostate cancers from the androgen-dependent to the more aggressive androgen-independent phenotypes, resistance to chemo- and radiation therapies, tumour metastasis, recurrence post-surgery, and significant overall poor prognosis for patients [118].
PTEN abnormalities extend far beyond cancer related diseases. Changes in PTEN cellular levels, and related cellular compartmentation, have also been implicated in prominent diseases such as diabetes and neurological disorders including Parkinson’s disease and Alzheimer’s disease [63, 105, 107, 119,120,121]. Inappropriate activation of the PI3K/Akt pathway, consequent to PTEN loss through gene deletions or mutations, especially those affecting the active site residues, has been suggested as a mechanism involved in adverse neuropsychiatric cell signalling [58]. Also suggested by Kitagishi and Matsuda [58] is the potential of targeting the PI3K signalling pathway in the treatment of neurological impairment such as that seen in Parkinson’s disease. PTEN haploinsufficiency also underlies profound insulin sensitivity resulting in predisposition to obesity and diabetes type II, as well as cancer [122]. A common PTEN variant, rs1102614, has been linked to peripheral insulin resistance and development of Type II diabetes [123].
Here, we have highlighted some of the more prominent diseases associated with PTEN mutations, however as more PTEN genetic data emerges, the importance of PTEN as a major checkpoint and regulator of disease will undoubtedly increase.
PTENP1 regulators in disease
Given their potential regulatory role in normal cellular function, it is not unconceivable that specific changes in pseudogene expression occur and contribute to disease progression. Examples of changing dynamics in pseudogene expression have been shown in some cancers [124, 125] and in diabetes [126], two major diseases of the developed world. The PTENP1 pseudogene, as a key player in PTEN regulation, has the potential to strongly influence tumour development and progression. Fluctuating levels of PTEN/PTENP1 are often correlated in prostate cancer samples and deletion of PTENP1 occurs frequently in some sporadic cancers such as endometrial, colon and prostate cancers, attributing a tumour suppressor function to PTENP1, that is independent of its regulation of PTEN [32, 127, 128]. A further example of the action of the PTENP1 antisense transcript is PTENP1(as) has been shown to alter doxorubicin sensitivity in cancer cells, a clinically actionable phenotype [69].
Cancer therapeutic potential of PTEN: Modulating RTK-dependent PI3K/Akt overactivation
Aberrations in the PI3K pathway are common to many cancer types and targeting the RTK/PI3K/Akt pathway continues to provide key opportunities for therapeutic intervention. Overactivation of the RTK pathway is endemic in cancer progression and tight downstream regulation of this pathway is enforced in the cell at many levels. The employment of RTK inhibitors as therapeutic agents has been a major breakthrough in the treatment of cancers such as melanoma (BCR-ABL, KIT, PDGFR), breast cancer (Herceptin 2: HER2), colorectal cancer (EGFR, VEGF) and non-small cell lung cancer (EGFR) [129], and, to-date, the Food and Drug Administration (FDA) have approved 26 kinase inhibitors for cancer treatment, of which 8 are TK inhibitors [130]. However, intrinsic (primary) and acquired (secondary) resistance to conventional drug regimes is the major challenge to overcome in cancer therapeutics. Each step in the RTK cascade is a potential cancer target. Understanding the signalling pathways associated with RTK signalling networks and targeting intermediates in the PI3K/PTEN pathway may be a step forward in diagnostics/prognostics and allow translatable approaches in new therapeutic designs to potentially overcome drug resistance.
Specific PI3K inhibitors are proving to be promising cancer targets though few have made it into successful clinical outcomes. One such inhibitor identified is the PI3Kδ inhibitor Idelalisib, currently approved for use in patients with chronic lymphocytic leukaemia, small lymphocytic lymphoma and follicular lymphoma [129]. There are a number of PI3K targeting drugs currently under development, and in various stages of clinical trials (phase II-III) from pan-class 1 PI3K inhibitors such as buparlisib (BMK120), Copanlisib (BAY80–6946) and pictilisib (GDC-094), which target all four PI3K isoforms, to PI3K isoform-specific inhibitors such as IPI-145 and Alpelisib [116]. PI3K inhibitors, BAY80–6946 (Copanlisib), GDC0032 and IPI145, which target PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ, are undergoing phase II–III trials for treatment of lymphoma, breast/uterine cancer and lymphocytic leukemia/lymphoma respectively [131, 132]. Furthermore, a number of the PI3K isoform-specific inhibitors are in stage I or II of clinical trial, including, but not limited to, NVP-BYL719 or Alpelisib (targeting PI3Kα, PI3Kβ and PI3Kγ), INK1117 or MLN1117, SAR260301, KIN-193 or AZD6482, GS-9820 (all targeting PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ), GSK2636771 (targeting PI3Kβ) and AMG319 (targeting PI3Kδ) [132,133,134,135]. Other inhibitors currently in preclinical trial are described in detail in [132,133,134,135].
