
Abstract
Bioactive lipid molecules have been proposed to play important roles linking obesity/metabolic syndrome and cancers. Studies reveal that aberrant lipid metabolic signaling can reprogram cancer cells and non-cancer cells in the tumor microenvironment, contributing to cancer initiation, progression, metastasis, recurrence, and poor therapeutic response. Existing evidence indicates that controlling lipid metabolism can be a potential strategy for cancer prevention and therapy. By reviewing the current literature on the lipid metabolism in various cancers, we summarized major lipid molecules including fatty acids and cholesterol as well as lipid droplets and discussed their critical roles in cancer cells and non-cancer in terms of either promoting- or anti-tumorigenesis. This review provides an overview of the lipid molecules in cellular entities and their tumor microenvironment, adding to the existing knowledge with lipid metabolic reprogramming in immune cells and cancer associated cells. Comprehensive understanding of the regulatory role of lipid metabolism in cellular entities and their tumor microenvironment will provide a new direction for further studies, in a shift away from conventional cancer research. Exploring the lipid-related signaling targets that drive or block cancer development may lead to development of novel anti-cancer strategies distinct from traditional approaches for cancer prevention and treatment.
Similar content being viewed by others
Introduction
Tumor cells are not isolated entities. In fact, the cellular microenvironment plays a critical role in carcinogenesis. For example, hypoxia and nutritional deficiency in the microenvironment usually contribute to a carcinogenic sequence [1, 2]. The crosstalk between tumor cells and the tumor microenvironment (TME) is considered by both basic scientists and clinicians as a pivotal feature of cancer that can be targeted for prevention and therapy [3]. In normal cells, a network system for the metabolism of lipids and glucose supports cell growth and function. In contrast to normal cells that use energy from respiration, most tumor cells depend primarily on fermentative glycolysis, referred to as the “Warburg effect”, even in the presence of oxygen [4]. Warburg effect is characterized by regeneration of NAD + from NADH in the pyruvate to lactate step that completes aerobic glycolysis. Warburg effect is not an efficient means of generating ATP, however the production of lactate from glucose metabolism through Warburg effect occurs 10–100 times faster than the complete oxidation of glucose in the mitochondria [5]. Therefore, Warburg effect is critical for the rapid biosynthesis to support cancer cell growth and proliferation, as a supply line not only generating ATP but also producing NADPH for lipid generation. The Warburg effect implications including growth advantages to cancer cells, chemo-resistant, and therapeutic targets have been reviewed previously [6,7,8], however biosynthetic programs of lipids in cancer cells are largely unknown. Further research is needed to elucidate the Warburg Effect functions in lipid biosynthesis in cancers. Lipid molecules, such as fatty acids, cholesterol, and lipid droplets, are used to provide energy and cell-synthetic materials for a variety of cellular processes. The process of lipid and glucose metabolism is also critical for maintaining tumor cell growth and function. However, in cancer cells, over-consumption of glucose, lipids, and amino acids is necessary to supply energy and cell-synthetic materials for pro-tumorigenic processes, including rapid proliferation, metastasis, and invasion of cancer cells [9, 10]. As lipid molecules provide not only the necessary materials but also energy for cancer cell growth, they may play key roles in carcinogenetic signaling during maladaptive transformation and development of tumor cells. In 1953, it was reported that tumor cells can synthesize lipids that are required for rapid tumor proliferation [11]. Since then, the role of lipid metabolism in carcinogenesis has evoked research interest. A large number of studies have demonstrated the correlation between abnormal lipid metabolism and various cancers, including breast cancer [12], prostate cancer [13], colon cancer [14], liver cancer [15], and etc. Thus, the connection between the lipid metabolism and the tumor microenvironment is highly significant. Increasing evidence points to several lipid signaling-associated biomarkers as potential diagnostic and therapeutic strategies to treat various cancers [16]. Cancer cells can also rewire lipid metabolism to mediate the development of acquired drug resistance [17], while targeting lipid metabolic enzymes (e.g., fatty acid synthase) re-sensitize breast cancer resistant to HER2-targeted therapies [18]. Several reviews have contributed greatly to the current knowledge on the roles of lipid metabolism, lipid biomarkers, diagnosis and therapeutic intervention, and drug resistance in cancers [19,20,21,22,23]. The approved cancer metabolic drugs and the small-molecule metabolic inhibitors in cancer clinical trials have been outlined and discussed in a recent review article [24]. In addition, lipid nanoparticles and lipid carriers are considered as a major research area for drug delivery and cancer cell targeting/killing, but this area is beyond the scope of the current review. This review, adding to the existing knowledge, focuses on the lipid-associated tumor microenvironment to discuss the roles of diverse types and multiple functions of lipid molecules in cancer and in the immune response to cancer.
Tumor microenvironment
Accumulating evidence indicates the active role of TME played in carcinogenetic initiation, progression, metastasis, recurrence, and therapeutic responses. In addition to the cancer cells, TME is populated by many highly heterogeneous groups of non-cancer cells, including endothelial cells, adipose cells, immune/inflammatory cells, myeloid-derived suppressor cells (MDSCs), as well as other tumor-associated cells such as cancer-associated fibroblasts (CAFs), cancer-associated adipocytes (CAAs), tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs) [25,26,27]. TME has been widely accepted as an arena where the tumor cells undergo a metabolic remodeling to meet their needs for growth and survival to compete or cooperate with other non-cancer cells for nutrients and cell-signaling molecules [28]. When TME becomes hypoxic because vasculatures are inadequate, cancer cells can bypass the bloodstream to acquire nutrients and self-growth signaling molecules from TME [29]. In the extracellular space, the communication between the cancer cells and non-cancer cells is manifested by a complex network through the soluble factors such as inflammatory chemokines, cytokines, matrix remodeling enzymes, and various growth factors [28, 29]. For example, fibroblasts, embedded within the fibrillar extracellular matrix (ECM), can be activated by the ECM-degrading proteases, showing a phenotype of CAFs which promote tumorigenesis and trigger chemoresistance through a paracrine manner interacting with the adjacent epithelial tissues [30, 31]. It has also been highlighted that dynamic crosstalk between adipocytes and cancer cells is mainly sustained by the steroid hormones, adipokines, and cytokines, leading to the reprogramming of adipocytes to generate CAAs, which affect cancer cells during all steps of tumor progression by releasing adipokines, growth factors, and metabolites [32, 33]. Nowadays, numerous studies have indicated that tumor-associated immune/inflammatory cells promote and accelerate metastasis by establishing an immunosuppressive microenvironment within primary lesions and suppressing tumor immune surveillance [34]. Notably, the terminology of TANs does not relate to a specific differentiation step and activation status [35] but neutrophils as the first mediators of inflammatory reactions can influence tumor progression, depending on the tumor microenvironment [36]. A study indicates that TANs recruit macrophages and Treg cells, contributing to an immunosuppressive microenvironment to promote HCC progression and chemoresistance [37]. In parallel, there are also coexisted heterogeneous macrophage populations which influence both tumor growth and immune response in the tumor compartment [38]. M1-like macrophages are the subpopulation for anti-tumor immunity, whereas M2-like TAMs promote carcinogenetic progression and suppress immune response [36]. As the aberrant lipid metabolism can reprogram not only the cancer cells but also the surrounding non-cancer cells, TME is becoming very important research field to study lipid molecules contributing to cellular lipid metabolism reprogramming.
