
Abstract
Neurofibromatosis type 2 (NF2) is a genetic condition marked by the development of multiple benign tumors in the nervous system. The most common tumors associated with NF2 are bilateral vestibular schwannoma, meningioma, and ependymoma. The clinical manifestations of NF2 depend on the site of involvement. Vestibular schwannoma can present with hearing loss, dizziness, and tinnitus, while spinal tumor leads to debilitating pain, muscle weakness, or paresthesias. Clinical diagnosis of NF2 is based on the Manchester criteria, which have been updated in the last decade. NF2 is caused by loss-of-function mutations in the NF2 gene on chromosome 22, leading the merlin protein to malfunction. Over half of NF2 patients have de novo mutations, and half of this group are mosaic. NF2 can be managed by surgery, stereotactic radiosurgery, monoclonal antibody bevacizumab, and close observation. However, the nature of multiple tumors and the necessity of multiple surgeries over the lifetime, inoperable tumors like meningiomatosis with infiltration of the sinus or in the area of the lower cranial nerves, the complications caused by the operation, the malignancies induced by radiotherapy, and inefficiency of cytotoxic chemotherapy due to the benign nature of NF-related tumors have led a march toward exploring targeted therapies. Recent advances in genetics and molecular biology have allowed identifying and targeting of underlying pathways in the pathogenesis of NF2. In this review, we explain the clinicopathological characteristics of NF2, its genetic and molecular background, and the current knowledge and challenges of implementing genetics to develop efficient therapies.
Similar content being viewed by others

Introduction
Neurofibromatosis (NF) is a multiple tumor predisposing syndrome classified into: type 1 (NF1), type2 (NF2), and schwannomatosis [1]. NF1 is the most common type caused by mutations in the tumor suppressor NF1 gene (OMIM: 613113) on chromosome 17. In comparison, schwannomatosis is caused by mutations of SMARCB1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1) (OMIM: 601607) and LZTR1 (Leucine zipper-like transcription regulator 1) (OMIM: 600574) genes on chromosome 22, encoding tumor suppressor proteins [1, 2]. The clinical description of NF2 was provided by Scottish surgeon JH Wishart in 1822 by dissecting a young male with multiple brain tumors originating from the skull [3]. NF2 is a mixed neuro-cutaneous genetic disease predisposing to the development of multiple benign tumors throughout the lifetime. The development of bilateral vestibular schwannoma (VS) (aka acoustic neuroma) is a characteristic of NF2. Hearing loss, tinnitus, and balance dysfunction are common symptoms of VS. Other common tumor characteristics include schwannomas of the cranial, spinal, and peripheral nerves as well as intracranial and intraspinal meningiomas (Fig. 1) [4].

Schematic illustration and magnetic resonance imaging (MRI) of tumors associate with neurofibromatosis type 2 (NF2). A Different clinical symptoms associated with NF2. B A schematic depiction of vestibular schwannoma arising from eighth cranial nerve. C MRI of bilateral vestibular schwannoma as a hallmark feature of NF2 patients. The figure is redrawn from refs [2, 13].
The incidence of NF2 is around 1 in 25,000 [5, 6]. The prevalence of diagnosed patients is 1 in 100,000 dues to different reasons, such as lack of medical resources, insidious onset of clinical manifestations, and high rate of novel and somatic mutations [5,6,7]. Despite benign behavior, NF2-associated tumors result in considerable morbidity and mortality rates [2]. The early signs of NF2 usually manifest in the late teens or early twenties. The survival period after diagnosis is about 15 years, and the average age at death is between 36 and 39 years, with a 10-year survival rate of 67% [5, 8]. These rates illustrate the importance of NF2 management, despite its benign features.
Currently, there is no established approach to prevent or cure NF2 [1]. Cytotoxic chemotherapeutics are generally ineffective due to the benign biology of NF2-associated tumors [9]. NF2 can be managed by surgery, stereotactic radiosurgery, monoclonal antibody bevacizumab, and close observation. Even though these approaches can provide good local control, their shortcomings impede their general application. Despite all care, surgery can be associated with complications due to direct nerve manipulation or vascular events during the intervention. On the other hand, the concern due to radiation-induced secondary malignancy can enormously affect the patients’ quality of life. Moreover, the application of surgery and radiotherapy might be limited for tumors in the context of NF2 because the tumors are typically multiple in these patients [10, 11]. These concerns and shortcomings have led scholars to approach systemic therapies targeting NF2-associated tumors.
The advent of molecular biology to scheme the pathogenesis of NF2 can guide to address of new therapeutic potentials for the effective prevention and treatment of NF2-related tumors. NF2 is caused by loss of function mutations, deletions, or epigenetic modifications of the NF2 gene (OMIM: 607379) [12]. The NF2 gene encodes a 70-kDa protein called merlin, mainly acting as a tumor suppressor and participating in diverse cell signaling pathways [10]. Tumorigenesis occurs upon merlin loss of function by modifying downstream signaling pathways. Being such, the collaborative proteins of merlin can serve as novel targets for NF2 treatment [13,14,15].
Genotype–phenotype correlation and the role of NF2 genetic investigation in differentiating NF-related disorders highlight the importance of conducting genetic testing to detect at-risk patients, prognostication, and treatment [7, 16]. Also, it helps to find new treatments by reversing pathologic genetic/epigenetic modifications to replace normal merlin function, decrease tumor burden, preserve hearing, and improve survival of patients with NF2.
The present review focuses on the clinical features, diagnostic criteria, and the genetic and epigenetic background of NF2. It then highlights the current position and future perspectives of NF2 treatment.
Genetic overview
Type 2 neurofibromatosis occurs due to the alterations of the NF2 gene, located on chromosome 22q12.2 [17]. The defective gene is dominantly inherited and has nearly 100% penetrance by 60 years of age [16, 18]. The inactivation of the two alleles of the NF2 gene is consistent with Knudson’s two-hit hypothesis. The NF2 gene has 17 exons and encodes a 595-amino acid protein. The ultimate result of the translation of this gene is a protein called merlin, also known as neurofibromin 2 or schwannomin, which is a cell membrane-related protein with tumor suppressor activities [19].
NF2 gene variants
Nearly half of the patients with NF2 have de novo mutations without a family history, and about 60% of patients with novel mutations are mosaic [20]. This event is the result of post-zygotic interruptions during embryo development. Therefore, only a small population of cells will obtain the defective NF2 gene [5]. This incident might challenge the detection of the mutations in peripheral blood analysis. In this condition, the molecular examination of tumor samples has more sensitivity [21]. Mosaic NF2 patients manifest milder to no symptoms depending on the nature of the mutations (Table 1) [22]. Typically, mosaic patients present with less severe hearing loss, fewer bilateral VS, CNS tumors, and the development of ocular signs at older ages. Patients with sporadic mosaic NF2 can exhibit clinical symptoms 7–8 years later than sporadic non-mosaic cases [23]. The risk of defective gene inheritance in mosaic patients is less than 50 percent; however, if inherited, offspring would develop more severe phenotypes in contrast with their parents because of a larger number of affected cells as a result of the transmission of the mutated gene to the next generation [24].
The type and severity of NF2 manifestations depend on the type of gene mutation. The variability within families is usually fewer than variabilities between families, implying a considerable influence of the underlying genotype. Evidence favors a substantial link between genotype and phenotype for NF2-related disorders [16, 25]. The types of pathogenic mutations can predict the number of intracranial meningiomas, spinal tumors, and tumors of peripheral nerves [7]. The UK NF2 Genetic Severity Score was developed—per clinical manifestations and genotypes—to categorize the patients based on severity into severe, moderate, and mild. [16] These groups are different in age at diagnosis and show different manifestations. The UK NF2 Genetic Severity Score has recently been validated in the Spanish NF2 cohort (Table 1) [26].
Genotype–phenotype correlation in NF2
Generally, NF2 due to truncating mutations (nonsense or frameshift) is more severe and appears at younger ages [27]. Evidence denotes that patients with truncating mutations are more likely to develop symptoms earlier (before 20 years), have a greater risk of developing at least two CNS tumors in addition to VS (before 30 years), and have a shorter average life expectancy [16]. Missense mutations and large deletions usually give rise to milder phenotypes. However, the underlying mechanisms linking large deletions and milder NF2 phenotypes are still unknown [27]. Unlike truncating and missense mutations, splice site mutations are associated with more diverse phenotypes (Table 1) [28].
The involved site also matters. Mutations in the amino-terminal domain of NF2 proteins are associated with early tumor onset with more severe disease progression [21]. It has been shown that exons 2 and 3 are necessary for merlin’s self-association, which is required for its tumor suppressor activity. In mouse models, the knockout of this part of the gene gave rise to higher tumorigenesis in Schwan cells [29, 30]. Patients with truncating variants of the 3’ region of exons 2 through 13 have more severe presentations with poor survival outcomes than the same variants involving exons 1, 14, and 15. Moreover, frameshift variants close to the NF2 translation initiation codon improve life expectancy [16, 28]. Re‐evaluation of missense variant classifications indicated that most NF2‐associated variants are located in exon 7, and minor variants are toward the C‐terminus of the NF2 protein [31].
Mutations at the amino-terminal domain of the NF2 are associated with more meningiomas, [32] especially in the intracranial space [33]. The Wishart phenotype, also known as the severe form, is associated with truncating mutations and alterations at the amino-terminal domain of merlin [34, 35]. In contrast, the Gardner phenotype, also known as the adult form of NF2, is associated with missense or splice site mutations, especially at the carboxy-terminal. Generally, the Gardner phenotype has a better prognosis with a lower risk of meningioma [32].
Epigenetic overview
Epigenetics, defined as heritable alterations in gene expression that do not result in permanent changes in DNA sequence, plays a crucial role in maintaining cell identity and phenotypic characteristics [36] Through epigenetics, cells can undergo differentiation and development into various cell lines. The main epigenetic mechanisms include DNA modifications, chromatin modifications, and non-coding RNA interactions. DNA modification, especially methylation, occurs at CpG islands of gene promoters [37].
Research on monozygotic twins with NF2 introduced epigenetic changes as one of the main factors in phenotypic heterogeneity [38]. Further research indicated the epigenetic modifications at the 5’ flanking region of the NF2 promoter [39]. Hyper-methylation in CpG islands leads to gene silencing by decreasing the mRNA expression of the NF2 gene [40, 41]. Later, several studies reported a low level of methylation in the promoter region of the NF2 gene in patients with sporadic VS. Therefore, NF2 methylation may not serve as a primary reason for developing VS. [42, 43] A methylation-specific PCR on schwannomas has shown aberrant methylation in tumor-related genes including THBS1, TP73, MGMT, and TIMP3. [41, 44] Lassaletta et al. indicated aberrant methylation status in twelve tumor-related genes of patients with VS, including RASSF1A, VHL, PTEN, TP16, CASP8, TIMP3, MGMT, DAPK, THBS1, HMLH1, TP73, and GSTP1. Among these genes, the RASSF1A methylation is inversely correlated with the clinical growth index, and methylation in CASP8 is associated with the patient's age and tumor size. The methylation of TP73 is associated with hearing loss. [45] TP73 is a tumor suppressor gene, mediating apoptosis in neural cells. However, the mechanism underlying its contribution to VS formation is yet to be determined. [46] Previous studies demonstrated the link between methylation of homeobox genes (HOX) and several malignancies, including leukemia and breast cancer. [47, 48] Genome-wide methylation analysis in VS demonstrated global hypomethylation at the HOX gene cluster [49]. Other epigenetic modifications pertaining to VS formation are post-transcriptional changes of NF2, alterations in lysine acetylation, and dysregulation of miRNA expression [50,51,52,53]. In meningiomas, epigenetics has been applied to categorize the subtypes. In fact, the DNA methylation-based classification and grading system of meningiomas has improved the prediction of tumor prognosis and recurrence by selecting clinically homogenous groups [54]. In the case of VS, however, this application is still in its infancy and requires further investigations. More research is required to delineate the genetic and epigenetic changes explaining the molecular and phenotypic differences between individuals with NF2.
Clinical features
Tumors in the context of NF2 can involve central and peripheral nervous systems (Fig. 1a) [2]. The clinical manifestations of NF2 are in a wide range, from no symptoms to life-threatening symptoms, depending on the involved nerves [2]. The bilateral VS is the hallmark feature of NF2 that originate from myelin-forming Schwann cells in the vestibulocochlear nerve (Fig. 1b, c) [13]. VS is the main reason for hearing impairment, balance dysfunction, and tinnitus in NF2 patients. VS growth can compress the adjacent facial nerve, leading to facial weakness, numbness, or paresis. In advanced cases, life-threatening intracranial difficulties (e.g., hydrocephalus) might happen upon brain stem or cerebellar compression.
At least two-thirds of patients with NF2 might develop spinal tumors presented as debilitating pain, muscle weakness, or paresthesias [55, 56]. The most common NF2-related spinal tumors are schwannomas. These arise from the dorsal root and can take on a characteristic dumbbell shape. Most persons with spinal cord involvement have multiple tumors [57].
Approximately half of NF2 cases have meningiomas that usually present as multiple meningiomas with considerable morbidity due to seizures, paralysis, and headaches [2]. The incidence of meningiomas increases with age, and lifetime risk may approach 80% [32]. Most cases are intracranial, although intradural and extramedullary spinal meningiomas are also reported. Orbital meningiomas can lead to visual loss by compressing the optic nerve. Those at the skull base may cause cranial neuropathy, brain stem compression, and hydrocephalus [57]. As such, the site of involvement determines the severity and type of symptoms of NF2-related meningioma.
Individuals with NF2 may develop visual impairment due to cataract, optic nerve meningiomas, retinal hamartomas, and the epiretinal membrane [5]. Cataracts are reported in 60–80 percent of patients and typically are manifested as posterior subcapsular lenticular opacities. Lens opacities may appear prior to the onset of symptoms of VS and can be seen in children [58]. It has been demonstrated that ophthalmic manifestations can get worse with an increased genetic severity score. Painter et al. demonstrated that the prevalence of cataracts, optic atrophy, epiretinal membranes, and combined hamartomas significantly increased with genetic severity score. The authors also found that greater genetic severity is associated with greater visual morbidity at an earlier age [35]. These findings reflect the positive correlation between genetic mutations and clinical manifestations in NF2.
The cutaneous manifestations of NF2 are diverse, including plaque-like lesions (usually pigmented with hair overgrowth), subcutaneous nodules (often palpable along the peripheral nerves), and intracutaneous tumors. The great majority of these tumors are schwannomas [5]. Cutaneous involvement of NF2 may precede neurological and ophthalmic symptoms by several years, thereby can contribute to the early diagnosis [59].
Neurogenic manifestations of NF2 are various [5, 60]. A recognized feature of NF2 is mononeuropathy, particularly in childhood [61], which usually involves the facial nerve and can precede the development of other NF2 manifestations. It also can present as foot or hand drop. A progressive polyneuropathy of adulthood not directly related to tumor masses is also recognized [62].
VS presents in 90% of patients with NF2, usually manifested as a progressive hearing impairment [2, 25]. The definite pathophysiology of hearing loss in patients with VS is yet to be determined [13]. One possible mechanism is through mechanical pressure of the growing tumor inside the bony space of the auditory canal. Six studies found an association between VS tumor size and the severity of hearing impairment [63].However, this hypothesis is challenged by the finding that there is no consistent correlation between tumor size and the severity of hearing loss [64]. Furthermore, Sakamoto et al. found no significant correlation between hearing loss speed and tumor size [65]. In support, Caye-Thomasen et al. found that gradual or sudden hearing loss may occur without a change in tumor size or configuration [66]. Hence, the association between VS tumor size and the severity of hearing impairment remains an open question. Another hypothesis pertains to cochlear ischemia due to the compressive effect of the growing tumor on the supplying vessels [67]. Despite clinical evidence for this hypothesis [68, 69], it may not be generalized to all patients because the histological vascular changes were detected in a subset of patients. An emerging hypothesis has considered tumor-secreting molecules, including ototoxic and neurotoxic agents, as the leading cause of cochlear damage [70]. The extracellular vesicles secreted by tumor cells (so-called exosomes) can mediate cochlear damage [71]. Whether these mechanisms are in-whole or in-part justify VS-associated hearing loss is still unclear and requires further dedicated studies.