Clinical trials with AKT inhibitors have shown limited clinical success, and miltefosine is currently the only approved therapy as a typical treatment for cutaneous breast cancer [136].
Targeting PTEN per se, as a cancer therapeutic strategy, is very problematic given its key role in cell regulation and proliferation and changes in PTEN expression can trigger profound biological effects. Therapeutic approaches to increase PTEN levels have anti-cancer benefits however increasing PTEN has a positive influence in tissue regeneration [137].
On the one hand, increasing functional dose/ levels of PTEN has been shown to promote its tumour suppressor activity, thus making PTEN a good candidate for cancer treatment. Insertion of PTEN protein in PTEN null prostate cancer cells (PC-3) [138] induced apoptosis and regression of PTEN-null xenograft tumors in mice [139]. Interestingly, introduction of additional exogenous PTEN expression by generating PTEN-transgenic mice, or “Super-PTEN” mice, reduced cancer susceptibility by altering cellular cells’ metabolic pathway, negatively impacting the ‘Warburg effect’, a metabolic feature of tumour cells [140]. Notably, these “Super-PTEN” mice, showed reduced body size and a decrease in cell number with a positive healthy metabolism [141]. Based on these findings, pharmaceutical delivery of functional dosage of PTEN through PTEN protein delivery, inhibition of PTEN-targeting miRNAs, and PTEN gene editing would benefit cancer patients.
On the other hand, as mentioned, decrease of functional PTEN dose increases cell growth and proliferation, which is shown to be useful in regenerative medicine for Alzheimer’s disease and ischemia however decreasing functional PTEN dose has the potential for tumourigenicity [5]. Conditional PTEN deletion leads to mTOR activation and stimulates and promotes axon regeneration as demonstrated in crush injuries in corticospinal neurons [142]. Cardiac specific deletion of the PTEN gene in a mouse model protected cardiac myocytes after cardiac ischemia/reperfusion injury by inhibiting anti-apoptotic signals [143]. In a recent study on cellular and animal models of Alzheimer's disease, it was illustrated that inhibition of PTEN saved the normal synaptic function and cognition [144]. Modulation strategies used for functional PTEN reduction include, direct protein inhibition through inhibition of PTEN phosphatase activity or inhibition of PTEN by protein-protein interaction, targeting of PTEN mRNA to reduce PTEN protein translation and gene editing through new technologies such as C2c2, CRISPR/Cas9, or Cpf1.
Understanding the roles of pseudogenes, such as PTENP1, which has come to the forefront as a modulator of PTEN, and regulatory functions thereof, may improve our current knowledge of tumour biology, providing a new perspective for the discovery of candidate drugs as opportunistic therapies as well as future biomarkers. There is accumulating evidence that lncRNA PTENP1 possesses a tumour suppressive role in several cancers and has been downregulated or deleted in numerous cancers such as prostate, gastric carcinoma, clear-cell renal carcinomas, lung cancer, melanoma and colon cancer [32, 128, 145,146,147]. The overexpression of PTENP1 in cell lines and in in vivo studies has been shown to regulate cell proliferation, reduce tumour growth, invasion, metastasis and apoptosis [147,148,149,150,151], further solidifying the importance of PTENP1 in regulating the biology of a cell by acting as a tumour suppressor, independent of PTEN.
Reduction in PTENP1 expression has been presented in numerous cancer studies and has been predicted to be a promising candidate as a future prognostic biomarker [32, 128, 145,146,147, 151]. A personalised medicine approach is possible in the distant future, however, before this can become a reality, a complete understanding of the multiple layers and complexity of the regulation of the regulator of the RKT-dependent PI3K/AKT pathway, PTEN, and its pseudogene (PTENP1), the regulator of PTEN, and its antisense transcripts, needs to be further investigated and understood. One of the major considerations in modulating PTEN/PTENP1 in cancer therapy is the majority of cancers are age related. Many diseases, which would not benefit from increased PTEN, such as reduction in cognitive functions, including Alzheimer's, are more prevalent with aging.
Summary and conclusion
PTEN is dysregulated in many human cancers, and recent studies highlight the complexity of regulation of PTEN expression. Ablation of PTEN can drive oncogenic PI3K signalling, leading to diverse phenotypic outcomes. The relative expression levels of PTEN, and its sense and antisense pseudogene transcripts may mediate this distinction whereby different levels of these transcripts are expressed in different tumour types or tumours of variable stages and histological grades. PTEN and its pseudogene transcripts have specific subcellular localisations and thus it is conceivable that compartmentalisation of PTEN, PTENP1(s) and PTENP1(as) may contribute to their observed downstream function. Further investigation of PTEN and PTENP1 transcript dysregulation within different cancer types may help define the highly dynamic and complex regulatory role the PTEN pseudogene lncRNAs play in tumourigenesis and determine whether miRNA-based treatments, or other alternative approaches will be effective cancer therapeutic strategies. Here, we have highlighted a framework for identification of intermediaries and downstream modulators in the RTK-dependent PI3K/Akt pathway which can be targeted for diagnosis, prognosis and treatment of cancer. The challenge is now to determine the pathways to intrinsic and acquired resistance and to identify potential candidate cancer-related intermediaries, such as the PTEN pseudogene, as potential biomarkers and therapeutic targets.