Fatty acid
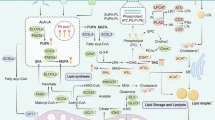
Most mammalian cells obtain lipolytic free fatty acids (FFAs) from the blood. When fatty acids (FAs) enter proliferative fibroblast cells such as HeLa and H460 cells [39], they combine with fatty acid binding proteins (FABPs). Generally, FAs are transported to various cellular organelles by binding to the FABPs [40, 41]. Overexpression of FABPs has been reported to be significantly related to the malignant degree of breast cancer and poor prognosis in patients [12, 42]. FABPs are reported to play a key role in tumor initiation and progression in ovarian cancer and glioblastoma [43, 44]. Studies show that upregulation of FABP5 can activate peroxisome proliferator-activated receptor-β/δ (PPARβ/δ) and increase FABP5 methylation levels in CPG islands to accelerate the proliferation of cancer cells [45,46,47]. Tumor cells can also increase the uptake of FAs from plasma by upregulating cell-surface receptors (e.g., cluster of differentiation 36 [CD36] [48, 49]) to facilitate the transport of FAs. CD36 has been widely reported as a prognostic marker in various cancers, including gliomas and breast, prostate, ovary, colon, and liver cancers [50]. It has been shown that the suppression of CD36 successfully inhibits the metastasis of tumor cells, whereas overexpression of CD36 reverses this inhibitory effect. In this regard, CD36, as a key receptor for tumor metastasis, has been suggested as a therapeutic target [51, 52]. Most importantly, cells can produce FAs from citric acid by de novo synthesis via enzymatic reaction using adenosine triphosphate [ATP] citrate lyase (ACLY), acetyl coenzyme A carboxylase (ACC), and fatty acid synthase (FASN). De novo FA synthesis is accepted as a defining characteristic of cancer cells [53]. Upregulated expression of FASN significantly increases FA production by de novo synthesis, leading to a deteriorated outcome of breast cancer [54]. In contrast, the viability of cancer cells notably decreases when they are treated with FASN inhibitors such as triclosan and orlistat [55]. Similarly, upregulation of other enzymes such as ACLY and ACC has also been demonstrated to promote proliferation of cancer cells in glioblastoma, colorectal cancer, lung cancer, liver cancer, prostate cancer, and other tumors [56, 57]. The FA cellular metabolic events as well as the upregulated carcinogenetic signals during metabolic processes are summarized in Fig. 1. Of note, de novo lipogenesis is not the cause of malignancy and the tumor metabolism relate to the ability of FASN cannot elicit its malignant capabilities. Because FA synthesis expends energy, this is not an advantage for the survival of cancer cells. Additional work is required to fully understand the regulatory actions of FA de novo synthesis in cancer cells.

FA metabolic events and upregulated carcinogenetic signals. Scheme showing cellular fatty acid metabolic events, including FA transport, FA storage, lipolysis, β-oxidation, and de novo FA synthesis. Uptake of extracellular FAs is mediated by FA transport protein (FATP), CD36, and FABPs. The intracellular FAs form FA pool, and the FAs are either stored as lipids droplets or activated by reaction with CoA to form fatty-acyl-CoA (FACoA) for β-oxidation in mitochondria. The de novo FA synthesis begins with acetyl-CoA which is either derived from mitochondria or converted from citrate by ACLY. Acetyl-CoA is then converted to malonyl-CoA by ACC, and malonyl-CoA is further converted to FA by FASN. Red pentagon: upregulated FA metabolic signals contribute to carcinogenesis
Epidemiological and experimental studies reveal the modulatory effects of FA on tumorigenesis [58, 59], while polyunsaturated fatty acids (PUFAs) are the extensively investigated FAs in various human cancer cells, e.g., colon cancer, glioblastoma, breast cancer, and prostate cancer. Previously, studies have demonstrated that gamma-linolenic acid, arachidonic acid, and eicosapentaenoic acid have selective tumoricidal actions by either augmentation of free-radical generation and lipid peroxidation or cytokine-mediated antitumor effects [60, 61]. As the precursors of prostaglandins, gammma-linolenic acid, dihomo-gamma-linolenic acid, and arachidonic acid are reported to not only prevent genetic damage but also augment immune responses and tumoricidal actions of macrophages [62]. Lately, accumulating evidence suggests that PUFAs suppress tumor growth by a variety of mechanisms depending on the cell types and the metabolism of PUFAs being handled by tumor cells. In human colon cancer cells, both n-3 PUFAs (α-linolenic acid, eicosapentaenoic acid and docosahexaenoic acid) and n-6 PUFAs (linoleic acid, gamma-linolenic acid and arachidonic acid) are demonstrated to trigger apoptosis through a mitochondrial pathway [63]. Supplementation of various n-3 PUFAs and n-6 PUFAs to human prostate cancer cells (RWPE-1 and PC-3) enhances the content of their long-chain metabolites and inhibits proliferation in these cells, but there was no correlation between inhibition of cell proliferation and free radical generation [64]. In human glioblastoma cell (T98G), arachidonic acid (n-6 PUFA) supplementation inhibits the growth of T98G cells though up-regulated level of arachidonoylethanolamide, a endogenous cannabinoid ligand, while eicosapentaenoic acid (n-3 PUFA) reduced the oleic acid (non-EFA) enhanced proliferation in breast cancer cells (MCF7) [65]. In mouse myeloma cells (SP2/0), alpha-linolenic and eicosapentaenoic acids induce suppression of SP2/0 cell proliferation which is dependent on the activities of cyclooxygenase, lipoxygenase, and superoxide [66]. The tumoricidal action of PUFAs (γ-linolenic acid, arachidonic acid and docosahexaenoic acid) is also found to associate with modulation of the expression of microRNAs and their targeted genes to trigger apoptosis in glioma cells [67]. In addition, studies suggest that PUFAs are capable to improve the therapeutic efficacy of chemotherapy on the drug-resistant cancer cells by enhancing drug uptake and reducing its efflux [68]. Exogenous FA is also reported to have tumoricidal action, for example, F-6 (a C-20 furanoic acid from Arabian Gulf catfish skin) can suppress proliferation and promote apoptotic cell death in leukemic and breast cancer cells [69]. Taken together, it is highly promising for FAs, particularly PUFAs, to be used as potential anti-cancer drug candidates for clinical patients.
