
Abstract
N6-methyladenosine (m6A), one of the most common RNA methylation modifications, has emerged in recent years as a new layer of the regulatory mechanism controlling gene expression in eukaryotes. As a reversible epigenetic modification, m6A not only occurs on mRNAs but also on Long non-coding RNAs (LncRNAs). As we all known, despite LncRNAs cannot encode proteins, they affect the expression of proteins by interacting with mRNAs or miRNAs, thus playing important roles in the occurrence and development of a variety of tumors. Up to now, it has been widely accepted that m6A modification on LncRNAs affects the fate of the corresponding LncRNAs. Interestingly, levels and functions of m6A modifications are also mediated by LncRNAs through affecting the m6A methyltransferases (METTL3, METTL14, WTAP, METTL16, etc.), demethylases (FTO, ALKBH5) and methyl-binding proteins (YTHDFs, YTHDCs, IGF2BPs, HNRNPs, etc.), which are collectively referred to as “m6A regulators”. In this review, we summarized the mutual regulation mechanisms between N6-methyladenosine modification and LncRNAs in cancer progression, metastasis, invasion and drug resistance. In detail, we focus on the specific mechanisms of m6A modification, which is mediated by methyltransferases and demethylases, involves in the regulation of LncRNA levels and functions in the first part. And section two intensively displays the mediation roles of LncRNAs in m6A modification via changing the regulatory proteins. At last part, we described the interaction effects between LncRNAs and methyl-binding proteins of m6A modification during various tumor occurrence and development.
Similar content being viewed by others

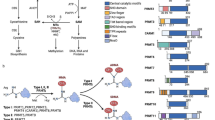
Background
Recently, the regulatory roles of epigenetic modifications in the occurrence and development of various diseases have attracted much attention [1]. To date, more than 170 RNA modifications have been identified and methylation modifications, including N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), N7-methylguanosine (m7G), are the most concerned [2]. Among these methylation modifications, m6A is the most abundant and important RNA modification in mammals [3]. First discovered on mRNA in 1974, m6A modification refers to methylation of the adenosine base at nitrogen-6 position [4]. Nowadays, a large number of studies confirmed that m6A exists not only on mRNAs but also on different non-coding RNAs, including Long non-coding RNAs (LncRNAs) [5, 6]. It has been widely accepted that m6A modification is a reversible methylation modification and is dynamically catalyzed by methyltransferases, demethylases and methyl-binding proteins, which are collectively referred to as m6A regulators [7]. Specifically, methyltransferases, which mainly include methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), Wilms’ tumor-associated protein (WTAP), methyltransferase-like 16 (METTL16) and so forth, catalyze the formation of m6A modification, while the demethylases including obesity-associated protein (FTO) and alkB homologue 5 (ALKBH5) erase the m6A modification. Methyl-binding proteins, which combine with m6A modification and mediate its biological function, generally included YT521-B homology (YTH) domain family, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) and heterogeneous nuclear ribonucleoproteins (hnRNPs) [4]. Under the simultaneous mediation of these regulatory proteins, RNA m6A modification widely involves in RNA generation, splicing, stabilization and degradation, thus participating in a variety of pathological and physiological functions [8]. Accumulating studies indicated that biological effects of m6A modification are closely associated with a variety of human diseases, particularly cancers, in which m6A modification can drive the progression, metastasis, invasion, and drug resistance of tumor cells [7, 9].
Long non-coding RNAs (LncRNAs) are transcripts of more than 200 nucleotides which generally lack protein-coding potential, while they could perform distinct biological functions through interacting with RNAs or proteins [10]. There are currently 172,216 human and 131,697 mouse transcripts annotated in the systematic NONCODEv5 database and 270,044 human LncRNAs in the curated knowledgebase LncBook, making LncRNAs become the most diverse class of regulatory ncRNAs [11]. At present, LncRNAs are abnormally expressed in various tumors and are widely accepted as desirable biomarkers for indicating progression, metastasis and invasion processes of cancer cells [12]. Recently, accumulating studies have found that LncRNAs were modified by m6A modifications, which conversely affect processing and function of corresponding LncRNAs [13]. In detail, m6A modification not only affects the cleavage, stability and degradation of corresponding LncRNAs but also influences the interactions between LncRNAs and miRNAs, mRNAs or proteins [14,15,16]. Interestingly, emerging researches have demonstrated that LncRNAs have critical roles in regulating m6A modification through mediating its methyltransferases demethylases and methyl-binding proteins [17,18,19]. At present, the mutual regulation between m6A modification and LncRNAs has attracted a lot of attention in tumor occurrence and development. Thus, in this review, we thoroughly summarized the recent advances of m6A modification on LncRNAs in the modulation of cancer cell progression, metastasis and invasion, as well as the mediation roles of LncRNAs in m6A modification levels and functions through affecting m6A regulators during the occurrence and development of various tumors, indicating that targeting the interaction between m6A modification and LncRNAs is expected to become a desirable strategy for cancer prevention and treatment.
LncRNAs regulated by m6A modification in various tumors
As mentioned above, m6A modification on LncRNAs has been well established to mediate the functions of corresponding LncRNAs. Recently, accumulating studies have confirmed that m6A modification, which is mediated by METTL3, METTL14, WTAP, FTO and ALKBH5, the widely known methyltransferases and demethylases, on LncRNAs can affect the level and biological function of corresponding LncRNAs, thus involving in the occurrence and development of various tumors, which originate from respiratory system, digestive system, reproductive system and so forth [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61].
m6A-modified LncRNAs mediated by methyltransferases
METTL3 mediates the m6A modification on LncRNAs
As a dynamic and reversible modification, m6A methylation is catalyzed by methyltransferase complex, in which METTL3 serves as the main catalytic core [9]. At present, METTL3 has been well evidenced to mediate various LncRNAs through catalyzing m6A modification. For example, the stability of LncRNA FAM225A is strengthened by m6A modification, which mediated by METTL3, thus up-regulating LncRNA FAM225A level in nasopharyngeal carcinoma cells. Mechanistically, FAM225A functioned as a competing endogenous RNA (ceRNA) for sponging miR-590-3p and miR-1275, leading to the upregulation of their target integrin β3 (ITGB3), finally activation of focal adhesion kinase (FAK)/phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT/PKB) signaling to promote nasopharyngeal carcinoma cells proliferation and invasion [16]. In addition, LncRNA small nucleolar host gene 1 (SNHG1) transcript was also modified by METTL3-mediated m6A modification, thus improving the stability of LncRNA SNHG1 and decreasing the rate of RNA degradation, which leads to upregulation of LncRNA SNHG1 in non-small cell lung cancer (NSCLC). Further, LncRNA SNHG1, as a competing endogenous RNA, was able to sponge miR-140-3p to increase ubiquitin-conjugating enzyme endometrial cancer consortium (E2C2) expression in NSCLC cell lines [5]. Interestingly, small nucleolar host gene 17 (SNHG17), a LncRNA homologous to SNHG1, has been also shown to play a regulatory role and closely relate to m6A modification in lung adenocarcinoma progression. Mechanistically, METTL3-mediated m6A modification could induce the upregulation of LncRNA SNHG17 by enhancing its RNA transcript stability [20]. Also in lung adenocarcinoma cells, the highly expressed LncRNA AC098934 facilitates the cell proliferation as well as invasion either in vitro or in vivo. Further exploration data displayed that LncRNA AC098934 promoted the malignant behavior of lung adenocarcinoma cells under the m6A modification induced by METTL3 [21]. Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a LncRNA highly correlate to the occurrence and development of lung cancer, has been also indicated that its level was increased due to a higher level of m6A modification mediated by METTL3 and meanwhile, METTL3/YTHDF3 complex is able to elevate the stability of MALAT1 in non-small cell lung carcinoma (NSCLC) cells [22]. Similar to that in lung adenocarcinoma, LncRNA MALAT1 is also methylated and delocalized by METTL3 in Thymic epithelial tumors [23]. Besides, LncRNA Lung Cancer Associated Transcript 3 (LCAT3) was also found to be up regulated in lung adenocarcinomas and its over-expression was closely associated with the poor prognosis of lung adenocarcinoma patients. Regarding the regulatory mechanisms, METTL3-mediated m6A modification on LncRNA LCAT3 transcript can enhance its stabilization and upregulate its level [24]. Similarly, the enhanced stability of LncRNA ABHD11 antisense RNA 1 (ABHD11-AS1), which ectopic overexpression closely associated with unfavorable prognosis of non-small cell lung carcinoma patients, was also manipulated by METTL3-mediated m6A modification and thus upregulation of LncRNA ABHD11-AS1 level [25]. In addition, high expression of LncRNA SVIL-AS1, of which expression is also upregulated by METTL3-mediated m6A modification, can promote the occurrence and development of lung adenocarcinoma [26]. Another study indicated that the level of LINC00958, which independently predicted poor overall survival of hepatocellular carcinoma patients, was also elevated by METTL3-mediated m6A modification [27]. In addition, LncRNA THAP7-AS1 has been detected as showing high expression and correlating with positive lymph node metastasis and poorer prognosis of gastric cancer. Further exploring results revealed that METTL3 catalyzes the formation of m6A modification on LncRNA THAP7-AS1 and enhanced its expression depending on the "reader" protein IGF2BP1 [28]. In colorectal cancer cells, Wu et al. found that LncRNA RP11 level was enhanced by METTL3-mediated m6A methylation and positively regulated the migration, invasion and epithelial mesenchymal transition (EMT) of colorectal cancer cells. Mechanistically, heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNPA2B1), as well known m6A methyl-binding protein, formed a complex with LncRNA RP11 to mediate the degradation E3 ligases, Siah1 and Fbxo45 mRNA in an m6A-dependent manner, thus preventing the proteasomal degradation of Zinc finger E-box-binding homeobox (ZEB1), of which upregulation was essential for LncRNA RP11-induced cell dissemination [15]. In addition, high expression of LncRNA plasmacytoma variant translocation 1 (PVT1) made prostate cancer cells more proliferative, migratory and invasive, whereas LncRNA PVT1 knockdown led to the opposite phenotype. In terms of specific mechanisms, the expression of LncRNA PVT1 upregulated by METTL3-mediated m6A modification on LncRNA PVT1, which subsequently sequestered miR-27b-3p within cells, thereby indirectly promoting the bloom syndrome protein expression [29].
