
Abstract
Across the world, oral cancer is a prevalent tumor. Over the years, both its mortality and incidence have grown. Oral cancer metastasis is a complex process involving cell invasion, migration, proliferation, and egress from cancer tissue either by lymphatic vessels or blood vessels. MicroRNAs (miRNAs) are essential short non-coding RNAs, which can act either as tumor suppressors or as oncogenes to control cancer development. Cancer metastasis is a multi-step process, in which miRNAs can inhibit or stimulate metastasis at all stages, including epithelial-mesenchymal transition, migration, invasion, and colonization, by targeting critical genes in these pathways. On the other hand, long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs), two different types of non-coding RNAs, can regulate cancer metastasis by affecting gene expression through cross-talk with miRNAs. We reviewed the scientific literature (Google Scholar, Scopus, and PubMed) for the period 2000–2023 to find reports concerning miRNAs and lncRNA/circRNA-miRNA-mRNA networks, which control the spread of oral cancer cells by affecting invasion, migration, and metastasis. According to these reports, miRNAs are involved in the regulation of metastasis pathways either by directly or indirectly targeting genes associated with metastasis. Moreover, circRNAs and lncRNAs can induce or suppress oral cancer metastasis by acting as competing endogenous RNAs to inhibit the effect of miRNA suppression on specific mRNAs. Overall, non-coding RNAs (especially miRNAs) could help to create innovative therapeutic methods for the control of oral cancer metastases.
Similar content being viewed by others
Introduction
Oral cancer is the 11th most frequent carcinoma worldwide, which has drawn interest from all across the world [1]. Oral cancer led to 177,757 new fatalities and 377,713 new cases in 2020 [2]. The majority of individuals are diagnosed with oral cancer at an advanced stage [3]. This late diagnosis leads to a poor prognosis and a greater prevalence of lymphatic metastasis [4, 5].
Differentiated oral squamous cell carcinoma (OSCC), which tends to spread to lymph nodes [6], is the 6th most frequent type of cancer in the world, with more than 200,000 new cases each year. OSCC occurs in three main anatomical sites: lip SCC (LSCC), tongue SCC (TSCC), and buccal mucosal SCC (BMSCC) [7, 8]. Males have morbidity and fatality rates of 6.6 and 3.1 per 100,000 persons, respectively, while females have rates of 2.9 and 1.4 per 100,000 persons. Also, the prevalence of OSCC is increasing in young caucasians between the ages of 18 and 44, especially in women [9, 10]. OSCC is a major problem for both individual health and socioeconomic well-being on account of risk factor exposure, limited treatment options, and high mortality.
Oral cancer carcinogenesis is influenced by a number of risk factors, like genetic variations, smoking, betel nut chewing, radiation exposure, and other lifestyle factors [11]. One additional potential risk factor for OSCC is infection with the human papillomavirus (HPV). As early as 2007, the International Agency for Research on Cancer recognized HPV16 as a risk factor for OSCC. OSCC has been linked to other sub-types of the virus such as HPV33, HPV35, that are also seen in cervical cancer [12]. Treatment for oral cancer may involve surgery, targeted therapy or chemotherapy depending on the tumor stage. Despite advances in treatment and detection methods over the last ten years, the five year survival rate, which averages between 45 and 50%, has not significantly increased [13]. Generally speaking, oral cancer still has a poor prognosis and a low survival rates [14]. It has a high likelihood of migration to nearby lymph nodes, adjacent tissues, and distant metastasis, and an unusually high chance of recurrence over the patient’s lifespan in those diagnosed with advanced-stage tumors [15].
MicroRNAs (miRNAs) are short (19–25 nt) single-stranded non-coding RNAs (ncRNAs) which specifically bind to the 3′ untranslated region (UTR) of the targeted gene mRNA to control its expression [16]. In light of the fact that numerous genes can be targeted by a single miRNA and a single target gene can have several miRNA binding sites, more than 60% of the genes in humans are thought to be controlled by miRNAs [17, 18]. Therefore, miRNAs play a critical role as regulators in almost all biology, including normal physiological and pathological processes, most notably cancer. It is accepted that miRNAs are crucial for preventing the growth and spread of cancer [19,20,21]. Additionally, increasing evidence suggests that the circRNA/lncRNA-miRNA-mRNA regulatory axis controls how oral cancer spreads [22,23,24,25,26]. In this overview, we summarize the role of specific miRNAs in the metastatic spread of oral cancer. Additionally, we discuss the function of lncRNA/circRNA-miRNA-mRNA networks in regulating signaling pathways and associated genes that are linked to the spread of oral cancer. These networks are becoming recognized as crucial regulators of carcinogenesis.
Cancer metastasis
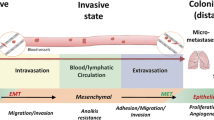
The process through which cancer cells detach themselves from the primary tumor, and go on to establish additional tumors at sites distant from the original tumor, is known as cancer metastasis. The initial tumor is generally not the principal reason for cancer death, but most fatalities are caused by metastasis. Around 9/10 of cancer deaths are caused by cancer metastasis, which also produces most morbidity [27, 28]. In 1889 Stephen Paget, a surgeon from the UK, first proposed the theory that the secondary sites of tumor dispersal are not due to chance alone, but instead, by an interaction between “seeds” (cancer cells) and the “congenial soil” (organs that certain tumor types spread to). This theory has now been widely accepted [29]. Up until recently, most cancer research was devoted to early tumor detection techniques and new therapeutic agents, as well as tumor growth inhibitors. If detected and treated early enough, the majority of solid tumors are now considered treatable or even curable, thanks to advances in early cancer detection and treatment. Howevert, once cancer has spread beyond the primary site, it is typically considered incurable and fatal [30, 31]. The mechanisms underlying the metastatic process are poorly understood, and in terms of preventing and controlling cancer metastasis, very little progress has yet been made.
There is still much to learn about the complex process of metastasis, which entails a number of sequential and connected steps as well as many biochemical reactions. The four crucial processes of detachment, invasion, migration, and adhesion all work togther in the formztion of metastases. Following their initial separation from the primary tumor, cancer cells initially migrate, invade, and travel through blood and lymphatic vessels to various locations. When the arrive at their destination, they settle (adhere), proliferate, and the secondary tumor spreads [32]. Multiple signaling mechanisms control metastatic growth, and the extracellular matrix (ECM) in the destination site has an efffect. The genes that respond to stress are now thought to act as metastatic genes to support inflammation, stress-induced wound healing, and angiogenesis [32, 33].
Prior to spreading, cells must initially detach from the primary tumor [28]. When normal cells endothelial or epithelial) become separated from their ECM, they undergo a type of cell death known as anoikis (cell death brought on by ECM detachment) [34]. Both the mitochondrial route of apoptosis and the death receptor pathways are engaged during anoikis [34]. Metastatic cancer cells are able create a defense against anoikis [35, 36]. The term “epithelial-mesenchymal transition” (EMT) describes the resistance of tumor cells to anoikis, as well as a number of other properties, including alterations in the adherence of cells and tissues, cell invasion and migration, and cell polarity. The majority of metastatic cells share the defining characteristics of EMT [36]. From being highly differentiated, polarized, and structured cells, epithelial cells are transformed into a mesenchymal-like immature state, as isolated cells with the potential for invasion and migration [37].
Two essential components of the metastatic cascade are the cell’s ability to migrate and invade. There are two different ways that metastatic cells can infiltrate through the ECM, mesenchymal (fibroblastoid) and amoeboid cellular migration. Protease-dependent enzyme activities are required for mesenchymal cell migration in order to break down the ECM structure and allow the cells to pass through. Mesenchymal cell migration can be stopped by blocking ECM-degrading proteases, e.g. matrix metalloproteinases (MMPs), which are important in wound healing. In the protease-independent method of amoeboid cell migration, cells use mechanical forces rather than enzymatic degradation to build a passageway. The ability to move chemotactically (to follow a chemokine gradient), total loss of cell polarity, and loose ECM attachment are characteristics of amoeboid cell invasion [36].
Single metastatic cancer cells or large clusters of cells can move throughout the body [36, 38]. In contrast to the collective migration of cell clusters, which only uses mesenchymal cell migration, both mesenchymal migration mediated by proteases and protease-independent amoeboid-like migration are capable of facilitating the movement of individual cancer cells. Epithelial cell polarity is lost and an EMT-induced mesenchymal state is produced in single cells, which leave a primary tumor from its periphery. A solid epithelial tumor may release one or more single cells as a result of EMT, which is characterized by interference with tight cell-cell junctions, adopting a fibroblastoid spindle-shaped morphology, increased cell-stromal interactions, and slower cell division rates and invasiveness [38].
Cells which migrate collectively maintain their cell-cell connections by ongoing production of adhesion molecules, in contrast to solitary cells that migrate on their own. They may migrate as unattached cell clusters or groups, and move as strands, tubes, sheets, or clusters (cohort migration), or they may keep their attachment to the parent tumor (coordinated invasion) [36]. Cancer cell migration in a clustered cohort appears to be very effective at obliterating lymphatic or blood vessels and preserving the cells under flow conditions. Mostly squamous cell carcinoma and basal cell carcinoma from various origins undergo collective cell migration [36, 38].
Throughout the past 30 years, major improvements have been made in understanding cancer metastasis at the molecular, cellular, and signaling pathway levels, thus opening up a variety of potential targets for preventing cancer metastasis. These may involve modifying the biochemical mechanisms and signaling pathways that control cell adhesion, dissociation, invasion, migration, and interaction with the tumor microenvironment [28]. The present review aims to provide a comprehensive explanation of how various miRNAs and other ncRNAs could regulate the process of invasion, migration, and adhesion in oral cancer cells.
MicroRNA biogenesis
The stability and translation of messenger RNAs (mRNA) are controlled by a class of endogenous ncRNAs called microRNAs (miRNAs), which are 19–25 nucleotides (nt) in length. Ambros et al. originally discovered miRNAs in 1993, completely overturning previous theories about mRNA translation [39]. Canonical miRNA processing starts with RNA polymerase II converting a miRNA gene into a stem-loop-structured primary miRNA (pri-miRNA). The Drosha and DGCR8 microprocessor complex cleaves this pri-miRNA using endonuclease activity to produce a hairpin miRNA precursor with a 70 nt length [40, 41]. Exportin-5 facilitates the cytoplasmic entry of pre-export miRNAs [42], plus within the cytoplasm, the TRBP and Dicer(double-stranded) complex converts the pre-miRNA into a miRNA duplex. The complex that induces silencing via RNA appears to contain either both mature miRNA duplex strands, according to in vitro experiments (RISC). One strand of this duplex must be destroyed to cause the intended mRNA to be repressed, possibly depending on thermodynamic stabilities [43]. The mature miRNA is directed to a target mRNA by the RISC complex, and this complex contains the key component Argonaute 2, after strand selection. Here, the interactions promote target mRNA destabilisation and translational repression by cleaving the target mRNA or deadenylating (shortening the 3′ poly-A tail) the target mRNA [44, 45].
Alterations in miRNA expression in cancer cells
George Calin, Carlo Croce, and associates reported the loss of miR-16 and miR-15a expression within B-cell chronic lymphocytic leukemia (CLL) in 2002, providing the first proof that human tumors express abnormal miRNAs [46]. There are no protein-coding genes in this area, which is lost in most B-cell CLL specimens. Since that first report miRNA expression in human cancers has been extensively studied to discover any new miRNAs associated with cancer. Many investigations have analyzed miRNA expression in various types of cancer, and reported miRNA profiless that differ in expression between healthy and malignant cells or tissues. In 334 samples of tumors and healthy tissue, Jun Lu, Todd Golub, et al. evaluated the expression of 217 miRNAs as well as 16,000 mRNAs. It was shown that a number of miRNAs were either up-regulated or down-regulated in cancer, and these miRNA expression patterns were more accurate in distinguishing different cancer types compared to mRNA expression patterns [47].
A variety of mechanisms, including chromosomal abnormalities, amplification, deletion, mutation, and translocation, can affect the production of miRNAs in human cancer. Activation or repression of transcription, epigenetic modifications, and structural flaws in the genes that produce miRNAs could also be involved. The Croce group examined the chromosomal position of various miRNAs, and found that miRNA genes are frequently located in those genetic regions that are altered in cancer [48]. These DNA sequences contain insertion sites or chromosomal translocation for tumor-associated viruses like HPV, regions of deletion that might encode tumor-suppressor genes, and regions of amplification that may contain oncogenes [49].
Chromosome 13q14 is where the miR-15a/miR-16-1 cluster is located, an area that is often eliminated in CLL. The family of miR-29 miRNAs, which is found in the areas 7q32 and 1q30 and is often lost in acute myeloid leukemia, is another example. Other factors in addition to genetic changes, are involved in the downregulation or deletion of particular miRNAs. For example, the mir-34 miRNA group was found to be epigenetically silenced via CpG island hypermethylation in a variety of cancer types, resulting in downregulation of miR-34 expression [49]. Unusual transcription factor activity may potentially contribute to deregulation of miRNA expression, which could involve either decreased or increased transcription from the miRNA gene. As one example, the tumor suppressor transcription factor p53, binds to the promoter for mir-34 and activates it. This may lead to miR-34 downregulation in those human cancers, where p53 is commonly altered or deleted. Moreover, p53 also favorably regulates the clusters miR-29 and miR-15a/miR-16-1 [50]. However, Oncoproteins which are frequently overexpressed or upregulated in human cancer, include certain transcription factors. For instance, the mir-29 group mir-26a and let-7a, every one of whom are downregulated in cancer, are transcriptionally repressed by MYC [49].
MiRNAs as either tumor suppressors or oncomiRs in cancer
By affecting the expression of tumor oncogenes or tumor suppressors respectively, miRNAs can either promote or suppress the cancer phenotype. Typically, tumor-suppressor miRNAs are underexpressed in cancer, while tumors generally exhibit an overexpression of oncogenic miRNAs (oncomiRs). Depending on the type of cancer and the particular miRNA that is affected, cancer cell invasion, progression and/or survival could be drastically affected when these tumor-suppressor or oncomiR miRNAs are suppressed or activated, respectively. It is also conceivable that cancers can become totally dependent on, or “addicted” to, a specific oncomiR, in which case suppressing the oncomiR could cause the tumor to completely regress [51]. As a result, miRNAs can be categorized as either tumor-promoters or tumor-suppressors, and this distiction would govern whether to alter their expression for therapeutic purposes.