Diagnosis
Over the past four decades, the diagnostic criteria of NF2 have been developed. Table 2 represents the previous and current diagnostic criteria for NF2. In 1987, the National Institutes of Health (NIH) published the first set of criteria [72]. Later, in 1992, the diagnostic criteria of NF2 were updated according to the Manchester consensus [73]. The NIH and Manchester criteria are merely based on the clinical features and significant family history. The diagnosis of NF2 is often postponed due to a wide clinical heterogeneity, particularly in cases without a significant family history or those with distinct manifestations prior to the VS development [74]. The advent of sequencing technologies has provided many opportunities to diagnose NF2 patients, especially for differential diagnosis and the mosaic form of NF2. In the 2016 revision of the Manchester criteria, the following updates were considered: patients with unilateral VS and nondermal schwannomas require testing for LZTR1 to rule out the schwannomatosis. In addition, the age limit of 70 years was considered for the development of VS, given the observation that up to 50% of individuals aged 70 and older may have bilateral VS without underlying mosaic or constitutional NF2 mutation [33]. In 2019, the consensus updated the diagnostic criteria as follows: “glioma” and “neurofibroma” were removed from the NF-associated lesions, and “ependymoma” was added to the list [75]. In addition, the siblings were not considered as the “first-degree relative” in the criteria because of the zero positive predictive value [75]. However, these criteria were misleading for patients with multiple schwannomas. In the recent update (2022 consensus), the role of genetic testing was highlighted to discriminate NF2 from schwannomatosis. In this criteria, the “NF2” term was updated to “NF2-related schwannomatosis”, and the previous “schwannomatosis” became updated per the relevant pathogenic variant: SMARCB1-related schwannomatosis, LZTR1-related schwannomatosis, 22q-related schwannomatosis, schwannomatosis-NOS (not otherwise specified), or schwannomatosis NEC (not elsewhere classified) [76]. Overall, during the past four decades, the diagnostic criteria of NF2 have evolved from purely clinical to clinical-genetic criteria.
The current diagnostic criteria for NF2 are based on clinical examinations and genetic tests. Genetic testing is suggested in all patients with suspected schwannomatosis predisposition syndromes [77]. The clinical assessments include family history, physical examinations such as cutaneous, ear, and eye examinations, and a contrast-enhanced MRI of the brain and whole spine [5]. Next-generation sequencing (NGS) offers an endorsed method to detect the NF2 gene variation, with a detection rate of up to 90 percent for patients within familial NF2. However, the sensitivity of NGS for sporadic NF2 decreases to 25–60 percent because of somatic mosaicism [78]. NGS is more sensitive than Sanger sequencing at detecting genetic variants present at a low-level allele fraction (< 50%) [79]. Along with NGS, multiplex ligation-dependent probes and high-resolution karyotyping complete the genetic analysis of patients [76]. In the first step, investigating NF2, SMARCB1, and LZTR1 in blood or saliva samples is recommended to detect the genetic background of patients with NF2 and the differential diagnosis. Thereafter, analysis of two independent tumor samples helps to detect second hit mutations. Finally, tumor tissue analysis for methylation patterns is suggested. New emerging evidence indicates the role of second hit mutation, epigenetic factor, and modifier genes in phenotypic variability within NF2 families [25, 76, 78].
Prognostic information, earlier intervention, and efficacious therapies are the robust benefits of early genetic testing in patients with suspected NF2-related diseases [2]. Moreover, understanding the genomic and molecular pathogenesis of NF2 variants will provide insights into better knowledge about tumorigenesis-related manifestations, such as meningioma, spinal schwannomas, ependymomas, and dermal schwannomas [25]. In addition, it can improve diagnostic precision to better discriminate NF2-related schwannomatosis from its differential diagnosis [76]. Finally, pre-and post-test genetic counseling allows patients and their relatives to make informed decisions and improves management.
Treatment
There is currently no cure for NF2, so management focuses on close observation and treating problems when they arise. The National Health Service recommends active monitoring for asymptomatic cases with annual brain MRI to find or follow the brain tumors, annual eye tests, and annual hearing tests [80].The significant impacts of NF2 on the patients’ quality of life and the severity of symptoms urge the need to explore effective therapies. Therefore, treatment recommendations for NF2-related tumors aim to preserve of the physiologic function and quality of life [5]. Hence, incidental identification of a tumor is not an indication for treatment per se, and the potential benefits must be compared against the risks of active intervention. Treatment is usually selected when there is a risk of brainstem compression, hearing impairment, or facial nerve dysfunction. The management of patients with NF2 is typically determined in a multidisciplinary setting, consisting of neurotologists, audiologists, neurologists, neurosurgeons, oncologists, ophthalmologists, and geneticists [2].
VS are usually managed surgically if treatment is indicated. Besides, first-line bevacizumab (a humanized anti-VEGF monoclonal antibody) therapy has indicated promising results in rapidly growing tumors. Currently, however, there is no FDA-approved targeted therapy for NF2 [13]. Bevacizumab has shown promise in the treatment of progressive VS in NF2 patients [27]. A meta-analysis of 161 patients with NF2-related VS (total of 196 VS tumors) reported that bevacizumab resulted in partial regression, stable disease, and progression in 41% (95% CI 31–51%), 47%, (95% CI 39–55%), and 7% (95% CI 1–15%), respectively. Hearing improvement was reported in 20% (95% CI 9–33%), stability in 69% (95% CI 51–85%), and additional loss in 6% (95% CI 1–15%) [81].
Similarly, the mainstay treatment of progressive meningiomas threatening functional loss is surgery. Regarding inoperable cases, radiotherapy is also used. Most meningiomas grow to a specific size and stop; therefore, they do not require special treatment [82]. Bevacizumab is indicated to repress tumor growth in recurrent WHO grades II/III meningiomas [83].
Usually, there is no need for intervention in NF2-related ependymomas due to their slow growth rate. Occasionally, where intervention is needed, surgery is recommended [84]. Emerging evidence suggests that bevacizumab can improve the symptoms of NF2-associated ependymomas [85, 86].
Surgical interventions can also be applied to improve hearing impairments due to NF2 tumors. To this end, cochlear or auditory brainstem implants are extensively applied worldwide [87, 88].
The multifocal nature of NF2-related tumors, their proximity to the vital structures (e.g., brain stem and internal carotid artery), or neural involvement (e.g., facial and auditory nerves) can limit surgical interventions. In patients with NF2, radiotherapy can increase the risk of developing additional benign tumors in the irradiated field and malignant transformation of existing benign tumors [89]. Therefore, it is only applied in inoperable cases. In addition, there is still a risk of tumor recurrence after surgery or radiotherapy, with a long-term tumor relapse rate of 40% [1, 90,91,92,93]. Unfortunately, cytotoxic chemotherapies have limited efficacy in these patients due to the benign nature of NF-related tumors [94]. Since merlin plays a role in various cellular pathways involved in cell growth, proliferation, and also cell–cell interactions, identifying signaling pathways and major mediators of NF2 pathogenesis can provide new therapeutic perspectives in the treatment of NF2-related tumors.
To summarize, the treatment of NF2 must start with one critical question: how much do the treatment benefits outweigh its risks? This concern originates from two points: (a) long-term quiescence of NF2-related tumors [95] and (b) significant adverse effects of treatments (neural damages in surgical interventions and risk of secondary malignancy due to radiotherapy). The multifocality of NF2-related tumors and their proximity to critical structures can further limit the administration of surgery and radiotherapy in these patients. As such, systemic treatments can overpass these limitations. However, the available systemic therapies are limited to a handful of choices (e.g., VEGF or mTOR inhibitors) with limited efficacy. As noted, NF2 is a genetic disease. Hence, exploring its genetic and epigenetic backgrounds and the involved signaling pathways can help to find better treatments. The following sections discuss the role, structure, and function of merlin in the pathology and treatment of NF2.
Molecular biology of merlin
Merlin is a member of the ERM protein family, and its name stands for ezrin-radixin-moesin-like protein. The ERM family members are highly conserved during the evolution, reflecting their crucial roles in human cells [96]. Generally, ERM family members provide cross-links between the plasma membrane and actin-based cytoskeleton, essential for plasma membrane maintenance and function. Nearly all ERM proteins have high similarities in their structure; however, slight differences result in different cell function [96].
Merlin binds to the actin cytoskeleton and is involved in the stabilization of the membrane cytoskeletal interface by interacting with PI3K (Phosphoinositide-3 kinase)/AKT (Ak strain transforming), Raf (Rapidly accelerated fibrosarcoma)/MEK (Mitogen-activated protein kinase)/ERK (Extracellular-signal-regulated kinase), Wnt/β-catenin, RTKs (Receptor tyrosine kinases), mTOR (Mechanistic target of rapamycin), and Hippo signaling pathway [97]. The interaction of merlin with the aforementioned biomarkers has another face. It has been demonstrated that merlin (as a tumor suppressor) has inhibitory effects on PI3K [98], Raf/ERK [99], Wnt/β-catenin [100], RTKs [1], and mTOR [101]. Hence, in patients with NF2, the inhibitory effect of merlin on these pathways is removed, and these pro-tumorigenic pathways become activated. This interaction can serve as an opportunity to design targeted therapies inhibiting the activated pathways in patients with NF2 (discussed in "Future therapeutic perspective" Section).
Merlin structure
The NF2 gene expresses ten different isoforms resulting from alternative splicing, among which isoforms I and II are the major ones and both have tumor suppressive function [102, 103]. Each isoform has a specific expression pattern throughout the body. Merlin is highly expressed during the embryo’s development, and specific isoforms (7 and 9) are absent in adult tissues. Generally, merlin can be primarily detected in neurons, Schwann cells, meningeal cells, and lens cells [104].
Merlin is a multidomain protein consisting of three parts (Fig. 2). The first part is at the N-terminus, which is highly conserved in ERM family members and is called FERM (band 4.1, ezrin, radixin, moesin) or N-terminal ERM association domain. FERM binds to the plasma membrane or plasma membrane proteins, including adherens junctions and cell surface receptors like integrins, RTKs, CD44, CD43, ICAMs, and scaffolding/effector proteins [105, 106]. The FERM itself has three subdomains (F1, F2, and F3). These subdomains form a tri-lobed, cloverleaf conformation. The stabilization of this structure lies within the biochemical properties of regions between each subdomain residing on this domain. The second part of merlin is the α-helical domain, consisting of three α-helices (α1H, α2H, and α3H) and a hinge between the latter two. The α-helical domains α2H and α3H form a coiled-coil conformation in the monomer state [106]. The third part of merlin is a short, mainly helical C-terminus domain (CTD) or C-terminal ERM-association domain, which has specific biological functions and structure in each ERM family member. The CTD part regulates protein–protein or membrane-protein interactions and enables interactions with the FERM domain. The CTD contains specific residues that are targets for critical post translational modifications (PTM) [107]. Merlin (and other ERM family members) links the cell membrane and the basal actin cytoskeleton and mediates membrane remodeling, membrane structure maintenance, and vesicle trafficking [2]. Merlin may produce distinct cellular effects depending on its conformation and modifications. The significant difference is that ERM proteins can bind to the actin cytoskeleton via their actin-binding domain located on their C-terminus; however, in merlin, the actin-binding domain is located in the FERM domain (N-terminus) [108, 109]. In addition, merlin is the only member of the ERM family manifesting tumor suppressor activity [110]. Besides, merlin may have intranuclear effects. Its NLS (nuclear localization sequence) addresses its role in signaling pathways regulating gene expression (Fig. 3) [111].

A schematic depiction of protein domain structure and states merlin. A Merlin is a multidomain protein and consists of three parts. The first part is at the N-terminus, which is highly conserved and is called FERM or N-terminal ERM association domain. The FERM itself has three subdomains F1, F2, and F3. The second part is the α- helical domain, which consists of three α- helices (α1H, α2H, and α3H). The third part is a short, mainly helical C- terminus domain (CTD) or C-terminal ERM association domain. B Merlin’s head-to-tail folding between FERM and CTD domains in monomer structure renders the protein in closed inactive conformation upon phosphorylation of Ser 518

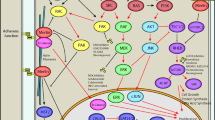
Merlin signaling pathways and potential therapeutic targets in NF2. A schematic depiction of the main intracellular pathways regulated by the protein product of the NF2 gene (merlin (is shown by golden)). Merlin regulates cell survival, proliferation, and cell–cell interaction in response to multiple proliferative signaling pathways at the plasma membrane and in the nucleus. Merlin can predominantly block the RTKs’ activity on their downstream targets, including RAS, PI3K, and Rac. Merlin inhibits Wnt/β-catenin signaling through inhibit translocation of β-catenin to the nucleus. At the nucleus (inhibition CRL4DCAF1) and cell cortex (promoting MST1/2), merlin can regulate Hippo signaling pathway. As a result, the expression of target genes of YAP will decrease. Merlin may also block LIN28B in the nucleus, reducing let-7 miRNA cluster repression and downregulating proto-oncogenic proteins including MYC and RAS. Diverse treatment options have been investigated for NF2 patients, including the suppression of merlin-regulated proteins and other cellular receptors. Pathogenic mutations in NF2 patients cause merlin function loss, which modulates downstream activity in each pathway, promoting cell growth, protein and fatty acid synthesis, proliferation, and survival. The five white boxes provided are current inhibitors of this pathway with their respective targets. Proteins are also illustrated in circular shapes, and each of them has been given a distinct color. the bilayer cellular plasma membrane with phospholipid compounds is presented at the top in pink. Also, the blue area in the cell represents the nucleus and the two blue straight lines within, represent genes involved in this pathway. Black arrows indicate the act of promotion and blocking lines indicate the act of suppression
The ‘open’ and ‘closed’ states
The function of ERM proteins is regulated by transitioning between the ‘open’ and ‘closed’ states, leading to activation and deactivation of the protein, respectively. Merlin and other ERM proteins can form a hetero- or homodimer conformation by constructing a head (being the FERM domain)-to-tail (being the CTD) conformation at intermolecular and intramolecular levels [112]. During FERM-CTD interactions, the CTD (shaped as a mono-layer surface) covers the hydrophobic parts of the F2 and F3 subdomains within the FERM domain (Fig. 2) [113]. This interaction is highly conserved in the ERM protein family, including merlin. In the open state, merlin can interact with other substances, including plasma membrane or integral proteins, and act as a scaffolding protein mediating the signaling cascades [114]. Deactivation occurs when the protein is in the closed state, and some residues are not accessible for other modifications or interactions; exceptionally, the closed state of merlin triggers its tumor suppressor activity [115]. The factors involved in transitioning between the two states and the outcome of their interactions require more investigation. It has been proposed that the binding of phosphatidylinositol 4,5-bisphosphate and the PTM on specific residues can induce the open form [116]. The open form provides sites on FERM and CTD domains to bind the partner proteins. For example, the FERM domain can bind to proteins residing on the membrane, including CD44, CD43, PSGL-1, MT1-MMP, ICAM-1, ICAM-3, and neprilysin, as well as proteins involved in the Hippo signaling [117,118,119].