In conclusion, an in-depth understanding of novel mechanisms of RTK/PI3K/Akt regulation may present new cancer therapeutic targets and opportunities through the targeting of key regulators of cell signalling downstream of RTKs, such as the PTEN/PTENP1 rheostat.
Abbreviations
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- AKT:
-
Protein kinase B
- ATF2:
-
Activating transcription factor 2
- BRRS:
-
Bannayan Riley Ruvalcaba
- CS:
-
Cowden Syndrome
- EGFR:
-
epidermal growth factor receptor
- EGR1:
-
early growth response transcriptional factor 1
- FDA:
-
Food and Drug Administration
- FGFR:
-
Fibroblast growth factor receptor
- GPCR:
-
G-protein-coupled receptor
- HGFR:
-
Hepatocyte growth factor receptor
- IGFR:
-
Insulin growth factor receptor
- IR:
-
Insulin receptor
- microRNA:
-
miRNA, miR
- mRNA:
-
messenger RNA
- MVP:
-
Major vault protein
- ncRNA:
-
Non-coding RNA
- NEDD4–1:
-
Neural precursor cell expressed developmental downregulated-4-1
- NLS:
-
Nuclear localisation signal
- Nt:
-
Nucleotide
- PBD:
-
PIP2-binding domain
- PDGFR:
-
Platelet derived growth factor receptor
- PDK1:
-
Phosphoinositide-dependent kinase 1
- PEST:
-
Proline, glutamic acid, serine, threonine
- PI3K:
-
Phosphoinositide 3-kinase
- PS:
-
Proteus Syndrome
- PtdIns(3,4,5)P3:
-
Phosphatidylinositol 3,4,5-trisphosphate
- PtdIns(4,5)P2:
-
Phosphatidylinositol-4,5-bisphosphate
- PTEN:
-
Phosphatase and tensin homologue
- PTENP1:
-
PTEN pseudogene
- PTENP1(AS):
-
PTENP1 antisense transcript
- PTPase:
-
Protein tyrosine phosphatase
- Ran:
-
Ras-related nuclear protein
- RTK:
-
receptor tyrosine kinase
- RNAi:
-
RNA interference
- SUMO:
-
Small ubiquitin-related modifiers (SUMO)
- Ub:
-
Ubiquitination
- UTR:
-
Untranslated region
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012;13:283–96.
Boosani CS, Agrawal DK. PTEN modulators: a patent review. Expert Opin Ther Pat. 2013;23:569–80.
Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–14.
Wang X, Jiang X. Post-translational regulation of PTEN. Oncogene. 2008;27:5454–63.
Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, et al. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010;42:454–8.
Planchon SM, Waite KA, Eng C. The nuclear affairs of PTEN. J Cell Biol. 2008;121:249–53.
Bassi C, Satmbolic V. PTEN, here, there, everywhere. Cell Death Differ. 2013;20:1595–6.
Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res. 1997;57:2124–9.
Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–62.
Furnari FB, Lin H, Huang H-JS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci U S A. 1997;94:12479–84.
Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A. 1998;95:13513–8.
Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM. Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science. 1998;280:1614–7.
Lee J-O, Yang H, Georgescu M-M, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its Phosphoinositide Phosphatase activity and membrane association. Cell. 1999;99:323–34.
Chalhoub N, Baker SJ. PTEN and the PI3-Kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–50.
Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014;83:641–69.
Rizo J, Südhof TC. C2-domains, structure and function of a universal Ca2+−binding domain. J Biol Chem. 1998;273:15879–82.
Georgescu MM, Kirsch KH, Akagi T, Shishido T, Hanafusa H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc Natl Acad Sci U S A. 1999;96:10182–7.
Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci U S A. 2003;100:7491–6.
Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–8.
Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus implications for PTEN stability to proteasome-mediated degradation. J Biol Chem. 2001;276:993–8.
Simpson L, Parsons R. PTEN: life as a tumor suppressor. Exp Cell Res. 2001;264:29–41.
Ming M, He Y-Y. PTEN in DNA damage repair. Cancer Lett. 2012;319:125–9.
Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH. PTEN enters the nucleus by diffusion. J Cell Biochem. 2005;96:221–34.
Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, et al. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell. 2007;128:141–56.
Peters R. Fluorescence microphotolysis to measure nucleocytoplasmic transport and intracellular mobility. Biochim Biophys Acta. 1986;864:305–59.
Gil A, Andres-Pons A, Fernandez E, Valiente M, Torres J, Cervera J, Pulido R. Nuclear localization of PTEN by a ran-dependent mechanism enhances apoptosis: involvement of an N-terminal nuclear localization domain and multiple nuclear exclusion motifs. Mol Biol Cell. 2006;17:4002–13.