Nonetheless, there are arguments in favor of caution in therapeutic approaches involving miRNAs. There are contradictory reports in the literature about whether or not any particular miRNA is a tumor-suppressor or tumor-promoter. Some miRNAs have repeatedly been demonstrated to be tumor suppressive in one situation yet carcinogenic in another. Given the wide array of genes that any specific miRNA can affect, the variety of consequences is not surprising. It implies that any designation of a miRNA as tumor suppressive or oncogenic should always be carefully examined because it may constitute an oversimplification [52].
MiRNAs as inhibitors of metastasis in oral cancer
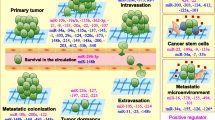
Table 1 provides a summary of the main miRNAs which have been implicated in the process of metastasis. In 2007, it was first discovered that miRNAs and metastasis were connected to each other. Li Ma, Robert Weinberg, and coworkers evaluated the miRNA expression profiles in breast cancer cells, both metastatic and non-metastatic, as well as in healthy human mammary epithelial cells. This resulted in the discovery of numerous miRNAs linked to metastasis [53]. One of them, miR-101 also prevented the spread of oral cancer by inhibiting migration and invasion [54, 55].
The CX chemokine receptor 7 (CXCR7) recognizes its ligand CXCL12, and affects cell adhesion, viability, and tumor growth [56]. Furthermore, the stimulation of the CXCR7 signaling pathway caused by CXCL12 increases the proliferation, invasion and metastasis of tumor cells. CXCR7 is broadly expressed in many types of cancer [57, 58]. CXCL12 and CXCR7 are both concurrently increased in rapidly proliferating oral cancer [59]. Hui et al. [60] discovered that miR-101 could act as a tumor suppressor in OSCC, by targeting CXCR7. According to their findings, OSCC cell lines and tissues showed strongly increased expression of CXCR7 and downregulation of miR-101. Furthermore, OSCC tumor metastasis and growth were inhibited in vivo by either deletion of CXCR7 or overexpression of miR-101. Moreover, both in vitro and in vivo, miR-101 inhibited invasion and migration, which lowered OSCC metastasis [60]. It was later discovered that the expression of exosomal miR-101-3p, which was produced from human bone marrow mesenchymal stem cells (hBMSCs), could inhibit oral cancer invasion and migration by downregulating COL10A1 (type X alpha 1 chain of collagen) [61]. Different cellular processes and reactions are controlled by TGF-β signaling pathways, including proliferation, differentiation, cell death, and migration. TGF-β signaling operates in a different way depending on the tumor stage. In pre-malignant cells, it initially acts as a tumor suppressor, but as the disease progresses, it promotes invasion and metastasis [62]. Recent studies have also shown that miR-101 may play a significant role in TGF-R1 activity, as a miR-101-mediated modulator of OSCC cell motility by inhibiting TGF-R1-induced OSCC cell invasion and metastasis [63]. Additionally, the ectopic expression of miR-101 might block the AKT/mTOR pathway to attenuate the EMT in OSCC, providing a theoretical foundation for targeted treatment of OSCC to lower metastasis and recurrence and improve patient cure rates [64].
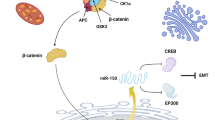
MiR-34a is the 2nd most investigated metastasis-associated miRNA, with a variety of functional roles that are related to metastasis (Table 1). According to reports, OSCC cells showed a dramatic downregulation of miR-34a expression [65,66,67]. On the other hand, increasing miR-34a expression could prevent OSCC cells from metastasizing by affecting MMP9 and MMP14 activity [66], as well as IL6R expression [67]. Interestingly, Li et al. [65] reported that oral cancer cells showed increased proliferation and metastasis as a result of miR-34a-5p exosome-mediated paracrine signaling in cancer-associated fibroblasts (CAFs). They discovered that exosomes produced from CAFs had much lower miR-34a-5p levels, and that fibroblasts could deliver exosomal miR-34a-5p into OSCC cells. Their findings showed that miR-34a-5p overexpression in CAFs could prevent OSCC cells from developing into tumors during mouse xenograft experiments. One direct downstream target of miR-34a-5p is AXL, to which it can bind in order to inhibit OSCC cell proliferation and metastasis. The miR-34a-5p/AXL axis could support the progression of OSCC via the AKT/GSK-3/β-catenin signaling pathway, and may promote the EMT to encourage the spread of oral cancer cells. Nuclear translocation of β-catenin was promoted by the miR-34a-5p/AXL axis, which also increased the Snail transcription factor. thus triggering MMP-9 and MMP-2. In oral cancer cells, the miR-34a-5p/AXL axis increased aggressiveness and could be a therapeutic target for OSCC by affecting AKT, GSK-3, β-catenin, and Snail signaling (Fig. 1) [65].

Exosomal miR-34a-5p can suppress metastasis of OSCC cells by targeting AXL. Transfer of CAF-derived exosomal miR-34a-5p to OSCC cells promotes by fibroblasts and miR-34a-5p inhibits metastasis and proliferation of OSCC cells through inhibiting EMT and MMP-2/9 activation by targeting AXL [65]
Recently, Dharavath et al. [68] reported that miR-944 showed potential anti-metastatic activity in vivo and in vitro. They observed that miR-944 inhibited migration, invasion, and EMT of TSCC cells by targeting MMP-10. Hence they suggested that this miRNA could be a therapeutic target in TSCC patients [68].
Cancer stem cells (CSCs), or tumor-initiating cells comprise a very small fraction of all the cells in a tumor. CSCs possess the ability of adult stem cells for self-renewal and differentiation. According to some theories, CSCs are mainly responsible for tumor growth, the initiation of invasion and metastasis, as well as recurrence [69, 70]. The development of both primary and metastatic cancers is caused by CSCs [71]. Tumorigenesis and metastasis have been reported to be inhibited by miRNAs which suppress CSC properties. In oral cancer, miR-495 could suppress tumor progression [72]. The regulation of miR-495 is disrupted in a variety of stem cells and cancer cells [73,74,75]. For example, in OSCC, miR-495 expression was markedly downregulated, and when it was expressed ectopically in CSCs, it reversed the EMT process, inhibited cellular migration, proliferation, and invasion, and promoted cell death via the HOXC6-mediated TGF-β signaling pathway [72].
As was previously mentioned, the mesenchymal-epithelial transition (MET), which is the opposite of the EMT process, encourages metastatic colonization in certain cancer types while facilitating tumor cell invasion and dissemination in carcinoma cells [76, 77]. According to several experimental studies, cancer cells interact with the chemokine (C-C motif) ligand 21 (CCL21)/chemokine (C-C motif) receptor type 7 (CCR7). This interaction (along with CXCL5/CXCR2) activates Snail and glycogen synthase kinase (GSk)-3 to promote the EMT [78]. Furthermore, it has been suggested that TGF-β interacts with the NF-kB signaling system to promote EMT in cancer cells [79]. In oral cancer, certain metastatic miRNAs control the EMT process. MiR-153-3p targets Snail in OSCC cells to block activation of EMT (Fig. 2) [80], while miR-532-3p targets CCR7 to reduce EMT, migration, and invasion [81]. Nevertheless, miR-940 overexpression prevented TSCC cells from metastasizing by inhibiting EMT, invasion and migration because it targeted the IL-8/CXCR2/NF-B pathway [82][83].

Regulation of EMT activation by miR-153-3p and WISP-1. WNT1-inducible-signaling pathway protein 1 (WISP-1) promotes the activation of EMT by upregulating Snail expression and regulating the integrin αvβ3/FAK/ILK/Akt signaling pathway. MiR-153-3p inhibits OSCC cell EMT and metastasis by targeting Snail, in return WISP-1 and induces metastasis [80]
The process of angiogenesis governs tumor development and metastasis, and significantly promotes cancer growth [84]. Neuropilin 1 (NRP-1) is a vascular endothelial growth factor (VEGF) co-receptor [85, 86] that shows increased expression in OSCC [87, 88], and esophageal carcinoma [89]. NRP1 overexpression has been found to affect immunity, angiogenesis, tumor invasion, and growth [90, 91]. The molecular pathways of NRP1 activity, however, have not yet been clarified. Recently, multiple studies showed that NRP1 expression in human cancer may be controlled by a variety of miRNAs [87, 90, 92]. MiR1247 was found to target NRP1 and reduce cell growth in pancreatic cancer models both in vivo and in vitro [90]. Cui et al. [93] found that by targeting NRP1, miR-9 and miR-181b prevented vascular endothelial cells in oral cancer from migrating, adhering to one another, and forming tubes. This report was consistent with Shang et al. [94] who reported that ectopic expression of miR-9 led to the induction of apoptosis, cell cycle arrest, and inhibited invasion of OSCC cells by targeting CDK 4/6 pathways [94]. Moreover, Wu et al. demonstrated that miR-320 also carried out the same function [87]. Liu and colleagues [88] discovered that miR-338 targeted NRP1 in OSCC cells, and that overexpression of NRP1 could significantly reduce the ability of miR-338 to prevent proliferation in these cells. Moreover, miR-338 overexpression prevented OSCC metastasis by inhibiting NRP1, suggesting that miR-338 could be a possible therapeutic target in OSCC [88].
Recent research suggests that miRNAs may also be involved in lymphangiogenesis, the creation of new mature lymphatic tubes from already-existing lymphatic vessels. MiR-126 is the best example of a lymphangiogenic miRNA. MiR-126 downregulation was shown to support oral cancer angiogenesis and lymphangiogenesis by increasing the expression of the genes for VEGF-A and VEGF-C, respectively [95]. Another example is miR-300 which can regulate the interaction between WISP-1 (protein 1 of the WNT1-inducible signaling pathway) and CCN4 (a member of the CCN family of cysteine-rich matrix proteins) [96]. It was reported that WISP-1 is responsible for downregulating miR-300, resulting in increased VEGF-C production [96]. As a result, OSCC lymphatic vessel formation will be increased, which would encourage cancer metastasis. In contrast, WISP-1 also controls VEGF-A [96]. Further investigation into the relationship between WISP-1 and miR-126 would be interesting [97].
Recently, niclosamide (an anthelmintic medication) has been shown to decrease vasculogenic mimicry (VM) by affecting miR-124 expression levels [98]. VM is the ability of an aggressive tumor to spontaneously produce blood vessels containing tumor cells but without any endothelial cells. This will guarantee that there is a sufficient blood supply for the growth and metastasis of oral cancer [99]. Niclosamide could inhibit VM by upregulating miR-124, which in turn has anti-tumor effects [98].
Mammalian species have five Notch ligands (Jagged 1 & 2, and Delta-like 4, 3, & 1), as well as four Notch receptors (Notch 1, 2, 3 & 4). When Notch ligands and receptors interact, the signaling pathway is triggered. Two important proteolytic enzymes are involved in this process. The first is an enzyme that converts tumor necrosis factor (also known as ADAM, a distintegrin and metalloprotease), which breaks down the receptor extracellular region. Then secretase catalyzes the second hydrolysis process to release the Notch intracellular domain (NICD) in the nucleus, which then attaches to the transcription factor CSL to activate downstream genes [100]. It was discovered that OSCC cells and tissues overexpress Notch1, and it has been found that this overexpression was related to the clinical stage, TNM-stage, differentiation, degree of invasion, metastasis to lymph nodes, and localized recurrence [101,102,103]. According to studies, Notch1 activation increases OSCC cell migration, invasion, and EMT, while inhibiting apoptosis, [101, 104]. These findings suggest that treatment of OSCC by specifically targeting Notch1 could be beneficial. Recently, Lv et al. [74] showed that in vitro invasion and proliferation of OSCC cells were inhibited by exogenous expression of miR-495. They investigated the inhibitory mechanism of miR-495 activity. They found that Notch1 was a direct functional target of miR495 in OSCC. By reactivating Notch1, the inhibitory effects of miR495 on OSCC cell invasion and proliferation were reversed. Their findings revealed that miR495 played a major role in the control of OSCC metastasis, in part by targeting Notch1 [74].
In order to spread to distant regions of the human body, epithelial tumor cells lose their characteristic markers, such as cytokeratins and E-cadherin, and instead express proteins found in mesenchymal cells, such as vimentin, N-cadherin and fibronectin in a process called the EMT [105]. After spreading to distant regions of the body, these mesenchymal cancer cells can then undergo the MET (reverse of EMT), which enables them to revert to expressing epithelial markers. The “c-MET tyrosine kinase” is encoded by the c-MET proto-oncogene, and promotes tumor migration and metastasis, [106, 107], This could be a significant factor in treatment resistance in oral cancer [108,109,110,111]. c-MET is often increased in a range of cancers to encourage the spread and growth of tumors. A poor prognosis and increased tumor metastasis have been associated with expression of the HGF receptor, also known as c-Met [112, 113]. It has been suggested that CD44v3 might encourage phosphorylation of c-Met in response to HGF. Additionally, OSCC has an upregulation of CD44 v3 and c-Met [114,115,116,117]. The ability of OSCC cell lines to invade and migrate is closely linked to the overexpression of miR-143 [118]. Xu et al. [119] reported that miR-143 overexpression in OSCC cell lines may inhibit invasion and migration while having little to no effect on proliferation. OSCC often exhibits a significant downregulation of miR-143 expression. They also found that inhibition of CD44v3 decreased migration in OSCC cells, which was directly linked to the activation of c-Met via the CD44 v3/HGF signaling pathway. Additionally, miR-143 may target CD44v3 to prevent phosphorylation of c-Met, which would inhibit OSCC cell invasion and migration. This investigation suggested that miR-143 might play a role in a new treatment approach for OSCC.
MiRNAs as promoters of metastasis during oral cancer
While some miRNAs promote metastasis, the bulk of those that have been investigated so far have inhibitory effects (Table 2).