Cholesterol
Cholesterol is an essential lipid molecule for the growth of all eukaryotic cells. The synthesis and metabolism of cholesterol are highly conserved in various organisms, from yeast to humans. Because de novo cholesterol synthesis is energetically expensive, most cells can take the premade cholesterol from circulating lipoproteins. As excessive cholesterol is harmful to the cell, cholesterol synthesis is therefore aimed at supplementing that exogenous supply based on demand, while elaborate mechanisms have evolved to tightly regulate cholesterol levels [70]. Cholesterol synthesis, in brief, begins with acetyl-coenzyme A derived from mitochondria and transported to the cytosol. In cytosol, one molecule of acetyl-coenzyme A and one molecule of acetoacetyl-CoA sare converted to HMG-CoA, being catalyzed by 3-hydroxy-3-methylglutaryl (HMG)-CoA synthase (HMGCS). The subsequent steps occur in the endoplasmic reticulum where HMG-CoA is reduced to mevalonate by HMG-CoA reductase (HMGCR). Mevalonate is phosphorylated to isopentyl pyrophosphate which is further converted to geranyl pyrophosphate. Two molecules of isopentyl pyrophosphate are condensed to form farnesyl pyrophosphate. Squalene synthase catalyzes the condensation of two molecules of farnesyl pyrophosphate to form squalene. Squalene is then cyclized to form lanosterol [71]. Subsequently, cholesterol is produced from lanosterol via the Bloch or Kandutsch-Russell pathway. Cholesterol synthesis is strictly controlled in normal cells, but not in tumor cells [72]. In fact, the pathways of cholesterol synthesis are closely associated with tumor development [73], for example, the mevalonate pathway has been reported to be a critical regulator of tumor progression and a therapeutic target [74]. Almost all the genes encoding cholesterol synthesis enzymes are transcribed through the regulation of sterol regulatory element-binding proteins (SREBPs), while the SREBP family of transcription factors is activated in response to low sterol status and helps coordinate the cholesterol synthesis [75]. SREBP-2 binds to the sterol regulatory elements in the promoters of genes such as HMGCR and mevalonate kinase (MVK) to activate and regulate the enzymes in mevalonate pathway, involving in the progression of various cancers [76]. Cholesterol- and steroid-dependent tumor cell proliferation has been widely reported [77]. MicroRNAs (miRNAs) have also been reported as transcriptional modulators of cholesterol metabolism and play a pivotal role in tumorigenesis [78, 79]. For example, miR-33a, an internal miRNA located in the gene encoding SREBF-2, mediates cholesterol metabolism and promotes tumor proliferation [80, 81]. Furthermore, multiple regulatory pathways for cholesterol signaling have been suggested as potential chemotherapy targets [82,83,84]. The metabolic intermediates of cholesterol are also major factors accounting for tumor initiation and progression [75]. Both cholesterol and metabolic intermediates for cholesterol are recognized to play a deteriorative role in various cancers. For example, 27-hydroxycholesterol (27HC), a cholesterol metabolite, is a selective estrogen receptor modulator [85]. In a murine breast cancer model, it was found that 27HC can accelerate carcinogenetic initiation and progression [85]. Alternatively, 27HC can activate the liver X receptor (LXR), which has a notable effect on malignancy [86, 87], e.g., promoting metastasis in pulmonary tumors [88]. The metabolic intermediates of cholesterol are also found to play a critical role in the emergence and development of prostate cancer [89, 90]. In addition, squalene monooxygenase, a rate-limiting enzyme in the metabolic process of cholesterol, has been reported to act as a reliable indicator of angiogenesis in prostate cancer [13]. The mevalonate pathway which is responsible for de novo cholesterol synthesis is significantly upregulated by mutant p53, while mutant p53 depletion phenotypically reverts breast cancer cells to a more acinar-like morphology, implicating down-regulation of cholesterol synthesis as a therapeutic target for tumors bearing mutations in p53 [91]. Targeting low-density lipoprotein receptor (LDLR) with LXR agonist causes inducible degrader of LDLR (IDOL)-mediated LDLR degradation and increased expression of the cholesterol efflux transporter to promote tumor cell death in glioblastoma [92]. On the other hand, the evidence is inconsistent in women; for example, cholesterol level is reported to be inversely related to the risk of gastric cancer among postmenopausal women [93]. Pathways of cholesterol synthesis and regulation of carcinogenetic signals are summarized in Fig. 2.

Pathways of cholesterol synthesis and regulation of carcinogenetic signals. Scheme showing cholesterol synthetic pathways, carcinogenetic signaling regulation in mevalonate pathway, and the cholesterol oxygenated metabolites feedback on cholesterol synthesis and efflux/influx. PI3K/AKT/mTOR and mutant p53 activate the mevalonate pathway via upregulation of SREBP, while Statins and LXR can inhibit cholesterol synthesis. 27HC negatively feedbacks on cholesterol synthesis and efflux/influx but miR-33a decreases cholesterol efflux. MVK mevalonate kinase; MVD mevalonate diphosphate decarboxylase; PMVK phosphomevalonate kinase; FPPS farnesylpyrophosphate synthase; SQS squalene synthase; SQLE squalene epoxidase; LSS lanosterol synthase
Lipid droplets
Lipid droplets (LDs), independent organelles produced by the endoplasmic reticulum and Golgi apparatus to store excess lipids, are wrapped around double lipid components [94,95,96,97,98]. Most lipids stored in LDs are neutral lipids, such as FFAs and cholesterol. The gradual fusion of small LDs contributes to larger LDs, and the release of long-chain FAs (LCFAs) and cholesterol from LDs occurs through an enzymatic reaction [94, 98]. Multiple components exist in LDs, and these components may vary among different types of cells. Triglyceride (TG) is the main component in LDs found in fatty cells, while cholesteryl ester is mainly located in macrophage LDs [99]. The concept of LDs has improved our understanding of the potential carcinogenesis in terms of cancer cell adaptability and resilience to microenvironmental stress [100]. The changes in LD components correspond to alterations in the tumor cell phenotype and the surrounding microenvironment [101, 102]. LDs are important energy resources, and many organs obtain energy from LDs via FA oxidation and beta oxidation in mitochondria [103, 104]. In tumor cells, the LD energy supply can become the main source of energy for most cancers, including colon, prostate, ovarian, and breast cancer [105, 106]. LCFAs are supplied from circulation as part of TGs and reach various tissues, as well as tumor cells [107]. Free LCFAs can be directly taken up through binding to cellular lipoproteins, while the albumin-combined LCFAs can be transported into the cytoplasm through the FA transport molecule CD36 located on the surface of cells. Once inside the cell, LCFAs can diffuse into LDs [108, 109]. Subsequently, the LCFAs in LDs provide energy through beta oxidation and are reported to play a fundamental role in the pathogenesis of melanoma, ovarian cancer, and breast cancer [110]. The carcinogenetic transformation in clear cell renal cell carcinoma (ccRCC) is also related to abnormal lipid storage in LDs [111]. Increased accumulation of cholesterol, via hypoxia-inducible factor-1-dependent LDLR, further escalates the formation of LDs, contributing to the initiation of ccRCC [112]. In addition, SREBPs regulate, through sterol regulatory elements in gene regulatory region, the activation of several genes including FASN and stearoyl-CoA desaturase (SCD), which are the key factors governing the formation of LDs [111]. In parallel, SCD-1 is essential for ccRCC cell growth, whereas inhibition of SCD-1 induces apoptosis in ccRCC. A recent study demonstrated a key role of autophagy in LD formation during ccRCC pathogenesis. An autophagy-related protein, microtubule-associated protein 1S (MAP1S), suppresses ccRCC tumorigenesis by negatively regulating LD formation [113]. In prostate cancer, both cell proliferation and migration are positively correlated with LD formation [114, 115]. Prostate cancer can be divided into androgen-dependent and non-androgen-dependent subtypes. The expression of androgens is related to SREBP-1 expression [114]. According to Raman spectrum analysis, the number of LDs increases with the elevation of androgen levels [101]. LDs have also been reported to affect other hormone-dependent breast and ovarian cancer cells. The formation of LDs in breast cancer is correlated with estrogen receptor and progesterone receptor [116,117,118], while the development of ovarian cancer is controlled by increased LD formation, which is mediated by FASN expression [119, 120]. In addition to hormone-dependent cancer cells, LDs in non-hormone-dependent cancer cells also play a crucial role in tumor development. For example, the number of LDs in colon cancer cells is much higher than that in normal cells, and the elevated number of LDs show a direct regulatory effect on the growth of colon cancer cells [14, 121]. Recently, extracellular vesicles (EVs), phospholipid lipid bilayer particles with abundant cholesterol and ceramide, have attracted much attention. Moreover, EVs may play central roles in the metabolism and signaling pathways of the tumor microenvironment [122]. For examples, EVs, as cargos, can deliver the parental cells’ genomic and proteomic information to the surrounding/distant recipient cells to modulate their behavior [123]. EVs are also novel drug resistance modulators that add to the complexity of resistance mechanisms [124]. The use of EVs as novel cancer therapeutics has potential to improve clinical outcomes in patients [125].