Denning G, Jean-Joseph B, Prince C, Durden DL, Vogt PK. A short N-terminal sequence of PTEN controls cytoplasmic localization and is required for suppression of cell growth. Oncogene. 2007;26:3930–40.
Chung JH, Ginn-Pease ME, Eng C. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res. 2005;65:4108–16.
Minaguchi T, Waite KA, Eng C. Nuclear localization of PTEN is regulated by ca(2+) through a tyrosil phosphorylation-independent conformational modification in major vault protein. Cancer Res. 2006;66:11677–82.
Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, Nuzzo CM, Vaccaro V, Vari S, Cognetti F, Ciuffreda L. PTEN: multiple functions in human malignant tumors. Front Oncol. 2015;5:24.
Meng F, Henson R, Lang M, Wehbe H, Maheshwari S, Mendell JT, Jiang J, Schmittgen TD, Patel T. Involvement of human micro-RNA in growth and response to chemotherapy in human cholangiocarcinoma cell lines. Gastroenterology. 2006;130:2113–29.
Poliseno L, Salmena L, Zhang J, Carver B, Haveman WJ, Pandolfi PP. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–8.
Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, Sportoletti P, Varmeh S, Egia A, Fedele G, et al. Identification of the miR-106b∼25 MicroRNA cluster as a proto-Oncogenic PTEN-targeting Intron that cooperates with its host gene MCM7 in transformation. Sci Signal. 2010;3:ra29.
Bonofiglio D, Gabriele S, Aquila S, Catalano S, Gentile M, Middea E, Giordano F, Ando S. Estrogen receptor alpha binds to peroxisome proliferator-activated receptor response element and negatively interferes with peroxisome proliferator-activated receptor gamma signaling in breast cancer cells. Clin Cancer Res. 2005;11:6139–47.
Patel L, Pass I, Coxon P, Downes CP, Smith SA, Macphee CH. Tumor suppressor and anti-inflammatory actions of PPARgamma agonists are mediated via upregulation of PTEN. Curr Biol. 2001;11:764–8.
Shen YH, Zhang L, Gan Y, Wang X, Wang J, LeMaire SA, Coselli JS, Wang XL. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem. 2006;281:7727–36.
Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, et al. PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms. Cancer Cell. 2003;3:117–30.
Nakanishi A, Kitagishi Y, Ogura Y, Matsuda S. The tumor suppressor PTEN interacts with p53 in hereditary cancer (review). Int J Oncol. 2014;44:1813–9.
Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27:462–7.
Uygur B, Abramo K, Leikina E, Vary C, Liaw L, Wu WS. SLUG is a direct transcriptional repressor of PTEN tumor suppressor. Prostate. 2015;75:907–16.
Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–83.
Guil S, Esteller M. RNA–RNA interactions in gene regulation: the coding and noncoding players. Trends Biochem Sci. 2015;40:248–56.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta stone of a hidden RNA language? Cell. 2011;146:353–8.
Tay Y, Kats L, Salmena L, Weiss D, Tan SM, Ala U, Karreth F, Poliseno L, Provero P, Di Cunto F, et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–57.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20.
Phelps M, Coss C, Wang H, Cook M. Registered report: coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. elife. 2016;5:1–34.
Mohammadi A, Mansoori B, Baradaran B. The role of microRNAs in colorectal cancer. Biomed Pharmacother. 2016;84:705–13.
Issabekova A, Berillo O, Regnier M, Anatoly I. Interactions of intergenic microRNAs with mRNAs of genes involved in carcinogenesis. Bioinformation. 2012;8:513–8.
An Y, Furber KL, Ji S. Pseudogenes regulate parental gene expression via ceRNA network. J Cell Mol Med. 2017;21:185–92.
Dong H, Lei J, Ding L, Wen Y, Ju H, Zhang X. MicroRNA: function, detection, and bioanalysis. Chem Rev. 2013;113:6207–33.
He L. Posttranscriptional regulation of PTEN dosage by noncoding RNAs. Sci Signal. 2010;3:pe39–45.
Poliseno L, Pandolfi PP. PTEN ceRNA networks in human cancer. Methods. 2015;77-78:41–50.
Tang J, Ning R, Zeng B, Li Y. Molecular evolution of PTEN Pseudogenes in mammals. PLoS One. 2016;11:e0167851.
Shi X, Nie F, Wang Z, Sun M. Pseudogene-expressed RNAs: a new frontier in cancers. Tumor Biol. 2016;37:1471–8.
Chandrasekaran C, Betrán E. Origins of new genes and pseudogenes. Nat Educ. 2008;1:181.
Fsihi H, De Rossi E, Salazar L, Cantoni R, Labò M, Riccardi G, Takiff HE, Eiglmeier K, Bergh S, Cole ST. Gene arrangement and organization in a~ 76 kb fragment encompassing the oriC region of the chromosome of mycobacterium leprae. Microbiology. 1996;142:3147–61.