One of the most well known tumour suppressors in squamous cell carcinoma is called phosphatase and tensin homolog deleted on chromosome 10 (PTEN), a phosphatase enzyme whose primary substrate is phosphatidylinositol 3,4,5-trisphosphate (PIP3) [163, 164]. PTEN controls the PI3K/AKT pathway via phosphatidylinositol 3-kinase [165]. When AKT is phosphorylated it becomes activated, and then controls cell death, migration and invasion [166]. PIP3 is dephosphorylated by PTEN, which converts it into phosphatidylinositol-4,5-bisphosphate (PIP2), which inactivates AKT by dephosphorylating it. This regulation of PTEN can have anti-cancer effects. According to earlier research, PTEN expression is suppressed by some miRNAs, which also contribute to the growth of cancer [167, 168]. Lizumi and associates [169] demonstrated that miR-142-5p increased the amount of active AKT by targeting PTEN (p-AKT). This shows that PTEN effectively inhibits PIP3 dephosphorylation to accelerate cancer growth by regulating the PI3K/AKT pathway. Additionally, in OSCC cells, PTEN expression was decreased when miR-142-5p was overexpressed which then stimulated proliferation and invasion. Additionally, PTEN-deficient OSCC cells exhibited the same behavior as cells that had been treated with a miR-142-5p mimic [169]. Various miRNAs stimulate the PTEN/AKT pathway to inhibit the invasion and growth of many cancer types [170]. According to additional research, increased miR-655 expression prevented OSCC cells from proliferating and invading while also preventing the PTEN/AKT pathway from becoming activated [124, 171]. Zheng et al showed that the miR-24 seed sequence bound to the 3’ UTR of PTEN mRNA, thereby inhibiting PTEN translation by activating the PI3K/Akt pathway. The AKT pathway is involved in the control of PTEN expression by miR-24. MiR-24 increases cell invasion, viability, and chemoresistance via targeting the PTEN/Akt pathway [172]. Moreover, OSCC patients have higher miR-155 levels, and BMSCs displaying increased expression of miR-155 might promote the proliferation and spread of OSCC by inhibiting PTEN12 [173].
While certain miRNAs, such as miR-29b-1-5p, target E-cadherin directly to promote the EMT, other miRNAs indirectly stimulate the EMT [174]. For example, miR-134 targets PDCD7 thereby decreasing E-cadherin expression, promoting EMT, and stimulating oral cancer metastastasis [175]. Li et al. [176] asked whether or not miR-424 or miR-19a could regulate the expression of the TGFBR3 gene (transforming growth factor beta receptor 3). They discovered that in CAL-27 cells, miR-19a and miR-424 overexpression facilitated migration and EMT. When CAL-27 cells were transfected with a plasmid expressing the TGFBR3 gene, this reversed the increased migration that was induced by miR-19a or miR-424, and prevented EMT from progressing by decreasing p-p65 expression in comparison to the control group. These findings demonstrated that miR-424 or miR-19a overexpression could trigger EMT and encourage cell migration by targeting TGFBR3 [176].
Dickkopf-1 (DKK1) is a transcriptional target of the Wnt/β-catenin pathway, and acts as a suppressor of Wnt signaling [177]. According to research, lithium chloride usually encourages the spread of cancer cells, while Wnt signaling suppression via DKK1 reduces invasion in many cancers [177]. Since antagonist molecules may negatively control the Wnt/β-catenin signalling pathway, miRNAs that target these molecules could act as EMT drivers. It was discovered that the DKK gene complex (DKK1-4) inhibited tumor migration and invasion via negatively regulating β–catenin [178, 179]. It was found that DKK1 was targeted by miR-373-3p in TSCC tissues. MiR-373-3p increased TSCC metastases caused by EMT and constitutively activated Wnt/β-catenin signaling by specifically targeting DKK1 [180]. Secreted frizzled-related protein 1 (SFRP1) is a Wnt signaling antagonist, which interacts with Wnt proteins via its CRD domain, in contrast to the transmembrane frizzled receptor [181]. It was discovered that miR-27a-3p targeted SFRP1 and initiated EMT in OSCC stem cells [182].
The metastasis suppressor gene known as metastasis suppressor-1 (MTSS1), also referred to as MIM (missing in metastasis), was first shown to be a tumor suppressor gene in non-metastatic bladder cancer cell lines. Its expression is restricted to chromosome 8q24.1 in humans [183]. In metastatic cells, MTSS1 expression is typically diminished, whereas its relative expression is unclear in primary cancers. For example, it was confirmed that MTSS1 expression is decreased in esophageal, ovarian, prostate, colorectal, and breast cancers [184, 185], although hepatocellular carcinoma and breast cancer were found to have higher levels of MTSS1 expression [186, 187]. Recent studies revealed that both bladder and kidney cancer displayed low or nonexistent MTSS1 protein staining [188, 189]. The MTSS1 gene was found to be down-regulated in TSCC tissues. According to functional and mechanistic studies, the MTSS1 protein might be associated with the spread of cancer to various organ sites, most likely by interaction with the actin cytoskeleton, or by being regulated by miRNAs [190,191,192]. Guo et al. [193] analyzed the effect of the MTSS1 gene on the proliferation and invasion of Tca8113 cells using MTT, scratch wound healing, and invasion assays. They also examined whether miR-96 targeted MTSS1 and how it affected the biological changes caused by the MTSS1 gene in Tca8113 cells. They discovered that Tca8113 cells and TSCC tissues both showed down-regulated MTSS1 expression. Moreover, forced expression of MTSS1 resulted in reduced numbers of migrating cells, slower wound healing, and hindered proliferation. Moreover, Tca8113 cell proliferation and spread may be regulated by miR-96 through the activity of MTSS1. MiR-96 was unable to fully reverse Tca8113 cell propensity to invade. Therefore, they hypothesized that miR-96 targeting and MTSS1 suppression may hasten the development of TSCC by evading the regulation of proliferation and metastasis. Another study looked at connections between MTSS1 and miR-182-5p in OSCC [194]. Higher TNM grades were linked to elevated miR-182-5p expression in OSCC, so miR-182-5p was proposed to be involved in invasion and migration of OSCC. MiR-182-5p directly targeted MTSS1, and by down-regulating MTSS1 expression levels it may promote invasion and migration in oral cancer [194].
Cyclin-dependent kinases (CDKs). are sequentially activated and inactivated during the cell cycle progression. Cyclin-CDK inhibitors inactivate CDKs and work together with positive regulators (cyclins) to activate them. One cyclin-CDK inhibitor is P27 (also known as CDKN1B, KIP1, or cyclin-dependent kinase inhibitor 1B), and prevents the cell cycle from entering the S-phase. Ubiquitin-mediated protein degradation controls the levels of p27 at the post-translation level [195]. The substrate recognition element that binds to and marks p27 for ubiquitination and eventual degradation was identified as the F-box protein SKP2 [195]. In certain malignancies, low levels of p27 have been linked to faster tumor growth and a poor prognosis [195, 196]. MiRNA-221 was found to target CDKN1B (cyclin-dependent kinase inhibitor 1B, also known as p27) [197][198]. Moreover, CDKN1B/p27 has been suggested to be a potential therapeutic target and prognostic marker in ovarian cancer [199,200,201]. Yang et al. [202], investigated how miR-222-3p affected cell division, invasion, migration and apoptosis. They found that compared to healthy tissues, OSCC tissues had higher levels of miR-222-3p. Also, they claimed that miR222-3p might inhibit cell division, migration, and invasion as well as cause the death of Tca-83 and SCC-15 cells. Moreover, testing with luciferase reporters showed that miR-222-3p specifically targeted CDKN1B in OSCC cells. As a result, OSCC cell migration, invasion, and proliferation were reduced while the rate of cell death was increased when CDKN1B was overexpressed. Overall, they demonstrated that miR-222-3p, which targeted CDKN1B, caused OSCC cells to invade and metastasize by acting as an oncomiR, and could be used as a predictive biomarker in OSCC patients [202]. Chen et al. [203] found that OSCC tissues showed overexpression of miR-222, and that by targeting CDKN1, it could promote invasion and metastasis [203].
The Fbxw7 protein, also known as Sel-10, hCdc4, or hAgo, is a substrate for the ubiquitin ligase complex called Skp1-Cul1-F-box protein-Rbx1 (SCF), which binds to its receptor [204]. SCF is an E3-ubiquitin ligase which ubiquitinates certain proteins and leads to proteasome degradation [204, 205]. Moreover, FBXW7 controls many biological processes, such as cell cycle progression, differentiation, and stemness of brain cells, maintenance of genomic stability, and cell proliferation [206]. In a number of human cancers, FBXW7 acts as a tumor suppressor. Recent studies suggested that FBXW7 contributes to tumor metastasis, because increasing FBXW7 reduces cancer metastasis and EMT [204, 207]. FBXW7 was recently discovered to be a target gene for some miRNAs in various cancers. For example, miR-223 controlled acute lymphoblastic leukemia by reducing FBXW7 expression [208]. MiR-27a increased lung cancer cell growth by inhibiting FBXW7, demonstrating that FBXW7 could act as a tumor suppressor [209]. In TSCC cells, proliferation, migration, and invasion were markedly reduced when miR-24 was inhibited. When FBXW7 was suppressed, this caused TSCC cells to proliferate, migrate, and invade more readily. Conversely, when FBXW7 was restored, it significantly reduced the oncogenic effect of miR-24. They concluded that miR-24 could target FBXW7 especially in TSCC cells. As a result, miR-24 can promote TSCC cell metastasis by targeting FBXW7 and increasing migration and invasion [210]. In a different investigation, Jiang et al. demonstrated that by reducing the expression of FBXW7 in OSCC cells, miR-223 could encourage OSCC cell migration [211].
CircularRNA/lncRNA/miRNA networks and oral cancer metastasis
Noncoding RNAs (ncRNAs) are crucial for most cell biology processes, and also contribute to the growth and spread of malignancies. The ncRNAs that control cancer cell proliferation, cell death, invasion, and metastasis can be divided into circular RNAs (circRNAs), long noncoding RNAs (lncRNAs), as well as miRNAs (discussed above) [236,237,238]. NcRNAs control the growth of cancer by affecting target gene e asxpression (mRNA) [239]. Both lncRNAs and circRNAs contain binding sites for specific complementary miRNAs, so they can act as competing endogenous RNAs or miRNA sponges, thereby enhancing the expression of the miRNA target genes. If the miRNAs are removed, they can no longer block the translation of mRNAs and silence their target gene expression. The regulatory networks for ceRNAs are associated with the biological basis of cancer [240, 241]. Additionally, circRNAs and lncRNAs may directly interact with various specific proteins to regulate gene transcription [242]. Several studies have examined the role of circRNA-lncRNA-miRNA-gene regulatory networks in oral cancer metastasis, which we summarize below. This research increases our understanding of oral cancer pathogenesis, and could provide new opportunities for less invasive early detection methods and improved therapeutic options.
LncRNA/miRNA networks
LncRNAs are RNA sequences that are over 200 nucleotides long and do not code for any specific proteins. LncRNAs are important for the growth, development, and metastasis of many different cancers, according to accumulating evidence. They are also becoming promising molecular biomarkers for the prognosis and early diagnosis of cancer patients [243, 244]. A variety of tumor types may have lncRNAs intriguing therapeutic targets in and the pathways that they affect (Fig. 3).

LncRNAs act as competitive endogenous RNAs (ceRNAs). The interactions between lncRNAs and miRNAs play critical roles in the regulation of oral cancer metastasis pathways [285]
A number of human cancers show aberrant expression of LncRNA-H19 [245, 246], which is typically connected to the spread of cancer, a poor prognosis, and cancer recurrence. H19 has also been shown to control the invasion, metastasis, and migration of different malignancies by functioning as a ceRNA [245, 247, 248]. An essential mechanism for how H19 controls OSCC metastasis is its interaction with certain specific miRNAs. Kou et al. [249] discovered that the expression of H19 was higher in metastatic TSCC tissues compared to non-metastatic TSCC tissues, and that H10 knockdown may reduce TSCC cell invasion and migration. According to the proposed mechanism, let-7 was targeted by H19 in order to enhance the expression of HMGA2, a crucial regulator of tumor metastasis. In contrast, TSCC cell migration and invasion were less inhibited by H19 knockdown when let-7a was inhibited [249]. According to a different study, the expression of H19 was inversely connected to overall survival and was favorably associated with the TNM stage. According to another study, H19 could sponge miR-138, thereby increasing the expression of its target gene EZH2, thus promoting OSCC cell invasion, migration, and EMT [32].
LncRNA-MEG3 mostly acts as a tumor suppressor, in contrast to H19. MEG3 is not highly expressed in an all types of cancer according to various studies [250, 251], but when it is expressed it inhibits their invasion, metastasis, and migration. By acting as a ceRNA, it has been shown that MEG3 could also prevent OSCC from migrating and invading. Tan et al. [252], showed that OSCC tissues generally had low MEG3 expression. In return, MEG3 overexpression promoted OSCC cell invasion and migration by downregulation of miR-548d-3p. MiR-548d-3p is inhibited by MEG3, which in turn promotes the production of SOCS5 (cytokine signaling suppressor 5), and SOCS6 (cytokine signaling suppressor 6). Secondly, the JAK/STAT (Janus kinase/signal transducer and activator of transcription) pathway is inhibited by SOCS5/SOCS6 to prevent OSCC invasion and migration [252]. According to a different study, miR-21 and MEG3 were inversely associated in OSCC tissues. The subsequent dual luciferase assay proved that MEG3 and miR-21 directly interacted with each other. Additionally, lowering miR-21 would reduce OSCC cell migration, but inhibiting MEG3 would partially undo the effect of miR-21 downregulation on the migration of OSCC cells [253]. They suggested that MEG3 may sponge miR-21 thereby preventing the OSCC cells from migrating. However, further investigations into the underlying molecular mechanisms are necessary, and confirming the role of MEG3 in OSCC metastasis will require more in vivo experiments.
LncRNAs-TUG1 is an lncRNA which has been intensively studied. TUG1 has been found to enhance the proliferation of OSCC cells by sponging some miRNAs which have an inhibitory effect on the cancer cells. According to Liu et al., OSCC cells showed high TUG1 expression levels which could increase the ability of these cells to migrate. TUG1 and DLX1 (distal-less homeobox 1) may compete with one another for binding to miR-524-5p and thus upregulating DLX1 expression [254]. Additionally, Yan et al. found that TUG1 overexpression boosted OSCC cell capacity to metastasis in vivo while TUG1 knockdown decreased the OSCC cells ability to migrate and invade. Further investigation revealed that TUG1 and miR-219 inhibited one another in a reciprocal fashion, suggesting that suppressing miR-219 in OSCC cells might counteract the inhibitory effects of TUG1 knockdown. They concluded that TUG1 encourages metastasis by functioning as a ceRNA to scavenge miR-219 and increase the expression of the miR-219 target FMNL2 (formin-like protein 2) [255].