The role of lipid metabolism in the immune response to cancer
Growing evidence has highlighted the key role of lipid metabolism as a major influence in immune responses to cancer. As reported previously, T cell subsets have different metabolic traits that direct T cell survival, proliferation, and effector functions [126]. For example, increased fatty acid oxidation (FAO) level is found in activated CD4+ T cells but not in CD8+ T cells, which rapidly produce adenosine triphosphate via glycolysis for energy supply [127, 128]. Cancer cells can rewrite T cell metabolic programs to create a suppressive tumor microenvironment; for example, PD-1-expressing cancer cells alter the metabolic program of tumor-infiltrating T cells (TILs) by enhancing FAO level and inhibiting glycolysis [129]. Cancer cells can also impair T cell metabolism and effector functions by competing for key nutrients such as glucose and glutamine to drive tumor progression [130]. Furthermore, tumor-derived regulatory T (Treg) cells can interact with responder T cells to trigger cell senescence by rewriting their metabolic programs [131]. Treg cells can also modulate lipid metabolism in tumor-associated macrophages (TAMs) to promote tumor suppression [132]. TAMs represent a subpopulation that acquires an M2-like tumor-promoting phenotype characterized by high levels of immunosuppressive markers such as Arginase 1 (ARG1) and Interleukin (IL)10 [133]. In fact, lipid metabolic traits of macrophages are reflected by their M1/M2 polarization state, a classical (M1) or alternative (M2) activation in response to microenvironmental signals, showing either anti- or pro-tumorigenic properties [134]. In macrophages, lipid metabolism is oriented toward fatty acid synthesis (FAS) in M1 polarization, while M2 polarization activates FAO to generate ATP and produce acetyl-CoA, which participates in the Krebs cycle (TCA cycle) and cholesterol biosynthesis [135]. FFAs can stimulate tissue-resident macrophages to release cytokines, which tune hematopoiesis to further engage immune cells into tissues [136]. Cancer cells drive the cells, represented by TAMs as well as MDSCs. In tumor-infiltrating MDSCs, the use of energy is shifted from glycolysis toward FAO, which is characterized by CD36-mediated fatty acid uptake and upregulated expression of FAO enzymes [137]. In addition, increased levels of FAS and intracellular TG storage are also found in tumor-associated dendritic cells [138, 139]. Modulating the key lipid molecular players of lipid metabolism appears to be a promising tool for tuning immune responses to boost the intrinsic anti-tumor activity of both adaptive and innate immune compartments.
Regarding the lipid molecules, FAs have been reported to interfere with immune responses such as the modulation of lymphocyte proliferation and natural killer activity [140, 141]. A recent review article discussed the regulatory roles of the fatty acid palmitate in controlling immune balance, in which palmitate could modulate innate immunity by not only regulating the activation of pattern recognition receptors in local innate immune cells but also coordinating the immunological activity in tissues [142]. High-fiber diets are associated with a decreased risk of colorectal cancer, but the anti-cancer mechanism is largely unknown [143, 144]. Studies have shown that short-chain fatty acids, the major microbial-derived metabolites, can modulate immune response through free fatty acid receptor 2 (FFAR2) which is highly expressed on immune cells, including myeloid cell populations [145] and Tregs [146]. In addition, FFAR2 can regulate macrophage cytokine expression [147], while dendritic cells from FFAR2-deficient mice are unable to promote promoted B-cell IgA production, resulting in unresolved inflammation and broken intestinal homeostasis [148]. Fatty acid-binding protein 5, a cellular chaperone of long-chain fatty acids being well-studied in various immune cells [149], is reported to regulate the commitment of dendritic cells and generation of Tregs in tumor microenvironment [150]. Cholesterol is also a key player for immune responses, e.g., the ATP-binding cassette transporter G1-dependent cholesterol efflux can suppress Tregs development [151]. The oxidized LDLs are found to be the main types of lipids accumulating in tumor-infiltrating lymphocytes [152], as a result of the immunosuppressive tumor microenvironment. For example, the CD36-mediated bad cholesterol uptake in CD8+ T lymphocytes can lead to their dysfunction and cancer progression [153]. Bioactive lipids are also played key roles in modulation of immune check point inhibitors, which has been discussed in a recent review article [154]. Therefore, comprehensively exploring the lipid metabolic profiles of immune cells and cancer cells will facilitate the development of novel strategies for cancer therapy. Lipid metabolism of immune cells and the immunosuppressive cells in tumor microenvironment is summarized in Fig. 3.

Lipid metabolism of immune cells and the immunosuppressive cells in tumor microenvironment. Scheme showing lipid and energy metabolism of immune cells and the immunosuppressive cells in tumor microenvironment. The tumor suppressive immune cells with warm-toned in cytosol are shown in left, while the tumor-promoting cells with cold-toned in cytosol are shown in right. Increased fatty acid synthesis in dendritic cells and increased fatty acid oxidation in CD4+ T cells are found in tumor microenvironment. The responder CD8+ T cells (cytotoxic T cells) produce ATP via glycolysis for energy supply, however, tumor cell can rewrite the lipid metabolic programs of TILs which decrease glycolysis but increase FAO for energy supply. Tumor cell can also concerting MDSCs and TAMs to immunosuppressive cells. Treg cells can trigger senescence of cytotoxic T cells. Increased FAO is also found in the immunosuppressive cells (MDSC, Treg, and TAM) as well as in M2-macrophage
Contribution of the current work to the existing knowledge
In comparison with other review articles, the current work adds to the existing knowledge in the following three aspects. (1) The canonical rationale for investigating lipid metabolism in cancer cells is to study the lipid requirements for plasma membrane synthesis and energy production. The current work emphasizes that abnormal lipid metabolism in cancer cells contributing to carcinogenic signaling within the tumor microenvironment. (2) The introduction of lipid metabolism has led to a paradigm change in the treatment of cancer. However, several challenges remain in exploring the carcinogenic roles of lipid molecules. In the current work, we summarize that the lipid molecules exhibit not only a tumorigenic effect but also an anti-tumorigenic effect, in the context of different cancer types; such contrasting effects merit further study. (3) We discuss the available studies on lipid metabolism in the immune response to cancer and suggest that key lipid molecular players can be promising tools for tuning such immune responses. Lipid metabolic reprogramming may help immune checkpoints restore effective anti-tumor immunity. Unlike previous studies that mostly focused on lipid metabolism in maintaining cellular structure and providing energy, the current work discusses the available studies on lipid metabolism related signaling targets in the tumor microenvironment that either promote or inhibit carcinogenesis. Exploring the lipid-related signaling targets that drive or block cancer development will provide a new direction for further studies, in a shift away from conventional cancer research. The limitations of the current work are as follows: (1) relationship between lipid molecules and oncogenes is not well discussed. There are functional links between lipid molecules and oncogenes. For examples, the growth factor receptor-driven cancers depend on membrane lipid remodeling for transduction of oncogenic signals [155]. Oncogene KRAS activates lipogenesis resulting in distinct proteomic and lipid signatures in lung adenocarcinoma [156]; (2) lipid signaling-based biomarkers for cancer diagnosis and treatment response are not discussed. Lipid molecules may be used as tumor-related biomarkers for diagnosis and treatment response; for example, positron emission tomography has been applied to observe the synthesis of tumor-active lipids using tracers [157]. Thus, further study is needed to explore these aspects.