Ramos-Onsins S, Aguadé M. Molecular evolution of the Cecropin multigene family in drosophila: functional genes vs. pseudogenes. Genetics. 1998;150:157–71.
Loguercio LL, Wilkins TA. Structural analysis of a hmg-coA-reductase pseudogene: insights into evolutionary processes affecting the hmgr gene family in allotetraploid cotton (Gossypium Hirsutum L.). Curr Genet. 1998;34:241–9.
Khachane AN, Harrison PM. Assessing the genomic evidence for conserved transcribed pseudogenes under selection. BMC Genomics. 2009;10:435.
Dahia PL, FitzGerald MG, Zhang X, Marsh DJ, Zheng Z, Pietsch T, von Deimling A, Haluska FG, Haber DA, Eng C. A highly conserved processed PTEN pseudogene is located on chromosome band 9p21. Oncogene. 1998;16:2403–6.
Li W, Yang W, Wang X-J. Pseudogenes: Pseudo or Real Functional Elements? J Genet Genomics. 2013;40:171–7.
Mighell AJ, Smith NR, Robinson PA, Markham AF. Vertebrate pseudogenes. FEBS Lett. 2000;468:109–14.
Kitagishi Y, Wada Y, Matsuda S. Roles of PI3K/AKT/PTEN pathway in the pathogenesis of parkinson’s disease and the neuropsychiatric symptoms. Int Neuropsychiatr Dis J. 2014;2:1–12.
McCarrey JR, Riggs AD. Determinator-inhibitor pairs as a mechanism for threshold setting in development: a possible function for pseudogenes. Proc Natl Acad Sci U S A. 1986;83:679–83.
He X, Huang Y, Li B, Gong C-X, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer's disease. Neurobiol Aging. 2010;31:398–408.
Zheng LL, Zhou KR, Liu S, Zhang DY, Wang ZL, Chen ZR, Yang JH, Qu LH. dreamBase: DNA modification, RNA regulation and protein binding of expressed pseudogenes in human health and disease. Nucleic Acids Res. 2017;46:D85–91.
Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792–8.
Fujii GH, Morimoto AM, Berson AE, Bolen JB. Transcriptional analysis of the PTEN/MMAC1 pseudogene, psiPTEN. Oncogene. 1999;18:1765–9.
Johnsson P, Ackley A, Vidarsdottir L, Lui WO, Corcoran M, Grander D, Morris KV. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat Struct Mol Biol. 2013;20:440–6.
Wu T, Du Y. LncRNAs: from basic research to medical application. Int J Biol Sci. 2017;13:295.
Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T. Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem. 2005;280:35195–202.
Zhang XC, Piccini A, Myers MP, Van Aelst L, Tonks NK. Functional analysis of the protein phosphatase activity of PTEN. Biochem J. 2012;444:457–64.
Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem. 2002;277:20336–42.
Cao J, Schulte J, Knight A, Leslie NR, Zagozdzon A, Bronson R, Manevich Y, Beeson C, Neumann CA. Prdx1 inhibits tumorigenesis via regulating PTEN/AKT activity. EMBO J. 2009;28:1505–17.
Okumura K, Mendoza M, Bachoo RM, DePinho RA, Cavenee WK, Furnari FB. PCAF modulates PTEN activity. J Biol Chem. 2006;281:26562–8.
Ding L, Chen S, Liu P, Pan Y, Zhong J, Regan KM, Wang L, Yu C, Rizzardi A, Cheng L, et al. CBP loss cooperates with PTEN haploinsufficiency to drive prostate cancer: implications for epigenetic therapy. Cancer Res. 2014;74:2050–61.
Ikenoue T, Inoki K, Zhao B, Guan KL. PTEN acetylation modulates its interaction with PDZ domain. Cancer Res. 2008;68:6908–12.
Chae HD, Broxmeyer HE. SIRT1 deficiency downregulates PTEN/JNK/FOXO1 pathway to block reactive oxygen species-induced apoptosis in mouse embryonic stem cells. Stem Cells Dev. 2011;20:1277–85.
Wang X, Shi Y, Wang J, Huang G, Jiang X. Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation. Biochem J. 2008;414:221–9.
Yang JM, Schiapparelli P, Nguyen HN, Igarashi A, Zhang Q, Abbadi S, Amzel LM, Sesaki H, Quinones-Hinojosa A, Iijima M. Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization. Oncogene. 2017;36:3673–85.
Gonzalez-Santamaria J, Campagna M, Ortega-Molina A, Marcos-Villar L, de la Cruz-Herrera CF, Gonzalez D, Gallego P, Lopitz-Otsoa F, Esteban M, Rodriguez MS, et al. Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 2012;3:e393.