METTL14 mediates the m6A modification on LncRNAs
METTL14, another important catalytic enzyme for m6A modification, has also been well established to regulate m6A modification on different LncRNAs in various cancers, including head and neck squamous cell carcinoma, oral squamous cell carcinoma, lung adenocarcinoma, colorectal cancer, renal cell carcinoma, hepatocellular carcinoma and so forth [30,31,32,33,34,35,36,37,38]. In detail, LncRNA LNCAROD overexpressed in head and neck squamous cell carcinoma and shortened overall survival of patients. The dysregulation of m6A modification on LncRNA-activating regulator of DKK1 (LNCAROD), which was mediated by METTL3 and METTL14, might account for enhancing the LNCAROD stability and elevating its level, thus promoting proliferation, mobility and tumorigenicity in head and neck squamous cells [30]. Li et al. indicated that both METTL14 and LncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) were elevated in oral squamous cell carcinoma tissues and cells. Silencing METTL14 repressed OSCC cell viability and colony formation while excitation of LncRNA MALAT1 was able to reverse the inhibition effects of silencing METTL14. Further experimental data demonstrated that METTL14 induced m6A modification on LncRNA MALAT1 thus increasing the level of LncRNA MALAT1, which acts as a sponge to absorb miR-224–5 and promote histone lysine demethylase 2A transcription [31]. In addition, LncRNA human leukocyte antigen complex group 11 (HCG11) served as a tumor suppressor to restrain tumor growth in lung adenocarcinoma. Mechanistically, the METTL14-mediated m6A modification on LncRNA HCG11 enhanced its nuclear exportation, thus recruiting IGF2BP2 to target Large Tumor Suppressor Kinase 1 (LATS1) mRNA to enhance the stability and promote the translation of LATS1, finally influencing the growth of lung adenocarcinoma [32]. The study from Yang et al. indicated that knockdown of METTL14 drastically enhanced proliferative and invasive ability in colorectal cancer cells. Further exploring data identified that LncRNA X inactive-specific transcript (XIST) was the downstream target of METTL14, of which low expression significantly attenuated m6A modification on LncRNA XIST and augmented LncRNA XIST expression through attenuating degradation [33]. In addition, studies have revealed that LncRNA ZFAS1/RAB22A in atherosclerosis [34], LINC01320 in gastric cancer [35], Lnc-LSG1 and LncRNA NEAT1 in renal cell carcinoma [36, 37], LncRNA MIR155HG in hepatocellular carcinoma were also regulated by METTL14-mediated m6A modification [38], indicating that METTL14 can regulate the modification of m6A on various LncRNAs, therefore participating in the genesis and development of various tumors.
WTAP mediates the m6A modification on LncRNAs
As a kind of adaptor protein of m6A methyltransferase complex, WTAP has been well established to involving in the mediation of m6A modification on LncRNAs, such as LncRNA DIAPH1-AS1 [39], LncRNA FOXD2-AS1 [40]. In detail, WTAP-mediated m6A level on DIAPH1-AS1 can enhance the stability of DLGAP1-AS1 in an IGF2BP2-dependent pathway, thus participating in the growth and metastasis of naso-pharyngeal carcinoma cells [39]. In addition, m6A-modified LncRNA FOXD2 adjacent opposite strand RNA 1 (FOXD2-AS1) has been also evidenced to involving in the occurrence and progression of osteosarcoma. Mechanistically, a remarkable number of m6A-modified sites were found on the 3'-UTR of FOXD2-AS1, thus enhancing the stability of FOXD2-AS1 transcript, on which m6A modification was promoted by WTAP [40].
Other methyltransferases mediate the m6A modification on LncRNAs
In addition to METTL3, METTL14 and WTAP, some RNA m6A modification methyltransferases, such as Zinc finger CCHC domain-containing protein 4 (ZCCHC4), KIAA1429 and METTL16, also catalyze m6A modification on LncRNAs [41,42,43]. In detail, ZCCHC4, one of latest identified m6A methyltransferases which primarily methylates human 28S rRNA and also interacts with a subset of mRNAs, was found to be inhibited DNA damage-induced apoptosis in hepatocellular carcinoma cells by interacting with LncRNA AL133467.2 Further exploring data revealed that knockout of ZCCHC4 promotes AL133467.2 and γH2AX interaction for enhancing chemosensitivity in hepatocellular carcinoma cells [41]. KIAA1429, another methyltransferase, was also involved in mediation of m6A modification on LncRNA LINC00958 in gastric cancer cells. Mechanistically, m6A-modified sites in LINC00958 have been identified by using methylated RNA immunoprecipitation sequencing (MeRIP-Seq). Moreover, KIAA1429 catalyzed the m6A modification on LINC00958, which interacted with glucose transporter type 1 (GLUT1) mRNA in an m6A-dependent manner to strengthen the stability of GLUT1 mRNA, thus promoting the gastric cancer cells' aerobic glycolysis [42]. In addition, originally thought to be a ribosomal RNA methyltransferase, METTL16 has now been shown to bind and methylate LncRNA RAB11B-AS1. In detail, METTL16 directly bound LncRNA RAB11B-AS1 and induced its m6A modification, which decreased the stability of LncRNA RAB11B-AS1 transcript thus resulting in the down-regulation of RAB11B-AS1. This reduction in LncRNA RAB11B-AS1 level caused by the elevation of METTL16 was correlated with poor prognosis of patients with hepatocellular carcinoma [43].
m6A-modified LncRNAs mediated by demethylases
FTO mediates the m6A modification on LncRNAs
Recently, emerging studies have demonstrated that FTO was sincerely involved in the mediation of m6A modification on LncRNAs, thus participating in the occurrence and development of various tumors [44,45,46]. Results from Cui et al. indicated that the elevation of FTO expression demethylated m6A modification on LINC00022 transcript, thus hindering the LINC00022 degradation mediated by YTHDC2 in esophageal squamous cell carcinoma [44]. Han et al. systematically assessed the m6A modification expression of 407 gastric cancer clinical samples based on 23 m6A regulators and comprehensively associated these genes with LncRNAs. Importantly, LncRNA AC026691.1 could inhibit both migration and proliferation of gastric cancer through FTO demethylation [45]. In cervical cancer, LncRNA HOXC13 antisense RNA (HOXC13-AS) was increased and promoted the malignant phenotype of cervical cancer cells. Mechanistically, LncRNA HOCX13-AS1 expression was augmented Frizzled class receptor 6 (FZD6) by cAMP-response element binding protein-binding protein (CBP)-modulated histone H3 on lysine 27 acetylation (H3K27ac). Additionally, FTO reduced m6A and stabilized HOXC13-AS thus up-regulating FZD6 and activating Wnt/β-catenin signaling to drive cervical cancer cell proliferation, invasion and epithelial mesenchymal transition, suggesting HOXC13-AS as a potential target for cervical cancer treatment [46].
ALKBH5 mediates the m6A modification on LncRNAs
As a demethylase of m6A modification, ALKBH5 directly reverses m6A modification on adenosine [9]. Recently, some studies have revealed that ALKBH5 detaches m6A modification on various LncRNAs and involves in the occurrence and development of various tumors [47,48,49,50,51,52,53,54,55,56,57,58,59,60]. As we all known, LncRNA nuclear enriched abundant transcript 1 (NEAT1) is a carcinogenic LncRNA and its level and function were closely related to ALKBH5-demethylated m6A modification [47,48,49,50,51]. Literature from Dong et al. indicated that m6A-modified LncRNA NEAT1 was involved in glioblastoma multiforme. Specifically, hypoxia-induced ALKBH5 removed m6A modification from the LncRNA NEAT1, enhancing its transcript stabilization and promoting NEAT1-mediated paraspeckle assembly, ultimately generating an immunosuppressive tumor microenvironment [47]. According to predictions in bioinformatics, Zhang et al. indicated that LncRNA NEAT1 is a potential binding LncRNA of ALKBH5. Further detection results demonstrated that both LncRNA NEAT1 and ALKBH5 were overexpressed in gastric cancer cells and tissues. ALKBH5 influences the expression of NEAT1 through removing m6A modifications on LncRNA NEAT1, of which high expression can subsequently lead to the enhancer of enhancer of zeste homolog 2 (EZH2) elevation. Specifically, NEAT1 can function as a scaffold by interacting with EZH2 to regulate the expression of EZH2 downstream genes, of which dysregulation is associated with invasion and metastasis of gastric cancer cells [48]. As same in gastric cancer, ALKBH5 also could up-regulate LncRNA NEAT1 expression by inhibiting m6A enrichment on LncRNA NEAT1 in hepatocellular carcinoma [49] and in colon cancer [50]. In addition to digestive system tumors, the elevation of LncRNA NEAT1 expression promoted by ALKBH5 demethylation is also observed in infantile hemangioma [51]. ALKBH5-mediated m6A modification on LncRNAs also plays a key role in the development of respiratory system tumors. Results from Li et al. indicated that LncRNA potassium voltage-gated channel subfamily Q member 1 opposite strand 1 (KCNQ1OT1) could directly bind to Homeobox A9 (HOXA9) to further regulate the proliferation, invasion and metastasis of Laryngeal squamous cell carcinoma cells. Further exploring data showed that ALKBH5 mediates LncRNA KCNQ1OT1 expression in an m6A-YTHDF2 dependent manner [52]. In esophageal squamous cell carcinoma tissues, the expression of LncRNA cancer susceptibility candidate 8 (CASC8) was higher than that in the control tissues and positively associated with the poor prognosis of patients. Further mechanism exploring data showed that the stability of LncRNA CASC8 transcript was enhanced by ALKBH5 mediated m6A demethylation [53]. ALKBH5-demethylated m6A modification on LncRNA RMRP was also identified in lung adenocarcinoma tissues [54]. Further, the cell proliferation, metastasis and cell cycle progression regulated by LncRNA TP53TG1 were closely related to m6A modification in gastric cancer cells. In detail, there were multiple m6A modification sites on LncRNA TP53 target 1 (TP53TG1), of which stabilization and expression were reduced by ALKBH5-demethylated m6A modification [55]. As a special LncRNA, long intergenic non-coding RNA LINC02551, a bona fide m6A target of ALKBH5, was downregulated by ALKBH5 in an m6A dependent manner in hepatocellular carcinoma [56]. LncRNA KCNK15-AS1 was able to effectively arrest proliferation, migration and EMT in pancreatic cancer cells. Data from mechanical experiments revealed that ALKBH5 was verified to increase m6A demethylation of LncRNA KCNK15-AS1 to control its elevation [57]. Also in pancreatic cancer, LncRNA DDIT4-AS1 was identified as one of the downstream targets of ALKBH5 through m6A-RNA immunoprecipitation and RNA sequencing in combination with bioinformatics analysis. Further experimental data revealed that stabilization of LncRNA DDIT4-AS1 was maintained by m6A-modified sites, which is essential for recruitment of Hu-Antigen R (HuR) to LncRNA DDIT4-AS1 [58]. Investigation from Chen et al. revealed that ALKBH5 could associate with LncRNA PVT1 and suppress LncRNA PVT1 degradation via erasing m6A modification in osteosarcoma. Mechanically, ALKBH5 decreased the m6A modification level on LncRNA PVT1, thus repressing the interaction between YTHDF2 and LncRNA PVT1 [59]. As same in osteosarcoma, knockdown of ALKBH5 contributed to reducing the stability of LncRNA PVT1 in lung cancer cells [60].