LncRNA-UCA1 was first identified in bladder cancer and has since been shown to facilitate bladder cancer cell invasion and migration [256, 257]. Recently, OSCC metastasis has also been linked to aberrant UCA1 expression. UCA1 is overexpressed in TSCC tissues, according to Fang et al., and its degree of expression was strongly linked with lymph node metastasis [258]. Similarly, Zhang et al. found a link between UCA1 overexpression and a poor prognosis in TSCC patients, especially the occurrence of lymph node metastasis and shorter survival. Unexpectedly, UCA1 could sponge miR-124 in TSCC cells and therefore negatively regulate itself. Further research suggested that UCA1 could activate the JAG1/Notch pathway by targeting miR-124 to increase the production of TGF-1, thus promoting TSCC cell invasion and EMT [259]. Additionally, in a different study, it was found that UCA1 competitively bound to miR-143-3p and affected MYO6 (myosin VI) expression thus enhancing TSCC cell invasion, EMT and migration [260].
CircRNA/miRNA networks
CircRNAs are a group of closed circular single-stranded RNA molecules that can regulate gene expression at both the transcriptional and posttranscriptional levels [261]. In 2012, Salzman et al. [262] reported that more than 10% of expressed genes have the potential to create circRNAs. Circular transcripts of the protein linked to cerebellar degeneration 1 (CDR1, also called ciRS-7) antisense RNA were discovered to act as miR-7 sponges, according to a 2013 study by Hansen et al., and Memczak et al. [263, 264]. These studies made circRNAs a new focus for scientific investigation in the noncoding RNA field. CircRNAs significantly contribute to signaling networks that promote the growth and spread of cancer. For example, lncRNA-WDFY3-AS2 suppressed OSCC cell metastasis by targeting the Wnt/β-catenin signaling pathway [265]. The migration, invasion, and metastasis of OSCC are likewise affected by dysregulated circRNA expression since these genes are regulated by sponging of various miRNAs (Table 3; Fig. 4) For instance, Xia et al. reported that circ-0001162 (circ-MMP9), a metastasis-associated circRNA, was elevated in OSCC samples [266]. Given its strong relationship with MMP9 expression, circ-MMP9 may serve as a sponge for miR-149 to target AUF1. This was shown to be correct both in vitro and in vivo. Circ-MMP could prevent OSCC from spreading by inhibiting MMP9 expression. CircUHRF1 may up-regulate c-Myc by acting as a miR-526b-5p sponge, which may enhance the transcription of ESRP1 and TGF-1. In OSCC, CircUHRF1 was markedly overexpressed [267]. Additionally, it was found that circUHRF1 could circularize and increase with the aid of ESRP1, creating a feedback loop between those two factors, as well as TGF-1, c-Myc, miR-526b-5p, and ESRP1 that working together could promote OSCC carcinogenesis and EMT [267]. Studies conducted in vitro have shown that inhibition of circUHRF1 could reduce the ability of OSCC cells to migrate, proliferate, invade, and undergo the EMT. Additionally, in vivo functional tests demonstrated that blocking circUHRF1 could effectively halt OSCC tumor growth. When circ-PKD2 was overexpressed the ability of miR-204 to promote cancer metastasis was dramatically reduced, because OSCC migration and and invasion was inhibited. An in vivo study showed that the size and weight of OSCC xenografted tumors were greatly reduced by overexpression of circ-PKD2 [267]. Dual-luciferase reporter analysis confirmed that miR-204-3p and circ-PKD2 directly interacted, and miR-204-3p targeted APC2 through a downstream signaling pathway [22]. The extracellular signal-regulated kinase 1/2, protein kinase B, and β-catenin pathways were all inhibited by circ-PKD2 because it up regulated APC2 and reduced the inhibitory effect of miR-204-3p. Moreover another circRNA called circDOCK1 was found to strongly expressed in OSCC cells, and it was discovered that circDOCK1 targeted miR-196a-5p [22].

Interaction between circIGHG and miR-142- 5p/IGF2BP3 in OSCC. CircIGHG is a circRNA derived from the IGHG locus. The expression of circIGHG is increased in OSCC cells and is positively correlated with poor prognosis in OSCC. MiR-142-5p can inhibit OSCC metastasis by targeting IGF2BP3, in return, circIGHG promotes EMT activation of OSCC cells by sponging miR-142-5p and upregulating IGF2BP3 [275]
Circ-0000140 showed markedly decreased expression in OSCC patient samples [268]. According to one study, low circ-0000140 expression might function as a biomarker for OSCC progression, because it is strongly associated with OSCC patient poor prognosis [23, 269]. However, further research into the function and mechanism of circ-0000140 in OSCC is needed. Recently, Peng et al. [269] reported that OSCC patients showed downregulation of circ 0000140, and low circ-0000140 expression was correlated with lymph node metastasis and more advanced TMN stage. Additionally, survival analysis revealed that OSCC patients with low circ-0000140 expression levels had a significantly worse 5-year survival rate. Mechanistic studies in vitro showed that OSCC cell proliferation, invasion and migration were all inhibited by the overexpression of circ-0000140. Further evidence that circ0000140 inhibited the EMT in OSCC cells was provided by the higher E-cadherin and lower N-cadherin protein levels observed in OSCC cells with overexpressed circ-0000140 [269]. When circ0000140 was overexpressed in a xenograft mouse model, in addition to shrinking the tumor, it also prevented lung metastasis of tumors grown from two separate OSCC cancer cell lines. Moreover, it was shown that overexpression of circ-0000140 caused a > 50% reduction in metastatic lung nodules in an in vivo model. Furthermore, circ-0000140 was found to bind to miR-31 and thus increase the expression of its target gene LATS2, which would have an effect on the EMT in OSCC cells [269]. LATS1/2 are significant regulators of both tumor-suppressor as well as oncogenic effectors during cancer spread [270]. Taken together, these findings showed that circ-0000140 inhibited OSCC metastasis via targeting the miR-31/LATS2 axis [269]. In accordance with the findings of Peng et al. [269], Guo and colleagues [23] found that OSCC showed low levels of circ-0000140 expression, and its overexpression prevented cell invasion and migration, and reduced glycolysis. When circ-0000140 was overexpressed it suppressed OSCC tumor growth in vivo. Moreover, they found that circ-0000140 sponged miR-182-5p which targeted CDC73. A miR-182-5p mimic reversed the inhibition of OSCC caused by circ 0000140 overexpression. They discovered that circ-0000140 may prevent OSCC from spreading by controlling the miR-182-5p/CDC73 axis [23].
Recently, Hei and colleagues found that the expression level of circ_0020377 was significantly upregulated in OSCC tissue samples and OSCC cell lines. Also, they found that miR-194-5p acted as tumor suppressor miRNA in OSSC cells by targeting KLF7. Furthermore, they observed that upregulated circ_0020377 promoted OSCC cancer development and metastasis. Mechanistically, they suggested that circ_0020377 promoted invasion and migration of OSCC cells by by sponging miR-194-5p and thus upregulating KLF7 [271]. These findings suggest that further investigation is required into the circRNA-miRNA networks which control signaling pathways and related genes that are relevant to the development and spread of oral cancer.
Conclusion
Metastatic cancer is still mostly incurable due to the lack of effective clinical treatment and our limited understanding of how oral cancer metastasizes. Our understanding of the roles of certain miRNAs in metastasis has advanced a lot in the last ten years. In order to treat oral cancer, many of these metastasis-inducing miRNAs could be attractive therapeutic targets. It should be emphasized that certain lncRNAs or circRNAs have the ability to regulate the actions of specific miRNAs. Moreover, a single miRNA (or a miRNA cluster/family) may play more than one role in the invasion-metastasis cascade. For example, miR-200 family members inhibit tumor cell EMT, migration, and infiltration, but they can also aid in metastasis by increasing the suitability of the OSCC microenvironment for invasion. Therefore, prior to entering clinical trials, the therapeutic potential of miRNA-based therapeutics should be rigorously assessed using relevant laboratory models and in real life clinical situations. A full understanding of the role of miRNA biogenesis and their mode of action in metastasis requires further study. Crosstalk between miRNAs and lncRNAs/circRNAs has recently become a fascinating study area (Table 3). Nonetheless, functional investigations have shown that miRNAs are an important pathway by which ceRNAs can control oncogenes and tumor suppressor genes. Therefore, future studies must pay greater attention to the circRNA/lncRNA-miRNA-mRNA networks in oral cancer metastasis.
Data Availability
Not applicable.
References
D’souza S, Addepalli V. Preventive measures in oral cancer: an overview. Biomed Pharmacother. 2018;107:72–80.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J Clin. 2021;71(3):209–49.
Van Zyl AW, Bunn BK. Clinical features of oral cancer: clinical review. South Afr Dent J. 2012;67(10):566–9.
Noguti J, De Moura CFG, De Jesus GPP, Da Silva VHP, Hossaka TA, Oshima CTF, et al. Metastasis from oral cancer: an overview. Cancer Genomics Proteomics. 2012;9(5):329–35.
Okura M, Aikawa T, Sawai NY, Iida S, Kogo M. Decision analysis and treatment threshold in a management for the N0 neck of the oral cavity carcinoma. Oral Oncol. 2009;45(10):908–11.
Chi AC, Day TA, Neville BW. Oral cavity and oropharyngeal squamous cell carcinoma—an update. Cancer J Clin. 2015;65(5):401–21.
Sankunny M, Parikh RA, Lewis DW, Gooding WE, Saunders WS, Gollin SM. Targeted inhibition of ATR or CHEK1 reverses radioresistance in oral squamous cell carcinoma cells with distal chromosome arm 11q loss. Genes Chromosom Cancer. 2014;53(2):129–43.
Pannone G, Santoro A, Papagerakis S, Lo Muzio L, De Rosa G, Bufo P. The role of human papillomavirus in the pathogenesis of head & neck squamous cell carcinoma: an overview. Infect agents cancer. 2011;6:1–11.
Patel SC, Carpenter WR, Tyree S, Couch ME, Weissler M, Hackman T, et al. Increasing incidence of oral tongue squamous cell carcinoma in young white women, age 18 to 44 years. J Clin Oncol. 2011;29(11):1488–94.
Mehrotra R, Yadav S. Oral squamous cell carcinoma: etiology, pathogenesis and prognostic value of genomic alterations. Indian J Cancer. 2006;43(2):60–6.
Kumar M, Nanavati R, Modi TG, Dobariya C. Oral cancer: etiology and risk factors: a review. J Cancer Res Ther. 2016;12(2):458–63.
Ramqvist T, Grün N, Dalianis T. Human papillomavirus and tonsillar and base of tongue cancer. Viruses. 2015;7(3):1332–43.
Bloebaum M, Poort L, Böckmann R, Kessler P. Survival after curative surgical treatment for primary oral squamous cell carcinoma. J Cranio-Maxillofacial Surg. 2014;42(8):1572–6.
Le Campion ACOV, Ribeiro CMB, Luiz RR, da Silva Júnior FF, Barros HCS et al. Dos Santos KdCB,. Low survival rates of oral and oropharyngeal squamous cell carcinoma. International journal of dentistry. 2017;2017.
Sarode G, Maniyar N, Sarode SC, Jafer M, Patil S, Awan KH. Epidemiologic aspects of oral cancer. Dis Mon. 2020;66(12):100988.
Shukla GC, Singh J, Barik S. MicroRNAs: processing, maturation, target recognition and regulatory functions. Mol Cell Pharmacol. 2011;3(3):83.
Friedman RC, Farh KK-H, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19(1):92–105.
Mousavi SM, Mahdian SMA, Ebrahimi MS, Taghizadieh M, Vosough M, Nahand JS et al. Microfluidics for detection of exosomes and microRNAs in cancer: state of the art. Mol Therapy-Nucleic Acids. 2022.
Erfanparast L, Taghizadieh M, Shekarchi AA. Non-coding RNAs and oral Cancer: small molecules with big functions. Front Oncol. 2022;12:914593.
Nahand JS, Shojaie L, Akhlagh SA, Ebrahimi MS, Mirzaei HR, Baghi HB, et al. Cell death pathways and viruses: role of microRNAs. Mol Therapy-Nucleic Acids. 2021;24:487–511.
Sadri Nahand J, Salmaninejad A, Mollazadeh S, Tamehri Zadeh SS, Rezaee M, Sheida AH et al. Virus, Exosome, and MicroRNA: New Insights into Autophagy. Cell Biology and Translational Medicine, Volume 17: Stem Cells in Tissue Differentiation, Regulation and Disease: Springer. 2022;97–162.
Gao L, Zhao C, Li S, Dou Z, Wang Q, Liu J, et al. circ-PKD2 inhibits carcinogenesis via the miR‐204‐3p/APC2 axis in oral squamous cell carcinoma. Mol Carcinog. 2019;58(10):1783–94.
Guo J, Su Y, Zhang M. Circ_0000140 restrains the proliferation, metastasis and glycolysis metabolism of oral squamous cell carcinoma through upregulating CDC73 via sponging miR-182-5p. Cancer Cell Int. 2020;20:1–12.
Zeng B, Li Y, Jiang F, Wei C, Chen G, Zhang W, et al. LncRNA GAS5 suppresses proliferation, migration, invasion, and epithelial-mesenchymal transition in oral squamous cell carcinoma by regulating the miR-21/PTEN axis. Exp Cell Res. 2019;374(2):365–73.
Yao Y, Liu Y, Jin F, Meng Z. LINC00662 promotes oral squamous cell Carcinoma Cell Growth and Metastasis through miR-144-3p/EZH2 Axis. Yonsei Med J. 2021;62(7):640–9.
Li X, Li Y, Jiang C, Chen L, Gan N. MicroRNA-144-3p inhibits tumorigenesis of oral squamous cell carcinoma by downregulating ERO1L. J Cancer. 2020;11(3):759–68.
Seyfried TN, Huysentruyt LC. On the origin of cancer metastasis. Crit Reviews™ Oncog. 2013;18:1–2.