Huang J, Yan J, Zhang J, Zhu S, Wang Y, Shi T, Zhu C, Chen C, Liu X, Cheng J, et al. SUMO1 modification of PTEN regulates tumorigenesis by controlling its association with the plasma membrane. Nat Commun. 2012;3:911.
Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341:395–9.
Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–91.
Schlessinger J. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb Perspect Biol. 2014;6:1–13.
Spangle JM, Roberts TM. Epigenetic regulation of RTK signaling. J Mol Med. 2017;95:791–8.
Sangwan V, Park M. Receptor tyrosine kinases: role in cancer progression. Curr Oncol. 2006;13:191–3.
Singh P, Alex JM, Bast F. Insulin receptor (IR) and insulin-like growth factor receptor 1 (IGF-1R) signaling systems: novel treatment strategies for cancer. Med Oncol. 2014;31:805.
Hagerstrand D, Lindh MB, Pena C, Garcia-Echeverria C, Nister M, Hofmann F, Ostman A. PI3K/PTEN/Akt pathway status affects the sensitivity of high-grade glioma cell cultures to the insulin-like growth factor-1 receptor inhibitor NVP-AEW541. Neuro-Oncology. 2010;12:967–75.
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol. 1997;7:261–9.
Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3, 4, 5-trisphosphate. J Biol Chem. 1998;273:13375–8.
Stambolic V, Suzuki A, De La Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39.
Sun H, Lesche R, Li D-M, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3, 4, 5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–204.
Shi Y, Wang J, Chandarlapaty S, Cross J, Thompson C, Rosen N, Jiang X. PTEN is a protein tyrosine phosphatase for IRS1. Nat Struct Mol Biol. 2014;21:522–7.
Zhang S, Huang W-C, Li P, Guo H, Poh S-B, Brady SW, Xiong Y, Tseng L-M, Li S-H, Ding Z. Combating trastuzumab resistance by targeting SRC, a common node downstream of multiple resistance pathways. Nat Med. 2011;17:461–9.
Tibarewal P, Zilidis G, Spinelli L, Schurch N, Maccario H, Gray A, Perera NM, Davidson L, Barton GJ, Leslie NR. PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor-suppressing phenotype, but not with AKT activity. Sci Signal. 2012;5:ra18.
Papa A, Wan L, Bonora M, Salmena L, Song MS, Hobbs RM, Lunardi A, Webster K, Ng C, Newton RH. Cancer-associated PTEN mutants act in a dominant-negative manner to suppress PTEN protein function. Cell. 2014;157:595–610.
Fang J, Ding M, Yang L, Liu L-Z, Jiang B-H. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell Signal. 2007;19:2487–97.
Graupera M, Guillermet-Guibert J, Foukas LC, Phng L-K, Cain RJ, Salpekar A, Pearce W, Meek S, Millan J, Cutillas PR, et al. Angiogenesis selectively requires the p110[agr] isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–6.
Smith JM, Kirk EP, Theodosopoulos G, Marshall GM, Walker J, Rogers M, Field M, Brereton JJ, Marsh DJ. Germline mutation of the tumour suppressor PTEN in Proteus syndrome. J Med Genet. 2002;39:937–40.
Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet. 1997;16:64–7.
Mitchell F. Diabetes: PTEN mutations increase insulin sensitivity and obesity. Nat Rev Endocrinol. 2012;8:698.
Mester JL, Moore RA, Eng C. PTEN germline mutations in patients initially tested for other hereditary cancer syndromes: would use of risk assessment tools reduce genetic testing? Oncologist. 2013;18:1083–90.
Butler MG, Dasouki MJ, Zhou X-P, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–21.
Chen HJ, Romigh T, Sesock K, Eng C. Characterization of cryptic splicing in germline PTEN intronic variants in Cowden syndrome. Hum Mutat. 2017;38:1372–7.
Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet A. 2007;143:589–93.
Tilot AK, Frazier TW 2nd, Eng C. Balancing proliferation and connectivity in PTEN-associated autism Spectrum disorder. Neurotherapeutics. 2015;12:609–19.
Eng C. PTEN: one gene, many syndromes. Hum Mutat. 2003;22:183–98.
Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301.
Nassif NT, Lobo GP, Wu X, Henderson CJA, Morrison CD, Eng C, Jalaludin B, Segelov E. PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene. 2004;23:617–28.
Louis DN, Ramesh V, Gusella JF. Neuropathology and molecular genetics of neurofibromatosis 2 and related tumors. Brain Pathol. 1995;5:163–72.
Ma H, Brosens LAA, Offerhaus GJA, Giardiello FM, de Leng WWJ, Montgomery EA. Pathology and genetics of hereditary colorectal cancer. Pathology. 2018;50:49–59.
Ngeow J, Sesock K, Eng C. Breast cancer risk and clinical implications for germline PTEN mutation carriers. Breast Cancer Res Treat. 2017;165:1–8.
Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat Rev. 2015;41:1–8.