In summary, accumulating studies have indicated that methyltransferases and demethylases influence the LncRNA levels and functions in various carcinogenic processes. We drew a schematic diagram, in which take METTL3 [16], METTL14 [31], WTAP [40], FTO [44] and ALKBH5 [48] as examples, to show the specific mechanisms of m6A modification involving in affecting LncRNA functions and levels (Fig. 1). Besides, the effects of m6A and its methyltransferases and demethylases on LncRNAs in different cancers are summarized and shown in Table 1.

M6A modification and its methyltransferases and demethylases affected LncRNA function and level in various carcinogenic process. a In nasopharyngeal carcinoma (NPC), METTL3 up-regulated LncRNA FAM225A level thus promoting cells proliferation, invasion and metastasis. b In oral squamous cell carcinoma (OSCC), METTL14 increased LncRNA MALAT1 level, leading to elevating cell viability and proliferation. c In osteosarcoma (OS), WTAP enhanced LncRNA DIAPH1-AS1 stability, thus involving in the tumor cell growth and metastasis. d In esophageal squamous (ESCC), FTO inhibited LINC00022 decay, thus resulting in promoting cell proliferation. e In gastric cancer(GC), ALKBH5 reduced m6A level of LncR NEAT1, thereby promoting invasion and metastasis
m 6 A modification regulated by LncRNAs in various tumors
LncRNAs play essential roles in regulation of gene transcription and mRNA translation, implicating that LncRNA may influence m6A modification level through controlling methyltransferases and demethylases. Recently, accumulating studies have confirmed that LncRNA does impact the level and biological function of m6A modification via mediating METTL3, METTL14, WTAP, FTO and ALKBH5, and these regulatory relationships involved in the occurrence and development of various tumors, which originate from respiratory system, digestive system, reproductive system and so forth [45, 61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76].
LncRNAs mediate m 6 A modification through changing methyltransferases
Regulatory effects of LncRNAs on METTL3
As the active center of the methyltransferase complex, METTL3 can be regulated by LncRNAs in directly or indirectly manner. In terms of direct regulation, METTL3 expression was directly interacted with LncRNA ARHGAP5-AS1 [61], LINC00470 [19, 62] and LncRNA SNHG4 [63] in different cancer cells. Zhu et al. have indicated that targeting ARHGAP5-AS1/ARHGAP5 axis may be a promising strategy to overcome chemoresistance in gastric cancer. In detail, LncRNA Rho GTPase activating protein 5 antisense RNA 1 (ARHGAP5-AS1) has been upregulated in chemoresistant gastric cancer cells and its knockdown reversed chemoresistance. M6A modification on LncRNA ARHGAP5 was significantly inhibited by depletion of METTL3, which was effectively recruited by LncRNA ARHGAP5-AS1 to facilitate m6A modification on ARHGAP5 mRNA [61]. Yan et al. revealed that LncRNA LINC00470 oncogenic functions on gastric cancer cell proliferation, migration and invasion were closely related to METTL3-mediated m6A modification on PTEN mRNA. In detail, an RIP assay results exhibited that METTL3 could remarkably enrich LINC00470 transcripts. Further confirmation data from RNA pull-down assay demonstrated an obvious interaction between LINC00470 and METTL3. Moreover, knockdown of LINC00470 significantly reduced the binding of METTL3 in Phosphatase and tensin homolog (PTEN) mRNA and depletion of METTL3 expression rescued the LINC00470-induced PTEN mRNA degradation, indicating LINC00470 positively regulated m6A modification on PTEN mRNA via relation to METTL3 [19]. This regulatory manner was also identified in chronic myelocytic leukemia (CML) cells, in which the alteration of LINC00470 had no effect on the luciferase activity of the PTEN promoter but affected the half-life of PTEN mRNA. Specifically, LINC00470 is a regulator of METTL3, which positively regulate the m6A modification on PTEN mRNA, thus enhancing the PTEN expression and stability. High expression of PTEN was able to promote protein kinase B (PKB/AKT) activity while inhibit hexokinase 1 (HK1) ubiquitination, thereby stimulating tumorigenesis of chronic myelocytic leukemia (CML) cells [62]. In addition, LncRNA SNHG4 overexpression facilitated cell proliferation and migration while inhibited cell apoptosis in neonatal pneumonia patients. Mechanistically, the enrichment of LncRNA SNHG4 in the METTL3 promoter region resulted in the downregulation of METTL3, of which interference can restrain the m6A modification on STAT2 mRNA, thus promoting STAT2 mRNA translation efficiency [63].
For the indirect regulation, LncRNAs mediate METTL3 expression mainly in a miRNA-dependent manner. In detail, Wang et al. reported that NUTM2A-AS1/miR-590-5p/METTL3 axis was involved in lung adenocarcinoma progression. Mechanically, miR-590-5p was predicted and verified as the direct target of LncRNA NUTM2A-AS1 according to bioinformatics analysis and a dual luciferase reporter assay. And further exploring data confirmed that miR-590-5p could target the METTL3 mRNA and cut down METTL3 expression in NCI-H23 and A549 cells [64]. In addtion, the functional role of LINC00240/miR-338-5p/METTL3 axis was investigated in regulating the aggressiveness of gastric cancer cells. Specifically, LINC00240 has been identified and validated to negatively regulate miR-338-5p, which could target METTL3 mRNA at 3'UTR to downregulate its protein translation [65].
Regulatory effects of LncRNAs on METTL14
Up to now, the regulatory effects of LncRNAs on METTL14 is mainly observed in breast cancer and acute myeloid leukemia (AML) [66,67,68,69]. In detail, results from Sun et al. uncovered a novel LNC942-METTL14-CXCR4/CYP1B1 signaling axis in breast cancer initiation and progression. They unveil that LINC00942 (LNC942) exerts its functions as an oncogene in promoting METTL14-mediated m6A modification and regulates the expression and stability of LNC942 downstream target genes C-X-C motif chemokine receptor 4 (CXCR4) and cytochrome P450 1B1 gene (CYP1B1) in breast cancer initiation and progression. Mechanistically, LNC942 directly recruits METTL14 protein by harboring the specific recognize sequence (+ 176- + 265), thereby stabilizing the CXCR4 and CYP1B1 mRNA in an m6A modification dependent manner thus finally promoting cell proliferation and colony formation of breast cancer cells [66]. LncRNA urothelial carcinoma-associated 1 (UCA1) also served as a risk factor for indicating poor prognosis of breast cancer patients and silencing LncRNA UCA1 could attenuate cell proliferation and invasion. Further exploring results showed that LncRNA UCA1 augmented the METTL14 expression through altering METTL14 promoter region methylation, which promote the METTL14 translation, thus mediating the miR-375 level in an m6A-dependent manner and increasing SOX12 expression levels and curbing the progression of breast cancer [67]. Besides breast cancer, overexpression of LncRNA UCA1 also accelerated Acute myeloid leukemia (AML) development by regulating METTL14-mediated CXCR4 and CYP1B1 mRNA [68].
Regulatory effects of LncRNAs on WTAP
As an important adaptor protein in m6A methyltransferase complex, WTAP can be also regulated by a variety of LncRNAs in a miRNA-dependent manner, therefore involving in the occurrence and development of various tumors [70,71,72,73,74,75]. In detail, results from Zhu et al. indicated that hypoxia upregulated the expressions of LncRNA EMS and WTAP as well as reduced the level of miR-758-3p in esophageal cancer cell line ECA-109. Further exploring data verified that targeting regulatory relationships between LncRNA EMS and miR-758-3p, as well as miR-758-3p and WTAP, implicating a critical role of LncRNA-EMS/miR-758-3p/WTAP axis in regulating hypoxia-mediated cisplatin resistance in esophageal cancer [69]. High expression of LncRNA PCGEM1 accelerates cell proliferation, migration and invasion but restrains cell apoptosis via sponging miR-433-3p to upregulate WTAP thus promoting the non-small cell lung cancer progression [70]. In hepatocellular carcinoma, functional interactions between LINC00839, miR-144-3p and WTAP were validated. In terms of specific mechanisms, LINC00839 served as a sponge to negatively regulate miR-144-3p activity, which contributed to elevation the WTAP expression [71]. A novel mechanistic role of LncRNA DUXAP8/miR-448/WTAP/Fak signaling axis has also been identified in pancreatic carcinoma [72]. In addition, Ge et al. confirmed that LncRNA SNHG10 could bind to miR-141-3p, which further targeted binding with WTAP, meaning LncRNA SNHG10 upregulated WTAP through decreasing miR-141-3p expression in osteosarcoma genesis [73].