Post translational modifications
Merlin has a multitasking feature since it functions both at the plasma membrane and in the nucleus (Fig. 3) [1]. This multitasking attribute results from merlin’s PTM by phosphorylation, cysteine modifications, ubiquitination, acetylation, or SUMOylation, resulting in structural and biochemical changes determining open or closed states (Fig. 2) [120, 121]. Most PTM occurs after the phosphorylation of specific residues of merlin domains, including Ser10, Ser13, Ser518, and Thr581. Ser10 and Ser13 are located in the merlin N-terminal domain. Ser10 shares a kinase partner with Ser518 and is phosphorylated by protein kinase A (PKA) in vivo. The phosphorylation of Ser10 is necessary for cell migration and regulating the morphology of the actin cytoskeleton [122].
Among these residues, Ser518 is of great interest. Ser518 residue is mainly located at the CTD. The phosphorylation of Ser518 residue can render the strength of interaction between FERM and CTD and mediates the formation of filopodia (the actin-rich protrusions from the cell surface sensing, migration, and cell–cell interaction) [123]. Ser518 is the target of PKA and PAK (P21 activated kinase) and results in the repression of merlin’s tumor suppressor activity and loss of contact inhibition [124]. Inactivation of merlin’s tumor suppressor activity can be inverted by an enzyme called myosin phosphatase MYPT1-PP1δ that dephosphorylates Ser518 residue (Fig. 2) [125].
Merlin concentration is regulated by AKT (also called protein kinase B) phosphorylation of residues Ser10, Ser315, and Thr230 leading to ubiquitin-mediated protein degradation. As a result, merlin cannot interact with its binding partners [126]. The exact consequences of these modifications and transitioning between the two states must be explored.
Merlin in signaling pathways
Merlin regulates cell survival and proliferation in response to multiple proliferative signaling pathways (Fig. 3). As a result, the inactivation of merlin can lead to uncontrolled cell proliferation and tumorigenesis, especially in the nervous system. The mechanisms underlying how merlin regulates cell proliferation are yet to be elucidated. Recent evidence demonstrated the role of merlin in various pathways, including mTOR, RTKs, Wnt/β-catenin, PI3K/AKT, and Hippo pathways [10, 13].
RTKs, as a member of the protein tyrosine kinases family, are a group of membrane receptors that dimerize after ligand binding. The phosphorylation of RTK’s intracellular domain activates cell proliferation, survival, and migration signaling pathways [127]. Merlin is essential for blocking RTK signaling pathways [1]. For instance, merlin binds to CD44 and restricts it from binding to hyaluronan, an extracellular matrix component, and inducing contact growth inhibition by blocking the CD44-Rac axis (Fig. 3) [110, 128]. Merlin interacts with RTKs, such as PDGFR (Platelet-derived growth factor receptor), EGFR (Epidermal growth factor receptor), HGFR (Hepatocyte growth factor receptor), VEGFR (Vascular endothelial growth factor receptor), and ErbB2/ErbB3 via its FERM domain. Merlin can predominantly block the RTKs’ activity on their downstream targets, including RAS, PI3K, and Rac (Fig. 3) [10, 13]. In patients with VS, EGFR expression level is correlated with increased tumor size and younger age onset [129]. In addition, the expression and activation of PDGFR-α and PDGFR-β are increased in VS compared with intact nerves [130].
Compared with healthy individuals, the PI3K/AKT/mTOR pathway is enhanced in patients with VS. [131] PIKE-L (Phosphatidylinositol 3-kinase enhancer-L), a human GTP (guanosine triphosphate)-binding protein, activates the PI3K and increases cell proliferation through PI3K/AKT pathway. Merlin regulates this pathway by inhibiting PIKE-L, and loss of merlin induces tumorigenesis in schwannoma and meningioma by activating PI3K/AKT pathway and increasing cell proliferation [132, 133]. The mTORC1 signaling is highly activated in NF2-deficient mesothelioma, schwannomas and meningiomas, thereby inducing tumor growth and rapamycin sensitivity [101]. Loss of merlin results in an integrin-associated activation of mTORC1 signaling via PAK1 and increases the expression of cyclin D1 to promote cell cycle progression (Fig. 3) [134].
At the nucleus and cell cortex, merlin can regulate the Hippo signaling pathway. The Hippo pathway is essential for regulating cell survival and proliferation [135]. In the cell cortex, merlin acts as an upstream effector where it binds to Lats1/2 (Large tumor suppressor kinases) via its FERM domain and elevates their concentration at the plasma membrane. This interaction results in the activation of Lats1/2 proteins leading to further inactivation of YAP (Yes-associated protein) and transcriptional coactivator with PDZ-binding motif (TAZ) [136]. Phosphorylation of YAP/TAZ is conducive to its degradation or cytoplasmic retention by binding to the 14.3.3 protein complex residing in the cytoplasm. With the absence of merlin, YAP/TAZ will be stabilized within the nucleus and bind to their protein partner TEAD (TEA domain transcription factor) and in turn, promote tumor cell survival and proliferation [136]. In patients with schwannoma, the expression of YAP and its transcription targets, including PDGFRβ, HER2, and HER3, are significantly elevated [137]. In the nucleus, unphosphorylated merlin binds to CRL4DCAF1(DDB1- and Cul4-Associated Factor 1), an E3 ubiquitin ligase, thereby inhibiting its role in YAP activation [97]. Moreover, binding to CRL4DCAF1 blocks the LATS1/2 degradation, which in turn increases YAP phosphorylation and its inactivation (Fig. 3) [138].
Wnt/β-catenin pathway counts as another target for merlin. Usually, Wnt signaling increases the nucleus translocation of β-catenin, enhances the expression of c-Myc and cyclin D1, and increases cell proliferation by TCF/LEF (T-cell factor/lymphoid enhancer factor) transcription factors [139]. The merlin connection to LRP6 (Low-density lipoprotein receptor-related proteins 6) by the FERM domain centralizes the β-catenin in the cytoplasm [100]. In addition, merlin in a complex with β-catenin, α-catenin, and E-cadherin localizes β-catenin near the plasma membrane [140]. Alternatively, merlin can prevent the transfer of β-catenin into the nucleus by inactivating Rac1 (Ras-related C3 botulinum toxin substrate 1). Usually, phosphorylated β-catenin by Rac1-activated JNK2 (c-Jun N-terminal kinase) is translocated to the nucleus (Fig. 3). [141].
The RNA-binding protein called Lin28B (Lin-28 homolog B), which regulates cellular growth and reprogramming, is another partner of merlin. Lin28B can suppress the generation of Let-7 (Lethal-7), a miRNA with a tumor suppressor activity that inhibits the expression of various proto-oncogenes, including MYC and RAS. Merlin can interact with Lin28B via its FERM domain, allowing an intact generation of Let-7 through Lin28B sequestration in the cytoplasm (Fig. 3) [142].
Future therapeutic perspective
A comprehensive understanding of molecular mechanisms in tumor progression of NF2-related tumors will provide opportunities for exploring more efficient treatments. Currently, many drugs are being examined targeting pathways involved in the pathogenesis of NF2 [14, 94, 143]. In this section, clinical trials evaluating the targeted therapies for NF-related tumors are discussed (Fig. 3, Table 3).
Targeting the PI3K/AKT/mTORC1 pathway
The PI3K/AKT/mTORC1 pathway serves as a hub in the intracellular signaling hierarchy by integrating signals from various upstream pathways and regulating cell proliferation, metabolism, and apoptosis [144]. As mentioned in Sect. 7.4, merlin can suppress the constitutively active mTORC1 effector. Therefore, mTORC1 can serve as a target for VS treatment [101]. In vivo evidence has shown an efficacy of a mTORC1 inhibitor (sirolimus). In this study on a mouse xenograft model, sirolimus (aka rapamycin) induced growth arrest in the growing VS tumors [145]. Everolimus, a rapamycin analog with an mTOR kinase inhibitor activity, has been evaluated in different clinical trial studies [146, 147]. In a phase II clinical trial on patients with VS in the context of NF2 (NCT01419639), 10 mg/day of everolimus on continuous daily dosing for 12 months resulted in stable disease in five out of nine patients (55.5%) and progressive disease in the remaining four (45.5%). No high-grade toxicities were recorded. Although no clinical response was reported, the significant disease control rate was considered encouraging for further studies [147]. Four-year follow-up of patients revealed that one patient remained with stable disease. In this study, three patients with progressive disease on everolimus were treated with bevacizumab. One out of three patients had stable disease after 8-month treatment. This study indicated that the progressive disease on everolimus may not preclude further treatment with bevacizumab [146].
Two VS features are slightly elevated AKT gene expression and marked AKT protein phosphorylation, which is necessary for proper AKT activity [15] AKT phosphorylation is primarily mediated by phosphoinositide-dependent kinase-1 (PDK1). An in vivo study demonstrated that AR-12 (OSU-03012), a PDK1 inhibitor, can proceed schwannoma cells to apoptosis by inhibiting AKT phosphorylation in VS and malignant schwannoma cells [15, 148]. Another study revealed that a novel histone deacetylase inhibitor, AR-42 (OSU-HDAC42), inhibits the downstream AKT and PDK1 expression, leading to G2 arrest and apoptosis in VS cells [149]. Two pilot studies of AR-42 in human NF2, VS, and meningiomas (NCT01129193 and NCT02282917) were safe and well tolerated, and no grade 3–4 toxicities were seen at the low dose 40 mg (pilot 2) three times weekly for three weeks of a 28-day cycle [150, 151]. A phase II/III randomized trial on 89 patients with NF2 or NF2-mutated recurrent meningiomas 12 years and older is recruiting (NCT05130866).
Targeting RTKs
The importance of different RTKs (including VEGFR, EGFR, PDGFR, and ErbB2/3) in the pathogenesis of NF2 has been established. Given the inhibitory effects of merlin on RTKs activity, several studies have examined the RTK inhibitors in patients with NF2.
VEGF-A/VEGFRs
VEGFRs activation stimulates the signaling cascade of angiogenesis upon binding to its substrate, VEGF. The expression of VEGF and VEGFR-1 is related to VS's growth rate, leading to an increased vessel density and abnormal cellular proliferation [152]. In a murine VS xenograft model, the inhibition of VEGF inhibited tumor growth (mean: 50%) and improved survival (more than 50%) [153].
Bevacizumab (an anti-VEGF antibody) has demonstrated encouraging results in patients with NF2-associated VS. [13, 14] In patients with progressive disease, it has successfully controlled the growth rate and helped to improve the hearing status [81, 154,155,156]. Different studies indicated a 36%–41% partial response of bevacizumab for NF2-related VS in patients 12 years and older [14]. The therapeutic effect of bevacizumab in pediatrics and adults with smaller and slow-growing tumors was less prominent [156,157,158]. The median treatment duration was 16 months, which does not produce a long-lasting effect. It led to severe toxicity in 17% (95% CI 10–26%), including amenorrhea, proteinuria, and hypertension [81]. The bevacizumab side effects are dose-dependent. A retrospective study reported 62% proteinuria and 58% hypertension in patients with NF2 treated with a 5 mg/kg, biweekly regimen [159]. In comparison, another study demonstrated that dose reduction to 2.5 mg/kg was associated with stable disease in all patients without significant side effects [160]. New evidence indicated hearing improvement and tumor volume reduction in safety and preliminary efficacy of the VEGFRs peptide vaccine in patients with progressive NF2 [161]. The DCE-MRI (Dynamic contrast-enhanced magnetic resonance imaging) findings showed that bevacizumab could improve vascular perfusion and oxygen supply by restoring the normal function of tumor vasculature. This function decreased tumor edema and improved the radiation effect in an NF2 schwannoma model [162]. It has been demonstrated that treatment of progressive meningioma (after surgery and radiotherapy) with bevacizumab led to disease stabilization with 6-month progression-free survival rates of 87, 77, and 46% in grades I to III meningiomas [83]. Studies on other inhibitors of VEGF (endostatin) and VEGFR (axitinib) did not demonstrate encouraging results [14, 163].
The VEGF level circulating in the peripheral blood can serve as a predictive biomarker to identify NF2 patients who benefit from anti-VEGF therapy [13]. The evidence showed that increased HGF level is a poor prognostic factor of hearing after bevacizumab therapy [154, 164]. This finding addresses that HGF/cMET (C-mesenchymal-epithelial transition) signaling pathway may underly the hearing loss in patients with VS and determines the response to bevacizumab [165].
HGFR
HGFR (c-MET) is an RTK participating in cancer progression [166] and (chemo) radiotherapy resistance [167]. Both HGF and HGFR are overexpressed in sporadic VS. [168] Several studies have evaluated the effects of combination RTK inhibitors in NF2-related VS cell lines. An in vitro study demonstrated that targeting VEGF-A reduced c-MET expression and targeting c-MET reduced VEGF-A expression. This finding suggests a crosstalk between c-MET and VEGFA in VS biology [165]. Therefore, the combination of RTK inhibitors is a potential treatment for NF2-related VS. An in vivo study indicated that crizotinib, a c-MET inhibitor, can improve radiosensitivity in NF2 schwannoma cells. It is demonstrated that low-dose radiation concurrent with crizotinib was as effective as high-dose radiation. Therefore, concurrent crizotinib can help to improve hearing with reduced radiation doses and fewer toxicities [169]. A phase II clinical trial of crizotinib for children and adults with NF2 and progressive VS is ongoing (NCT04283669). In an in vitro study on merlin-deficient mouse schwannoma cells, the combination therapy with cabozantinib (c-MET inhibitor) and saracatinib (Src inhibitor) suppressed the growth of the cells and promoted caspase-dependent apoptosis [170].
ErbB
The ErbB family is a group of RTKs consisting of four members, including ErbB1 (EGFR/HER1), ErbB2 (HER2/neu), ErbB3 (HER3), and ErB4 (HER4). Activation of ErbB receptors requires dimerization upon binding to the ligand. All four members of the ErbB family can form heterodimers. It has been evidenced that ErbB family members participate in Schwann cell differentiation and proliferation [171]. Therefore, they can serve as targets to halt VS growth in patients with NF2. EGFR and ErbB2 heterodimers are the most common ErbB receptor dimerization type reported in VS. [172] Lapatinib has a dual inhibitory impact on EGFR/ErbB2 [129]. A phase II trial on NF2 patients with progressive VS indicated that lapatinib resulted in improved hearing in 4/13 patients (30.7%) and radiographic response in 4/17 patients (23.5%) [173]. It has been demonstrated that lapatinib continuation has the potential to arrest or reduce the growth of NF2-related meningiomas [174]. A phase II study on the efficacy and safety of icotinib, an oral EGFR, indicated minimal toxicity and also radiographic and hearing responses in patients with NF2 and progressive VS. [175] In a nude mouse model, both erlotinib (an EGFR inhibitor) and trastuzumab (a HER2/neu inhibitor) significantly inhibited the development of VS xenografts [176]. However, a clinical study on eleven patients with NF2-related VS demonstrated poor hearing and radiographic responses with erlotinib (150 mg daily) [177].
PDGFR
Imatinib mesylate inhibits PDGFRs and their downstream signaling pathways in VS cells. This action enhances apoptosis and decreases cell viability in a dose-dependent manner [178]. In addition, imatinib can prevent angiogenesis in both sporadic and NF2-related VS. The dual inhibitory effect of imatinib on tumorigenesis and angiogenesis has made it a promising drug for further trials on schwannoma [179]. Nilotinib, a second-generation RTK inhibitor, has a similar mechanism of action and structure to imatinib, with greater lipophilicity. This feature improves its tissue penetration with lower toxicity and higher efficacy [180]. The suppression of PDGFRs and their downstream signaling mediators (AKT and mTOR) can justify their anti-tumorigenic action [180]. Ponatinib has been gaining more attention for its potential in treating VS. In merlin-deficient human Schwann cells, ponatinib promotes G1 arrest by inhibiting the phosphorylation of PDGFRα/β, AKT, MEK1/2, ERK1/2, STAT3, and p70S6K [181].