Ciccarese C, Massari F, Iacovelli R, Fiorentino M, Montironi R, Di Nunno V, Giunchi F, Brunelli M, Tortora G. Prostate cancer heterogeneity: discovering novel molecular targets for therapy. Cancer Treat Rev. 2017;54:68.
Abounader R. Interactions between PTEN and receptor tyrosine kinase pathways and their implications for glioma therapy. Expert Rev Anticancer Ther. 2009;9:235–45.
Uzoh CC, Perks CM, Bahl A, Holly JM, Sugiono M, Persad RA. PTEN-mediated pathways and their association with treatment-resistant prostate cancer. BJU Int. 2009;104:556–61.
Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101.
Li A, Qiu M, Zhou H, Wang T, Guo W. PTEN, insulin resistance and cancer. Curr Pharm Des. 2017;23:3667–76.
Sonoda Y, Mukai H, Matsuo K, Takahashi M, Ono Y, Maeda K, Akiyama H, Kawamata T. Accumulation of tumor-suppressor PTEN in Alzheimer neurofibrillary tangles. Neurosci Lett. 2010;471:20–4.
Pal A, Barber TM, Van de Bunt M, Rudge SA, Zhang Q, Lachlan KL, Cooper NS, Linden H, Levy JC, Wakelam MJ, et al. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N Engl J Med. 2012;367:1002–11.
Grinder-Hansen L, Ribel-Madsen R, Wojtaszewski JF, Poulsen P, Grunnet LG, Vaag A. A common variation of the PTEN gene is associated with peripheral insulin resistance. Diabetes Metab. 2016;42:280–4.
Suo G, Han J, Wang X, Zhang J, Zhao Y, Zhao Y, Dai J. Oct4 pseudogenes are transcribed in cancers. Biochem Biophys Res Commun. 2005;337:1047–51.
Zou M, Baitei EY, Alzahrani AS, Al-Mohanna F, Farid NR, Meyer B, Shi Y. Oncogenic activation of MAP kinase by BRAF pseudogene in thyroid tumors. Neoplasia. 2009;11:57–65.
Chiefari E, Iiritano S, Paonessa F, Le Pera I, Arcidiacono B, Filocamo M, Foti D, Liebhaber SA, Brunetti A. Pseudogene-mediated posttranscriptional silencing of HMGA1 can result in insulin resistance and type 2 diabetes. Nat Commun. 2010;1:40.
Ioffe YJ, Chiappinelli KB, Mutch DG, Zighelboim I, Goodfellow PJ. Phosphatase and tensin homolog (PTEN) pseudogene expression in endometrial cancer: a conserved regulatory mechanism important in tumorigenesis? Gynecol Oncol. 2012;124:340–6.
Poliseno L, Haimovic A, Christos PJ, Vega Y Saenz de Miera EC, Shapiro R, Pavlick A, Berman RS, Darvishian F, Osman I. Deletion of PTENP1 pseudogene in human melanoma. J Invest Dermatol. 2011;131:2497–500.
Gschwind A, Fischer OM, Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer. 2004;4:361.
Gharwan H, Groninger H. Kinase inhibitors and monoclonal antibodies in oncology: clinical implications. Nat Rev Clin Oncol. 2016;13:209–28.
Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res. 2015;21:1537–42.
Wang X, Ding J, Meng LH. PI3K isoform-selective inhibitors: next-generation targeted cancer therapies. Acta Pharmacol Sin. 2015;36:1170–6.
LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34:3803–15.
Zhao W, Qiu Y, Kong D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm Sin B. 2017;7:27–37.
Massacesi C, Di Tomaso E, Urban P, Germa C, Quadt C, Trandafir L, Aimone P, Fretault N, Dharan B, Tavorath R, Hirawat S. PI3K inhibitors as new cancer therapeutics: implications for clinical trial design. OncoTargets Ther. 2016;9:203–10.
Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos DΑ, Libra M, Tsatsakis AM. Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use. Int J Oncol. 2016;48:869–85.
McLoughlin NM, Mueller C, Grossmann TN. The therapeutic potential of PTEN modulation: targeting strategies from gene to protein. Cell Chem Biol. 2018;25:19–29.
Altinoglu SA, Wang M, Li KQ, Li Y, Xu Q. Intracellular delivery of the PTEN protein using cationic lipidoids for cancer therapy. Biomater Sci. 2016;4:1773–80.
Hopkins BD, Hodakoski C, Barrows D, Mense SM, Parsons RE. PTEN function: the long and the short of it. Trends Biochem Sci. 2014;39:183–90.
Tanaka M, Grossman HB. In vivo gene therapy of human bladder cancer with PTEN suppresses tumor growth, downregulates phosphorylated Akt, and increases sensitivity to doxorubicin. Gene Ther. 2003;10:1636–42.
Garcia-Cao I, Song Min S, Hobbs Robin M, Laurent G, Giorgi C, de Boer Vincent CJ, Anastasiou D, Ito K, Sasaki Atsuo T, Rameh L, et al. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62.
Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–81.
Ruan H, Li J, Ren S, Gao J, Li G, Kim R, Wu H, Wang Y. Inducible and cardiac specific PTEN inactivation protects ischemia/reperfusion injury. J Mol Cell Cardiol. 2009;46:193–200.
Knafo S, Sanchez-Puelles C, Palomer E, Delgado I, Draffin JE, Mingo J, Wahle T, Kaleka K, Mou L, Pereda-Perez I, et al. PTEN recruitment controls synaptic and cognitive function in Alzheimer's models. Nat Neurosci. 2016;19:443–53.
Yu G, Yao W, Gumireddy K, Li A, Wang J, Xiao W, Chen K, Xiao H, Li H, Tang K. Pseudogene PTENP1 functions as a competing endogenous RNA to suppress clear-cell renal cell carcinoma progression. Mol Cancer Ther. 2014;13:3086–97.
Sacco JJ, Yau TY, Darling S, Patel V, Liu H, Urbe S, Clague MJ, Coulson JM. The deubiquitylase Ataxin-3 restricts PTEN transcription in lung cancer cells. Oncogene. 2014;33:4265–72.
Guo X, Deng L, Deng K, Wang H, Shan T, Zhou H, Liang Z, Xia J, Li C. Pseudogene PTENP1 suppresses gastric cancer progression by modulating PTEN. Anti Cancer Agents Med Chem. 2016;16:456–64.
Chen CL, Tseng YW, Wu JC, Chen GY, Lin KC, Hwang SM, Hu YC. Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials. 2015;44:71–81.
Gao L, Ren W, Zhang L, Li S, Kong X, Zhang H, Dong J, Cai G, Jin C, Zheng D, Zhi K. PTENp1, a natural sponge of miR-21, mediates PTEN expression to inhibit the proliferation of oral squamous cell carcinoma. Mol Carcinog. 2017;56:1322–34.
Gong T, Zheng S, Huang S, Fu S, Zhang X, Pan S, Yang T, Sun Y, Wang Y, Hui B, et al. PTENP1 inhibits the growth of esophageal squamous cell carcinoma by regulating SOCS6 expression and correlates with disease prognosis. Mol Carcinog. 2017;56:2610–19.
Liu J, Xing Y, Xu L, Chen W, Cao W, Zhang C. Decreased expression of pseudogene PTENP1 promotes malignant behaviours and is associated with the poor survival of patients with HNSCC. Sci Rep. 2017;7:41179–90.
Yao Y, Ma J, Xue Y, Wang P, Li Z, Liu J, Chen L, Xi Z, Teng H, Wang Z, et al. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015;359:75–86.
Li N, Miao Y, Shan Y, Liu B, Li Y, Zhao L, Jia L. MiR-106b and miR-93 regulate cell progression by suppression of PTEN via PI3K/Akt pathway in breast cancer. Cell Death Dis. 2017;8:e2796.
Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross-talk, and therapies. Oral Dis. 2015;21:815–25.
Carico C, Nuno M, Mukherjee D, Elramsisy A, Dantis J, Hu J, Rudnick J, Yu JS, Black KL, Bannykh SI, Patil CG. Loss of PTEN is not associated with poor survival in newly diagnosed glioblastoma patients of the temozolomide era. PLoS One. 2012;7:e33684.
Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19.
Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, Carey M, Hu Z, Guan Y, Sahin A, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91.
Carden CP, Stewart A, Thavasu P, Kipps E, Pope L, Crespo M, Miranda S, Attard G, Garrett MD, Clarke PA, et al. The association of PI3 kinase signaling and chemoresistance in advanced ovarian cancer. Mol Cancer Ther. 2012;11:1609–17.
Marsit CJ, Zheng S, Aldape K, Hinds PW, Nelson HH, Wiencke JK, Kelsey KT. PTEN expression in non-small-cell lung cancer: evaluating its relation to tumor characteristics, allelic loss, and epigenetic alteration. Hum Pathol. 2005;36:768–76.
Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–510.
Zhang J, Roberts TM, Shivdasani RA. Targeting PI3K signaling as a therapeutic approach for colorectal cancer. Gastroenterology. 2011;141:50–61.
Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene. 2005;24:7465–74.
Acknowledgements
The study is supported by a grant from the Centre for Health Technologies, University of Technology Sydney. NH is the recipient of a UTS International Research Scholarship (UTS IRS), University of Technology Sydney.
Author information
Authors and Affiliations
Contributions
EM, NH, NN conceived and designed the content and structure of the review. EM, NH, YL, GT, and NN drafted, and edited the review. EM, NH, YL, and NN revised and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Haddadi, N., Lin, Y., Travis, G. et al. PTEN/PTENP1: ‘Regulating the regulator of RTK-dependent PI3K/Akt signalling’, new targets for cancer therapy. Mol Cancer 17, 37 (2018). https://doi.org/10.1186/s12943-018-0803-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-018-0803-3