LncRNAs mediate m 6 A modification through demethylases
Regulatory effects of LncRNAs on FTO
As the first identified m6A demethylase, FTO plays important roles in regulating the LncRNA levels and functions [45,46,47]. Interestingly, recent studies have found that the level of FTO is conversely regulated by LncRNAs, including LncRNA JPX [74], Lnc-H2AFV-1 [75], LncRNA CASC15 [76] and LncRNA AC026691.1 [45]. In detail, LncRNA Just Proximal to the X-inactive specific transcript (JPX) has been proven to be involved in glioblastoma multiforme through the FTO/PDK1 axis. Mechanistically, FTO interaction with LncRNA JPX, which forms a complex with phosphoinositide dependent kinase-1 (PDK1) mRNA, can enhance FTO-mediated PDK1 mRNA demethylation, thus promoting stability and translation of PDK1 mRNA [74]. Lnc-H2AFV-1 was found to be upregulated in head and neck squamous cell carcinoma tissues, in which the expression of FTO was contrary to that of lnc-H2AFV-1. Further results revealed that lnc-H2AFV-1 overexpression led to the elevated expression and maximal m6A methylation of intraflagellar transport (IFT) 80 by regulating the FTO to promote HNSCC progression [75]. Qin et al. reported that inhibition of FTO expression significantly restrained proliferative and anti-apoptotic effects, which were mediated by LncRNA CASC15. Further exploring data indicated that LncRNA CASC15 promoted esophageal squamous cell carcinoma carcinogenesis by decreasing single minded 2 (SIM2) stability via FTO-regulated demethylation [76]. LncRNA AC026691.1 and FTO were intimately associated with the regulation of m6A RNA methyladenine in gastric cancer. Combined effect of LncRNA AC026691.1 and FTO might suppress gastric cancer via downregulation of m6A level [45].
Regulatory effects of LncRNAs on ALKBH5
Up to now, two studies have reported that LncRNA CASC11 and LncRNA GAS5-AS1 can mediate the m6A modification through affecting ALKBH5 in hepatocellular carcinoma and cervical cancer, respectively [77, 78]. In detail, LncRNA CASC11 is involved in the regulation of cell proliferation, migration as well as invasion through the upregulation of Ubiquitin-conjugating enzyme E2T (UBE2T) in an m6A-dependent manner in hepatocellular carcinoma. Specificly, LncRNA CASC11 stabilized UBE2T mRNA through reducing UBE2T mRNA m6A modification via recruiting ALKBH5. Moreover, LncRNA CASC11 also interfered the interaction between UBE2T mRNA and YTHDF2, thereby influencing proliferation and metastasis of hepatocellular carcinoma cells [77]. Interestingly, LncRNA GAS5-AS1, the antisense RNA of GAS5, has been detected to interact with GAS5 and enhanced its stability through decreasing ALKBH5-mediated GAS5 m6A modification, therefore reducing the proliferation, migration and invasion of cervical cancer cells [78].
Taken together, current studies have reported that LncRNAs influenced m6A modification through mediating catalytic enzymes, including METTL3, METTL14, WTAP, FTO and ALKBH5, during the occurrence and development of various tumors. Although specific mechanisms underlying LncRNA mediation m6A modification are complex, there are some similarities in LncRNA regulating m6A modification. We drew a schematic diagram, which takes LINC00470 [62], LncRNA SNHG10 [73], Lnc-CASC15 [76] and Lnc-CASC11 [77] as examples, to show the specific mechanisms of LncRNAs regulating the m6A modification through affecting m6A catalytic enzymes (Fig. 2). Additionally, the regulatory mechanisms of LncRNAs in m6A modification and its catalytic enzymes in different tumors are summarized and shown in Table 2.

Mechanisms underlying the LncRNAs influencing m6A modification in multiple tumors. a LINC00470 reduced the PTEN expression by regulating METTL3 and promoting proliferation of chronic myeloid leukemia (CML) cells. b LncRNA SNHG10 sponged miR-141-3p thus upregulating WTAP expression and resulting in promoting cell proliferation in osteosarcoma genesis (OS). c In glioblastoma multiforme (GMB), LncRNA interacted with FTO and enhanced FTO-mediated PDK1 mRNA demethylation, thus involving in increasing proliferation and inhibiting apoptosis. d In hepatocellular carcinoma (HCC), Lnc-CASC11 decreased UBE2T m6A modification via recruiting ALKBH5, thereby promoting proliferation, migration and invasion
Interaction between LncRNAs and methyl-binding proteins of m 6 A modification
As universally acknowledged m6A binding proteins, including YTHDFs, YTHDCs, IGF2BPs and hnRNPs, not only participate in multiple processes of LncRNA metabolism but also involve in a variety of tumorigenesis through binding to the m6A modification sites of target LncRNAs [79,80,81]. Nowadays, the interaction between LncRNAs and methyl-binding proteins of m6A modification has been widely considered as an important biological event during the occurrence and progression of various tumors [53, 82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111].
Mutual regulatory effects between LncRNAs and YTH family members
YTH family members were originally identified as m6A methyl-binding proteins and classified into five categories, including YTHDC1, YTHDC2, YTHDF1, YTHDF2 and YTHDF3. Most of these YTH family members have been well established to interact with LncRNAs, thus involving in various cancers [82,83,84,85,86,87,88,89,90,91,92,93,94,95].
Interaction between YTHDC1 and LncRNAs in tumors
YTHDC1, mainly located in the nucleus, has been proven to be related to the transport, stability maintenance and degradation of various LncRNAs during tumor occurrence and development [82]. In detail, super-resolution imaging from Wang et al. revealed that the concatenated m6A residues on LncRNA MALAT1 acted as a scaffold for recruiting YTHDC1 to nuclear speckles. Further investigation data revealed that the recognition of m6A-modified MALAT1 by YTHDC1 played an essential role in maintaining the composition and genomic binding sites of nuclear speckles, which regulate the expression of several key oncogenes. More importantly, artificially tethering YTHDC1 onto m6A-deficient MALAT1 largely rescues the metastatic potential of cancer cells [83]. Results from Chen et al. indicated that the m6A-modified LncRNA TERRA formed R-loops and promoted homologous recombination, which was essential for the alternative lengthening of telomeres pathway in cancer cells. Mechanically, reduction of m6A modification on LncRNA TERRA by abating METTL3 or inhibition of YTHDC1 expression enhanced degradation of LncRNA TERRA, indicating the interaction between m6A modification and LncRNA TERRA [84]. Loss-of-function experiment demonstrated that knockdown of LINC00857 restrained cell viability, proliferation and migration as well as epithelial mesenchymal transition and strengthened cell apoptosis in colorectal cancer. Further exploring data showed that LINC00857 can be effectively bound by YTHDC1 due to the presence of m6A modification. Interestingly, YTHDC1 ultimately combined with solute carrier family 7 member 5 (SLC7A5) and increased SLC7A5 mRNA stability to promote the proliferation and migration of colorectal cancer cells, implicating the critical role of LINC00857/YTHDC1/SLC7A5 axis in colorectal cancer progression [85]. In clear cell renal cell carcinoma cells, m6A-modified Lnc-LSG1, which was identified as a target of METTL14 via high-throughput methylated RNA immunoprecipitation sequencing (MeRIP-seq), can directly bind to epithelial splicing regulatory protein 2 (ESRP2) protein and then facilitate ESRP2 ubiquitination, finally resulting in ESRP2 degradation by interaction with YTHDC1 [37].
Interaction between YTHDF1 and LncRNAs in tumors
YTHDF1, as another YTH family member, has been evidenced to preferentially recognize m6A modification on LncRNAs, thus affecting the level and biological function of LncRNAs [86]. Recently research revealed that YTHDF1 involved in stability and degradation of multiple LncRNAs, such as LncRNA HCP5 [87] and LncRNA DLGAP1-AS2 [88], thus participating in regulating the progression of esophageal squamous cell carcinoma and non-small cell lung cancer, respectively. In detail, YTHDF1-regulated LncRNA HCP5 has been evidenced to involve in progression of esophageal squamous cell carcinoma. Regarding the mechanisms, LncRNA HCP5 was able to directly interact with YTHDF1, thereby strengthening the binding of YTHDF1 to HK2 mRNA in an m6A- dependent manner, leading to enhancing the stability and expression of HK2 mRNA, of which high expression is mainly responsible for carcinogenicity of esophageal squamous cell carcinoma [87]. Moreover, the interaction between LncRNA DLGAP1 antisense RNA 2 (DLGAP1-AS2) and YTHDF1 has been established to involve in non-small cell lung cancer. Further exploring data indicated that m6A sites on LncRNA DLGAP1-AS2 were added by METTL3 and LncRNA DLGAP1-AS2 interacted with YTHDF1 to enhance the stability of c-Myc mRNA through DLGAP1-AS2/YTHDF1/m6A/c-Myc axis [88].
Interaction between YTHDF2 and LncRNAs in tumors
Up to now, LncRNA FENDRR [89], LncRNA FGF14-AS2 [90], LncRNA THOR [91], LncRNA-CBSLR [92], and LncRNA STEAP3-AS1 [93], have been evidenced to interplay with YTHDF2, thus regulating the proliferation, metastasis, invasion and ferroptosis of various cancer cells. Specifically, the expression level of LncRNA FENDRR was decreased in cancerous tissues of endometrioid endometrial carcinoma patients, while the m6A methylation levels on LncRNA FENDRR were elevated. Further detection results demonstrated that m6A-modified LncRNA FENDRR was recognized by YTHDF2, which mediated LncRNA FENDRR degradation, thus promoting cell proliferation by elevating SOX4 expression in endometrioid endometrial carcinoma [89]. LncRNA FGF14-AS2 is an essential inhibitor in breast cancer metastasis and patients with high YTHDF2 and low FGF14-AS2 expression levels showed worse distant metastasis-free survival. The exploring data showed that LncRNA FGF14-AS2 is repressed by YTHDF2-regulated RNA degradation in an m6A-dependent manner [90]. Interestingly, LncRNA THOR can also be simultaneously combined by YTHDF1 and YTHDF2. In detail, m6A sites are highly enriched on LncRNA THOR transcripts and specific m6A readers YTHDF1 and YTHDF2 can read the m6A motifs and regulate the stability of the LncRNA THOR, thus mediating the proliferation, migration and invasion of cancer cells [91]. It is widely accepted that the hostile hypoxic microenvironment takes primary responsibility for the rapid expansion of tumors. A shypoxia-inducible LncRNA, LncRNA CBSLR has been identified in gastric cancer and high expression of LncRNA CBSLR protected gastric cancer cells from ferroptosis. Mechanically, LncRNA CBSLR interacted with YTHDF2 to form LncRNA CBSLR/YTHDF2/CBS signaling axis, of which activation was able to reduce the stability of CBS mRNA by enhancing the binding of YTHDF2 to the m6A-modified coding sequence (CDS) of cystathionine beta-synthase (CBS) mRNA [92]. Zhou et al. identified that upregulation of LncRNA STEAP3-AS1 facilitated the proliferation and metastasis of colorectal cancer cells both in vitro and in vivo. Mechanistically, LncRNA STEAP3 antisense RNA 1 (STEAP3-AS1) interacted competitively with the YTHDF2, resulting in the disassociation of YTHDF2 with Six-transmembrane epithelial antigen of the prostate 3 (STEAP3) mRNA, which escaped the fate of degradation mediated by m6A modification and elevated the protein expression of STEAP3, thus finally activation of Wnt/β-catenin signaling to promote colorectal cancer progression [93].