Conclusion
Lipid molecules, LDs, and metabolic intermediates for lipid biosynthesis are extremely critical for signaling between cancer cells and their surrounding microenvironment. Changes in lipid metabolism in cancer cells as well as in immune cells can affect initiation and progression of cancer in a subtle way. The proliferation of cancer cells depends on not only the energy and nutrient supplies, but also on the communication with their microenvironment, in which lipids inevitably function as prominent modulators. Comprehensively understanding the regulatory role of lipid metabolism in carcinogenesis would provide a novel strategy for cancer prevention and treatment.
Availability of data and materials
Not applicable.
Abbreviations
- FFAs:
-
Free fatty acids
- FAs:
-
Fatty acids
- FATP:
-
FA transport protein
- FABPs:
-
Fatty acid binding proteins
- CD36:
-
Cluster of differentiation 36
- ATP:
-
Adenosine triphosphate
- FACoA:
-
Fatty-acyl-CoA
- ACLY:
-
ATP citrate lyase
- ACC:
-
Acetyl coenzyme A carboxylase
- FASN:
-
Fatty acid synthase
- MVK:
-
Mevalonate kinase
- MVD:
-
Mevalonate diphosphate decarboxylase
- PMVK:
-
Phosphomevalonate kinase
- FPPS:
-
Farnesylpyrophosphate synthase
- SQS:
-
Squalene synthase
- SQLE:
-
Squalene epoxidase
- LSS:
-
Lanosterol synthase
- HMGCS:
-
3-Hydroxy-3-methylglutaryl (HMG)-CoA synthase
- HMGCR:
-
The enzyme 3-hydroxy-3-methylglutaryl-CoA reductase
- SREBPs:
-
Sterol regulatory element-binding proteins
- 27HC:
-
27-Hydroxycholesterol
- miRNAs:
-
MicroRNAs
- LXR:
-
Liver X receptor
- LDLR:
-
Low-density lipoprotein receptor
- IDOL:
-
Inducible degrader of LDLR
- LCFAs:
-
Long-chain FAs
- ccRCC:
-
Clear cell renal cell carcinoma
- TG:
-
Triglyceride
- SCD:
-
Stearoyl-CoA desaturase
- MAP1S:
-
Microtubule-associated protein 1S
- EVs:
-
Extracellular vesicles
- FAO:
-
Fatty acid oxidation
- Treg:
-
Regulatory T cell
- TME:
-
Tumor microenvironment
- CAFs:
-
Cancer-associated fibroblasts
- CAAs:
-
Cancer-associated adipocytes
- TANs:
-
Tumor-associated neutrophils
- ECM:
-
Extracellular matrix
- TAMs:
-
Tumor-associated macrophages
- ARG1:
-
Arginase 1
- IL:
-
Interleukin
- FAS:
-
Fatty acid synthesis
- MDSCs:
-
Myeloid-derived suppressor cells
- FFAR2:
-
Free fatty acid receptor 2
References
Qiu B, Simon MC. Oncogenes strike a balance between cellular growth and homeostasis. Semin Cell Dev Biol. 2015;43:3–10.
Boroughs LK, Deberardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–30.
Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–33.
Tyagi K, Mandal S, Roy A. Recent advancements in therapeutic targeting of the Warburg effect in refractory ovarian cancer: a promise towards disease remission. Biochim Biophys Acta Rev Cancer. 2021;1876(1): 188563.
Liu C, Jin Y, Fan Z. The mechanism of Warburg effect-induced chemoresistance in cancer. Front Oncol. 2021;11: 698023.
Lemasters JJ. Metabolic implications of non-electrogenic ATP/ADP exchange in cancer cells: a mechanistic basis for the Warburg effect. Biochim Biophys Acta Bioenerg. 2021;1862(7): 148410.
Gaglio D, Metallo CM, Gameiro PA, Hiller K, Danna LS, Balestrieri C, Alberghina L, Stephanopoulos G, Chiaradonna F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523.
Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyhchang N. Glutamine supports pancreatic cancer growth through a Kras-regulated metabolic pathway. Nature. 2013;496(7443):101–5.
Medes G, Thomas A, Weinhouse S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 1953;13(1):27.
Thumser AE, Moore JB, Plant NJ. Fatty acid binding proteins: tissue-specific functions in health and disease. Curr Opin Clin Nutr Metab Care. 2014;17(2):124–9.
Stopsack KH, Gerke TA, Sinnott JA, Penney KL, Tyekucheva S, Sesso HD, Andersson SO, Andrén O, Cerhan JR, Giovannucci EL. Cholesterol metabolism and prostate cancer lethality. Can Res. 2016;76(16):4785–90.
Penrose H, Heller S, Cable C, Makboul R, Chadalawada G, Chen Y, Crawford SE, Savkovic SD. Epidermal growth factor receptor mediated proliferation depends on increased lipid droplet density regulated via a negative regulatory loop with FOXO3/Sirtuin6. Biochem Biophys Res Commun. 2016;469(3):370–6.
Liu X, Ping Z, Martin RC, Cui G, Wang G, Yi T, Lu C, Lv G, Yan L. Lack of fibroblast growth factor 21 accelerates metabolic liver injury characterized by steatohepatities in mice. Am J Cancer Res. 2016;6(5):1011.
Robey RB, Weisz J, Kuemmerle NB, Salzberg AC, Berg A, Brown DG, Kubik L, Palorini R, Almulla F, Altemaimi R. Metabolic reprogramming and dysregulated metabolism: cause, consequence and/or enabler of environmental carcinogenesis? Carcinogenesis. 2015;36(Suppl 1):S203.
Feng WW, Wilkins O, Bang S, Ung M, Li J, An J, Del Genio C, Canfield K, DiRenzo J, Wells W, et al. CD36-mediated metabolic rewiring of breast cancer cells promotes resistance to HER2-targeted therapies. Cell Rep. 2019;29(11):3405-3420 e3405.