Guan X. Cancer metastases: challenges and opportunities. Acta Pharm sinica B. 2015;5(5):402–18.
Damsky W, Theodosakis N, Bosenberg M. Melanoma metastasis: new concepts and evolving paradigms. Oncogene. 2014;33(19):2413–22.
Meirson T, Gil-Henn H, Samson AO. Invasion and metastasis: the elusive hallmark of cancer. Oncogene. 2020;39(9):2024–6.
Scuoppo C, Wang J, Persaud M, Mittan SK, Basso K, Pasqualucci L et al. Repurposing dasatinib for diffuse large B cell lymphoma. Proceedings of the National Academy of Sciences. 2019;116(34):16981-6.
Weber GF. Why does cancer therapy lack effective anti-metastasis drugs? Cancer Lett. 2013;328(2):207–11.
Chakravarty D, Ratnani P, Huang L, Dovey Z, Sobotka S, Berryhill R et al. Association between Incidental Pelvic inflammation and aggressive prostate Cancer. Cancers (Basel). 2022;14(11).
Sakamoto S, Kyprianou N. Targeting anoikis resistance in prostate cancer metastasis. Mol Aspects Med. 2010;31(2):205–14.
Eccles SA, Welch DR. Metastasis: recent discoveries and novel treatment strategies. The Lancet. 2007;369(9574):1742–57.
Alizadeh AM, Shiri S, Farsinejad S. Metastasis review: from bench to bedside. Tumor biology. 2014;35:8483–523.
Brabletz T, Kalluri R, Nieto MA, Weinberg RA. EMT in cancer. Nat Rev Cancer. 2018;18(2):128–34.
Spano D, Heck C, De Antonellis P, Christofori G, Zollo M, editors. Molecular networks that regulate cancer metastasis. Seminars in cancer biology. Elsevier; 2012.
Lee RC, Feinbaum RL, Ambros VJc. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. 1993;75(5):843–54.
Bartel DPJc. MicroRNAs: genomics, biogenesis, mechanism, and function. 2004;116(2):281–97.
Winter J, Jung S, Keller S, Gregory RI, Diederichs SJNcb. Many roads to maturity: microRNA biogenesis pathways and their regulation. 2009;11(3):228–34.
Yi R, Qin Y, Macara IG, Cullen BRJG, editors. development. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. 2003;17(24):3011-6.
Yoshida T, Asano Y, Ui-Tei KJN-cR. Modulation of MicroRNA Processing by Dicer via its Associated dsRNA. Binding Proteins. 2021;7(3):57.
O’Brien J, Hayder H, Zayed Y, Peng CJFie. Overview of microRNA biogenesis, mechanisms of actions, and circulation. 2018;9:402.
Nejad C, Stunden HJ. Gantier MPJTFj. A guide to miRNAs in inflammation and innate immune responses. 2018;285(20):3695 – 716.
Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, Noch E et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proceedings of the national academy of sciences. 2002;99(24):15524-9.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8.
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proceedings of the National Academy of Sciences. 2004;101(9):2999–3004.
Ma L. MicroRNA and metastasis. Adv Cancer Res. 2016;132:165–207.
He X, He L, Hannon GJ. The guardian’s little helper: microRNAs in the p53 tumor suppressor network. Cancer Res. 2007;67(23):11099–101.
Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010;467(7311):86–90.
Svoronos AA, Engelman DM, Slack FJ. OncomiR or tumor suppressor? The duplicity of microRNAs in cancer. Cancer Res. 2016;76(13):3666–70.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449(7163):682–8.
Zheng M, Jiang Y-p, Chen W, Liu X, Gao S-y, Feng H, et al. Snail and slug collaborate on EMT and tumor metastasis through miR-101-mediated EZH2 axis in oral tongue squamous cell carcinoma. Oncotarget. 2015;6(9):6794.
Wu B, Lei D, Wang L, Yang X, Jia S, Yang Z, et al. MiRNA-101 inhibits oral squamous-cell carcinoma growth and metastasis by targeting zinc finger E-box binding homeobox 1. Am J cancer Res. 2016;6(6):1396.
Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203(9):2201–13.
Keeley EC, Mehrad B, Strieter RM. CXC chemokines in cancer angiogenesis and metastases. Adv Cancer Res. 2010;106:91–111.
Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29(4):709–22.
Xia J, Wang J, Chen N, Dai Y, Hong Y, Chen X, et al. Expressions of CXCR7/ligands may be involved in oral carcinogenesis. J Mol Histol. 2011;42:175–80.
Hui Y, Li Y, Jing Y, Feng JQ, Ding Y. miRNA-101 acts as a tumor suppressor in oral squamous cell carcinoma by targeting CX chemokine receptor 7. Am J translational Res. 2016;8(11):4902–11.
Xie C, Du L-Y, Guo F, Li X, Cheng B. Exosomes derived from microRNA-101-3p-overexpressing human bone marrow mesenchymal stem cells suppress oral cancer cell proliferation, invasion, and migration. Mol Cell Biochem. 2019;458:11–26.
Xie F, Ling L, van Dam H, Zhou F, Zhang L. TGF-β signaling in cancer metastasis. Acta Biochim Biophys Sin (Shanghai). 2018;50(1):121–32.
Wang Y, Jia RZ, Diao S, He J, Jia L. miRNA-101 targets TGF-βR1 to Retard the progression of oral squamous cell carcinoma. Oncol Res. 2020;28(2):203–12.
Long F, Wang N, Wang J, Zheng Y. miR-101 inhibits AKT/mTOR and attenuates oral cancer cell colony formation and epithelial-mesenchymal transition. J Biomaterials Tissue Eng. 2020;10(10):1436–40.
Li Y-y, Tao Y-w, Gao S, Li P, Zheng J-m, Zhang S-e, et al. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine. 2018;36:209–20.
Jia L-f, Wei S-b, Mitchelson K, Gao Y, Zheng Y-f, Meng Z, et al. miR-34a inhibits migration and invasion of tongue squamous cell carcinoma via targeting MMP9 and MMP14. PLoS ONE. 2014;9(9):e108435.
Li T, Li L, Li D, Wang S, Sun J. MiR-34a inhibits oral cancer progression partially by repression of interleukin-6-receptor. Int J Clin Exp Pathol. 2015;8(2):1364–73.
Dharavath B, Butle A, Pal A, Desai S, Upadhyay P, Rane A, et al. Role of miR-944/MMP10/AXL-axis in lymph node metastasis in tongue cancer. Commun Biology. 2023;6(1):57.
Shiozawa Y, Nie B, Pienta KJ, Morgan TM, Taichman RS. Cancer stem cells and their role in metastasis. Pharmacol Ther. 2013;138(2):285–93.
Liao WT, Ye YP, Deng YJ, Bian XW, Ding YQ. Metastatic cancer stem cells: from the concept to therapeutics. Am J stem cells. 2014;3(2):46–62.
Larzabal L, El-Nikhely N, Redrado M, Seeger W, Savai R, Calvo A. Differential effects of drugs targeting cancer stem cell (CSC) and non-CSC populations on lung primary tumors and metastasis. PLoS ONE. 2013;8(11):e79798.
You X, Zhou Z, Chen W, Wei X, Zhou H, Luo W. MicroRNA-495 confers inhibitory effects on cancer stem cells in oral squamous cell carcinoma through the HOXC6-mediated TGF-β signaling pathway. Stem Cell Res Ther. 2020;11(1):1–14.
Hwang-Verslues W, Chang P, Wei P, Yang C, Huang C, Kuo W, et al. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011;30(21):2463–74.
Lv L, Wang Q, Yang Y, Ji H. MicroRNA–495 targets Notch1 to prohibit cell proliferation and invasion in oral squamous cell carcinoma. Mol Med Rep. 2019;19(1):693–702.
Ajemian I. Seminar on Canadian Medical Education in Palliative Care–A Report. 7th International Congress on Care of the terminally ill, Montreal, October 20, 1988. J Palliat Care. 1989;5(1):50–1.
Tsai JH, Yang J. Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–206.
Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22(6):725–36.
Zhou S-L, Zhou Z-J, Hu Z-Q, Li X, Huang X-W, Wang Z, et al. CXCR2/CXCL5 axis contributes to epithelial–mesenchymal transition of HCC cells through activating PI3K/Akt/GSK-3β/Snail signaling. Cancer Lett. 2015;358(2):124–35.
Ma H, Gao L, Li S, Qin J, Chen L, Liu X, et al. CCR7 enhances TGF-β1-induced epithelial-mesenchymal transition and is associated with lymph node metastasis and poor overall survival in gastric cancer. Oncotarget. 2015;6(27):24348.
Chang A-C, Lien M-Y, Tsai M-H, Hua C-H, Tang C-H. WISP-1 promotes epithelial-mesenchymal transition in oral squamous cell carcinoma cells via the miR-153-3p/Snail axis. Cancers. 2019;11(12):1903.
Feng C, So HI, Yin S, Su X, Xu Q, Wang S, et al. MicroRNA-532-3p suppresses malignant behaviors of tongue squamous cell carcinoma via regulating CCR7. Front Pharmacol. 2019;10:940.
Ma T, Zhao Z, Wang Z, Wang C, Zhang L. MiR-940 inhibits migration and invasion of tongue squamous cell carcinoma via regulatingCXCR2/NF-κB system-mediated epithelial–mesenchymal transition. Naunyn Schmiedebergs Arch Pharmacol. 2019;392:1359–69.
Li D, Liu K, Li Z, Wang J, Wang X. miR-19a and miR-424 target TGFBR3 to promote epithelial-to-mesenchymal transition and migration of tongue squamous cell carcinoma cells. Cell Adhes Migr. 2018;12(3):236–46.
Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–70.
Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J Biol Chem. 2000;275(35):26690–5.
Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, et al. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem. 2007;282(33):24049–56.
Wu Y-Y, Chen Y-L, Jao Y-C, Hsieh I-S, Chang K-C, Hong T-M. miR-320 regulates tumor angiogenesis driven by vascular endothelial cells in oral cancer by silencing neuropilin 1. Angiogenesis. 2014;17:247–60.
Liu C, Wang Z, Wang Y, Gu W. MiR-338 suppresses the growth and metastasis of OSCC cells by targeting NRP1. Molecular and cellular biochemistry. 2015;398:115–22.
Alattar M, Omo A, Elsharawy M, Li J. Neuropilin-1 expression in squamous cell carcinoma of the oesophagus. Eur J Cardiothorac Surg. 2014;45(3):514–20.
Shi S, Lu Y, Qin Y, Li W, Cheng H, Xu Y, et al. miR-1247 is correlated with prognosis of pancreatic cancer and inhibits cell proliferation by targeting neuropilins. Curr Mol Med. 2014;14(3):316–27.
Prud’homme GJ, Glinka Y. Neuropilins are multifunctional coreceptors involved in tumor initiation, growth, metastasis and immunity. Oncotarget. 2012;3(9):921.
Peng Y, Liu Y-M, Li L-C, Wang L-L, Wu X-L. MicroRNA-338 inhibits growth, invasion and metastasis of gastric cancer by targeting NRP1 expression. PLoS ONE. 2014;9(4):e94422.
Cui Y, Han Z, Hu Y, Song G, Hao C, Xia H, et al. MicroRNA-181b and microRNA‐9 mediate arsenic‐induced angiogenesis via NRP1. J Cell Physiol. 2012;227(2):772–83.
Shang A, Lu W-Y, Yang M, Zhou C, Zhang H, Cai Z-X et al. miR-9 induces cell arrest and apoptosis of oral squamous cell carcinoma via CDK 4/6 pathway. Artificial Cells, Nanomedicine, and Biotechnology. 2018;46(8):1754-62.
Sasahira T, Kurihara M, Bhawal U, Ueda N, Shimomoto T, Yamamoto K, et al. Downregulation of miR-126 induces angiogenesis and lymphangiogenesis by activation of VEGF-A in oral cancer. Br J Cancer. 2012;107(4):700–6.
Lin C-C, Chen P-C, Lein M-Y, Tsao C-W, Huang C-C, Wang S-W, et al. WISP-1 promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-300 in human oral squamous cell carcinoma cells. Oncotarget. 2016;7(9):9993–10005.
Chuang J-Y, Chen P-C, Tsao C-W, Chang A-C, Lein M-Y, Lin C-C, et al. WISP-1, a novel angiogenic regulator of the CCN family, promotes oral squamous cell carcinoma angiogenesis through VEGF-A expression. Oncotarget. 2015;6(6):4239.
Li X, Yang Z, Han Z, Wen Y, Ma Z, Wang Y. Niclosamide acts as a new inhibitor of vasculogenic mimicry in oral cancer through upregulation of miR-124 and downregulation of STAT3. Oncol Rep. 2018;39(2):827–33.
Qiao L, Liang N, Zhang J, Xie J, Liu F, Xu D, et al. Advanced research on vasculogenic mimicry in cancer. J Cell Mol Med. 2015;19(2):315–26.
Gordon WR, Arnett KL, Blacklow SC. The molecular logic of notch signaling–a structural and biochemical perspective. J Cell Sci. 2008;121(19):3109–19.
Gan R-H, Wei H, Xie J, Zheng D-P, Luo E-L, Huang X-Y, et al. Notch1 regulates tongue cancer cells proliferation, apoptosis and invasion. Cell Cycle. 2018;17(2):216–24.
Yoshida R, Nagata M, Nakayama H, Niimori-Kita K, Hassan W, Tanaka T, et al. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab Invest. 2013;93(10):1068–81.
Wang S, Fan H, Xu J, Zhao E. Prognostic implication of NOTCH1 in early stage oral squamous cell cancer with occult metastases. Clin Oral Invest. 2018;22:1131–8.
Zhong R, Bao R, Faber PW, Bindokas VP, Bechill J, Lingen MW, et al. Notch1 activation or loss promotes HPV-Induced oral TumorigenesisParadoxical Notch1 activity drives primary HPV tumors. Cancer Res. 2015;75(18):3958–69.
Fares J, Fares MY, Khachfe HH, Salhab HA, Fares Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct Target therapy. 2020;5(1):28.
Organ SL, Tsao M-S. An overview of the c-MET signaling pathway. Therapeutic Adv Med Oncol. 2011;3(1suppl):7–S19.