Interaction between YTHDF3 and LncRNAs in tumors
YTHDF3, another m6A binding protein, also interacts with different LncRNAs in NSCLC, colorectal cancer and prostatic cancer. In detail, YTHDF3 has maintained the stability of LncRNA MALAT1 in an m6A-dependent manner in NSCLC cells. Further detecting results indicated that m6A modification on LncRNA MALAT1 has facilitated formation by METTL3 [94]. YTHDF3 also has been indicated to bind to m6A-modified LncRNA GAS5 and facilitate LncRNA GAS5 degradation in a methylation-dependent manner, indicating a new insight into LncRNA GAS5 in colorectal cancer progression [14]. Glycolysis is a pivotal process in metabolic reprogramming of tumorigenesis. Bio-information analysis indicated that LncRNA DICER1-AS1 was downregulated in prostatic cancer and negatively correlated with glycolytic gene expression. Mechanistically, enhanced interaction between m6A reader YTHDF3 and LncRNA DICER1-AS1 led to degradation of DICER1-AS1 in response to glucose depletion [95].
Mutual regulatory effects between LncRNAs and IGF2BPs in tumors
IGF2BPs, generally including IGF2BP1, IGF2BP2 and IGF2BP3, have also been identified as m6A methyl-binding proteins and they are involved in the stability and degradation of various LncRNAs during the tumor occurrence and development. As a binding protein of the same type, IGF2BP1, IGF2BP2 and IGF2BP3 interact with same LncRNAs in the same tumor and has the same function. For example, regulation of LncRNA FGF13-AS1 accelerated cell proliferation, migration and invasion by impairing glycolysis and stemness properties in breast cancer. Specifically, LncRNA FGF13-AS1 not only reduced the half-life of c-Myc mRNA by binding to IGF2BP1/2/3 but also disrupted the interaction between IGF2BP1/2/3 and Myc mRNA, of which high efficiency of protein translation conversely repressed LncRNA FGF13-AS1, thus forming a feedback loop of FGF13-AS1/IGF2BPs/Myc /FGF13-AS1 [96]. In addition, LncRNA MTAR1 enhanced binding between IGF2BP1/2/3 and PABP1, thereby promoting Myc mRNA stability [97]. Interestingly, IGF2BP1, IGF2BP2, and IGF2BP3 can also interact with different LncRNAs in various tumors and their effects on LncRNAs has great diversities [98,99,100,101,102,103,104,105,106,107].
Interaction between IGF2BP1 and LncRNAs in tumors
Xia et al. discovered that the global loss of LncRNA lnc-CTHCC restrained the occurrence and development of HCC. The high expression of LncRNA CTHCC in HCC benefited from increased stability, which was enhanced by METTL3-mediated m6A modification in an IGF2BP1/IGF2BP3-dependent manner [98]. LncRNA AC004812.2 was a protective factor in osteosarcoma and low expression of AC004812.2 predicted worse overall survival. Overexpression of AC004812.2 increased the expression levels of YTHDF1 and IGF2BP1 inhibiting 143B cell proliferation [99]. LncRNA KB1980E6.3 could facilitate breast cancer stem cells’ self-renewal and tumorigenesis under a hypoxic microenvironment both in vitro and in vivo. Mechanistically, LncRNA KB-1980E6.3 recruited IGF2BP1 to form a LncRNA KB-1980E6.3/IGF2BP1/c-Myc signaling axis that retained the stability of c-Myc mRNA through increasing binding of IGF2BP1 with m6A-modified c-Myc coding region instability determinant (CRD) mRNA [100]. As a tumor suppressor in multiple cancers, LINC00261 was downregulated in pancreatic cancer tissues and cell lines and high expression of LINC00261 induced cell cycle arrest and apoptosis. Specifically, LINC00261 could sequester IGF2BP1 thus inhibiting c-myc expression [101].
Interaction between IGF2BP2 and LncRNAs in tumors
Previous literature has reported that IGF2BP2 involves in the occurrence and development of tumors through enhancing the stability of lncRNA DANCR [103,104,105,106]. In detail, results from Hu et al. indicated that IGF2BP2 and LncRNA DANCR work together to promote cancer stemness-like properties and pancreatic cancer pathogenesis. Mechanistically, m6A-modified LncRNA DANCR was essential to the interaction between IGF2BP2 and DANCR, on which m6A modification was specifically recognized by IGF2BP2, thus promoting the LncRNA DANCR stabilization [102]. This appearance which IGF2BP2 promoted DANCR stability in an m6A modification dependent manner also has been evidenced in glioblastoma cells [103]. Besides LncRNA DANCR, IGF2BP2 also combined with LncRNA-PACERR, thus promoting the stability of Kruppel-like factor 12 (KLF12) and c-Myc mRNA, finally involving in elevating the number of M2-polarized cells and facilizing cell proliferation, invasion, and migration in vitro and in vivo [104]. In colorectal cancer tissues, Wang et al. reported that LncRNA LINRIS was also upregulated and LncRNA LINRIS inhibition led to impairing proliferative capability of colorectal cancer cell line. Specifically, high expression of LncRNA LINRIS promoted the IGF2BP2 stability via blocking IGF2BP2 K139 ubiquitination, which inhibited the degradation of IGF2BP2 through the autophagy-lysosome pathway [21]. In testicular cancer, data from Ye et al. indicated that LncRNA MALAT1 and IGF2BP2 were highly expressed. In terms of mechanisms, LncRNA MALAT1 contributed to testicular cancer progression through the upregulation of IGF2BP2 by binding to miR-204 [105].
Interaction between IGF2BP3 and LncRNAs in tumors
A novel LncRNA DMDRMR facilitated tumor growth and metastasis in clear cell renal cell carcinoma. Mechanistically, LncRNA DMDRMR bound IGF2BP3 to stabilize target genes, such as Cyclin-dependent kinases 4 (CDK4), Collagen type VI alpha 1 (COL6A1), Tumour-Derived Laminin α5 (LAMA5)and Fibronectin 1 (FN1), in an m6A-dependent manner [106]. LncRNA KCNMB2-AS1 served as a competing endogenous RNA to abundantly sponge miR-130b-5p and miR-4294, resulting in the upregulation of IGF2BP3 in cervical cancer. Moreover, LncRNA KCNMB2-AS1 and IGF2BP3 formed a positive regulatory circuit that enlarged the tumorigenic effect of KCNMB2-AS1 in cervical cancer [107].
Mutual regulatory effects between LncRNAs and hnRNPs
In addition to regulation of translation or RNA stability, the m6A modification can also lead to profound changes in the mRNA or LncRNA secondary structure, thus altering their interaction with proteins or RNAs, a process known as an “m6A switch”. As early as 2015, heterogeneous nuclear ribonucleoprotein C (hnRNPC) was identified as an “m6A switch” [108]. In detail, hnRNPC does not directly bind to the m6A modification sites but recognizes the RNA structure changed due to the presence of m6A modification [109]. The latest research shows that LncRNA DDX11-AS1 interacted with hnRNPC to promote Wnt/β-catenin and AKT pathways, thus promoting glioma cell proliferation and migration [110]. Subsequently, literature from Zhu et al. indicated that LncRNA cytoskeleton regulator (CYTOR) is highly expressed in Oral squamous cell carcinoma cells, and CYTOR can promote migration, invasion and epithelial mesenchymal transition in oral cancer cells. Mechanistically, nuclear-localized CYTOR interacts with hnRNPC, resulting in stabilization of Zinc finger E-box-binding homeobox (ZEB1) mRNAs by inhibiting the non-degradative ubiquitination of hnRNPC [111]. Another literature reported that m6A methylation was involved in the upregulation of LncRNA RP11. Mechanistically, a high level of m6A modification is able to increase binding between LncRNA RP11 and hnRNPA2B1, thus enhancing the stability of nascent LncRNA RP11 and elevating its expression in colorectal cancer cells [15]. LncRNA cancer susceptibility candidate 8 (CASC8) was highly overexpressed in esophageal squamous cell carcinoma tissues in an ALKBH5-mediated m6A modification dependent manner. Mechanistically, LncRNA CASC8 interacted with hnRNPL and inhibited its polyubiquitination and proteasomal degradation, thus stabilizing hnRNPL protein levels and activating the Bcl-2/caspase-3 pathway [53].
In summary, emerging studies have demonstrated that the interaction between LncRNAs and methyl-binding proteins of m6A modification is progressively critical to the occurrence and development of various tumors. We drew a schematic diagram, which takes Lnc-DLGAP1-AS2 [88], Lnc-CTHCC [98], Lnc-KCNMB2-AS1 [107], and Lnc-STEAP3-AS1 [93] as examples, to show the specific mechanisms of the interaction between LncRNAs and methyl-binding proteins in different tumors (Fig. 3). Furthermore, the interaction mechanisms of methyl-binding proteins of m6A modification and LncRNAs in diverse tumors are summarized and shown in Table 3.