Combination therapy with a dual mTORC1/2 kinase inhibitor vistusertib (AZD2014) and dasatinib (a multi-kinase inhibitor) demonstrated encouraging results as a novel therapeutic strategy for VS. [182] Notably, the synergic effect of AZD2014 and dasatinib results in fewer toxicities because lower doses can be administered. Moreover, the clinical trial (NCT03071874) has administered vistusertib to treat recurrent grade II or III NF2-mutated meningiomas to inhibit the mTORC1/C2 complexes. [14].
Targeting hippo pathway
The Hippo pathway, an evolutionarily conserved mechanism that controls tissue homeostasis, is one of the most well-known merlin-regulated pathways [136]. Multiple research projects have been conducted to examine the impact of targeting the Hippo pathway on NF2-related tumors. For instance, YAP knockdown halted tumorigenesis initiation and induced a decrease in cell proliferation of NF2-deficient meningioma and mesothelioma cell lines [183,184,185].
YAP1/TAZ's prooncogenic abilities are assumed to be mediated by TEADs (discussed in Sect. 7.4), thus disrupting their interaction can be a therapeutic target. Therefore, many compounds have been developed and assessed to produce novel anti-cancer drugs related to this interaction. The first small compound demonstrated to impede YAP-TEAD binding was verteporfin, a photosensitizer utilized therapeutically in photodynamic treatment for neovascular macular degeneration [186]. This small molecule has been shown to inhibit different forms of carcinomas such as hepatoma and glioblastoma and retinoblastoma [187]. In addition, it was demonstrated that verteporfin inhibits YAP activity as well as the viability, invasion, and tumor sphere formation of mesothelioma cell lines [188]. IAG933, another inhibitor of the interaction YAP-TEAD, is ongoing in a multi-center phase I clinical trial on patients with mesothelioma, NF2 mutated tumors, and tumors with functional YAP/TAZ fusions (NCT04857372). Furthermore, IK-930, a small molecule inhibitor of TEAD, is undergoing a phase I clinical trial on adult patients with advanced or metastatic solid tumors (NCT05228015). IK-930 prevents palmitate binding and thereby interrupts improper TEAD-dependent transcription [189].
Mammalian vestigial-like 4 (VGLL4) has previously been discovered as a natural YAP antagonist that binds to TEADs via its Tondu (TDU) domain and the VGLL4 TDU region is sufficient to block YAP activity. Jiao et al. indicated that disruption of YAP-TEADs interaction by a VGLL4-mimicking peptide may be a promising therapeutic strategy for YAP-driven human cancers [190].
Hippo signaling has been associated with the cellular metabolic state, including cellular responses to glucose restriction (energy stress) and the mevalonate cascade [191]. Glucose shortage lowers cellular ATP levels and activates AMP-dependent protein kinase (AMPK), a sensor of cellular energy stress. AMPK has been reported to decrease YAP activity through a range of processes, including reducing nuclear YAP levels and YAP-TEAD interactions, which provide new avenues to target this pathway more efficiently [192]. A recent study evaluated the metabolic aspect of YAP/TAZ-depleted in NF2-deficient schwannoma [193]. This study indicated that YAP/TAZ depletion reduces glycolysis-dependent growth and elevates mitochondrial respiration and reactive oxygen species buildup, resulting in oxidative stress-induced cell death. Moreover, they showed lysosome-mediated cAMP-PKA/EPAC-dependent activation of RAF-MEK-ERK signaling as a resistance mechanism to YAP/TAZ inhibition [193].
Targeting other pathways
Selumetinib, a MEK inhibitor, is the first FDA approved drug for NF1-associated plexiform neurofibromas in 2020. It showed 66% response rate for inoperable or progressive plexiform neurofibromas in children two years and older with a duration of response more than one year in 82% [194]. Phase II clinical trial of selumetinib for NF2-related tumors is underway (NCT03095248) [14].
Brigatinib, an anaplastic lymphoma kinase (ALK) inhibitor, is a potential choice for NF2-related tumors. It has been demonstrated that brigatinib causes tumor shrinkage in both NF2-deficient meningioma and schwannoma by inhibiting multiple tyrosine kinases, including EphA2, Fer, and focal adhesion kinase 1 (FAK1). Brigatinib can also inhibit multiple RTKs frequently activated in these tumors but not ALK [195]. Brigatinib is under investigation in an undergoing phase II clinical trial involving 80 patients (NCT04374305).
Another FAK inhibitor, GSK2256098, was evaluated in recurrent or progressive grade I-III meningiomas as part of the first genomically driven phase II trial. Patients with NF2 mutations were treated with GSK2256098 (750 mg orally twice daily) until progressive disease. It was well tolerated and improved progression-free survival at 6 months [196].
In summary, if we put the clinical studies of VS into account, evidence on bevacizumab is more than other targeted therapies. It could provide 88% clinical benefit (response rate 41%) out of 196 VS tumors [81]. Bevacizumab can also help to alleviate the compression effects on critical structures (by reducing edema) and can be applied as a radiosensitizer by enhancing tissue oxygenation [162]. In the second place is everolimus (a mTORC inhibitor) with a clinical benefit of 55% (all as stable diseases) [147]. This option makes bevacizumab available for progressive cases (clinical benefit rate 33%) [146]. Another available choice for VS is lapatinib (an ErbB2 inhibitor). It has improved hearing in 30% of cases with progressive VS with a clinical response of 23% [173].Lapatinib can also be applied in NF2-related meningioma [174]. The following sections present emerging and potential treatment choices based on the other aspects of NF2 pathophysiology.
Targeting proinflammatory mediators
The growing evidence emphasizes the importance of the tumor microenvironment (TME) in the progression and development of VS pathology, and inflammation counts as the most important factor in tumors' growth [197]. B and T lymphocytes and macrophages, especially a particular type of macrophage called tumor-associated macrophages (TAMs), are among the most important immune cell infiltrating the VS tissue [198,199,200].TAMs originate from circulating bone marrow-derived monocytes and are categorized into two main types, pro-inflammatory M1-type and pro-tumorigenic M2- type, which control tumor cells’ viability, proliferation, invasion, and angiogenesis [197]. A study on VS specimen showed a positive relationship between the expression of M2 macrophage marker CD163 and tumor growth and microvessel density. This finding indicates that M2-type macrophages in VS is associated with angiogenesis and volumetric tumor growth [198]. In another experimental study, de Vries et al. found that VS tissue had higher expression of macrophage colony-stimulating factor (M-CSF) and IL-34 (two cytokines regulating macrophage infiltration). In addition, the study demonstrated that the fast-growing VS and cystic tumors had significantly higher M-CSF expression. These findings address the positive link between M-CSF and VS progression [201]. In support, Lewis et al. in a composed imaging and neuropathology study, indicated that macrophages, rather than Schwann cells, are the dominant proliferating cells in the growing sporadic VS tumors [199].
The current understanding of the association between NF2-related VS and inflammation is limited to a handful of studies. Schulz et al. found that CD68+ macrophages are present in 90% [9/10] of NF2-related VS tumors, mainly with M2 phenotype [200]. Moreover, a higher expression of the macrophage marker CD68, T lymphocyte markers CD3 and CD8, and B lymphocytes marker CD20 was demonstrated in NF2-associated meningioma and schwannoma TME [202].
Another specified group of cells existing in the TME of NF2-related VS are myeloid-derived suppressor cells (MDSCs). These cells support tumor cells’ growth and survival by inhibiting the tumor-specific T cells [203]. Wang et al. demonstrated that MDSCs inhibit CD8+ T cells and induce their transformation into regulatory T cells by secreting TGF-β [203].
Detecting and quantifying the intratumoral inflammation biomarkers can provide new prospects for earlier detection and also allow specific targeted therapies. An in vivo study on nineteen VS patients with different degrees of growth (static, growing, and shrinking) aimed to find whether there is any difference between groups in terms of inflammation (using 11C-(R)-PK11195 PET scan) and vascular permeability (using dynamic contrast-enhanced MRI). The study showed that growing tumors (versus static tumors) had significantly increased inflammation and vascular permeability. The author introduced the 11C-(R)-PK11195 specific binding and DCE-MRI-derived parameters as imaging biomarkers of inflammation and vascular permeability in VS [199]. In a study of sporadic and NF2-related VS, Breun et al. showed the overexpression of C-X-C chemokine receptor type 4 (CXCR4) and also indicated the feasibility of CXCR4-directed PET/CT imaging of VS using the radiolabeled chemokine ligand [68 Ga]Pentixafor [204, 205].
A comparison between sporadic VS tissue and normal vestibular nerve showed a higher expression of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α [206]. In alignment with these results, a study on secreted factors from human VS demonstrated higher levels of TNF-α secretion correlating with poorer hearing among patients and also can induce cellular loss in murine cochlear explants [70].
The nuclear factor kappa-B (NF-κB) is one of the main mediators of inflammation. NF-κB is a transcription factor participating in physiologic cellular processes like cell growth, apoptosis, inflammation as well as malignant transformation [197]. The inhibitory role of merlin on NF-κB has been proven in murine fibroblasts, rat glioma cells, and human schwannoma cell lines [207, 208]. Bioinformatic studies put forward NF-kB as a determining factor of VS pathogenesis [209]. To confirm this notion, Dilwali et al. examined whether selective inhibition of NF-kB (using siRNA and curcumin) has an inhibitory effect on the VS cells. The authors found that NF-kB inhibitors reduced VS cells’ proliferation and increased their death [209]. A computational drug repositioning platform to match known drug-gene interactions showed that mifepristone has the potential to treat VS [210]. Mifepristone is a progesterone and glucocorticoid receptor antagonist that can cross the blood–brain barrier. Since it is well tolerated when taken orally, it can also be utilized for the palliative benefits of glioblastoma [211]. Mifepristone can decrease VS cells' metabolic and proliferative activities and promote cytotoxicity in a dose-dependent manner, regardless of whether the NF2 gene is mutated [210].Ingenuity Pathway Analysis showed that mifepristone targets NF-κB [210] as a pro-inflammatory transcription factor that participates in VS proliferation [209].
The NLRP3 gene (NLR family pyrin domain containing 3) is a member of neuroinflammation-related signaling that mediates VS progression. The NLRP3 gene product in a multi-protein complex triggers caspase-1 leading to the production of inflammatory cytokines, such as IL-1β and IL-18 [212]. So far, the mutated NLRP3 was reported as a reason for cochlear autoinflammation and syndromic and nonsyndromic hearing loss DFNA34. Anakinra, a nonglycosylated recombinant version of the human IL-1 receptor antagonist, reduced hearing loss in NLRP3 mutated patients [213]. Comprehensive pathway analysis using gene expression on VS microarray data together with validating this finding at the gene and protein expression level indicated higher expression of NLRP3 inflammasome in VS. Moreover, this overexpression is associated with reduced hearing loss in VS patients [214]. This finding suggests the therapeutic role of IL-1β blockade in patients with hearing loss secondary to VS.
The COX-2 (Cyclooxygenase-2) expression has been linked to VS proliferation in some studies [215, 216]. In NF2 patients, YAP activation (in the context of the Hippo pathway) enhances COX-2 production, which in turn catalyzes the prostaglandin E2 (PGE2) production. It has been demonstrated that PGE2 improves survival and proliferation and inhibits apoptosis in NF2-related schwannoma [184]. This finding shows that COX-2 inhibitors (such as aspirin) can diminish the progression of VS [217]. The inhibitory effect of aspirin on VS growth was demonstrated in a retrospective cohort. In this study on eighty-six patients, after an 11-year follow-up, aspirin significantly prevented VS growth (odds ratio [OR] 0.32, 95% CI 0.11–0.91) [218]. However, this study did not show a consistent correlation between VS growth and aspirin intake. Another two studies found no consistent correlation between aspirin administration and VS growth [219, 220]. Despite these controversies, aspirin is recommended by the Congress of Neurological Surgeons for VS patients [221]. To clarify this controversy, a phase II double-blind, randomized trial using aspirin on NF2-related or sporadic VS is underway (NCT03079999). Notably, the interaction between COX-2 and NF-κB pathway has been reported and aspirin modifies both NF-kB signaling and COX-2 expression [222].
A greater understanding of the inflammatory processes involved in NF2-related VS can assist in introducing novel targeted therapies.
Immunotherapy
The last decades have witnessed a revolution in medical oncology with the development of immunotherapy. This approach aims to activate the immune system against tumor cells. Immunotherapy encompasses different modalities—including immune checkpoint inhibitors (ICIs), T-cell therapy, cancer vaccines, oncolytic virus therapy, and non-specific cytokines.
The first immunotherapy experience in NF2-related VS likely belongs to the study that applied VEGFR-specific cytotoxic T lymphocytes in patients with progressive tumors. This study demonstrated the safety and clinical efficacy of this approach [161]. A phase I/II clinical trial using antigen-specific T cells (CAR-T) and engineered immune effector cytotoxic T cells modified by immunoregulatory genes and immune-modified dendritic cell vaccine (DCvac) in the treatment of neurofibromatosis or schwannoma is underway [14].
Among all immunotherapy modalities, the most expansive body of research belongs to ICIs, especially anti-programmed cell death protein-1 (PD-1) or its ligand (PD-L1). These antibodies block the PD-1 and PD-L1 binding, thereby activating the immune cells against tumor cells [223]. Therefore, tumors with high expression of PD-L1 have higher responses to anti-PD-(L)1 antibody.
In addition, the type and rate of intratumoral lymphocyte infiltration are predictive factors of response to immunotherapies. Tumors with high infiltration of CD8+ T cells are good candidates for immunotherapy [224]. More responses can be expected if this condition coincides with high PD-L1 expression on tumor cells. The histopathologic examination of NF2-related VS reveals PD-L1 overexpression in up to 70–100% of the specimens [202]. This finding provides hope to apply anti-PD-(L)1 antibody for NF2-related VS.
Interestingly, PD-L1 expression may have a prognostic value in sporadic VS. In 2019, Perry et al. realized that PD-L1 expression on VS cells is associated with more tumor progression, poor facial nerve function, and poor tumor control. [225] This issue may be rooted in the association between inflammation and PD-L1 expression on tumor cells [226]. Dissecting the mechanisms behind the PD-L1 expression and their association with inflammation can be helpful for the future of immunotherapy in NF2-related VS.
Rutland et al. demonstrated a positive correlation between NF2 gene mutation and the extent of tumor-infiltrating lymphocytes in meningioma tissues. The authors demonstrated that NF2 mutations were present in 40% of meningiomas with 1–25 scattered lymphocytes and 54% of meningiomas that had even more scattered lymphocytes. On the other hand, NF2 mutation was not detected in meningiomas with no tumoral infiltration [227]. This finding put forward immunotherapies an interesting choice for treating NF2-related meningioma.
Gene therapy
Gene-based therapies have provided novel opportunities for treating different diseases, such as retinal dystrophies, genetic hearing loss, and spinal muscular atrophy. In the case of retinal dystrophies, gene therapy has made a substantial contribution to therapeutics. In 2018, the FDA approved RPE65 gene delivery by adeno-associated virus (AAV) for the treatment of Leber's congenital amaurosis [228].
Different research groups have focused on inner ear gene therapy methods, including viral and non-viral vectors, gene replacement, and gene suppression by RNA-based therapies and CRISPR/Cas9-based genome editing [229, 230].
However, when the outer and inner ear is targeted, gene therapy may become quite challenging due to the existing barriers and the question of how long viral vectors can continue their expression in target cells. In recent years, gene therapy implicated in hearing loss caused by genetic mutations has shown promising results. For instance, using the Anc80 vector, wild-type harmonin was effectively delivered into the inner ear of a mouse model of type I Usher syndrome caused by mutations in Ush1c gene encoding the protein harmonin. In addition to restoring mechanotransduction, the therapy led to remarkable improvements in complex audiovestibular functioning to approximately wild-type levels for a minimum of six months [231]. Another interesting case is the dual transduction of viral vectors containing otoferlin cDNA that led to partial recovery of hearing function and restoration of protein expression to 30% of levels in the wild-type in mouse models [232, 233].