Sun Z, Liu Q, Ye D, Ye K, Yang Z, Li D. Role of c-Met in the progression of human oral squamous cell carcinoma and its potential as a therapeutic target. Oncol Rep. 2018;39(1):209–16.
Kim ES, Salgia R. MET pathway as a therapeutic target. J Thorac Oncol. 2009;4(4):444–7.
Ramisetty S, Kulkarni P, Bhattacharya S, Nam A, Singhal SS, Guo L, et al. A Systems Biology Approach for addressing Cisplatin Resistance in Non-Small Cell Lung Cancer. J Clin Med. 2023;12(2):599.
Goel H, Rahul E, Gupta I, Chopra A, Ranjan A, Gupta AK, et al. Molecular and genomic landscapes in secondary & therapy related acute myeloid leukemia. Am J blood Res. 2021;11(5):472–97.
Mohania D, Chandel S, Kumar P, Verma V, Digvijay K, Tripathi D et al. Ultraviolet radiations: skin defense-damage mechanism. Ultrav Light Hum Health Dis Environ. 2017:71–87.
Salgia R, editor. Editor role of c-Met in cancer: emphasis on lung cancer. Seminars in oncology. Elsevier; 2009.
Takeuchi H, Bilchik A, Saha S, Turner R, Wiese D, Tanaka M, et al. c-MET expression level in primary colon cancer: a predictor of tumor invasion and lymph node metastases. Clin Cancer Res. 2003;9(4):1480–8.
Klosek SK, Nakashiro K-I, Hara S, Li C, Shintani S, Hamakawa H. Constitutive activation of Stat3 correlates with increased expression of the c-Met/HGF receptor in oral squamous cell carcinoma. Oncol Rep. 2004;12(2):293–6.
Freudlsperger C, Alexander D, Reinert S, Hoffmann J. Prognostic value of c-Met expression in oral squamous cell carcinoma. experimental and therapeutic medicine. 2010;1(1):69–72.
Chou Y-E, Hsieh M-J, Hsin C-H, Chiang W-L, Lai Y-C, Lee Y-H, et al. CD44 gene polymorphisms and environmental factors on oral cancer susceptibility in Taiwan. PLoS ONE. 2014;9(4):e93692.
Bidaud P, Chasle J, Sichel F, Rousseau S, Petit P, Pottier D et al. Expression of p53 family members and CD44 in oral squamous cell carcinoma (OSCC) in relation to tumorigenesis. Histol Histopathol. 2010.
Sun X, Zhang L. MicroRNA-143 suppresses oral squamous cell carcinoma cell growth, invasion and glucose metabolism through targeting hexokinase 2. Biosci Rep. 2017;37(3).
Xu P, Li Y, Yang S, Yang H, Tang J, Li M. Micro-ribonucleic acid 143 (MiR-143) inhibits oral squamous cell carcinoma (OSCC) cell migration and invasion by downregulation of phospho-c-Met through targeting CD44 v3. Oral surgery, oral medicine, oral pathology and oral radiology. 2015;120(1):43–51.
Ma Y, Gao J, Guo H. miR-23a-3p Regulates Runx2 to Inhibit the Proliferation and Metastasis of Oral Squamous Cell Carcinoma. Journal of Oncology. 2022;2022.
Kim S, Park S, Oh J-H, Lee SS, Lee Y, Choi J. MicroRNA-18a regulates the metastatic properties of oral squamous cell carcinoma cells via HIF-1α expression. BMC Oral Health. 2022;22(1):1–12.
Wu K, Wang X, Yu H, Yu Z, Wang D, Xu X. LINC00460 facilitated tongue squamous cell carcinoma progression via the miR-320b/IGF2BP3 axis. Oral Dis. 2022;28(6):1496–508.
Lee SS, Choi JH, Lim SM, Kim GJ, Lee SK, Jeon YK. Alteration of Pituitary Tumor transforming gene 1 by MicroRNA-186 and 655 regulates Invasion ability of human oral squamous cell carcinoma. Int J Mol Sci. 2021;22(3):1021.
Wang Q, Lv L, Li Y, Ji H. MicroRNA–655 suppresses cell proliferation and invasion in oral squamous cell carcinoma by directly targeting metadherin and regulating the PTEN/AKT pathway retraction in/10.3892/mmr. 2022.12897. Mol Med Rep. 2018;18(3):3106–14.
Kang Y, Zhang Y, Sun Y. MicroRNA–198 suppresses tumour growth and metastasis in oral squamous cell carcinoma by targeting CDK4. Int J Oncol. 2021;59(1):1–13.
Tzeng HE, Tang CH, Tsai CH, Chiu CH, Wu MH, Yen Y. ET-1 promotes epithelial-mesenchymal transition in oral squamous cell carcinoma cells via the microRNA-489-3p /TWIST Axis. OncoTargets and therapy. 2021;14:5005–18.
Fang R, Lu Q, Xu B. Hsa–miR–5580–3p inhibits oral cancer cell viability, proliferation and migration by suppressing LAMC2. Mol Med Rep. 2021;23(6):1–11.
Wang C, Wang Z, Zhang L, Lin X. MiR-29c inhibits the metastasis of oral squamous cell carcinoma and promotes its cell cycle arrest by targeting SERPINH1. Annals of translational medicine. 2021;9(18):1423.
Chen L, Zhu Q, Lu L, Liu Y. MiR-132 inhibits migration and invasion and increases chemosensitivity of cisplatin-resistant oral squamous cell carcinoma cells via targeting TGF-β1. Bioengineered. 2020;11(1):91–102.
Wang X, Chang K, Gao J, Wei J, Xu G, Xiao L, et al. MicroRNA-504 functions as a tumor suppressor in oral squamous cell carcinoma through inhibiting cell proliferation, migration and invasion by targeting CDK6. Int J Biochem Cell Biol. 2020;119:105663.
Jin Y, Li Y, Wang X, Yang Y. Dysregulation of MiR-519d affects oral squamous cell carcinoma invasion and metastasis by targeting MMP3. J Cancer. 2019;10(12):2720.
Luo K, He J, Yu D, Açil Y. MiR-149-5p regulates cisplatin chemosensitivity, cell growth, and metastasis of oral squamous cell carcinoma cells by targeting TGFβ2. Int J Clin Exp Pathol. 2019;12(10):3728.
Chen J, Chen J, Li W. MicroRNA–149 targets specificity protein 1 to suppress human tongue squamous cell carcinoma cell proliferation and motility. Oncol Lett. 2017;13(2):851–6.
Huang W-C, Jang T-H, Tung S-L, Yen T-C, Chan S-H, Wang L-H. A novel miR-365-3p/EHF/keratin 16 axis promotes oral squamous cell carcinoma metastasis, cancer stemness and drug resistance via enhancing β5-integrin/c-met signaling pathway. J Experimental Clin Cancer Res. 2019;38:1–17.
Na C, Li X, Zhang J, Han L, Li Y, Zhang H. miR-107 targets TRIAP1 to regulate oral squamous cell carcinoma proliferation and migration. Int J Clin Exp Pathol. 2019;12(5):1820.
Zhu X, Gao J, Wu Y, Yang B, Ji M, Shu M. MiR-320a suppresses cell migration and invasion by targeting MRP2 in oral tongue squamous cell carcinoma. Int J Clin Exp Pathol. 2017;10(2):1263–72.
Kang Y, Zhang Y, Sun Y, Wen Y, Sun F. MicroRNA-300 suppresses metastasis of oral squamous cell carcinoma by inhibiting epithelial-to-mesenchymal transition. OncoTargets and therapy. 2018:5657–66.
Wang Z, Wang J, Chen Z, Wang K, Shi L. MicroRNA-1-3p inhibits the proliferation and migration of oral squamous cell carcinoma cells by targeting DKK1. Biochem Cell Biol. 2018;96(3):355–64.
Li X, He J, Shao M, Cui B, Peng F, Li J, et al. Downregulation of mir-218-5p promotes invasion of oral squamous cell carcinoma cells via activation of CD44-ROCK signaling. Biomed Pharmacother. 2018;106:646–54.
Cao ZH, Cheng JL, Zhang Y, Bo CX, Li YL. MicroRNA–375 inhibits oral squamous cell carcinoma cell migration and invasion by targeting platelet–derived growth factor–A. Mol Med Rep. 2017;15(2):922–8.
Wang K, Jin J, Ma T, Zhai H. MiR-376c-3p regulates the proliferation, invasion, migration, cell cycle and apoptosis of human oral squamous cancer cells by suppressing HOXB7. Biomed Pharmacother. 2017;91:517–25.
Chang Wm, Shiah SG, Hsiao M. MiR-376c inhibits Metastasis by Controlling RUNX2‐INHBA Axis in oral squamous cell carcinoma. FASEB J. 2015;29:4174.
Rastogi B, Kumar A, Raut SK, Panda NK, Rattan V, Joshi N, et al. Downregulation of miR-377 promotes oral squamous cell carcinoma growth and migration by targeting HDAC9. Cancer Invest. 2017;35(3):152–62.
Wang T, Ren Y, Liu R, Ma J, Shi Y, Zhang L et al. miR-195-5p suppresses the proliferation, migration, and invasion of oral squamous cell carcinoma by targeting TRIM14. BioMed research international. 2017;2017.
Kuribayashi N, Uchida D, Kinouchi M, Koshiji C, Izumi S, Hakata K, et al. Regulation of metabotropic glutamate receptor 5 expression by the miR-30 downregulation induced by the SDF-1/CXCR4 system in oral cancer cells. Cancer Res. 2016;76(14Supplement):1925.
Hui Y, Li Y, Jing Y, Feng J-Q, Ding Y. miRNA-101 acts as a tumor suppressor in oral squamous cell carcinoma by targeting CX chemokine receptor 7. Am J translational Res. 2016;8(11):4902.
Hu J, Ge W, Xu J. HPV 16 E7 inhibits OSCC cell proliferation, invasion, and metastasis by upregulating the expression of miR-20a. Tumor Biology. 2016;37:9433–40.
Wang X, Li F, Zhou X. Mir-204-5p regulates cell proliferation and metastasis through inhibiting CXCR4 expression in OSCC. Biomed Pharmacother. 2016;82:202–7.
Yu CC, Chen PN, Peng CY, Yu CH, Chou MY. Suppression of miR-204 enables oral squamous cell carcinomas to promote cancer stemness, EMT traits, and lymph node metastasis. Oncotarget. 2016;7(15):20180–92.
Fukumoto I, Koshizuka K, Hanazawa T, Kikkawa N, Matsushita R, Kurozumi A, et al. The tumor-suppressive microRNA-23b/27b cluster regulates the MET oncogene in oral squamous cell carcinoma. Int J Oncol. 2016;49(3):1119–29.
Li L, Ma HQ. MicroRNA–216a inhibits the growth and metastasis of oral squamous cell carcinoma by targeting eukaryotic translation initiation factor 4B. Mol Med Rep. 2015;12(2):3156–62.
Wang X-c, Ma Y, Meng P-s, Han J-l, Yu H-y. Bi L-j. miR-433 inhibits oral squamous cell carcinoma (OSCC) cell growth and metastasis by targeting HDAC6. Oral Oncol. 2015;51(7):674–82.
Deng L, Liu H. MicroRNA-506 suppresses growth and metastasis of oral squamous cell carcinoma via targeting GATA6. Int J Clin Exp Med. 2015;8(2):1862–70.
Wang Y, Jia R-Z, Diao S, He J, Jia L. MiRNA-101 targets TGF-βR1 to retard the progression of oral squamous cell carcinoma. Oncol Res Featuring Preclinical Clin Cancer Ther. 2020;28(2):203–12.
Yao Y, Xu Q, Yan L, Jiao Y, Su Q, Li X, et al. MiRNA-128 and MiRNA-142 regulate tumorigenesis and EMT in oral squamous cell carcinoma through HOXA10. Cancer Manage Res. 2020;12:9987.
He T, Li X, Lu D, Tian L, Xu B. MiR-137 silencing of BRD4 suppresses oral squamous cell carcinoma cells proliferation, migration and invasion. Int J Clin Exp Pathol. 2017;10(1):409–16.
Manasa VG, Thomas S, Kannan S. MiR-144/451a cluster synergistically modulates growth and metastasis of oral carcinoma. Oral Dis. 2021.
Ruan P, Tao Z, Tan A. Low expression of miR-30a-5p induced the proliferation and invasion of oral cancer via promoting the expression of FAP. Biosci Rep. 2018;38(1).
Jia L-F, Huang Y-P, Zheng Y-F, Ming-Yue L, Wei S-B, Meng Z, et al. miR-29b suppresses proliferation, migration, and invasion of tongue squamous cell carcinoma through PTEN–AKT signaling pathway by targeting Sp1. Oral Oncol. 2014;50(11):1062–71.
Ouyang J, Hu C, Zhang X, Wu Q. miRNA-200a regulating proliferation, Migration, and infiltration of Tongue squamous cell carcinoma cells by targeting DEK Proto-Oncogene. J Biomaterials Tissue Eng. 2021;11(3):492–9.
Li T, Wu Q, Liu D, Wang X. miR-27b suppresses Tongue squamous cell carcinoma epithelial-mesenchymal transition by targeting ITGA5. OncoTargets and therapy. 2020;13:11855–67.
Sun L, Yao Y, Liu B, Lin Z, Lin L, Yang M, et al. MiR-200b and miR-15b regulate chemotherapy-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting BMI1. Oncogene. 2012;31(4):432–45.
Bonneau D, Longy M. Mutations of the human PTEN gene. Hum Mutat. 2000;16(2):109–22.
Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9(4):125–8.
Georgescu M-M. PTEN tumor suppressor network in PI3K-Akt pathway control. Genes & cancer. 2010;1(12):1170–7.
Chan C-H, Jo U, Kohrman A, Rezaeian AH, Chou P-C, Logothetis C, et al. Posttranslational regulation of akt in human cancer. Cell & bioscience. 2014;4:1–9.
Zou Z-J, Fan L, Wang L, Xu J, Zhang R, Tian T, et al. miR-26a and miR-214 down-regulate expression of the PTEN gene in chronic lymphocytic leukemia, but not PTEN mutation or promoter methylation. Oncotarget. 2015;6(2):1276.