The interaction between LncRNAs and methyl-binding proteins in cancers. a In non-small cell lung cancer (NSCC), m6A modification enhanced the DLGAP1-AS2 stabilization, thus promoting Aerobic glycolysis and cell proliferation. b In hepatocellular carcinoma (HCC), METTL3-mediated m6A modification increased the stability of Lnc-CTHCC, resulting in elevating tumour growth and cell metastasis. c In cervical cancer (CC), Lnc-KCNMB2-AS1 up-regulation IGF2BP3 by sponging miR-130b-5p and miR-4294, resulting in increasing proliferation and restraining apoptosis. d In colorectal cancer (CRC), Lnc-MIR100HG competitively with YTHDF2 to elevating the protein expression of STEAP3, thus increasing cell proliferation, metastasis
The feedback loop regulation between m 6 A modification and LncRNAs in tumors
In most studies, the attentions are on the roles of m6A modification regulating LncRNAs or LncRNAs mediating m6A modification during tumor progression, as summarized in the three sections above. Interestingly, few studies revealed that the feedback loop of m6A modification and LncRNAs was identified in specific cancers [112,113,114]. In detail, highly expression of LncRNA DLGAP1-AS1 was closely correlated to poorer clinical prognosis of breast cancer patients. Further experiment results showed that LncRNA DLGAP1-AS1 promotes ADR-resistance of breast cancer cells through a WTAP/DLGAP1-AS1/miR-299-3p/WTAP feedback loop. Mechanistically, m6A methyltransferase WTAP bound to the m6A modified sites on the LncRNA DLGAP1-AS1 and enhanced the stability of DLGAP1-AS1, which further sponged miR-299-3p and thus relieving the repression of WTAP mRNA [112]. Also in the breast cancer, a KIAA1429/m6A/LINC00667/miR-556-5p/KIAA1429 feedback loop was identified and involved in promoting the development of proliferation and migration of breast cancer cells. In detail, KIAA1429, one of the components of m6A methyltransferase complex, catalyzed the formation of m6A modification on LINC00667 and enhanced its stability. Further data revealed that highly expression of LINC00667 positively regulated KIAA1429 via sponging miR-556-5p, forming a KIAA1429/m6A/LINC00667/miR-556-5p/KIAA1429 feedback loop [113]. In addition, LncRNA MIR100HG has been identified as a positive regulator of EMT in colorectal cancer cells, in which hnRNPA2B1, a recognized m6A binding protein, was further identified as a binding partner of MIR100HG. Mechanistically, MIR100HG maintained mRNA stability of Transcription factor 7 like 2 (TCF7L2), by interacting with hnRNPA2B, which recognized the m6A site of TCF7L2 mRNA in the presence of MIR100HG that may contribute to cetuximab resistance [114].
We also drew a schematic diagram to show the specific mechanisms of the the feedback loop regulation between m6A modification and LncRNAs in breast cancer and colorectal cancer (Fig. 4).

The feedback loop regulation between m6A modification and LncRNAs in tumors. a In breast cancer (BC), LncRNA DLGAP1-AS1 promotes ADR-resistance of breast cancer cells through a WTAP/DLGAP1-AS1/miR-299-3p/WTAP feedback loop, resulting in promoting cell proliferation. b LINC00667 positively regulated KIAA1429 via sponging miR-556-5p, forming a KIAA1429/m6A/LINC00667/miR-556-5p/KIAA1429 feedback loop in breast cancer, thus increasing cell proliferation and metastasis. c In colorectal cancer (CRC), Lnc-MIR100HG bounded hnRNPA2B1 to maintaining the TCF7L2 mRNA stability via a MIR100HG/hnRNPA2B1/TCF7L2 regulatory axis, thereby increasing cell invasion, metastasis
In summary, m6A modification drives cancer cell progression, metastasis and invasion by affecting LncRNAs cleavage, stability and degradation. Meanwhile, LncRNAs have critical roles in regulating m6A modification through mediating its regulators during the occurrence and development of various tumors. A set of these regulatory proteins have been summarized in Fig. 5. Additionally, based on the interaction between m6A modification and LncRNAs, we summarized the change trends and regulation relationships between m6A regulatory proteins and LncRNAs in different cancers, as shown in Fig. 6.

The mutual regulation between m6A modification and LncRNAs during tumor occurrence and development

Mutual regulation between m6A modifications and LncRNAs. The red and green arrows respectively indicate the level rise and fall. The purple arrows point to the regulated object
Conclusion and prospect
In this review, we comprehensively summarize the mutual regulation mechanisms between m6A modification and LncRNAs in cancer progression, metastasis, invasion and drug resistance. Firstly, m6A modification on LncRNAs has been shown to control corresponding LncRNA levels and functions. In detail, m6A modification not only affects the cleavage, stability and degradation of corresponding LncRNAs but also influences the interactions between LncRNAs and miRNAs, mRNAs or proteins, thereby regulating a variety of tumorigenesis events, such as proliferation, metastasis, invasion and apoptosis. Interestingly, LncRNAs have the potential to manipulate m6A modification level and biological effects through changing methyltransferases, demethylases and methyl-binding proteins of m6A modification, thereby affecting the occurrence and development of tumors. To be specific, LncRNA does impact the level and biological function of m6A modification via mediating METTL3, METTL14, WTAP, FTO and ALKBH5, and these regulatory relationships are involved in the occurrence and development of various tumors. The interactions between m6A modification and LncRNAs provide a new direction for exploring the potential regulatory mechanisms during carcinogenesis and contributes to targeting therapy of various tumors. Nevertheless, the present understanding of the crosstalk mechanisms between m6A modifications and LncRNAs may be only the tip of iceberg due to the diversity of LncRNA types and regulatory proteins of m6A modification. Given the critical roles of m6A modification and LncRNAs in tumors, further study of m6A and LncRNAs and their mutual regulation relationship in cancers will be worth further exploring.
Availability of data and materials
Not applicable.
References
Sun L, Zhang H, Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. 2022;13(12):877–919.
Wiener D, Schwartz S. The epitranscriptome beyond m(6)A. Nat Rev Genet. 2021;22(2):119–31.
Chen M, Wong CM. The emerging roles of N6-methyladenosine (m6A) deregulation in liver carcinogenesis. Mol Cancer. 2020;19(1):44.
Zhao Y, Shi Y, Shen H, et al. m(6)A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13(1):35.
Ruszkowska A. METTL16, methyltransferase-like protein 16: current insights into structure and function. Int J Mol Sci. 2021;22(4):2176.
Oerum S, Meynier V, Catala M, et al. A comprehensive review of m6A/m6A mRNA methyltransferase structures. Nucleic Acids Res. 2021;49(13):7239–55.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21(1):14.
Huang H, Weng H, Chen J. The biogenesis and precise control of RNA m6A methylation. Trends Genet. 2020;36(1):44.
Deng LJ, Deng WQ, Fan SR, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer. 2022;21(1):52.
Adnane S, Marino A, Leucci E. LncRNAs in human cancers: signal from noise. Trends Cell Biol. 2022;32(7):565–73.
Fang S, Zhang L, Guo J, et al. NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018;46(D1):D308–14.
Liu SJ, Dang HX, Lim DA, et al. Long noncoding RNAs in cancer metastasis. Nat Rev Cancer. 2021;21(7):446–60.
Chen DH, Zhang JG, Wu CX, et al. Non-coding RNA m6A modification in cancer: mechanisms and therapeutic targets. Front Cell Dev Biol. 2021;9: 778582.
Ni W, Yao S, Zhou Y, et al. Long noncoding RNA GAS5 inhibits progression of colorectal cancer by interacting with and triggering YAP phosphorylation and degradation and is negatively regulated by the m(6)A reader YTHDF3. Mol Cancer. 2019;18(1):143.
Wu Y, Yang X, Chen Z, et al. m(6)A-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of Zeb1. Mol Cancer. 2019;18(1):87.
Zheng ZQ, Li ZX, Zhou GQ, et al. Long noncoding RNA FAM225A promotes nasopharyngeal carcinoma tumorigenesis and metastasis by acting as ceRNA to sponge miR-590-3p/miR-1275 and upregulate ITGB3. Cancer Res. 2019;79(18):4612–26.
Cui X, Wang Z, Li J, et al. Cross talk between RNA N6-methyladenosine methyltransferase-like 3 and miR-186 regulates hepatoblastoma progression through Wnt/beta-catenin signalling pathway. Cell Prolif. 2020;53(3): e12768.
Wang Y, Lu JH, Wu QN, et al. LncRNA LINRIS stabilizes IGF2BP2 and promotes the aerobic glycolysis in colorectal cancer. Mol Cancer. 2019;18(1):174.
Yan J, Huang X, Zhang X, et al. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem Biophys Res Commun. 2020;521(4):887–93.
Zhang H, Wang SQ, Wang L, et al. m6A methyltransferase METTL3-induced lncRNA SNHG17 promotes lung adenocarcinoma gefitinib resistance by epigenetically repressing LATS2 expression. Cell Death Dis. 2022;13(7):657.
Huang S, Jin M, Lan X, et al. LncRNA AC098934 promotes proliferation and invasion in lung adenocarcinoma cells by combining METTL3 and m6A modifications. J Cancer. 2022;13(8):2662–72.
Jin D, Guo J, Wu Y, et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2019;12(1):135.
Iaiza A, Tito C, Ianniello Z, et al. METTL3-dependent MALAT1 delocalization drives c-Myc induction in thymic epithelial tumours. Clin Epigenetics. 2021;13(1):173.
Qian X, Yang J, Qiu Q, et al. LCAT3, a novel m6A-regulated long non-coding RNA, plays an oncogenic role in lung cancer via binding with FUBP1 to activate c-MYC. J Hematol Oncol. 2021;14(1):112.
Xue L, Li J, Lin Y, et al. m(6)A transferase METTL3-induced lncRNA ABHD11-AS1 promotes the Warburg effect of non-small-cell lung cancer. J Cell Physiol. 2021;236(4):2649–58.
Hu Z, Zhu L, Zhang Y, et al. N6-methyladenosine-induced SVIL antisense RNA 1 restrains lung adenocarcinoma cell proliferation by destabilizing E2F1. Bioengineered. 2022;13(2):3093–107.