The non-viral approaches are a potent alternate delivery treatment method that applies engineered non-viral delivery vehicles to satisfy the exact therapeutic need that is less immunogenic. In an in vivo study, delivery of cre-recombinase and genome editing agents through lipid compounds led to 90% recombination and 20% genome editing in newborn mouse OHCs-hair cells [234]. Furthermore, in a mouse model of dominant genetic hearing loss, injection into the cochlea of newborn mice reduced progressive hearing loss and increased hair cell survival in vivo. This was done by cationic lipid nanoparticles that incorporated Tmc1-targeting CRISPR/Cas-9 complexes [235]. Recently, tumor-penetrating nanocomplexes containing TNF-a short interfering RNAs (siRNA) were employed to target primary vestibular schwannoma cells in vitro actively; this method enables nanoparticles to be tumor tissue-specific with a systematic administration [236].
Several preclinical studies have indicated promising results with gene therapy in treating NF2. Merlin re-expression via gene replacement in NF2-null schwannomas led to increased apoptosis and tumor regression [237]. In a mouse model of schwannoma, direct injection of an AAV1 vector expressing caspase-1, under the control of Schwann-cell specific promoter, resulted in tumor regression [238]. In another study, AAV1 expressing ASC (Apoptosis-associated speck-like protein containing a CARD [caspase recruitment domain]) in human xenograft and murine allograft schwannoma models decreased tumor growth and resolved tumor-associated pain without detectable toxicity [239]. Peptide-based nanoparticles have been used to transport genetic materials to primary human VS cultures in vitro. This process was accomplished by coating the nanoparticle surface with a peptide that targets Schwann cells, reducing the release of an ototoxic inflammatory cytokine from tumor cells [240]. New evidence has put forward antisense therapy for the personalized therapy of NF2. In an in vitro study, antisense oligonucleotides targeting exon 11 rescued the NF2 phenotype [241]. It is still challenging to formulate gene or drug delivery methods that are effective for therapeutic purposes. Advances in the injection procedure through the round-window membrane will improve the injected medication's pharmacokinetics and pharmacodynamics within the inner ear [242].
NF2 models in preclinical research
To date, the restricted availability of tumor tissues and, more importantly, the absence of in vitro and in vivo model systems are barriers to clinical studies of NF2-associated tumors and deciphering the biological mechanisms related to their progression [13, 243].
Currently, there is only one transformed and immortalized VS cell line harboring HPV E6 and E7 oncogenes (HEI-193), which due to long-term passaging, shows malignant and aggressive growth characteristics; therefore, they may not correctly depict the biology of NF2-associated tumors. This cell line serves as the basis for several NF2 drug-testing investigations [178, 244].
Because of the broad spectrum of clinical symptoms associated with NF2, reliable animal models that reflect the histology and biochemical variations seen in patients are more demanding.
While NF2 genetically engineered mouse (GEM) models are beneficial for studying the disease pathogenesis [30, 245], due to the prolonged timescales, intensive breeding requirements, and challenges in producing synchronized carcinogenesis, GEM models pose a challenge to conducting reliable drug testing [246].
Lately, patient-derived xenograft (PDX) models have emerged as an essential platform for evaluating novel pharmacotherapies; however, attempts to generate PDX for schwannoma have mainly unsuccessful.
A few studies have demonstrated the macroscopic development of transplanted human schwannoma tissue in naked mice. Nevertheless, these xenografts have limited effectiveness in preclinical research due to their slower growth and absence of transplantation capability [247, 248]. For instance, in two studies, HEI-93 cells were xenografted into the mouse sciatic nerve (one of them with luciferase activity), and both showed less accurate anatomical features compared to NF2 characteristics but had fast tumor development [64, 249]. Moreover, Periostin-Cre NF2flox/flox mice were shown to be authentic transgenic models of NF2 and VS but had complications in sustaining and obtaining synchronous tumors [245]. Furthermore, other models have shown no relevance to NF2 phenotypes despite their minimal advantages [249,250,251,252].
Interestingly, in a recent study, researchers developed PDX and cell lines resembling NF2 characteristics regarding histopathology, morphology and molecular biology. Their model was able to preserve patient NF2 mutations, gene expression patterns mimicking patient tumor profiles, and many critical signaling pathways that are often dysregulated in human schwannomas [253].
New findings in human stem cell biology, organoid, and genome-editing techniques have provided the possibility to model nervous system tumors. However, we still face to lack of NF2 stem cell/organoid but models of inner ear organoids with full sensory circuits and myelinating Schwann cells raise new perspectives for paving the way for the development of the NF2 model [243]. Overall, establishing a reproducible experimental model of NF2-associated schwannoma has become a primary objective for the development of innovative and successful treatment methods, and there is a critical demand to fill this gap with further research.
Conclusions
NF2 is a hereditary complex neuro-cutaneous disease with significant morbidities. Over the past decades, unprecedented steps have been taken toward a better understanding of NF2 pathophysiology. The researches have made significant progress, but some ambiguous issues still need to be clarified. For instance, despite widely accepted management options, like long-term monitoring, surgery, and radiation therapy, there is still no efficient treatment for NF2.
This review summarized the literature on NF2 regarding its clinical features, diagnostic criteria, genetic and epigenetic background, merlin biology function, and the available treatments and future perspectives. Applying the NF2 gene as a target for gene therapy remains an open question. We believe that future studies can remove the veil of ignorance and answer some obscure aspects; for example, it is unclear how epigenetic modulations or modifier genes affect the variability in phenotype manifestation between individuals. We still need to learn more about how merlin participates in several intracellular pathways for the maintenance and proper function of cells or how the pathways manipulation can create new therapeutic perspectives for NF2 treatment. We believe that foreseeable investigations will shed light on merlin protein and its contribution to the disease process. This can, in turn, provide valuable information about its biological aspects, which will pave the way to utilize them effectively for therapeutic purposes.
Availability of data and materials
Not applicable.
Abbreviations
- NF2:
-
Neurofibromatosis type 2
- NF1:
-
Neurofibromatosis type 1
- SMARCB1:
-
SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1
- LZTR1:
-
Leucine zipper-like transcription regulator 1
- VS:
-
Vestibular schwannoma
- NIH:
-
National Institutes of Health
- Schwannomatosis-NOS:
-
Schwannomatosis not otherwise specified
- Schwannomatosis NEC:
-
Schwannomatosis not elsewhere classified
- ERM:
-
Ezrin/radixin/moesin
- NGS:
-
Next-generation sequencing
- HOX:
-
Homeobox genes
- PI3K:
-
Phosphoinositide-3 kinase
- AKT:
-
Ak strain transforming
- Raf:
-
Rapidly accelerated fibrosarcoma
- MEK:
-
Mitogen-activated protein kinase
- ERK:
-
Extracellular signal regulated kinase
- RTK:
-
Receptor tyrosine kinase signaling
- mTOR:
-
Mechanistic target of rapamycin
- FERM:
-
Band 4.1 ezrin radixin moesin
- CTD:
-
C- terminus domain
- PTM:
-
Post translational modifications
- NLS:
-
Nuclear localization sequence
- PKA:
-
Protein kinase A
- PAK:
-
P21 activated kinase
- PDGFR:
-
Platelet-derived growth factor receptor
- EGFR:
-
Epidermal growth factor receptor
- HGFR:
-
Hepatocyte growth factor receptor
- VEGFR:
-
Vascular endothelial growth factor receptor
- PIKE-L:
-
Phosphatidylinositol 3-kinase enhancer-L
- GTP:
-
Guanosine triphosphate
- Lats1/2:
-
Large tumor suppressor kinases
- YAP:
-
Yes-associated protein
- TAZ:
-
Transcriptional coactivator with PDZ-binding motif
- TEAD:
-
TEA domain transcription factor,
- CRL4DCAF1:
-
DDB1- and Cul4-associated factor 1
- TCF/LEF:
-
T-cell factor/lymphoid enhancer factor
- LRP6:
-
Low-density lipoprotein receptor-related proteins 6
- Rac1:
-
Ras-related C3 botulinum toxin substrate 1
- JNK2:
-
C-Jun N-terminal kinase
- LIN28B:
-
Lin-28 homolog B
- Let-7:
-
Lethal-7
- HGF:
-
Hepatocyte growth factor
- PKD1:
-
Phosphoinositide-dependent kinase-1
- DCE-MRI:
-
Dynamic contrast-enhanced magnetic resonance imaging
- cMET:
-
C-mesenchymal-epithelial transition
- VGLL4:
-
Mammalian vestigial-like 4
- AMPK:
-
AMP-dependent protein kinase
- ALK:
-
Anaplastic lymphoma kinase
- TME:
-
Tumor microenvironment
- TAMs:
-
Tumor-associated macrophages
- M-CSF:
-
Macrophage colony-stimulating factor
- MDSCs:
-
Myeloid-derived suppressor cells
- CXCR4:
-
C-X-C chemokine receptor type 4
- NF-κB:
-
Nuclear factor-kappa B
- NLRP3:
-
NLR family pyrin domain containing 3
- ICIs:
-
Immune checkpoint inhibitors
- PD-1:
-
Programmed cell death protein-1
- PD-L1:
-
Programmed cell death ligand siRNAs: short interfering RNAs
- AAV:
-
Adeno-associated virus
- ASC:
-
Apoptosis-associated speck-like protein containing a CARD
- CARD:
-
Caspase recruitment domain
- GEM:
-
Genetically engineered mouse
- PDX:
-
Patient-derived xenograft
References
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci. 2021;22(11):5850.
Bachir S, Shah S, Shapiro S, Koehler A, Mahammedi A, Samy RN, et al. Neurofibromatosis type 2 (NF2) and the implications for vestibular schwannoma and meningioma pathogenesis. J Int J Mol Sci. 2021;22(2):690.
Wishart J. Case of tumours in the skull, dura mater, and brain. Edinburgh Med Surg J. 1822;18(72):393.
Mautner VF, Lindenau M, Baser ME, Hazim W, Tatagiba M, Haase W, et al. The neuroimaging and clinical spectrum of neurofibromatosis 2. Neurosurgery. 1996;38(5):880–5.
Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93–7.
Baser ME, Kuramoto L, Joe H, Friedman JM, Wallace AJ, Gillespie JE, et al. Genotype-phenotype correlations for nervous system tumors in neurofibromatosis 2: a population-based study. Am J Hum Genet. 2004;75(2):231–9.
Picry A, Bonne NX, Ding J, Aboukais R, Lejeune JP, Baroncini M, et al. Long-term growth rate of vestibular schwannoma in neurofibromatosis 2: a volumetric consideration. Laryngoscope. 2016;126(10):2358–62.
Ammoun S, Hanemann CO. Emerging therapeutic targets in schwannomas and other merlin-deficient tumors. Nat Rev Neurol. 2011;7(7):392–9.
Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. J Oncogene. 2016;35(5):537–48.
Gugel I, Grimm F, Liebsch M, Zipfel J, Teuber C, Kluwe L, et al. Impact of surgery on long-term results of hearing in neurofibromatosis type-2 associated vestibular schwannomas. Cancers. 2019;11(9):1376.
Kluwe L, Friedrich RE, Hagel C, Lindenau M, Mautner VF. Mutations and allelic loss of the NF2 gene in neurofibromatosis 2-associated skin tumors. J Invest Dermatol. 2000;114(5):1017–21.
Ren Y, Chari DA, Vasilijic S, Welling DB, Stankovic KM. New developments in neurofibromatosis type 2 and vestibular schwannoma. Neuro-Oncol Adv. 2021;3(1):vdaa153.
Roman Souza G, Abdalla A, Mahadevan D. Clinical trials targeting neurofibromatoses-associated tumors: a systematic review. Neuro-Oncol Adv. 2022;4(1):vdac005.
Long J, Zhang Y, Huang X, Ren J, Zhong P, Wang B. A review of drug therapy in vestibular schwannoma. Drug Des Dev Ther. 2021;15:75–85.
Halliday D, Emmanouil B, Pretorius P, MacKeith S, Painter S, Tomkins H, et al. Genetic severity score predicts clinical phenotype in NF2. J Med Genet. 2017;54(10):657–64.
Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363(6429):515–21.
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Teare D, et al. A genetic study of type 2 neurofibromatosis in the United Kingdom. I. prevalence, mutation rate, fitness, and confirmation of maternal transmission effect on severity. J Med Genet. 1992;29(12):841–6.
Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes Dev. 2003;17(9):1090–100.
Evans DG, Hartley CL, Smith PT, King AT, Bowers NL, Tobi S, et al. Incidence of mosaicism in 1055 de novo NF2 cases: much higher than previous estimates with high utility of next-generation sequencing. Genet Med. 2020;22(1):53–9.
Selvanathan S, Shenton A, Ferner R, Wallace A, Huson S, Ramsden R, et al. Further genotype–phenotype correlations in neurofibromatosis 2. J Clin Genet. 2010;77(2):163–70.
Evans DG, Wallace AJ, Wu CL, Trueman L, Ramsden RT, Strachan T. Somatic mosaicism: a common cause of classic disease in tumor-prone syndromes? Lessons from type 2 neurofibromatosis. Am J Hum Genet. 1998;63(3):727–36.
Baser M, Wallace A, Strachan T, Evans D. Clinical and molecular correlates of somatic mosaicism in neurofibromatosis 2. J J Med Genet. 2000;37(7):542–3.
Evans DG, Ramsden RT, Shenton A, Gokhale C, Bowers NL, Huson SM, et al. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification. J Med Genet. 2007;44(7):424–8.
Dinh CT, Nisenbaum E, Chyou D, Misztal C, Yan D, Mittal R, et al. Genomics, epigenetics, and hearing loss in neurofibromatosis type 2. Otol Neurotol. 2020;41(5):e529.
Catasús N, García B, Galvan I, Plana A, Negro A, Rosas I, et al. Revisiting the UK Genetic Severity Score for NF2: a proposal for the addition of a functional genetic component. J Med Genet. 2022;59(7):678–686. https://doi.org/10.1136/jmedgenet-2020-107548. Epub 2021 Aug 4.
Halliday J, Rutherford SA, McCabe MG, Evans DG. An update on the diagnosis and treatment of vestibular schwannoma. Expert Rev Neurother. 2018;18(1):29–39.
Evans DG, Maher ER, Baser ME. Age related shift in the mutation spectra of germline and somatic NF2 mutations: hypothetical role of DNA repair mechanisms. J Med Genet. 2005;42(8):630–2.
Giovannini M, Robanus-Maandag E, Niwa-Kawakita M, van der Valk M, Woodruff JM, Goutebroze L, et al. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13(8):978–86.
Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev. 2000;14(13):1617–30.
Sadler KV, Rowlands CF, Smith PT, Hartley CL, Bowers NL, Roberts NY, et al. Re-evaluation of missense variant classifications in NF2. Hum Mutat. 2022;43(5):643–54.
Smith MJ, Higgs JE, Bowers NL, Halliday D, Paterson J, Gillespie J, et al. Cranial meningiomas in 411 neurofibromatosis type 2 (NF2) patients with proven gene mutations: clear positional effect of mutations, but absence of female severity effect on age at onset. J Med Genet. 2011;48(4):261–5.
Smith MJ, Bowers NL, Bulman M, Gokhale C, Wallace AJ, King AT, et al. Revisiting neurofibromatosis type 2 diagnostic criteria to exclude LZTR1-related schwannomatosis. Neurology. 2017;88(1):87–92.
Cooper J, Giancotti FG. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014;588(16):2743–52.
Painter SL, Sipkova Z, Emmanouil B, Halliday D, Parry A, Elston JS. Neurofibromatosis type 2-related eye disease correlated with genetic severity type. J Neuroophthalmol. 2019;39(1):44–9.