Folini M, Gandellini P, Longoni N, Profumo V, Callari M, Pennati M, et al. miR-21: an oncomir on strike in prostate cancer. Mol Cancer. 2010;9(1):1–12.
Iizumi S, Uchida F, Nagai H, Takaoka S, Fukuzawa S, Kanno NI, et al. MicroRNA 142-5p promotes tumor growth in oral squamous cell carcinoma via the PI3K/AKT pathway by regulating PTEN. Heliyon. 2021;7(10):e08086.
Li L, Zhu X, Shou T, Yang L, Cheng X, Wang J, et al. MicroRNA-28 promotes cell proliferation and invasion in gastric cancer via the PTEN/PI3K/AKT signalling pathway. Mol Med Rep. 2018;17(3):4003–10.
Jiang J, Lv Q, Yi Y, Liao J, Wang X, Zhang W. MicroRNA-200a promotes proliferation and invasion of ovarian cancer cells by targeting PTEN. Eur Rev Med Pharmacol Sci. 2018;22(19):6260–7.
Zheng X, Li J, Peng C, Zhao J, Chi J, Meng X, et al. MicroRNA-24 induces cisplatin resistance by targeting PTEN in human tongue squamous cell carcinoma. Oral Oncol. 2015;51(11):998–1003.
Ma G, Hou J, Qiu J, Fan J, Yang J. Bone marrow mesenchymal stem cells (BMSCs) with high expression miR-155 affect the proliferation and metastasis of oral squamous cell carcinoma by regulating phosphatase and Tensin Homolog 12 (PTEN12). J Biomaterials Tissue Eng. 2021;11(8):1571–5.
Kurihara-Shimomura M, Sasahira T, Shimomura H, Nakashima C, Kirita T. The oncogenic activity of miR-29b-1-5p induces the epithelial-mesenchymal transition in oral squamous cell carcinoma. J Clin Med. 2019;8(2):273.
Peng SY, Tu HF, Yang CC, Wu CH, Liu CJ, Chang KW, et al. miR-134 targets PDCD7 to reduce E‐cadherin expression and enhance oral cancer progression. Int J Cancer. 2018;143(11):2892–904.
Li D, Liu K, Li Z, Wang J, Wang X. miR-19a and miR-424 target TGFBR3 to promote epithelial-to-mesenchymal transition and migration of tongue squamous cell carcinoma cells. Cell Adh Migr. 2018;12(3):236–46.
Lei Y, Chen L, Zhang G, Shan A, Ye C, Liang B, et al. MicroRNAs target the Wnt/β–catenin signaling pathway to regulate epithelial–mesenchymal transition in cancer. Oncol Rep. 2020;44(4):1299–313.
Hoang BH, Kubo T, Healey JH, Yang R, Nathan SS, Kolb EA, et al. Dickkopf 3 inhibits invasion and motility of Saos-2 osteosarcoma cells by modulating the wnt-beta-catenin pathway. Cancer Res. 2004;64(8):2734–9.
Barrantes Idel B, Montero-Pedrazuela A, Guadaño-Ferraz A, Obregon MJ, Martinez de Mena R, Gailus-Durner V, et al. Generation and characterization of dickkopf3 mutant mice. Mol Cell Biol. 2006;26(6):2317–26.
Weng J, Zhang H, Wang C, Liang J, Chen G, Li W, et al. Mir-373-3p targets DKK1 to promote EMT-Induced Metastasis via the Wnt/β-Catenin pathway in Tongue squamous cell carcinoma. Biomed Res Int. 2017;2017:6010926.
Elzi DJ, Song M, Hakala K, Weintraub ST, Shiio Y. Wnt antagonist SFRP1 functions as a secreted mediator of senescence. Mol Cell Biol. 2012;32(21):4388–99.
Qiao B, He BX, Cai JH, Tao Q, King-Yin Lam A. MicroRNA-27a-3p modulates the Wnt/β-Catenin signaling pathway to promote epithelial-mesenchymal transition in oral squamous carcinoma stem cells by targeting SFRP1. Sci Rep. 2017;7:44688.
Lee Y-G, Macoska JA, Korenchuk S, Pienta KJ. MIM, a potential metastasis suppressor gene in bladder cancer. Neoplasia. 2002;4(4):291–4.
Xie F, Ye L, Chen J, Wu N, Zhang Z, Yang Y, et al. The impact of metastasis Suppressor-1, MTSS1, on oesophageal squamous cell carcinoma and its clinical significance. J Translational Med. 2011;9(1):1–10.
Isaksson HS, Sorbe B, Nilsson TK. Whole genome expression profiling of blood cells in ovarian cancer patients-prognostic impact of the CYP1B1, MTSS1, NCALD, and NOP14 genes. Oncotarget. 2014;5(12):4040.
Parr C, Jiang WG. Metastasis suppressor 1 (MTSS1) demonstrates prognostic value and anti-metastatic properties in breast cancer. Eur J Cancer. 2009;45(9):1673–83.
Ma S, Guan X-Y, Lee TK, Chan KW. Clinicopathological significance of missing in metastasis B expression in hepatocellular carcinoma. Hum Pathol. 2007;38(8):1201–6.
Du P, Ye L, Ruge F, Yang Y, Jiang WG. Metastasis suppressor-1, MTSS1, acts as a putative tumour suppressor in human bladder cancer. Anticancer Res. 2011;31(10):3205–12.
Du P, Ye L, Li H, Yang Y, Jiang WG. The tumour suppressive role of metastasis suppressor-1, MTSS1, in human kidney cancer, a possible connection with the SHH pathway. J Exp Ther Oncol. 2012;10(2).
Hirata H, Ueno K, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, et al. MicroRNA-182-5p promotes cell invasion and proliferation by down regulating FOXF2, RECK and MTSS1 genes in human prostate cancer. PLoS ONE. 2013;8(1):e55502.
Woodings JA, Sharp SJ, Machesky LM. MIM-B, a putative metastasis suppressor protein, binds to actin and to protein tyrosine phosphatase delta. Biochem J. 2003;371(2):463–71.
Wu W, Wang Z, Yang P, Yang J, Liang J, Chen Y, et al. MicroRNA-135b regulates metastasis suppressor 1 expression and promotes migration and invasion in colorectal cancer. Mol Cell Biochem. 2014;388:249–59.
Guo Y, Ren MS, Shang C, Zhu L, Zhong M. MTSS1 gene regulated by miR-96 inhibits cell proliferation and metastasis in tongue squamous cellular carcinoma Tca8113 cell line. Int J Clin Exp Med. 2015;8(9):15441–9.
Guo Y, Qiao X, Zhu L, Song R. MicroRNA-182-5p modulates oral squamous cell carcinoma migration and invasion via targeting MTSS1 gene. Pathol Oncol Res. 2020;26:1007–13.
Yang Q, Al-Hendy A. The emerging role of p27 in development of diseases. Cancer Stud Mol medicine: open J. 2018;4(1):e1.
Abukhdeir AM, Park BH. P21 and p27: roles in carcinogenesis and drug resistance. Expert Rev Mol Med. 2008;10:e19.
Hu XH, Zhao ZX, Dai J, Geng DC, Xu YZ. MicroRNA-221 regulates osteosarcoma cell proliferation, apoptosis, migration, and invasion by targeting CDKN1B/p27. J Cell Biochem. 2019;120(3):4665–74.
Li X, Tang M. Exosomes released from M2 macrophages transfer mir-221‐3p contributed to EOC progression through targeting CDKN1B. Cancer Med. 2020;9(16):5976–88.
Zhang Y-W, Morita I, Ikeda M, Ma K-W, Murota S. Connexin43 suppresses proliferation of osteosarcoma U2OS cells through post-transcriptional regulation of p27. Oncogene. 2001;20(31):4138–49.
Ren S-s, Yuan F, Liu Y-h, Zhou L-t, Li J. Effect of p27 gene combined with Pientzehuang on tumor growth in osteosarcoma-bearing nude mice. Chin J Integr Med. 2015;21:830–6.
Li Y, Nakka M, Kelly AJ, Lau CC, Krailo M, Barkauskas DA, et al. p27 is a candidate prognostic biomarker and metastatic promoter in OsteosarcomaThe Role of Mislocalized p27 in Osteosarcoma. Cancer Res. 2016;76(13):4002–11.
Yang K, Chen Y, Cui Z, Chen H, Yang L, Zhao J, et al. MicroRNA-222‐3p participates in the development of oral squamous cell carcinoma by targeting CDKN1B. J Oral Pathol Med. 2020;49(7):621–9.
Chen Q, Wang W, Wang Y. MiR-222 regulates the progression of oral squamous cell carcinoma by targeting CDKN1B. Am J translational Res. 2022;14(7):5215–27.
Yumimoto K, Nakayama KI. Fbxw7 suppresses cancer metastasis by inhibiting niche formation. Oncoimmunology. 2015;4(8):e1022308.
Yeh C-H, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer. 2018;17:1–19.
Cheng Y, Li G. Role of the ubiquitin ligase Fbw7 in cancer progression. Cancer Metastasis Rev. 2012;31:75–87.
Cai Y, Zhang M, Qiu X, Wang B, Fu Y, Zeng J et al. Upregulation of FBXW7 suppresses renal cancer metastasis and epithelial mesenchymal transition. Disease Markers. 2017;2017.
Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, et al. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013;210(8):1545–57.
Wang Q, Li D, Li Z, Liu C, Xiao Y, Zhang B, et al. Upregulation of miR-27a contributes to the malignant transformation of human bronchial epithelial cells induced by SV40 small T antigen. Oncogene. 2011;30(36):3875–86.
Zhao J, Hu C, Chi J, Li J, Peng C, Yun X, et al. miR-24 promotes the proliferation, migration and invasion in human tongue squamous cell carcinoma by targeting FBXW7. Oncol Rep. 2016;36(2):1143–9.
Jiang L, Liu X, Jiang X, Yin Q, Hao Y, Xiao L. MiR-223 promotes oral squamous cell carcinoma proliferation and migration by regulating FBXW7. Cancer Biomarkers. 2019;24(3):325–34.
Chang L, Wang D, Kan S, Hao M, Liu H, Yang Z et al. Ginsenoside Rd inhibits migration and invasion of tongue cancer cells through H19/miR-675-5p/CDH1 axis. J Appl Oral Sci. 2022;30.
Pang F, Liu C, Cui Y, Gong K, Liu G, Bian Y, et al. Mir-17-5p promotes proliferation and migration of CAL-27 human tongue squamous cell carcinoma cells involved in autophagy inhibition under hypoxia. Int J Clin Exp Pathol. 2019;12(6):2084.
Cui SH, Hu XD, Yan Y. Wnt/β-catenin signaling pathway participates in the effect of miR‐626 on oral squamous cell carcinoma by targeting RASSF4. J Oral Pathol Med. 2021;50(10):1005–17.
Wei Z, Lyu B, Hou D, Liu X. Mir-5100 mediates proliferation, migration and invasion of oral squamous cell carcinoma cells via targeting SCAI. J Invest Surg. 2021;34(8):834–41.
Zheng J, Wang J, Jia Y, Liu T, Duan Y, Liang X, et al. microRNA-211 promotes proliferation, migration, and invasion ability of oral squamous cell carcinoma cells via targeting the bridging integrator 1 protein. J Cell Biochem. 2019;120(3):4644–53.
Jung JE, Lee JY, Park HR, Kang JW, Kim YH, Lee JH. MicroRNA-133 targets phosphodiesterase 1 C in Drosophila and human oral Cancer cells to regulate epithelial-mesenchymal transition. J Cancer. 2021;12(17):5296–309.
Guo Y, Qiao X, Zhu L, Song R. MicroRNA-182-5p modulates oral squamous cell carcinoma migration and invasion via targeting MTSS1 gene. Pathol Oncol Res. 2020;26(2):1007–13.
Li K, Zhou Z, Li J, Xiang R. miR-146b functions as an oncogene in oral squamous cell carcinoma by targeting HBP1. Technol Cancer Res Treat. 2020;19:1533033820959404.
Wu M, Duan Q, Liu X, Zhang P, Fu Y, Zhang Z, et al. MiR-155-5p promotes oral cancer progression by targeting chromatin remodeling gene ARID2. Biomed Pharmacother. 2020;122:109696.
Qin W, Wang W, Wang X, Zhang X, Du J. MiR-1290 targets CCNG2 to promote the metastasis of oral squamous cell carcinoma. Eur Rev Med Pharmacol Sci. 2019;23(23):10332–42.
Yang C-N, Deng Y-T, Tang J-Y, Cheng S-J, Chen S-T, Li Y-J, et al. MicroRNA-29b regulates migration in oral squamous cell carcinoma and its clinical significance. Oral Oncol. 2015;51(2):170–7.
Kawakubo-Yasukochi T, Morioka M, Hazekawa M, Yasukochi A, Nishinakagawa T, Ono K, et al. miR‐200c‐3p spreads invasive capacity in human oral squamous cell carcinoma microenvironment. Mol Carcinog. 2018;57(2):295–302.
Lu M, Wang C, Chen W, Mao C, Wang J. Mir-654-5p targets GRAP to promote proliferation, metastasis, and chemoresistance of oral squamous cell carcinoma through Ras/MAPK signaling. DNA Cell Biol. 2018;37(4):381–8.
Xu D, Gu M, Liu H. MicroRNA-625-3p promotes cell migration of oral squamous cell carcinoma by regulating SCAI expression. Eur Rev Med Pharmacol Sci. 2019;23(2):641–8.
Hou YY, You JJ, Yang CM, Pan HW, Chen HC, Lee JH, et al. Aberrant DNA hypomethylation of miR-196b contributes to migration and invasion of oral cancer. Oncol Lett. 2016;11(6):4013–21.
Lu Y-C, Chang JT, Liao C-T, Kang C-J, Huang S-F, Chen I-H, et al. OncomiR-196 promotes an invasive phenotype in oral cancer through the NME4-JNK-TIMP1-MMP signaling pathway. Mol Cancer. 2014;13:1–14.
Shen L, Liu L, Ge L, Xie L, Liu S, Sang L, et al. miR-448 downregulates MPPED2 to promote cancer proliferation and inhibit apoptosis in oral squamous cell carcinoma. Experimental and therapeutic medicine. 2016;12(4):2747–52.