Zuo X, Chen Z, Gao W, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5.
Liu HT, Zou YX, Zhu WJ, et al. lncRNA THAP7-AS1, transcriptionally activated by SP1 and post-transcriptionally stabilized by METTL3-mediated m6A modification, exerts oncogenic properties by improving CUL4B entry into the nucleus. Cell Death Differ. 2022;29(3):627–41.
Chen B, Liu C, Long H, et al. N(6)-methyladenosine-induced long non-coding RNA PVT1 regulates the miR-27b-3p/BLM axis to promote prostate cancer progression. Int J Oncol. 2023;62(1):1.
Ban Y, Tan P, Cai J, et al. LNCAROD is stabilized by m6A methylation and promotes cancer progression via forming a ternary complex with HSPA1A and YBX1 in head and neck squamous cell carcinoma. Mol Oncol. 2020;14(6):1282–96.
Li J, Momen-Heravi F, Wu X, et al. Mechanism of METTL14 and m6A modification of lncRNA MALAT1 in the proliferation of oral squamous cell carcinoma cells. Oral Dis. 2022. https://doi.org/10.1111/odi.14220.
Mao J, Qiu H, Guo L. LncRNA HCG11 mediated by METTL14 inhibits the growth of lung adenocarcinoma via IGF2BP2/LATS1. Biochem Biophys Res Commun. 2021;580:74–80.
Yang X, Zhang S, He C, et al. METTL14 suppresses proliferation and metastasis of colorectal cancer by down-regulating oncogenic long non-coding RNA XIST. Mol Cancer. 2020;19(1):46.
Gong C, Fan Y, Liu J. METTL14 mediated m6A modification to LncRNA ZFAS1/ RAB22A: A novel therapeutic target for atherosclerosis. Int J Cardiol. 2021;328:177.
Hu N, Ji H. N6-methyladenosine (m6A)-mediated up-regulation of long noncoding RNA LINC01320 promotes the proliferation, migration, and invasion of gastric cancer via miR495-5p/RAB19 axis. Bioengineered. 2021;12(1):4081–91.
Liu T, Wang H, Fu Z, et al. Methyltransferase-like 14 suppresses growth and metastasis of renal cell carcinoma by decreasing long noncoding RNA NEAT1. Cancer Sci. 2022;113(2):446–58.
Shen D, Ding L, Lu Z, et al. METTL14-mediated Lnc-LSG1 m6A modification inhibits clear cell renal cell carcinoma metastasis via regulating ESRP2 ubiquitination. Mol Ther Nucleic Acids. 2022;27:547–61.
Peng L, Pan B, Zhang X, et al. Lipopolysaccharide facilitates immune escape of hepatocellular carcinoma cells via m6A modification of lncRNA MIR155HG to upregulate PD-L1 expression. Cell Biol Toxicol. 2022. https://doi.org/10.1007/s10565-022-09718-0.
Li ZX, Zheng ZQ, Yang PY, et al. WTAP-mediated m(6)A modification of lncRNA DIAPH1-AS1 enhances its stability to facilitate nasopharyngeal carcinoma growth and metastasis. Cell Death Differ. 2022;29(6):1137–51.
Ren Z, Hu Y, Sun J, et al. N(6)-methyladenosine methyltransferase WTAP-stabilized FOXD2-AS1 promotes the osteosarcoma progression through m(6)A/FOXM1 axis. Bioengineered. 2022;13(4):7963–73.
Zhu H, Chen K, Chen Y, et al. RNA-binding protein ZCCHC4 promotes human cancer chemoresistance by disrupting DNA-damage-induced apoptosis. Signal Transduct Target Ther. 2022;7(1):240.
Yang D, Chang S, Li F, et al. m(6)A transferase KIAA1429-stabilized LINC00958 accelerates gastric cancer aerobic glycolysis through targeting GLUT1. IUBMB Life. 2021;73(11):1325–33.
Dai Y, Liu Y, Li J, et al. METTL16 promotes hepatocellular carcinoma progression through downregulating RAB11B-AS1 in an m6A-dependent manner. Cell Mol Biol Lett. 2022;27(1):41.
Cui Y, Zhang C, Ma S, et al. RNA m6A demethylase FTO-mediated epigenetic up-regulation of LINC00022 promotes tumorigenesis in esophageal squamous cell carcinoma. J Exp Clin Cancer Res. 2021;40(1):294.
Han T, Xu D, Zhu J, et al. Identification of a robust signature for clinical outcomes and immunotherapy response in gastric cancer: based on N6-methyladenosine related long noncoding RNAs. Cancer Cell Int. 2021;21(1):432.
Wang T, Li W, Ye B, et al. FTO-stabilized lncRNA HOXC13-AS epigenetically upregulated FZD6 and activated Wnt/beta-catenin signaling to drive cervical cancer proliferation, invasion, and EMT. J BUON. 2021;26(4):1279–91.
Dong F, Qin X, Wang B, et al. ALKBH5 facilitates hypoxia-induced paraspeckle assembly and il8 secretion to generate an immunosuppressive tumor microenvironment. Cancer Res. 2021;81(23):5876–88.
Zhang J, Guo S, Piao HY, et al. ALKBH5 promotes invasion and metastasis of gastric cancer by decreasing methylation of the lncRNA NEAT1. J Physiol Biochem. 2019;75(3):379–89.
Yeermaike A, Gu P, Liu D, et al. LncRNA NEAT1 sponges miR-214 to promoted tumor growth in hepatocellular carcinoma. Mamm Genome. 2022;33(3):525–33.
Guo T, Liu DF, Peng SH, et al. ALKBH5 promotes colon cancer progression by decreasing methylation of the lncRNA NEAT1. Am J Transl Res. 2020;12(8):4542–9.
Peng K, Xia RP, Zhao F, et al. ALKBH5 promotes the progression of infantile hemangioma through regulating the NEAT1/miR-378b/FOSL1 axis. Mol Cell Biochem. 2022;477(5):1527–40.
Li Y, Yan B, Wang X, et al. ALKBH5-mediated m6A modification of lncRNA KCNQ1OT1 triggers the development of LSCC via upregulation of HOXA9. J Cell Mol Med. 2022;26(2):385–98.
Wu Q, Zhang H, Yang D, et al. The m6A-induced lncRNA CASC8 promotes proliferation and chemoresistance via upregulation of hnRNPL in esophageal squamous cell carcinoma. Int J Biol Sci. 2022;18(13):4824–36.
Yu H, Zhang Z. ALKBH5-mediated m6A demethylation of lncRNA RMRP plays an oncogenic role in lung adenocarcinoma. Mamm Genome. 2021;32(3):195–203.
Fang D, Ou X, Sun K, et al. m6A modification-mediated lncRNA TP53TG1 inhibits gastric cancer progression by regulating CIP2A stability. Cancer Sci. 2022. https://doi.org/10.1111/cas.15581.
Zhang H, Liu Y, Wang W, et al. ALKBH5-mediated m(6)A modification of lincRNA LINC02551 enhances the stability of DDX24 to promote hepatocellular carcinoma growth and metastasis. Cell Death Dis. 2022;13(11):926.
He Y, Yue H, Cheng Y, et al. ALKBH5-mediated m(6)A demethylation of KCNK15-AS1 inhibits pancreatic cancer progression via regulating KCNK15 and PTEN/AKT signaling. Cell Death Dis. 2021;12(12):1121.
Zhang Y, Liu X, Wang Y, et al. The m(6)A demethylase ALKBH5-mediated upregulation of DDIT4-AS1 maintains pancreatic cancer stemness and suppresses chemosensitivity by activating the mTOR pathway. Mol Cancer. 2022;21(1):174.
Chen S, Zhou L, Wang Y. ALKBH5-mediated m(6)A demethylation of lncRNA PVT1 plays an oncogenic role in osteosarcoma. Cancer Cell Int. 2020;20:34.
Shen W, Pu J, Zuo Z, et al. The RNA demethylase ALKBH5 promotes the progression and angiogenesis of lung cancer by regulating the stability of the LncRNA PVT1. Cancer Cell Int. 2022;22(1):353.
Zhu L, Zhu Y, Han S, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019;10(6):383.
Lai X, Wei J, Gu XZ, et al. Dysregulation of LINC00470 and METTL3 promotes chemoresistance and suppresses autophagy of chronic myelocytic leukaemia cells. J Cell Mol Med. 2021;25(9):4248–59.
Li SX, Yan W, Liu JP, et al. Long noncoding RNA SNHG4 remits lipopolysaccharide-engendered inflammatory lung damage by inhibiting METTL3 - Mediated m(6)A level of STAT2 mRNA. Mol Immunol. 2021;139:10–22.
Wang J, Zha J, Wang X. Knockdown of lncRNA NUTM2A-AS1 inhibits lung adenocarcinoma cell viability by regulating the miR-590-5p/METTL3 axis. Oncol Lett. 2021;22(5):798.
Wang G, Zhang Z, Xia C. Long non-coding RNA LINC00240 promotes gastric cancer progression via modulating miR-338-5p/METTL3 axis. Bioengineered. 2021;12(2):9678–91.
Sun T, Wu Z, Wang X, et al. LNC942 promoting METTL14-mediated m(6)A methylation in breast cancer cell proliferation and progression. Oncogene. 2020;39(31):5358–72.
Zhao C, Ling X, Xia Y, et al. LncRNA UCA1 promotes SOX12 expression in breast cancer by regulating m(6)A modification of miR-375 by METTL14 through DNA methylation. Cancer Gene Ther. 2022;29(7):1043–55.
Li J, Li Z, Bai X, et al. LncRNA UCA1 promotes the progression of AML by upregulating the expression of CXCR4 and CYP1B1 by affecting the stability of METTL14. J Oncol. 2022;2022:2756986.
Zhu ZJ, Pang Y, Jin G, et al. Hypoxia induces chemoresistance of esophageal cancer cells to cisplatin through regulating the lncRNA-EMS/miR-758-3p/WTAP axis. Aging (Albany NY). 2021;13(13):17155–76.