Carter B, Zhao K. The epigenetic basis of cellular heterogeneity. Nat Rev Genet. 2021;22(4):235–50.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22.
Baser ME, Ragge NK, Riccardi VM, Janus T, Gantz B, Pulst SM. Phenotypic variability in monozygotic twins with neurofibromatosis 2. Am J Med Genet. 1996;64(4):563–7.
Welling DB, Akhmametyeva EM, Daniels RL, Lasak JM, Zhu L, Miles-Markley BA, et al. Analysis of the human neurofibromatosis type 2 gene promoter and its expression. Otolaryngol Head Neck Surg. 2000;123(4):413–8.
Kino T, Takeshima H, Nakao M, Nishi T, Yamamoto K, Kimura T, et al. Identification of the cis-acting region in the NF2 gene promoter as a potential target for mutation and methylation-dependent silencing in schwannoma. Genes cells. 2001;6(5):441–54.
Gonzalez-Gomez P, Bello MJ, Alonso ME, Lomas J, Arjona D, Campos JM, et al. CpG island methylation in sporadic and neurofibromatis type 2-associated schwannomas. Clin Cancer Res Off J Am Assoc Cancer Res. 2003;9(15):5601–6.
Koutsimpelas D, Ruerup G, Mann WJ, Brieger J. Lack of neurofibromatosis type 2 gene promoter methylation in sporadic vestibular schwannomas. ORL J Oto-Rhino-Laryngol Relat Spec. 2012;74(1):33–7.
Kullar PJ, Pearson DM, Malley DS, Collins VP, Ichimura K. CpG island hypermethylation of the neurofibromatosis type 2 (NF2) gene is rare in sporadic vestibular schwannomas. Neuropathol Appl Neurobiol. 2010;36(6):505–14.
Bello MJ, Martinez-Glez V, Franco-Hernandez C, Pefla-Granero C, de Campos JM, Isla A, et al. DNA methylation pattern in 16 tumor-related genes in schwannomas. Cancer Genet Cytogenet. 2007;172(1):84–6.
Lassaletta L, Bello MJ, Del Río L, Alfonso C, Roda JM, Rey JA, et al. DNA methylation of multiple genes in vestibular schwannoma: relationship with clinical and radiological findings. Otol Neurotol. 2006;27(8):1180–5.
Ahmad ZK, Altuna X, Lopez JP, An Y, Wang-Rodriguez J, Juneja VR, et al. p73 expression and function in vestibular schwannoma. Arch Otolaryngol Head Neck Surg. 2009;135(7):662–9.
Jelinek J, Gharibyan V, He R, Saraf A, Lau S, Prchal J, Issa JP. DNA methylation of HOX genes in leukemia and myeloproliferative disorders. Cancer Res. 2007;67(9 Supplement):235.
Tommasi S, Karm DL, Wu X, Yen Y, Pfeifer GP. Methylation of homeobox genes is a frequent and early epigenetic event in breast cancer. Breast Cancer Res. 2009;11(1):1–17.
Torres-Martín M, Lassaletta L, de Campos JM, Isla A, Pinto GR, Burbano RR, et al. Genome-wide methylation analysis in vestibular schwannomas shows putative mechanisms of gene expression modulation and global hypomethylation at the HOX gene cluster. Genes Chromosom Cancer. 2015;54(4):197–209.
Chang LS, Akhmametyeva EM, Wu Y, Zhu L, Welling DB. Multiple transcription initiation sites, alternative splicing, and differential polyadenylation contribute to the complexity of human neurofibromatosis 2 transcripts. Genomics. 2002;79(1):63–76.
Cioffi JA, Yue WY, Mendolia-Loffredo S, Hansen KR, Wackym PA, Hansen MR. MicroRNA-21 overexpression contributes to vestibular schwannoma cell proliferation and survival. Otol Neurotol. 2010;31(9):1455–62.
Torres-Martin M, Lassaletta L, de Campos JM, Isla A, Gavilan J, Pinto GR, et al. Global profiling in vestibular schwannomas shows critical deregulation of microRNAs and upregulation in those included in chromosomal region 14q32. PLoS ONE. 2013;8(6):e65868.
Petrilli A, Bott M, Fernández-Valle C. Inhibition of SIRT2 in merlin/NF2-mutant Schwann cells triggers necrosis. Oncotarget. 2013;4(12):2354–65.
Sahm F, Schrimpf D, Stichel D, Jones DT, Hielscher T, Schefzyk S, et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 2017;18(5):682–94.
Patronas NJ, Courcoutsakis N, Bromley CM, Katzman GL, MacCollin M, Parry DM. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology. 2001;218(2):434–42.
Dow G, Biggs N, Evans G, Gillespie J, Ramsden R, King A. Spinal tumors in neurofibromatosis type 2 Is emerging knowledge of genotype predictive of natural history? J Neurosurg Spine. 2005;2(5):574–9.
Evans DG, et al. Neurofibromatosis 2. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, et al., editors. GeneReviews Seattle (WA): University of Washington, Seattle Copyright © University of Washington Seattle. Gene Reviews is a registered trademark of the all rights reserved. Seattle: University of Washington; 1993. p. 1993–2022.
Gaudioso C, Listernick R, Fisher MJ, Campen CJ, Paz A, Gutmann DH. Neurofibromatosis 2 in children presenting during the first decade of life. Neurology. 2019;93(10):e964–7.
Legoupil S, Bessis D, Picard F, Mallet S, Mazereeuw J, Phan A, et al. Dermatologic manifestations in paediatric neurofibromatosis type 2: a cross sectional descriptive multicentric study. Orphanet J Rare Dis. 2022;17(1):242.
Godel T, Bäumer P, Farschtschi S, Gugel I, Kronlage M, Hofstadler B, et al. Peripheral nervous system alterations in infant and adult neurofibromatosis type 2. Neurology. 2019;93(6):e590–8.
Evans DG, Birch JM, Ramsden RT. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child. 1999;81(6):496–9.
Sperfeld AD, Hein C, Schröder JM, Ludolph AC, Hanemann CO. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain J Neurol. 2002;125(Pt 5):996–1004.
Gan J, Zhang Y, Wu J, Lei D, Zhang F, Zhao H, et al. Current understanding of hearing loss in sporadic vestibular schwannomas: a systematic review. Front Oncol. 2021;11:687201.
McClatchey AI, Giovannini M. Membrane organization and tumorigenesis–the NF2 tumor suppressor. Merlin Genes Dev. 2005;19(19):2265–77.
Sakamoto T, Fukuda S, Inuyama Y. Hearing loss and growth rate of acoustic neuromas in follow-up observation policy. Auris Nasus Larynx. 2001;28(Suppl):S23–7.
Caye-Thomasen P, Dethloff T, Hansen S, Stangerup SE, Thomsen J. Hearing in patients with intracanalicular vestibular schwannomas. Audiol Neurootol. 2007;12(1):1–12.
Kanzaki J, Ogawa K, Inoue Y, Shiobara R, Toya S. Quality of hearing preservation in acoustic neuroma surgery. Am J Otol. 1998;19(5):644–8.
Sughrue ME, Kaur R, Kane AJ, Rutkowski MJ, Yang I, Pitts LH, et al. Intratumoral hemorrhage and fibrosis in vestibular schwannoma: a possible mechanism for hearing loss. J Neurosurg. 2011;114(2):386–93.
Badie B, Pyle GM, Nguyen PH, Hadar EJ. Elevation of internal auditory canal pressure by vestibular schwannomas. Otol Neurotol. 2001;22(5):696–700.
Dilwali S, Landegger LD, Soares VY, Deschler DG, Stankovic KM. Secreted factors from human vestibular schwannomas can cause cochlear damage. Sci Rep. 2015;5:18599.
Soares VY, Atai NA, Fujita T, Dilwali S, Sivaraman S, Landegger LD, et al. Extracellular vesicles derived from human vestibular schwannomas associated with poor hearing damage cochlear cells. Neuro Oncol. 2016;18(11):1498–507.
Neurofibromatosis. Conference statement. National institutes of health consensus development conference. Arch Neurol. 1988;45(5):575–8. https://doi.org/10.1001/archneur.1988.00520290115023
Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603–18.
Anand G, Vasallo G, Spanou M, Thomas S, Pike M, Kariyawasam DS, et al. Diagnosis of sporadic neurofibromatosis type 2 in the paediatric population. Arch Dis Child. 2018;103(5):463–9.
Evans DG, King AT, Bowers NL, Tobi S, Wallace AJ, Perry M, et al. Identifying the deficiencies of current diagnostic criteria for neurofibromatosis 2 using databases of 2777 individuals with molecular testing. Genet Med. 2019;21(7):1525–33.
Plotkin SR, Messiaen L, Legius E, Pancza P, Avery RA, Blakeley JO, et al. Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: an international consensus recommendation. Genet Med. 2022. https://doi.org/10.1016/j.gim.2022.05.007.
Evans DG, Wen PY. NF2-related schwannomatosis (formerly neurofibromatosis type 2). In: UpToDate, Post TW (Ed), UpToDate, Waltham, MA. (last updated: Oct 28, 2022)
Louvrier C, Pasmant E, Briand-Suleau A, Cohen J, Nitschké P, Nectoux J, et al. Targeted next-generation sequencing for differential diagnosis of neurofibromatosis type 2, schwannomatosis, and meningiomatosis. Neuro Oncol. 2018;20(7):917–29.
Halliday D, Emmanouil B, Evans DGR. Updated protocol for genetic testing, screening, and clinical management of individuals at risk of NF2-related schwannomatosis. Clin Genet. 2023. https://doi.org/10.1111/cge.14310.
National Health Service (NHS). Treatment of Neurofibromatosis type 2. 2023. https://www.nhs.uk/conditions/neurofibromatosis-type-2/treatment/.
Lu VM, Ravindran K, Graffeo CS, Perry A, Van Gompel JJ, Daniels DJ, et al. Efficacy and safety of bevacizumab for vestibular schwannoma in neurofibromatosis type 2: a systematic review and meta-analysis of treatment outcomes. J Neurooncol. 2019;144(2):239–48.
Wentworth S, Pinn M, Bourland JD, Deguzman AF, Ekstrand K, Ellis TL, et al. Clinical experience with radiation therapy in the management of neurofibromatosis-associated central nervous system tumors. Int J Radiat Oncol Biol Phys. 2009;73(1):208–13.
Grimm SA, Kumthekar P, Chamberlain MC, Schiff D, Wen PY, Iwamoto FM, et al. Phase II trial of bevacizumab in patients with surgery and radiation refractory progressive meningioma. J Clin Oncol. 2015;33(15 Suppl):2055.
Kalamarides M, Essayed W, Lejeune JP, Aboukais R, Sterkers O, Bernardeschi D, et al. Spinal ependymomas in NF2: a surgical disease? J Neurooncol. 2018;136(3):605–11.
Farschtschi S, Merker VL, Wolf D, Schuhmann M, Blakeley J, Plotkin SR, et al. Bevacizumab treatment for symptomatic spinal ependymomas in neurofibromatosis type 2. Acta Neurol Scand. 2016;133(6):475–80.
Snyder MH, Ampie L, DiDomenico JD, Asthagiri AR. Bevacizumab as a surgery-sparing agent for spinal ependymoma in patients with neurofibromatosis type II: systematic review and case. J Clin Neurosci. 2021;86:79–84.
Deep NL, Patel EJ, Shapiro WH, Waltzman SB, Jethanamest D, McMenomey SO, et al. Cochlear implant outcomes in neurofibromatosis type 2: implications for management. Otol Neurotol. 2021;42(4):540–8.
Neff BA, Wiet RM, Lasak JM, Cohen NL, Pillsbury HC, Ramsden RT, et al. Cochlear implantation in the neurofibromatosis type 2 patient: long-term follow-up. Laryngoscope. 2007;117(6):1069–72.
Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: substantial risks in certain tumour prone syndromes. J Med Genet. 2006;43(4):289–94.
Dewan R, Pemov A, Kim HJ, Morgan KL, Vasquez RA, Chittiboina P, et al. Evidence of polyclonality in neurofibromatosis type 2-associated multilobulated vestibular schwannomas. Neuro Oncol. 2015;17(4):566–73.
Evans DG, Baser ME, O’Reilly B, Rowe J, Gleeson M, Saeed S, et al. Management of the patient and family with neurofibromatosis 2: a consensus conference statement. Br J Neurosurg. 2005;19(1):5–12.
Chung LK, Nguyen TP, Sheppard JP, Lagman C, Tenn S, Lee P, et al. A systematic review of radiosurgery versus surgery for neurofibromatosis type 2 vestibular schwannomas. World Neurosurg. 2018;109:47–58.
Rowe JG, Radatz MW, Walton L, Soanes T, Rodgers J, Kemeny AA. Clinical experience with gamma knife stereotactic radiosurgery in the management of vestibular schwannomas secondary to type 2 neurofibromatosis. J Neurol Neurosurg Psychiatry. 2003;74(9):1288–93.
Sanchez LD, Bui A, Klesse LJ. Targeted therapies for the neurofibromatoses. Cancers. 2021;13(23):6032.
Dirks MS, Butman JA, Kim HJ, Wu T, Morgan K, Tran AP, et al. Long-term natural history of neurofibromatosis type 2-associated intracranial tumors. J Neurosurg. 2012;117(1):109–17.
Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72(5):791–800.
Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4DCAF1 in the nucleus. J Cell. 2010;140(4):477–90.
Petrilli AM, Fuse MA, Donnan MS, Bott M, Sparrow NA, Tondera D, et al. A chemical biology approach identified PI3K as a potential therapeutic target for neurofibromatosis type 2. Am J Trans Res. 2014;6(5):471–93.
Lim JY, Kim H, Jeun SS, Kang SG, Lee KJ. Merlin inhibits growth hormone-regulated Raf-ERKs pathways by binding to Grb2 protein. Biochem Biophys Res Commun. 2006;340(4):1151–7.
Kim M, Kim S, Lee S, Kim W, Sohn M, Kim H, et al. Merlin inhibits Wnt/β-catenin signaling by blocking LRP6 phosphorylation. J Cell Death Differ. 2016;23(10):1638–47.
James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29(15):4250–61.
Zhang Y, Long J, Ren J, Huang X, Zhong P, Wang B. Potential molecular biomarkers of vestibular schwannoma growth: progress and prospects. Front Oncol. 2021;11:731441.
Zoch A, Mayerl S, Schulz A, Greither T, Frappart L, Rübsam J, et al. Merlin isoforms 1 and 2 both act as tumour suppressors and are required for optimal sperm maturation. PLoS ONE. 2015;10(8):e0129151.
Pećina-Šlaus N. Merlin, the NF2 gene product. Pathol Oncol Res. 2013;19(3):365–73.
Turunen O, Sainio M, Jääskeläinen J, Carpén O, Vaheri A. Structure-function relationships in the ezrin family and the effect of tumor-associated point mutations in neurofibromatosis 2 protein. Biochim Biophys Acta. 1998;1387(1–2):1–16.
Li Q, Nance MR, Kulikauskas R, Nyberg K, Fehon R, Karplus PA, et al. Self-masking in an intact ERM-merlin protein: an active role for the central alpha-helical domain. J Mol Biol. 2007;365(5):1446–59.
Gary R, Bretscher A. Ezrin self-association involves binding of an N-terminal domain to a normally masked C-terminal domain that includes the F-actin binding site. Mol Biol Cell. 1995;6(8):1061–75.
McClatchey AI, Fehon RG. Merlin and the ERM proteins–regulators of receptor distribution and signaling at the cell cortex. Trends Cell Biol. 2009;19(5):198–206.
Xu HM, Gutmann DH. Merlin differentially associates with the microtubule and actin cytoskeleton. J Neurosci Res. 1998;51(3):403–15.
Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 2001;15(8):968–80.
Bretscher A, Edwards K, Fehon RG. ERM proteins and merlin: integrators at the cell cortex. Nat Rev Mol Cell Biol. 2002;3(8):586–99.
Nguyen R, Reczek D, Bretscher A. Hierarchy of merlin and ezrin N- and C-terminal domain interactions in homo- and heterotypic associations and their relationship to binding of scaffolding proteins EBP50 and E3KARP. J Biol Chem. 2001;276(10):7621–9.
Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101(3):259–70.
Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochim Biophys Acta. 2008;1785(1):32–54.
Yogesha SD, Sharff AJ, Giovannini M, Bricogne G, Izard T. Unfurling of the band 41, ezrin, radixin, moesin (FERM) domain of the merlin tumor suppressor. Protein Sci. 2011;20(12):2113–20.
Fievet BT, Gautreau A, Roy C, Del Maestro L, Mangeat P, Louvard D, et al. Phosphoinositide binding and phosphorylation act sequentially in the activation mechanism of ezrin. J Cell Biol. 2004;164(5):653–9.
Mori T, Kitano K, Terawaki S, Maesaki R, Fukami Y, Hakoshima T. Structural basis for CD44 recognition by ERM proteins. J Biol Chem. 2008;283(43):29602–12.
Hamada K, Shimizu T, Yonemura S, Tsukita S, Tsukita S, Hakoshima T. Structural basis of adhesion-molecule recognition by ERM proteins revealed by the crystal structure of the radixin-ICAM-2 complex. Embo j. 2003;22(3):502–14.
Takai Y, Kitano K, Terawaki S, Maesaki R, Hakoshima T. Structural basis of the cytoplasmic tail of adhesion molecule CD43 and its binding to ERM proteins. J Mol Biol. 2008;381(3):634–44.
Tang X, Jang SW, Wang X, Liu Z, Bahr SM, Sun SY, et al. Akt phosphorylation regulates the tumour-suppressor merlin through ubiquitination and degradation. Nat Cell Biol. 2007;9(10):1199–207.
Ye K. Phosphorylation of merlin regulates its stability and tumor suppressive activity. Cell Adh Migr. 2007;1(4):196–8.
Laulajainen M, Muranen T, Carpén O, Grönholm M. Protein kinase A-mediated phosphorylation of the NF2 tumor suppressor protein merlin at serine 10 affects the actin cytoskeleton. Oncogene. 2008;27(23):3233–43.
Surace EI, Haipek CA, Gutmann DH. Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene. 2004;23(2):580–7.
Alfthan K, Heiska L, Grönholm M, Renkema GH, Carpén O. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. J Biol Chem. 2004;279(18):18559–66.
Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006;442(7102):576–9.
Laulajainen M, Muranen T, Nyman TA, Carpén O, Grönholm M. Multistep phosphorylation by oncogenic kinases enhances the degradation of the NF2 tumor suppressor merlin. Neoplasia. 2011;13(7):643–52.
Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. 2018;17(1):58.
Bai Y, Liu YJ, Wang H, Xu Y, Stamenkovic I, Yu Q. Inhibition of the hyaluronan-CD44 interaction by merlin contributes to the tumor-suppressor activity of merlin. Oncogene. 2007;26(6):836–50.
Ammoun S, Cunliffe CH, Allen JC, Chiriboga L, Giancotti FG, Zagzag D, et al. ErbB/HER receptor activation and preclinical efficacy of lapatinib in vestibular schwannoma. Neuro Oncol. 2010;12(8):834–43.
Mukherjee J, Kamnasaran D, Balasubramaniam A, Radovanovic I, Zadeh G, Kiehl TR, et al. Human schwannomas express activated platelet-derived growth factor receptors and c-kit and are growth inhibited by Gleevec (Imatinib Mesylate). Cancer Res. 2009;69(12):5099–107.
Agnihotri S, Gugel I, Remke M, Bornemann A, Pantazis G, Mack SC, et al. Gene-expression profiling elucidates molecular signaling networks that can be therapeutically targeted in vestibular schwannoma. J J Neurosurg. 2014;121(6):1434–45.
Jacob A, Lee TX, Neff BA, Miller S, Welling B, Chang L-S. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. J Otol Neurotol. 2008;29(1):58–68.
Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci USA. 2004;101(52):18200–5.
López-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol Cell Biol. 2009;29(15):4235–49.
Lee S, Karas PJ, Hadley CC, Bayley VJ, Khan AB, Jalali A, et al. The role of merlin/NF2 loss in meningioma biology. Cancers (Basel). 2019. https://doi.org/10.3390/cancers11111633.
Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014;94(4):1287–312.
Boin A, Couvelard A, Couderc C, Brito I, Filipescu D, Kalamarides M, et al. Proteomic screening identifies a YAP-driven signaling network linked to tumor cell proliferation in human schwannomas. J Neuro Oncol. 2014;16(9):1196–209.
Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4DCAF1-mediated inhibition of the Hippo pathway kinases Lats1 and 2 in the nucleus. J Cancer cell. 2014;26(1):48–60.
Zhou L, Ercolano E, Ammoun S, Schmid MC, Barczyk MA, Hanemann CO. Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/β-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. J Neoplasia. 2011;13(12):1101-IN2.
Morrow KA, Das S, Meng E, Menezes ME, Bailey SK, Metge BJ, et al. Loss of tumor suppressor Merlin results in aberrant activation of Wnt/β-catenin signaling in cancer. J Oncotarget. 2016;7(14):17991.
Bosco EE, Nakai Y, Hennigan RF, Ratner N, Zheng Y. NF2-deficient cells depend on the Rac1-canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. J Oncogene. 2010;29(17):2540–9.
Hikasa H, Sekido Y, Suzuki A. Merlin/NF2-Lin28B-let-7 is a tumor-suppressive pathway that is cell-density dependent and hippo independent. J Cell reports. 2016;14(12):2950–61.
Blakeley J. Development of drug treatments for neurofibromatosis type 2-associated vestibular schwannoma. Curr Opin Otolaryngol Head Neck Surg. 2012;20(5):372–9.
Zou Z, Tao T, Li H, Zhu X. mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31.
Giovannini M, Bonne NX, Vitte J, Chareyre F, Tanaka K, Adams R, et al. mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro Oncol. 2014;16(4):493–504.
Goutagny S, Giovannini M, Kalamarides M. A 4-year phase II study of everolimus in NF2 patients with growing vestibular schwannomas. J Neurooncol. 2017;133(2):443–5.
Goutagny S, Raymond E, Esposito-Farese M, Trunet S, Mawrin C, Bernardeschi D, et al. Phase II study of mTORC1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. J Neurooncol. 2015;122(2):313–20.
Lee TX, Packer MD, Huang J, Akhmametyeva EM, Kulp SK, Chen CS, et al. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. Eur J Cancer. 2009;45(9):1709–20.
Bush ML, Oblinger J, Brendel V, Santarelli G, Huang J, Akhmametyeva EM, et al. AR42, a novel histone deacetylase inhibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro Oncol. 2011;13(9):983–99.
Welling DB, Collier KA, Burns SS, Oblinger JL, Shu E, Miles-Markley BA, et al. Early phase clinical studies of AR-42, a histone deacetylase inhibitor, for neurofibromatosis type 2-associated vestibular schwannomas and meningiomas. Laryngoscope Investig Otolaryngol. 2021;6(5):1008–19.
Collier KA, Valencia H, Newton H, Hade EM, Sborov DW, Cavaliere R, et al. A phase 1 trial of the histone deacetylase inhibitor AR-42 in patients with neurofibromatosis type 2-associated tumors and advanced solid malignancies. Cancer Chemother Pharmacol. 2021;87(5):599–611.
Cayé-Thomasen P, Werther K, Nalla A, Bøg-Hansen TC, Nielsen HJ, Stangerup SE, et al. VEGF and VEGF receptor-1 concentration in vestibular schwannoma homogenates correlates to tumor growth rate. Otol Neurotol. 2005;26(1):98–101.
Wong HK, Lahdenranta J, Kamoun WS, Chan AW, McClatchey AI, Plotkin SR, et al. Anti-vascular endothelial growth factor therapies as a novel therapeutic approach to treating neurofibromatosis-related tumors. Cancer Res. 2010;70(9):3483–93.
Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, Halpin C, Padera TP, Tyrrell A, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358–67.
Plotkin SR, Merker VL, Halpin C, Jennings D, McKenna MJ, Harris GJ, et al. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: a retrospective review of 31 patients. Otol Neurotol. 2012;33(6):1046–52.
Plotkin SR, Duda DG, Muzikansky A, Allen J, Blakeley J, Rosser T, et al. Multicenter, prospective, phase II and biomarker study of high-dose bevacizumab as induction therapy in patients with neurofibromatosis type 2 and progressive vestibular schwannoma. J Clin Oncol. 2019;37(35):3446–54.
Gugel I, Kluwe L, Zipfel J, Teuber C, Tatagiba M, Mautner VF, et al. Minimal effect of bevacizumab treatment on residual vestibular schwannomas after partial resection in young neurofibromatosis type 2 patients. Cancers (Basel). 2019. https://doi.org/10.3390/cancers11121862.
Renzi S, Michaeli O, Salvador H, Alderete D, Ponce NF, Zapotocky M, et al. Bevacizumab for NF2-associated vestibular schwannomas of childhood and adolescence. Pediatr Blood Cancer. 2020;67(5):e28228.
Slusarz KM, Merker VL, Muzikansky A, Francis SA, Plotkin SR. Long-term toxicity of bevacizumab therapy in neurofibromatosis 2 patients. Cancer Chemother Pharmacol. 2014;73(6):1197–204.
Farschtschi S, Kollmann P, Dalchow C, Stein A, Mautner VF. Reduced dosage of bevacizumab in treatment of vestibular schwannomas in patients with neurofibromatosis type 2. Eur Arch Otorhinolaryngol. 2015;272(12):3857–60.
Tamura R, Fujioka M, Morimoto Y, Ohara K, Kosugi K, Oishi Y, et al. A VEGF receptor vaccine demonstrates preliminary efficacy in neurofibromatosis type 2. Nat Commun. 2019;10(1):5758.
Gao X, Zhao Y, Stemmer-Rachamimov AO, Liu H, Huang P, Chin S, et al. Anti-VEGF treatment improves neurological function and augments radiation response in NF2 schwannoma model. Proc Natl Acad Sci USA. 2015;112(47):14676–81.
Phadnis S, Hagiwara M, Yaffe A, Mitchell C, Nicolaides T, Akshintala S, et al. NFB-08. Phase II study of axitinib in patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro Oncol. 2020;22(Suppl 3):iii419.
Blakeley JO, Ye X, Duda DG, Halpin CF, Bergner AL, Muzikansky A, et al. Efficacy and biomarker study of bevacizumab for hearing loss resulting from neurofibromatosis type 2-associated vestibular schwannomas. J Clin Oncol. 2016;34(14):1669–75.
Dilwali S, Roberts D, Stankovic KM. Interplay between VEGF-A and cMET signaling in human vestibular schwannomas and schwann cells. Cancer Biol Ther. 2015;16(1):170–5.
Konstorum A, Lowengrub JS. Activation of the HGF/c-Met axis in the tumor microenvironment: a multispecies model. J Theor Biol. 2018;439:86–99.
Delitto D, Vertes-George E, Hughes SJ, Behrns KE, Trevino JG. c-Met signaling in the development of tumorigenesis and chemoresistance: potential applications in pancreatic cancer. World J Gastroenterol. 2014;20(26):8458–70.
Xing F, Liu Y, Sharma S, Wu K, Chan MD, Lo HW, et al. Activation of the c-Met pathway mobilizes an inflammatory network in the brain microenvironment to promote brain metastasis of breast cancer. Cancer Res. 2016;76(17):4970–80.
Zhao Y, Liu P, Zhang N, Chen J, Landegger LD, Wu L, et al. Targeting the cMET pathway augments radiation response without adverse effect on hearing in NF2 schwannoma models. Proc Natl Acad Sci USA. 2018;115(9):E2077–84.
Fuse MA, Plati SK, Burns SS, Dinh CT, Bracho O, Yan D, et al. Combination therapy with c-Met and Src inhibitors induces caspase-dependent apoptosis of merlin-deficient schwann cells and suppresses growth of schwannoma cells. Mol Cancer Ther. 2017;16(11):2387–98.
Lyons DA, Pogoda H-M, Voas MG, Woods IG, Diamond B, Nix R, et al. erbb3 and erbb2 are essential for schwann cell migration and myelination in zebrafish. Curr Biol. 2005;15(6):513–24.
Ahmad ZK, Brown CM, Cueva RA, Ryan AF, Doherty JK. ErbB expression, activation, and inhibition with lapatinib and tyrphostin (AG825) in human vestibular schwannomas. Otol Neurotol. 2011;32(5):841–7.
Karajannis MA, Legault G, Hagiwara M, Ballas MS, Brown K, Nusbaum AO, et al. Phase II trial of lapatinib in adult and pediatric patients with neurofibromatosis type 2 and progressive vestibular schwannomas. Neuro Oncol. 2012;14(9):1163–70.
Osorio DS, Hu J, Mitchell C, Allen JC, Stanek J, Hagiwara M, et al. Effect of lapatinib on meningioma growth in adults with neurofibromatosis type 2. J Neurooncol. 2018;139(3):749–55.
Zhao F, Li SW, Zhang S, Li P, Zhao C, Zhao XB, et al. Phase II trial of icotinib in adult patients with neurofibromatosis type 2 and progressive vestibular schwannoma. J Neurosurg. 2022. https://doi.org/10.3171/2022.9.JNS22699.
Clark JJ, Provenzano M, Diggelmann HR, Xu N, Hansen SS, Hansen MR. The ErbB inhibitors trastuzumab and erlotinib inhibit growth of vestibular schwannoma xenografts in nude mice: a preliminary study. Otol Neurotol. 2008;29(6):846–53.
Plotkin SR, Halpin C, McKenna MJ, Loeffler JS, Batchelor TT, Barker FG 2nd. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135–43.
Altuna X, Lopez JP, Yu MA, Arandazi MJ, Harris JP, Wang-Rodriguez J, et al. Potential role of imatinib mesylate (Gleevec, STI-571) in the treatment of vestibular schwannoma. Otol Neurotol. 2011;32(1):163–70.
Yener U, Avsar T, Akgün E, Şeker A, Bayri Y, Kılıç T. Assessment of antiangiogenic effect of imatinib mesylate on vestibular schwannoma tumors using in vivo corneal angiogenesis assay. J Neurosurg. 2012;117(4):697–704.
Ammoun S, Schmid MC, Triner J, Manley P, Hanemann CO. Nilotinib alone or in combination with selumetinib is a drug candidate for neurofibromatosis type 2. Neuro Oncol. 2011;13(7):759–66.
Petrilli AM, Garcia J, Bott M, Klingeman Plati S, Dinh CT, Bracho OR, et al. Ponatinib promotes a G1 cell-cycle arrest of merlin/NF2-deficient human schwann cells. Oncotarget. 2017;8(19):31666–81.
Sagers JE, Beauchamp RL, Zhang Y, Vasilijic S, Wu L, DeSouza P, et al. Combination therapy with mTOR kinase inhibitor and dasatinib as a novel therapeutic strategy for vestibular schwannoma. Sci Rep. 2020;10(1):4211.
Mizuno T, Murakami H, Fujii M, Ishiguro F, Tanaka I, Kondo Y, et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene. 2012;31(49):5117–22.
Guerrant W, Kota S, Troutman S, Mandati V, Fallahi M, Stemmer-Rachamimov A, et al. YAP mediates tumorigenesis in neurofibromatosis type 2 by promoting cell survival and proliferation through a COX-2–EGFR signaling axis. Can Res. 2016;76(12):3507–19.
Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia. 2008;10(11):1204–12.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300–5.
Al-Moujahed A, Brodowska K, Stryjewski TP, Efstathiou NE, Vasilikos I, Cichy J, et al. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci Rep. 2017;7(1):7602.
Zhang WQ, Dai YY, Hsu PC, Wang H, Cheng L, Yang YL, et al. Targeting YAP in malignant pleural mesothelioma. J Cell Mol Med. 2017;21(11):2663–76.
Tolcher AW, Lakhani NJ, McKean M, Lingaraj T, Victor L, Sanchez-Martin M, et al. A phase 1, first-in-human study of IK-930, an oral TEAD inhibitor targeting the Hippo pathway in subjects with advanced solid tumors. Am Soc Clin Oncol. 2022. https://doi.org/10.1200/JCO.2022.40.16_suppl.TPS3168.
Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell. 2014;25(2):166–80.
Santinon G, Pocaterra A, Dupont S. Control of YAP/TAZ activity by metabolic and nutrient-sensing pathways. Trends Cell Biol. 2016;26(4):289–99.
Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, et al. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol. 2015;17(4):500–10.
White SM, Avantaggiati ML, Nemazanyy I, Di Poto C, Yang Y, Pende M, et al. YAP/TAZ inhibition induces metabolic and signaling rewiring resulting in targetable vulnerabilities in NF2-deficient tumor cells. Dev Cell. 2019;49(3):425-43.e9.
Casey D, Demko S, Sinha A, Mishra-Kalyani PS, Shen YL, Khasar S, et al. FDA approval summary: selumetinib for plexiform neurofibroma. Clin Cancer Res. 2021;27(15):4142–6.
Chang LS, Oblinger JL, Smith AE, Ferrer M, Angus SP, Hawley E, et al. Brigatinib causes tumor shrinkage in both NF2-deficient meningioma and schwannoma through inhibition of multiple tyrosine kinases but not ALK. PLoS ONE. 2021;16(7):e0252048.
Brastianos PK, Twohy EL, Gerstner ER, Kaufmann TJ, Iafrate AJ, Lennerz J, et al. Alliance A071401: phase II trial of focal adhesion kinase inhibition in meningiomas with somatic NF2 mutations. J Clin Oncol. 2022. https://doi.org/10.1200/JCO.21.02371.
Hannan CJ, Lewis D, O’Leary C, Donofrio CA, Evans DG, Roncaroli F, et al. The inflammatory microenvironment in vestibular schwannoma. Neuro Oncol Adv. 2020;2(1):023.
de Vries M, Briaire-de Bruijn I, Malessy MJ, de Bruïne SF, van der Mey AG, Hogendoorn PC. Tumor-associated macrophages are related to volumetric growth of vestibular schwannomas. Otol Neurotol. 2013;34(2):347–52.
Lewis D, Roncaroli F, Agushi E, Mosses D, Williams R, Li KL, et al. Inflammation and vascular permeability correlate with growth in sporadic vestibular schwannoma. Neuro Oncol. 2019;21(3):314–25.
Schulz A, Büttner R, Hagel C, Baader SL, Kluwe L, Salamon J, et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathol. 2016;132(2):289–307.
de Vries WM, Briaire-de Bruijn IH, van Benthem PPG, van der Mey AGL, Hogendoorn PCW. M-CSF and IL-34 expression as indicators for growth in sporadic vestibular schwannoma. Virchows Archiv. 2019;474(3):375–81.
Wang S, Liechty B, Patel S, Weber JS, Hollmann TJ, Snuderl M, et al. Programmed death ligand 1 expression and tumor infiltrating lymphocytes in neurofibromatosis type 1 and 2 associated tumors. J Neurooncol. 2018;138(1):183–90.
Wang Y, Li P, Wang B, Wang S, Liu P. Identification of myeloid-derived suppressor cells that have an immunosuppressive function in NF2 patients. J Cancer Res Clin Oncol. 2019;145(2):523–33.
Breun M, Monoranu CM, Kessler AF, Matthies C, Löhr M, Hagemann C, et al. [(68)Ga]-Pentixafor PET/CT for CXCR4-mediated imaging of vestibular schwannomas. Front Oncol. 2019;9:503.
Breun M, Schwerdtfeger A, Martellotta DD, Kessler AF, Perez JM, Monoranu CM, et al. CXCR4: a new player in vestibular schwannoma pathogenesis. Oncotarget. 2018;9(11):9940–50.
Taurone S, Bianchi E, Attanasio G, Di Gioia C, Ierinó R, Carubbi C, et al. Immunohistochemical profile of cytokines and growth factors expressed in vestibular schwannoma and in normal vestibular nerve tissue. Mol Med Rep. 2015;12(1):737–45.
Kim JY, Kim H, Jeun SS, Rha SJ, Kim YH, Ko YJ, et al. Inhibition of NF-kappaB activation by merlin. Biochem Biophys Res Commun. 2002;296(5):1295–302.
Ammoun S, Provenzano L, Zhou L, Barczyk M, Evans K, Hilton DA, et al. Axl/Gas6/NFκB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene. 2014;33(3):336–46.
Dilwali S, Briët MC, Kao SY, Fujita T, Landegger LD, Platt MP, et al. Preclinical validation of anti-nuclear factor-kappa B therapy to inhibit human vestibular schwannoma growth. Mol Oncol. 2015;9(7):1359–70.
Sagers JE, Brown AS, Vasilijic S, Lewis RM, Sahin MI, Landegger LD, et al. Publisher correction: computational repositioning and preclinical validation of mifepristone for human vestibular schwannoma. Sci Rep. 2018;8(1):17449.
Check JH, Wilson C, Cohen R, Sarumi M. Evidence that mifepristone, a progesterone receptor antagonist, can cross the blood brain barrier and provide palliative benefits for glioblastoma multiforme grade IV. Anticancer Res. 2014;34(5):2385–8.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26.
Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci USA. 2017;114(37):E7766–75.
Sagers JE, Sahin MI, Moon I, Ahmed SG, Stemmer-Rachamimov A, Brenner GJ, et al. NLRP3 inflammasome activation in human vestibular schwannoma: Implications for tumor-induced hearing loss. Hear Res. 2019;381:107770.
Dilwali S, Kao SY, Fujita T, Landegger LD, Stankovic KM. Nonsteroidal anti-inflammatory medications are cytostatic against human vestibular schwannomas. Transl Res. 2015;166(1):1–11.
Hong B, Krusche CA, Schwabe K, Friedrich S, Klein R, Krauss JK, et al. Cyclooxygenase-2 supports tumor proliferation in vestibular schwannomas. Neurosurgery. 2011;68(4):1112–7.
Kandathil CK, Dilwali S, Wu CC, Ibrahimov M, McKenna MJ, Lee H, et al. Aspirin intake correlates with halted growth of sporadic vestibular schwannoma in vivo. Otol Neurotol. 2014;35(2):353–7.
Kandathil CK, Cunnane ME, McKenna MJ, Curtin HD, Stankovic KM. Correlation between aspirin intake and reduced growth of human vestibular schwannoma: volumetric analysis. Otol Neurotol. 2016;37(9):1428–34.
MacKeith S, Wasson J, Baker C, Guilfoyle M, John D, Donnelly N, et al. Aspirin does not prevent growth of vestibular schwannomas: a case-control study. Laryngoscope. 2018;128(9):2139–44.
Behling F, Ries V, Skardelly M, Gepfner-Tuma I, Schuhmann M, Ebner FH, et al. COX2 expression is associated with proliferation and tumor extension in vestibular schwannoma but is not influenced by acetylsalicylic acid intake. Acta Neuropathol Commun. 2019;7(1):105.
Van Gompel JJ, Agazzi S, Carlson ML, Adewumi DA, Hadjipanayis CG, Uhm JH, et al. Congress of neurological surgeons systematic review and evidence-based guidelines on emerging therapies for the treatment of patients with vestibular schwannomas. Neurosurgery. 2018;82(2):E52–4.
Ma J, Cai Z, Wei H, Liu X, Zhao Q, Zhang T. The anti-tumor effect of aspirin: what we know and what we expect. Biomed Pharmacother. 2017;95:656–61.
Akbari H, Taghizadeh-Hesary F, Bahadori M. Mitochondria determine response to anti-programmed cell death protein-1 (anti-PD-1) immunotherapy: An evidence-based hypothesis. Mitochondrion. 2022;62:151–8.
Taghizadeh-Hesary F, Akbari H, Bahadori M, Behnam B. Targeted anti-mitochondrial therapy: the future of oncology. Genes. 2022. https://doi.org/10.3390/genes13101728.
Perry A, Graffeo CS, Carlstrom LP, Raghunathan A, Driscoll CLW, Neff BA, et al. Predominance of M1 subtype among tumor-associated macrophages in phenotypically aggressive sporadic vestibular schwannoma. J Neurosurg. 2019. https://doi.org/10.3171/2019.7.JNS19879.
Munir S, Lundsager MT, Jørgensen MA, Hansen M, Petersen TH, Bonefeld CM, et al. Inflammation induced PD-L1-specific T cells. Cell Stress. 2019;3(10):319–27.
Rutland JW, Gill CM, Loewenstern J, Arib H, Pain M, Umphlett M, et al. NF2 mutation status and tumor mutational burden correlate with immune cell infiltration in meningiomas. Cancer Immunol Immunother. 2021;70(1):169–76.
Bennett J, Wellman J, Marshall KA, McCague S, Ashtari M, DiStefano-Pappas J, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388(10045):661–72.
Delmaghani S, El-Amraoui A. Inner ear gene therapies take off: current promises and future challenges. J of Clin Med. 2020. https://doi.org/10.3390/jcm9072309.
Farhadi M, Razmara E, Balali M, Hajabbas Farshchi Y, Falah M. How Transmembrane Inner Ear (TMIE) plays role in the auditory system: a mystery to us. J Cell Mol Med. 2021;25(13):5869–83.
Pan B, Askew C, Galvin A, Heman-Ackah S, Asai Y, Indzhykulian AA, et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat Biotechnol. 2017;35(3):264–72.
Akil O, Dyka F, Calvet C, Emptoz A, Lahlou G, Nouaille S, et al. Dual AAV-mediated gene therapy restores hearing in a DFNB9 mouse model. Proc Natl Acad Sci USA. 2019;116(10):4496–501.
Al-Moyed H, Cepeda AP, Jung S, Moser T, Kügler S, Reisinger E. A dual-AAV approach restores fast exocytosis and partially rescues auditory function in deaf otoferlin knock-out mice. EMBO Mol Med. 2019. https://doi.org/10.15252/emmm.201809396.
Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33(1):73–80.
Gao X, Tao Y, Lamas V, Huang M, Yeh WH, Pan B, et al. Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature. 2018;553(7687):217–21.
Ren Y, Sagers JE, Landegger LD, Bhatia SN, Stankovic KM. Tumor-penetrating delivery of siRNA against TNFα to human vestibular schwannomas. Sci Rep. 2017;7(1):12922.
Prabhakar S, Beauchamp RL, Cheah PS, Yoshinaga A, Haidar EA, Lule S, et al. Gene replacement therapy in a schwannoma mouse model of neurofibromatosis type 2. Mol Ther Methods Clin Dev. 2022;26:169–80.
Prabhakar S, Taherian M, Gianni D, Conlon TJ, Fulci G, Brockmann J, et al. Regression of schwannomas induced by adeno-associated virus-mediated delivery of caspase-1. Hum Gene Ther. 2013;24(2):152–62.
Ahmed SG, Abdelnabi A, Maguire CA, Doha M, Sagers JE, Lewis RM, et al. Gene therapy with apoptosis-associated speck-like protein, a newly described schwannoma tumor suppressor, inhibits schwannoma growth in vivo. Neuro Oncol. 2019;21(7):854–66.
Ren Y, Landegger LD, Stankovic KM. Gene therapy for human sensorineural hearing loss. Front Cell Neurosci. 2019;13:323.
Catasus N, Rosas I, Bonache S, Negro A, Plana A, Salvador H, et al. Antisense oligonucleotides targeting exon 11 are able to partially rescue the Neurofibromatosis Type 2 phenotype in vitro. Mol Ther Nucleic Acids. 2022. https://doi.org/10.1016/j.omtn.2022.10.026.
Plontke SK, Hartsock JJ, Gill RM, Salt AN. Intracochlear drug injections through the round window membrane: measures to improve drug retention. Audiol Neurotol. 2016;21(2):72–9.
Duan J, Wang Y. Modeling nervous system tumors with human stem cells and organoids. Cell Regen. 2023;12(1):4.
Hung G, Li X, Faudoa R, Xeu Z, Kluwe L, Rhim JS, et al. Establishment and characterization of a schwannoma cell line from a patient with neurofibromatosis 2. Int J Oncol. 2002;20(3):475–82.
Gehlhausen JR, Park SJ, Hickox AE, Shew M, Staser K, Rhodes SD, et al. A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Hum Mol Genet. 2015;24(1):1–8.
Chen J, Landegger LD, Sun Y, Ren J, Maimon N, Wu L, et al. A cerebellopontine angle mouse model for the investigation of tumor biology, hearing, and neurological function in NF2-related vestibular schwannoma. Nat Protoc. 2019;14(2):541–55.
Chang LS, Jacob A, Lorenz M, Rock J, Akhmametyeva EM, Mihai G, et al. Growth of benign and malignant schwannoma xenografts in severe combined immunodeficiency mice. Laryngoscope. 2006;116(11):2018–26.
Stidham KR, Roberson JB Jr. Human vestibular schwannoma growth in the nude mouse: evaluation of a modified subcutaneous implantation model. Am J Otol. 1997;18(5):622–6.
Fernandez-Valle C, Tang Y, Ricard J, Rodenas-Ruano A, Taylor A, Hackler E, et al. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nat Genet. 2002;31(4):354–62.
Welling DB, Guida M, Goll F, Pearl DK, Glasscock ME, Pappas DG, et al. Mutational spectrum in the neurofibromatosis type 2 gene in sporadic and familial schwannomas. Hum Genet. 1996;98(2):189–93.
Roosli C, Linthicum FH Jr, Cureoglu S, Merchant SN. Dysfunction of the cochlea contributing to hearing loss in acoustic neuromas: an underappreciated entity. Otol Neurotol. 2012;33(3):473–80.
Nadol JB Jr, Diamond PF, Thornton AR. Correlation of hearing loss and radiologic dimensions of vestibular schwannomas (acoustic Neuromas). Am J Otol. 1996;17(2):312–6.
Zhao F, Chen Y, Li SW, Zhang J, Zhang S, Zhao XB, et al. Novel patient-derived xenograft and cell line models for therapeutic screening in NF2-associated schwannoma. J Pathol. 2022;257(5):620–34.
Acknowledgements
We thank the staff of the ENT and Head & Neck Research Center, Tehran, Iran, for their contribution.
Funding
This work was supported by Iran University of Medical Sciences (Grant Number: 1400-2-22-21687).
Author information
Authors and Affiliations
Contributions
MAG, performed literature search, drafted the primary manuscript, and prepared the figures; AA, revised the manuscript, confirmed the final manuscript, and supervised the whole study as the expert; MF, revised the manuscript, and confirmed the final manuscript; FTH, prepared the tables, revised the manuscript, and confirmed the final manuscript; MG, revised the manuscript, confirmed the final manuscript, and supervised the whole study as the expert; MF, contributed to study design, performed literature search, drafted the primary manuscript, prepared the figures and tables, confirmed the final manuscript, supervised the whole study as the expert. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ghalavand, M.A., Asghari, A., Farhadi, M. et al. The genetic landscape and possible therapeutics of neurofibromatosis type 2. Cancer Cell Int 23, 99 (2023). https://doi.org/10.1186/s12935-023-02940-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-023-02940-8