Peng H-Y, Jiang S-S, Hsiao J-R, Hsiao M, Hsu Y-M, Wu G-H, et al. IL-8 induces mir-424-5p expression and modulates SOCS2/STAT5 signaling pathway in oral squamous cell carcinoma. Mol Oncol. 2016;10(6):895–909.
Lin SC, Kao SY, Chang JC, Liu YC, Yu EH, Tseng SH, et al. Up-regulation of miR-187 modulates the advances of oral carcinoma by targeting BARX2 tumor suppressor. Oncotarget. 2016;7(38):61355–65.
Zeng Q, Tao X, Huang F, Wu T, Wang J, Jiang X, et al. Overexpression of miR-155 promotes the proliferation and invasion of oral squamous carcinoma cells by regulating BCL6/cyclin D2. Int J Mol Med. 2016;37(5):1274–80.
He S, Lai R, Chen D, Yan W, Zhang Z, Liu Z et al. Downregulation of miR-221 inhibits cell migration and invasion through targeting methyl-CpG binding domain protein 2 in human oral squamous cell carcinoma cells. BioMed research international. 2015;2015.
Kinouchi M, Uchida D, Kuribayashi N, Tamatani T, Nagai H, Miyamoto Y. Involvement of miR-518c-5p to growth and metastasis in oral cancer. PLoS ONE. 2014;9(12):e115936.
Kawakita A, Yanamoto S, Yamada S-i, Naruse T, Takahashi H, Kawasaki G, et al. MicroRNA-21 promotes oral cancer invasion via the Wnt/β-catenin pathway by targeting DKK2. Pathol Oncol Res. 2014;20:253–61.
Weng J, Zhang H, Wang C, Liang J, Chen G, Li W et al. miR-373-3p targets DKK1 to promote EMT-induced metastasis via the Wnt/-catenin pathway in tongue squamous cell carcinoma. BioMed Research International. 2017;2017.
Jiang M-C, Ni J-J, Cui W-Y, Wang B-Y, Zhuo W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am J cancer Res. 2019;9(7):1354.
Liu SJ, Dang HX, Lim DA, Feng FY, Maher CA. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21(7):446–60.
Nahand JS, Jamshidi S, Hamblin MR, Mahjoubin-Tehran M, Vosough M, Jamali M, et al. Circular RNAs: new epigenetic signatures in viral infections. Front Microbiol. 2020;11:1853.
Hao N-B, He Y-F, Li X-Q, Wang K, Wang R-L. The role of miRNA and lncRNA in gastric cancer. Oncotarget. 2017;8(46):81572.
Yu Y, Gao F, He Q, Li G, Ding G. lncRNA UCA1 functions as a ceRNA to promote prostate cancer progression via sponging miR143. Mol Therapy-Nucleic Acids. 2020;19:751–8.
Wang ZY, Duan Y, Wang P. SP1-mediated upregulation of lncRNA SNHG4 functions as a ceRNA for miR‐377 to facilitate prostate cancer progression through regulation of ZIC5. J Cell Physiol. 2020;235(4):3916–27.
Zeng X, Xiao J, Bai X, Liu Y, Zhang M, Liu J et al. Research progress on the circRNA/lncRNA–miRNA–mRNA axis in gastric cancer. Pathology-Research and Practice. 2022:154030.
Gao N, Li Y, Li J, Gao Z, Yang Z, Li Y, et al. Long non-coding RNAs: the regulatory mechanisms, research strategies, and future directions in cancers. Front Oncol. 2020;10:598817.
Abbasi-Kolli M, Nahand JS, Kiani SJ, Khanaliha K, Khatami AR, Taghizadieh M et al. The expression patterns of MALAT-1, NEAT-1, THRIL, and mir-155-5p in the acute to the post-acute phase of COVID-19 disease. Brazilian J Infect Dis. 2022;26.
Zhou Y, Zhang Y. Inhibition of LncRNAH19 has the effect of anti-tumour and enhancing sensitivity to Gefitinib and Chemotherapy in non‐small‐cell lung cancer in vivo. J Cell Mol Med. 2020;24(10):5811–6.
Yoshimura H, Matsuda Y, Yamamoto M, Michishita M, Takahashi K, Sasaki N, et al. Reduced expression of the H19 long non-coding RNA inhibits pancreatic cancer metastasis. Lab Invest. 2018;98(6):814–24.
Zhou W, Ye X-l, Xu J, Cao M-G, Fang Z-Y, Li L-Y, et al. The lncRNA H19 mediates breast cancer cell plasticity during EMT and MET plasticity by differentially sponging miR-200b/c and let-7b. Sci Signal. 2017;10(483):eaak9557.
Yang X, Liu L, Shen X, Shi L, Liu W. Dysregulation and implications of lncRNAs and miRNAs in oral tongue squamous cell carcinoma: in reply with emphasis on the role of ceRNAs. Oral Oncol. 2023;136:106277.
Kou N, Liu S, Li X, Li W, Zhong W, Gui L, et al. H19 facilitates tongue squamous cell carcinoma migration and invasion via sponging mir-let-7. Oncol Res Featuring Preclinical Clin Cancer Ther. 2019;27(2):173–82.
Zhou H, Tang K, Liu H, Zeng J, Li H, Yan L, et al. Regulatory network of two tumor-suppressive noncoding RNAs interferes with the growth and metastasis of renal cell carcinoma. Mol Therapy-Nucleic Acids. 2019;16:554–65.
Zhu Y, Chen P, Gao Y, Ta N, Zhang Y, Cai J, et al. MEG3 activated by vitamin D inhibits colorectal cancer cells proliferation and migration via regulating clusterin. EBioMedicine. 2018;30:148–57.
Tan J, Xiang L, Xu G. LncRNA MEG3 suppresses migration and promotes apoptosis by sponging miR-548d‐3p to modulate JAK–STAT pathway in oral squamous cell carcinoma. IUBMB Life. 2019;71(7):882–90.
Zhang L, Hu D, Zou L. Low expression of lncRNA MEG3 promotes the progression of oral squamous cell carcinoma by targeting miR-21. Eur Rev Med Pharmacol Sci. 2018;22(23):8315–23.
Liu S, Liu LH, Hu WW, Wang M. Long noncoding RNA TUG1 regulates the development of oral squamous cell carcinoma through sponging mir-524‐5p to mediate DLX1 expression as a competitive endogenous RNA. J Cell Physiol. 2019;234(11):20206–16.
Yan G, Wang X, Yang M, Lu L, Zhou Q. Long non-coding RNA TUG1 promotes progression of oral squamous cell carcinoma through upregulating FMNL2 by sponging miR-219. Am J cancer Res. 2017;7(9):1899.
Xue M, Chen W, Xiang A, Wang R, Chen H, Pan J, et al. Hypoxic exosomes facilitate bladder tumor growth and development through transferring long non-coding RNA-UCA1. Mol Cancer. 2017;16:1–13.
Wang X-S, Zhang Z, Wang H-C, Cai J-L, Xu Q-W, Li M-Q, et al. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006;12(16):4851–8.
Fang Z, Wu L, Wang L, Yang Y, Meng Y, Yang H. Increased expression of the long non-coding RNA UCA1 in tongue squamous cell carcinomas: a possible correlation with cancer metastasis. Oral surgery, oral medicine, oral pathology and oral radiology. 2014;117(1):89–95.
Zhang Th, Lz L, Xl L, Jn Wu, Su K, Chen J, et al. LncRNA UCA1/miR-124 axis modulates TGFβ1‐induced epithelial‐mesenchymal transition and invasion of tongue cancer cells through JAG1/Notch signaling. J Cell Biochem. 2019;120(6):10495–504.
Duan Q, Xu M, Wu M, Zhang X, Gan M, Jiang H. Long noncoding RNA UCA1 promotes cell growth, migration, and invasion by targeting mir-143‐3p in oral squamous cell carcinoma. Cancer Med. 2020;9(9):3115–29.
Chen L-L, Yang L. Regulation of circRNA biogenesis. RNA Biol. 2015;12(4):381–8.
Salzman J, Gawad C, Wang PL, Lacayo N, Brown PO. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE. 2012;7(2):e30733.
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–8.
Hansen TB, Jensen TI, Clausen BH, Bramsen JB, Finsen B, Damgaard CK, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–8.
Lin X, Ding J-M, Zheng X-Z, Chen J-G. Immunity-related long noncoding RNA WDFY3-AS2 inhibited cell proliferation and metastasis through Wnt/β-catenin signaling in oral squamous cell carcinoma. Arch Oral Biol. 2023:105625.
Xia B, Hong T, He X, Hu X, Gao Y. A circular RNA derived from MMP9 facilitates oral squamous cell carcinoma metastasis through regulation of MMP9 mRNA stability. Cell Transplant. 2019;28(12):1614–23.
Zhao W, Cui Y, Liu L, Qi X, Liu J, Ma S, et al. Splicing factor derived circular RNA circUHRF1 accelerates oral squamous cell carcinoma tumorigenesis via feedback loop. Cell Death & Differentiation. 2020;27(3):919–33.
Zhao S-Y, Wang J, Ouyang S-B, Huang Z-K, Liao L. Salivary circular RNAs Hsa_Circ_0001874 and Hsa_Circ_0001971 as novel biomarkers for the diagnosis of oral squamous cell carcinoma. Cell Physiol Biochem. 2018;47(6):2511–21.
Peng Q-S, Cheng Y-N, Zhang W-B, Fan H, Mao Q-H, Xu P. circRNA_0000140 suppresses oral squamous cell carcinoma growth and metastasis by targeting miR-31 to inhibit Hippo signaling pathway. Cell Death Dis. 2020;11(2):112.
Furth N, Aylon Y. The LATS1 and LATS2 tumor suppressors: beyond the Hippo pathway. Cell Death & Differentiation. 2017;24(9):1488–501.
Hei N, Liu P, Jin L, Peng S, Bao Y. Circular hsa_circ_0020377 regulates KLF7 by targeting mir-194-5p to facilitate tumor cell malignant behaviors and glycolysis in oral squamous cell carcinoma progression. Funct Integr Genom. 2023;23(1):52.
Chen H, Zhang Y, Wu K, Qiu X. circVAPA promotes the proliferation, migration and invasion of oral cancer cells through the miR-132/HOXA7 axis. J Int Med Res. 2021;49(6):03000605211013207.
Jiang X, Liu J, Li S, Jia B, Huang Z, Shen J, et al. CCL18-induced LINC00319 promotes proliferation and metastasis in oral squamous cell carcinoma via the miR-199a-5p/FZD4 axis. Cell Death Dis. 2020;11(9):777.
Wang X, Guo Y, Wang C, Wang Q, Yan G, Long Noncoding. RNA ZEB1-AS1 downregulates miR-23a, promotes Tumor Progression, and predicts the survival of oral squamous cell carcinoma patients. OncoTargets and therapy. 2021;14:2699–710.
Liu J, Jiang X, Zou A, Mai Z, Huang Z, Sun L, et al. circIGHG-induced epithelial-to-mesenchymal transition promotes oral squamous cell carcinoma progression via miR-142-5p/IGF2BP3 signaling. Cancer Res. 2021;81(2):344–55.
Liu M, Liu Q, Fan S, Su F, Jiang C, Cai G, et al. LncRNA LTSCCAT promotes tongue squamous cell carcinoma metastasis via targeting the miR-103a-2-5p/SMYD3/TWIST1 axis. Cell Death Dis. 2021;12(2):1–16.
Chen P-Y, Hsieh P-L, Peng C-Y, Liao Y-W, Yu C-H, Yu C-C. LncRNA MEG3 inhibits self-renewal and invasion abilities of oral cancer stem cells by sponging miR-421. J Formos Med Assoc. 2021;120(4):1137–42.
Li Y, Wan Q, Wang W, Mai L, Sha L, Mashrah M, et al. LncRNA ADAMTS9-AS2 promotes tongue squamous cell carcinoma proliferation, migration and EMT via the miR-600/EZH2 axis. Biomed Pharmacother. 2019;112:108719.
Wu Z, He X, Chen S. Oncogenic circDHTKD1 promotes tumor growth and metastasis of oral squamous cell carcinoma in vitro and in vivo via upregulating miR-326-mediated GAB1. Braz J Med Biol Res. 2021;54.
Yu L, Huo L, Shao X, Zhao J. lncRNA SNHG5 promotes cell proliferation, migration and invasion in oral squamous cell carcinoma by sponging miR–655–3p/FZD4 axis. Oncol Lett. 2020;20(6):1.
Wu K, Jiang Y, Zhou W, Zhang B, Li Y, Xie F, et al. Long noncoding RNA RC3H2 facilitates cell proliferation and invasion by targeting MicroRNA-101-3p/EZH2 axis in OSCC. Mol Therapy-Nucleic Acids. 2020;20:97–110.
Liu D, Jian X, Xu P, Zhu R, Wang Y. Linc01234 promotes cell proliferation and metastasis in oral squamous cell carcinoma via miR-433/PAK4 axis. BMC Cancer. 2020;20:1–10.
Jin N, Jin N, Bu W, Li X, Liu L, Wang Z, et al. Long non-coding RNA TIRY promotes tumor metastasis by enhancing epithelial-to-mesenchymal transition in oral cancer. Experimental Biology and Medicine. 2020;245(7):585–96.
Peng Q-S, Cheng Y-N, Zhang W-B, Fan H, Mao Q-H, Xu P. circRNA_0000140 suppresses oral squamous cell carcinoma growth and metastasis by targeting miR-31 to inhibit Hippo signaling pathway. Cell Death Dis. 2020;11(2):1–18.
(2021) Long Noncoding RNAs in the Metastasis of Oral Squamous Cell Carcinoma Frontiers in Oncology 1010.3389/fonc.2020.616717
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Meghdad Eslami, Saba Khazeni, Xaniar Mohammadi Khanaghah, Mohammad Hossein Asadi, Mohamad Amin Ansari, Javad Hayati Garjan, Mohammad Hassan Lotfalizadeh, Mobina Bayat, Mohammad Taghizadieh, Seyed Pouya Taghavi, Michael R Hamblin, Javid Sadri Nahand contributed to the conception, design, and drafting of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Eslami, M., Khazeni, S., Khanaghah, X.M. et al. MiRNA-related metastasis in oral cancer: moving and shaking. Cancer Cell Int 23, 182 (2023). https://doi.org/10.1186/s12935-023-03022-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-03022-5