Puig T, Aguilar H, Cufi S, Oliveras G, Turrado C, Ortega-Gutierrez S, Benhamu B, Lopez-Rodriguez ML, Urruticoechea A, Colomer R. A novel inhibitor of fatty acid synthase shows activity against HER2+ breast cancer xenografts and is active in anti-HER2 drug-resistant cell lines. Breast Cancer Res. 2011;13(6):R131.
Maan M, Peters JM, Dutta M, Patterson AD. Lipid metabolism and lipophagy in cancer. Biochem Biophys Res Commun. 2018;504(3):582–9.
Larrouy-Maumus G. Lipids as biomarkers of cancer and bacterial infections. Curr Med Chem. 2019;26(11):1924–32.
Butler LM, Perone Y, Dehairs J, Lupien LE, de Laat V, Talebi A, Loda M, Kinlaw WB, Swinnen JV. Lipids and cancer: emerging roles in pathogenesis, diagnosis and therapeutic intervention. Adv Drug Deliv Rev. 2020;159:245–93.
Pakiet A, Kobiela J, Stepnowski P, Sledzinski T, Mika A. Changes in lipids composition and metabolism in colorectal cancer: a review. Lipids Health Dis. 2019;18(1):29.
Feng WW, Kurokawa M. Lipid metabolic reprogramming as an emerging mechanism of resistance to kinase inhibitors in breast cancer. Cancer Drug Resist. 2020. https://doi.org/10.20517/cdr.2019.100.
Stine ZE, Schug ZT, Salvino JM, Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. 2022;21(2):141–62.
Bui TM, Yalom LK, Sumagin R. Tumor-associated neutrophils: orchestrating cancer pathobiology and therapeutic resistance. Expert Opin Ther Targets. 2021;25(7):573–83.
Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129(8):3006–17.
Cheng HS, Lee JXT, Wahli W, Tan NS. Exploiting vulnerabilities of cancer by targeting nuclear receptors of stromal cells in tumor microenvironment. Mol Cancer. 2019;18(1):51.
Maman S, Witz IP. A history of exploring cancer in context. Nat Rev Cancer. 2018;18(6):359–76.
Finicle BT, Jayashankar V, Edinger AL. Nutrient scavenging in cancer. Nat Rev Cancer. 2018;18(10):619–33.
Chatterjee S, Bhat V, Berdnikov A, Liu J, Zhang G, Buchel E, Safneck J, Marshall AJ, Murphy LC, Postovit LM, et al. Paracrine crosstalk between fibroblasts and ER(+) breast cancer cells creates an IL1beta-enriched niche that promotes tumor growth. iScience. 2019;19:388–401.
Chandra Jena B, Kanta Das C, Banerjee I, Das S, Bharadwaj D, Majumder R, Mandal M. Paracrine TGF-beta1 from breast cancer contributes to chemoresistance in cancer associated fibroblasts via upregulation of the p44/42 MAPK signaling pathway. Biochem Pharmacol. 2021;186: 114474.
Qureshi R, Picon-Ruiz M, Aurrekoetxea-Rodriguez I, de Paiva VN, D’Amico M, Yoon H, Radhakrishnan R, Morata-Tarifa C, Ince T, Lippman ME, et al. The major pre- and postmenopausal estrogens play opposing roles in obesity-driven mammary inflammation and breast cancer development. Cell Metab. 2020;31(6):1154-1172 e1159.
Hopkins BD, Goncalves MD, Cantley LC. Obesity and cancer mechanisms: cancer metabolism. J Clin Oncol. 2016;34(35):4277–83.
Lopez-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of metastasis by NK cells. Cancer Cell. 2017;32(2):135–54.
Nicolas-Avila JA, Adrover JM, Hidalgo A. Neutrophils in homeostasis, immunity, and cancer. Immunity. 2017;46(1):15–28.
Russo M, Nastasi C. Targeting the tumor microenvironment: a close up of tumor-associated macrophages and neutrophils. Front Oncol. 2022;12: 871513.
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646-1658 e1617.
He Z, Zhang S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol. 2021;12: 741305.
Yao CH, Grider RF, Mahieu NG, Liu GY, Chen YJ, Wang R, Singh M, Potter GS, Gross RW, Schaefer J. Exogenous fatty acids are the preferred source of membrane lipids in proliferating fibroblasts. Cell Chem Biol. 2016;23(4):483–93.
Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat Rev Drug Discov. 2008;7(6):489–503.
Hotamisligil GS, Bernlohr DA. Metabolic functions of FABPs—mechanisms and therapeutic implications. Nat Rev Endocrinol. 2015;11(10):592.
Liu RZ, Graham K, Glubrecht DD, Germain DR, Mackey JR, Godbout R. Association of FABP5 expression with poor survival in triple-negative breast cancer: implication for retinoic acid therapy. Am J Pathol. 2011;178(3):997.
Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buellgutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat Med. 2011;17(11):1498.
Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9(1):349.
Adamson J, Morgan EA, Beesley C, Mei Y, Foster CS, Fujii H, Rudland PS, Smith PH, Ke Y. High-level expression of cutaneous fatty acid-binding protein in prostatic carcinomas and its effect on tumorigenicity. Oncogene. 2003;22(18):2739–49.
Rasanen K, Sriswasdi S, Valiga A, Tang HY, Zhang G, Perego M, Somasundaram R, Li L, Speicher K, Klein-Szanto AJ, et al. Comparative secretome analysis of epithelial and mesenchymal subpopulations of head and neck squamous cell carcinoma identifies S100A4 as a potential therapeutic target. Mol Cell Proteom. 2013;12(12):3778–92.
Grigalavicius M, Juraleviciute M, Kwitniewski M, Juzeniene A. The influence of photodynamic therapy with 5-aminolevulinic acid on senescent skin cancer cells. Photodiagn Photodyn Ther. 2017;17:29–34.
Shiro K, Yohei M. Lipid droplets: a key cellular organelle associated with cancer cell survival under normoxia and hypoxia. Int J Mol Sci. 2016;17(9):1430.
Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM. Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol Cancer Ther. 2011;10(3):427.
Enciu AM, Radu E, Popescu ID, Hinescu ME, Ceafalan LC. Targeting CD36 as biomarker for metastasis prognostic: how far from translation into clinical practice? Biomed Res Int. 2018;2018:7801202.
Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini SO, Berenguer A, Prats N, Toll A, Hueto JA. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541(7635):41.
Li Z, Kang Y. Lipid metabolism fuels cancer’s spread. Cell Metab. 2017;25(2):228–30.
Santos CR, Schulze A. Lipid metabolism in cancer. FEBS J. 2012;279(15):2610–23.
Esslimanisahla M, Thezenas S, Simonylafontaine J, Kramar A, Lavaill R, Chalbos D, Rochefort H. Increased expression of fatty acid synthase and progesterone receptor in early steps of human mammary carcinogenesis. Int J Cancer. 2007;120(2):224–9.
Sadowski MC, Pouwer RH, Gunter JH, Lubik AA, Quinn RJ, Nelson CC. The fatty acid synthase inhibitor triclosan: repurposing an anti-microbial agent for targeting prostate cancer. Oncotarget. 2014;5(19):9362–81.