Weng L, Qiu K, Gao W, et al. LncRNA PCGEM1 accelerates non-small cell lung cancer progression via sponging miR-433-3p to upregulate WTAP. BMC Pulm Med. 2020;20(1):213.
Zhou X, Chang Y, Zhu L, et al. LINC00839/miR-144-3p/WTAP (WT1 Associated protein) axis is involved in regulating hepatocellular carcinoma progression. Bioengineered. 2021;12(2):10849–61.
Li JR, Liu L, Luo H, et al. Long noncoding RNA DUXAP8 promotes pancreatic carcinoma cell migration and invasion via pathway by miR-448/WTAP/Fak signaling axis. Pancreas. 2021;50(3):317–26.
Ge J, Liu M, Zhang Y, et al. SNHG10/miR-141-3p/WTAP axis promotes osteosarcoma proliferation and migration. J Biochem Mol Toxicol. 2022;36(6): e23031.
Li XD, Wang MJ, Zheng JL, et al. Long noncoding RNA just proximal to X-inactive specific transcript facilitates aerobic glycolysis and temozolomide chemoresistance by promoting stability of PDK1 mRNA in an m6A-dependent manner in glioblastoma multiforme cells. Cancer Sci. 2021;112(11):4543–52.
Chen X, Liu Y, Sun D, et al. Long noncoding RNA lnc-H2AFV-1 promotes cell growth by regulating aberrant m6A RNA modification in head and neck squamous cell carcinoma. Cancer Sci. 2022;113(6):2071–84.
Qin B, Dong M, Wang Z, et al. Long non-coding RNA CASC15 facilitates esophageal squamous cell carcinoma tumorigenesis via decreasing SIM2 stability via FTO-mediated demethylation. Oncol Rep. 2021;45(3):1059–71.
Chen F, Li M, Wang L. LncRNA CASC11 promotes hepatocellular carcinoma progression via upregulation of UBE2T in a m6A-dependent manner. Front Oncol. 2021;11: 772671.
Wang X, Zhang J, Wang Y. Long noncoding RNA GAS5-AS1 suppresses growth and metastasis of cervical cancer by increasing GAS5 stability. Am J Transl Res. 2019;11(8):4909–21.
Jiang X, Liu B, Nie Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74.
Lan Y, Liu B, Guo H. The role of M(6)A modification in the regulation of tumor-related lncRNAs. Mol Ther Nucleic Acids. 2021;24:768–79.
Yao ZT, Yang YM, Sun MM, et al. New insights into the interplay between long non-coding RNAs and RNA-binding proteins in cancer. Cancer Commun (Lond). 2022;42(2):117–40.
Widagdo J, Anggono V, Wong JJ. The multifaceted effects of YTHDC1-mediated nuclear m(6)A recognition. Trends Genet. 2022;38(4):325–32.
Wang X, Liu C, Zhang S, et al. N(6)-methyladenosine modification of MALAT1 promotes metastasis via reshaping nuclear speckles. Dev Cell. 2021;56(5):702–15.
Chen L, Zhang C, Ma W, et al. METTL3-mediated m6A modification stabilizes TERRA and maintains telomere stability. Nucleic Acids Res. 2022;50(20):11619–34.
Tang S, Liu Q, Xu M. LINC00857 promotes cell proliferation and migration in colorectal cancer by interacting with YTHDC1 and stabilizing SLC7A5. Oncol Lett. 2021;22(2):578.
Dai XY, Shi L, Li Z, et al. Main N6-Methyladenosine Readers: YTH Family Proteins in Cancers. Front Oncol. 2021;11: 635329.
Wang Y, Yu Z, Shi W, et al. HLA complex P5 upregulation is correlated with poor prognosis and tumor progression in esophageal squamous cell carcinoma. Bioengineered. 2022;13(4):9301–11.
Zhang Q, Zhang Y, Chen H, et al. METTL3-induced DLGAP1-AS2 promotes non-small cell lung cancer tumorigenesis through m(6)A/c-Myc-dependent aerobic glycolysis. Cell Cycle. 2022;21(24):2602–14.
Shen J, Feng XP, Hu RB, et al. N-methyladenosine reader YTHDF2-mediated long noncoding RNA FENDRR degradation promotes cell proliferation in endometrioid endometrial carcinoma. Lab Invest. 2021;101(6):775–84.
Zhang M, Wang J, Jin Y, et al. YTHDF2-mediated FGF14-AS2 decay promotes osteolytic metastasis of breast cancer by enhancing RUNX2 mRNA translation. Br J Cancer. 2022;127(12):2141–53.
Liu H, Xu Y, Yao B, et al. A novel N6-methyladenosine (m6A)-dependent fate decision for the lncRNA THOR. Cell Death Dis. 2020;11(8):613.
Yang H, Hu Y, Weng M, et al. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J Adv Res. 2022;37:91–106.
Zhou L, Jiang J, Huang Z, et al. Hypoxia-induced lncRNA STEAP3-AS1 activates Wnt/β-catenin signaling to promote colorectal cancer progression by preventing m6A-mediated degradation of STEAP3 mRNA. Mol Cancer. 2022;21(1):1–22.
Jin D, Guo J, Wu Y, et al. m(6)A mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2021;14(1):32.
Hu Y, Tang J, Xu F, et al. A reciprocal feedback between N6-methyladenosine reader YTHDF3 and lncRNA DICER1-AS1 promotes glycolysis of pancreatic cancer through inhibiting maturation of miR-5586-5p. J Exp Clin Cancer Res. 2022;41(1):69.
Ma F, Liu X, Zhou S, et al. Long non-coding RNA FGF13-AS1 inhibits glycolysis and stemness properties of breast cancer cells through FGF13-AS1/IGF2BPs/Myc feedback loop. Cancer Lett. 2019;450:63–75.
Gao Y, Jiang M, Guo F, et al. A novel lncRNA MTAR1 promotes cancer development through IGF2BPs mediated post-transcriptional regulation of c-MYC. Oncogene. 2022;41(42):4736–53.
Xia A, Yuan W, Wang Q, et al. The cancer-testis lncRNA lnc-CTHCC promotes hepatocellular carcinogenesis by binding hnRNP K and activating YAP1 transcription. Nat Cancer. 2022;3(2):203–18.
Zheng D, Yu L, Wei Z, et al. N6-methyladenosine-related lncRNAs are potential prognostic biomarkers and correlated with tumor immune microenvironment in osteosarcoma. Front Genet. 2021;12: 805607.
Zhu P, He F, Hou Y, et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene. 2021;40(9):1609–27.
Zhai S, Xu Z, Xie J, et al. Epigenetic silencing of LncRNA LINC00261 promotes c-myc-mediated aerobic glycolysis by regulating miR-222-3p/HIPK2/ERK axis and sequestering IGF2BP1. Oncogene. 2021;40(2):277–91.
Hu X, Peng WX, Zhou H, et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2020;27(6):1782–94.
Han J, Yu X, Wang S, et al. IGF2BP2 induces U251 glioblastoma cell chemoresistance by inhibiting FOXO1-mediated PID1 expression through stabilizing lncRNA DANCR. Front Cell Dev Biol. 2021;9: 659228.
Liu Y, Shi M, He X, et al. LncRNA-PACERR induces pro-tumour macrophages via interacting with miR-671-3p and m6A-reader IGF2BP2 in pancreatic ductal adenocarcinoma. J Hematol Oncol. 2022;15(1):52.
Ye M, Dong S, Hou H, et al. Oncogenic role of long noncoding RNAMALAT1 in thyroid cancer progression through regulation of the miR-204/IGF2BP2/m6A-MYC signaling. Mol Ther Nucleic Acids. 2021;23:1–12.
Gu Y, Niu S, Wang Y, et al. DMDRMR-mediated regulation of m 6 A-modified CDK4 by m6A reader IGF2BP3 drives ccRCC progression. Cancer Res (Chicago, Ill). 2021;81(4):923.
Zhang Y, Wang D, Wu D, et al. Long noncoding RNA KCNMB2-AS1 stabilized by N(6)-methyladenosine modification promotes cervical cancer growth through acting as a competing endogenous RNA. Cell Transplant. 2020;29:2138957086.
Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;7540(518):560–4.
Feng J, Zhou J, Lin Y, et al. hnRNP A1 in RNA metabolism regulation and as a potential therapeutic target. Front Pharmacol. 2022;13: 986409.
Xiang Z, Lv Q, Zhang Y, et al. Long non-coding RNA DDX11-AS1 promotes the proliferation and migration of glioma cells by combining with HNRNPC. Mol Ther Nucleic Acids. 2022;28:601–12.
Zhu W, Wang J, Liu X, et al. lncRNA CYTOR promotes aberrant glycolysis and mitochondrial respiration via HNRNPC-mediated ZEB1 stabilization in oral squamous cell carcinoma. Cell Death Dis. 2022;13(8):703.
Huang T, Cao L, Feng N, et al. N(6)-methyladenosine (m(6)A)-mediated lncRNA DLGAP1-AS1enhances breast canceradriamycin resistance through miR-299-3p/WTAP feedback loop. Bioengineered. 2021;12(2):10935–44.
Ren S, Zhang Y, Yang X, et al. N6-methyladenine-induced LINC00667 promoted breast cancer progression through m6A/KIAA1429 positive feedback loop. Bioengineered. 2022;13(5):13462–73.
Liu H, Li D, Sun L, et al. Interaction of lncRNA MIR100HG with hnRNPA2B1 facilitates m6A-dependent stabilization of TCF7L2 mRNA and colorectal cancer progression. Mol Cancer. 2022;21(1):74.
Acknowledgements
Not applicable.
Funding
This research was supported by grants from the National Natural Science Foundation of China (NO. 82060585; NO. 82260631) to Shiyan Gu and Zuoshun HE.
Author information
Authors and Affiliations
Contributions
NZ and YS contributed to writing the article, checking the content and sorting out the literature; ZM contributed to reference collection, induction and verification. ZH and SG revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, N., Sun, Y., Mei, Z. et al. Novel insights into mutual regulation between N6-methyladenosine modification and LncRNAs in tumors. Cancer Cell Int 23, 127 (2023). https://doi.org/10.1186/s12935-023-02955-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02955-1