Khwairakpam AD, Shyamananda MS, Sailo BL, Rathnakaram SR, Padmavathi G, Kotoky J, Kunnumakkara AB. ATP citrate lyase (ACLY): a promising target for cancer prevention and treatment. Curr Drug Targets. 2015;16(2):156–63.
Beckers A, Organe S, Timmermans L, Scheys K, Peeters A, Brusselmans K, Verhoeven G, Swinnen JV. Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Can Res. 2007;67(17):8180–7.
Eynard AR. Potential of essential fatty acids as natural therapeutic products for human tumors. Nutrition. 2003;19(4):386–8.
Mukerjee S, Saeedan AS, Ansari MN, Singh M. Polyunsaturated fatty acids mediated regulation of membrane biochemistry and tumor cell membrane integrity. Membranes. 2021. https://doi.org/10.3390/membranes11070479.
Das UN. Gamma-linolenic acid, arachidonic acid, and eicosapentaenoic acid as potential anticancer drugs. Nutrition. 1990;6(6):429–34.
Das UN. Tumoricidal action of cis-unsaturated fatty acids and their relationship to free radicals and lipid peroxidation. Cancer Lett. 1991;56(3):235–43.
Das UN. Nutrients, essential fatty acids and prostaglandins interact to augment immune responses and prevent genetic damage and cancer. Nutrition. 1989;5(2):106–10.
Zhang C, Yu H, Shen Y, Ni X, Shen S, Das UN. Polyunsaturated fatty acids trigger apoptosis of colon cancer cells through a mitochondrial pathway. Arch Med Sci. 2015;11(5):1081–94.
Meng H, Shen Y, Shen J, Zhou F, Shen S, Das UN. Effect of n-3 and n-6 unsaturated fatty acids on prostate cancer (PC-3) and prostate epithelial (RWPE-1) cells in vitro. Lipids Health Dis. 2013;12:160.
Gaston R, Maria Eugenia P, Das UN, Eynard AR. Polyunsaturated fatty acids differentially modulate cell proliferation and endocannabinoid system in two human cancer lines. Arch Med Res. 2017;48(1):46–54.
Sravan Kumar G, Das UN. Cytotoxic action of alpha-linolenic and eicosapentaenoic acids on myeloma cells in vitro. Prostaglandins Leukot Essent Fatty Acids. 1997;56(4):285–93.
Farago N, Feher LZ, Kitajka K, Das UN, Puskas LG. MicroRNA profile of polyunsaturated fatty acid treated glioma cells reveal apoptosis-specific expression changes. Lipids Health Dis. 2011;10:173.
Das UN, Madhavi N. Effect of polyunsaturated fatty acids on drug-sensitive and resistant tumor cells in vitro. Lipids Health Dis. 2011;10:159.
Al-Hassan JM, Fang Liu Y, Khan MA, Yang P, Guan R, Wen XY, Afzal M, Oommen S, Paul BM, Nair D, et al. Furanoic lipid F-6, a novel anti-cancer compound that kills cancer cells by suppressing proliferation and inducing apoptosis. Cancers. 2019. https://doi.org/10.3390/cancers11070960.
Afonso MS, Machado RM, Lavrador MS, Quintao ECR, Moore KJ, Lottenberg AM. Molecular pathways underlying cholesterol homeostasis. Nutrients. 2018. https://doi.org/10.3390/nu10060760.
Porter TD. Electron transfer pathways in cholesterol synthesis. Lipids. 2015;50(10):927–36.
Silvente-Poirot S, Poirot M. Cancer. Cholesterol and cancer, in the balance. Science. 2014;343(6178):1445–6.
Roizen MF. Hallmarks of cancer: the next generation. Yearb Anesthesiol Pain Manag. 2012;2012:13.
Gobel A, Rauner M, Hofbauer LC, Rachner TD. Cholesterol and beyond—the role of the mevalonate pathway in cancer biology. Biochim Biophys Acta Rev Cancer. 2020;1873(2): 188351.
Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. 2013;288(26):18707–15.
Xue L, Qi H, Zhang H, Ding L, Huang Q, Zhao D, Wu BJ, Li X. Targeting SREBP-2-regulated mevalonate metabolism for cancer therapy. Front Oncol. 2020;10:1510.
Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, Tsichlis PN, Liu XS, Struhl K. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010;17(4):348.
Huang S, He X. The role of microRNAs in liver cancer progression. Br J Cancer. 2011;104(2):235–40.
Moore KJ, Rayner KJ, Suárez Y, Fernándezhernando C. microRNAs and cholesterol metabolism. Trends Endocrinol Metab. 2010;21(12):699–706.
Rayner KJ, Suárez Y, Dávalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernándezhernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328(5985):1570.
Ibrahim AF, Weirauch U, Thomas M, Grünweller A, Hartmann RK, Aigner A. MicroRNA replacement therapy for miR-145 and miR-33a is efficacious in a model of colon carcinoma. Can Res. 2011;71(15):5214.
Sorrentino G, Ruggeri N, Specchia V, Cordenonsi M, Mano M, Dupont S, Manfrin A, Ingallina E, Sommaggio R, Piazza S. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat Cell Biol. 2014;16(4):357–66.
Yue S, Li J, Lee SY, Lee HJ, Shao T, Song B, Cheng L, Masterson TA, Liu X, Ratliff TL. Cholesteryl Ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014;19(3):393–406.
Gabitova L, Restifo D, Gorin A, Manocha K, Handorf E, Yang DH, Cai KQ, Kleinszanto AJ, Cunningham D, Kratz LE. Endogenous sterol metabolites regulate growth of EGFR/KRAS-dependent tumors via LXR. Cell Rep. 2015;12(11):1927.
Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342(6162):1094–8.
Bovenga F, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015;21(4):517–26.
Lin CY, Gustafsson JÅ. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer. 2015;15(4):216.
Wu Q, Ishikawa T, Sirianni R, Tang H, Mcdonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013;5(3):637.
Leon CG, Locke JA, Adomat HH, Etinger SL, Twiddy AL, Neumann RD, Nelson CC, Guns ES, Wasan KM. Alterations in cholesterol regulation contribute to the production of intratumoral androgens during progression to castration-resistant prostate cancer in a mouse xenograft model †. Prostate. 2010;70(4):390–400.
Lee BH, Taylor MG, Robinet P, Smith JD, Schweitzer J, Sehayek E, Falzarano SM, Magigalluzzi C, Klein EA, Ting AH. Dysregulation of cholesterol homeostasis in human prostate cancer through loss of ABCA1. Can Res. 2013;73(3):1211.
Freed-Pastor WA, Mizuno H, Zhao X, Langerã DA, Moon SH, Rodriguez-Barrueco R, Barsotti A, Chicas A, Li W, Polotskaia A. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148(1–2):244–58.
Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1(5):442–56.
Lim JH, Shin CM, Han K, Yoo J, Jin EH, Choi YJ, Lee DH. Nationwide cohort study: cholesterol level is inversely related with the risk of gastric cancer among postmenopausal women. Gastric Cancer. 2021. https://doi.org/10.1007/s10120-021-01241-1.
Olofsson SO, Boström P, Lagerstedt J, Andersson L, Adiels M, Perman J, Rutberg M, Li L, Borén J. The lipid droplet: a dynamic organelle, not only involved in the storage and turnover of lipids. Berlin: Springer; 2009.
Farese RV, Walther TC. Lipid droplets finally get a little R-E-S-P-E-C-T. Cell. 2009;139(5):855.
Beller M, Thiel K, Thul PJ, Jäckle H. Lipid droplets: a dynamic organelle moves into focus. FEBS Lett. 2010;584(11):2176–82.
Brasaemle DL, Wolins NE. Packaging of fat: an evolving model of lipid droplet assembly and expansion. J Biol Chem. 2012;287(4):2273.
Wilfling F, Haas JT, Walther TC, et al. Lipid droplet biogenesis. Curr Opin Cell Biol. 2014;29(1):39–45.
Marcel YL, Ouimet M, Wang MD. Cellular lipid traffic and lipid transporters: regulation of efflux and HDL formation. Berlin: Springer; 2009.
Petan T, Jarc E, Jusovic M. Lipid droplets in cancer: guardians of fat in a stressful world. Molecules. 2018. https://doi.org/10.3390/molecules23081941.
Potcoava MC, Futia GL, Aughenbaugh J, Schlaepfer IR, Gibson EA. Raman and coherent anti-Stokes Raman scattering microscopy studies of changes in lipid content and composition in hormone-treated breast and prostate cancer cells. J Biomed Opt. 2014;19(11): 111605.
Pan X, Wilson M, McConville C, Arvanitis TN, Griffin JL, Kauppinen RA, Peet AC. Increased unsaturation of lipids in cytoplasmic lipid droplets in DAOY cancer cells in response to cisplatin treatment. Metabolomics. 2013;9(3):722–9.
Donohoe DR, Collins LB, Wali A, Bigler R. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–26.
Zhang L, Keung W, Samokhvalov V, Wang W, Lopaschuk GD. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochem Biophys Acta. 2010;1801(1):1–22.
Wu X, Daniels G, Lee P, Monaco ME. Lipid metabolism in prostate cancer. Am J Clin Exp Urol. 2014;2(2):111.
Camarda R, Zhou AY, Kohnz RA, Balakrishnan S, Mahieu C, Anderton B, Eyob H, Kajimura S, Tward A, Krings G. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med. 2016;22(4):427.
Nakashima C, Shingo K, Fujiwara-Tani R, Luo Y, Kawahara I, Goto K, Sasaki T, Fujii K, Ohmori H, Kuniyasu H. Expression of long-chain fatty acid receptor GPR40 is associated with cancer progression in colorectal cancer: a retrospective study. Oncol Lett. 2018;15(6):8641–6.
Glatz JFC, Brinkmann JFF, Bonen A, Vusse GJVD, Luiken JJFP. Uptake of fatty acids by parenchymal cells: role of FAT/CD36. Adv Mol Cell Biol. 2003;33(33):89–98.
Su X, Abumrad NA. Cellular fatty acid uptake: a pathway under construction. Trends Endocrinol Metab. 2009;20(2):72–7.
Nomura DK. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. 2010;140(1):49–61.
Drabkin HA, Gemmill RM. Obesity, cholesterol, and clear-cell renal cell carcinoma (RCC). Adv Cancer Res. 2010;107(107):39–56.
Sundelin JP, Ståhlman M, Lundqvist A, Levin M, Parini P, Johansson ME, Borén J. Increased expression of the very low-density lipoprotein receptor mediates lipid accumulation in clear-cell renal cell carcinoma. PLoS ONE. 2012;7(11): e48694.
Xu G, Jiang Y, Xiao Y, Liu XD, Yue F, Li W, Li X, He Y, Jiang X, Huang H. Fast clearance of lipid droplets through MAP1S-activated autophagy suppresses clear cell renal cell carcinomas and promotes patient survival. Oncotarget. 2016;7(5):6255–65.
Huang WC, Li X, Jian L, Lin J, Chung LWK. Activation of androgen receptor, lipogenesis and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. 2012;10(1):133.
Gang X, Yang Y, Jian Z, Jiang K, Pan Y, Karnes RJ, Zhang J, Xu W, Wang G, Huang H. P300 acetyltransferase regulates fatty acid synthase expression, lipid metabolism and prostate cancer growth. Oncotarget. 2016;7(12):15135–49.
Schlaepfer IR, Hitz CA, Gijón MA, Bergman BC, Eckel RH, Jacobsen BM. Progestin modulates the lipid profile and sensitivity of breast cancer cells to docetaxel. Mol Cell Endocrinol. 2012;363(1–2):111–21.
Oba T, Ono M, Iesato A, Hanamura T, Watanabe T, Ito T, Kanai T, Maeno K, Ito K, Tateishi A. Lipid-rich carcinoma of the breast that is strongly positive for estrogen receptor: a case report and literature review. Oncotargets Ther. 2016;9(Issue 1):1641.
Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat. 2010;122(3):661–70.
Mukherjee A, Wu J, Barbour S, Fang X. Lysophosphatidic acid activates lipogenic pathways and de novo lipid synthesis in ovarian cancer cells. J Biol Chem. 2012;287(30):24990–5000.
Pyragius CE, Fuller M, Ricciardelli C, Oehler MK. Aberrant lipid metabolism: an emerging diagnostic and therapeutic target in ovarian cancer. Int J Mol Sci. 2013;14(4):7742–56.
Accioly MT, Pacheco P, Maya-Monteiro CM, Carrossini N, Robbs BK, Oliveira SS, Kaufmann C, Morgado-Diaz JA, Bozza PT, Viola JPB. Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Can Res. 2008;68(6):1732–40.
Record M, Carayon K, Poirot M, Silvente-Poirot S. Exosomes as new vesicular lipid transporters involved in cell–cell communication and various pathophysiologies. Biochem Biophys Acta. 2014;1841(1):108–20.
Yekula A, Yekula A, Muralidharan K, Kang K, Carter BS, Balaj L. Extracellular vesicles in glioblastoma tumor microenvironment. Front Immunol. 2019;10:3137.
Maacha S, Bhat AA, Jimenez L, Raza A, Haris M, Uddin S, Grivel JC. Extracellular vesicles-mediated intercellular communication: roles in the tumor microenvironment and anti-cancer drug resistance. Mol Cancer. 2019;18(1):55.
Boomgarden AC, Sheehan C, D’Souza-Schorey C. Extracellular vesicles in the tumor microenvironment: various implications in tumor progression. Adv Exp Med Biol. 2020;1259:155–70.
Zeng H, Chi H. Metabolic control of regulatory T cell development and function. Trends Immunol. 2015;36(1):3–12.
Angela M, Endo Y, Asou HK, Yamamoto T, Tumes DJ, Tokuyama H, Yokote K, Nakayama T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARgamma directs early activation of T cells. Nat Commun. 2016;7:13683.
Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–8.
Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, Karoly ED, Freeman GJ, Petkova V, Seth P, et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692.
Kouidhi S, Elgaaied AB, Chouaib S. Impact of metabolism on T-cell differentiation and function and cross talk with tumor microenvironment. Front Immunol. 2017;8:270.
Liu X, Mo W, Ye J, Li L, Zhang Y, Hsueh EC, Hoft DF, Peng G. Regulatory T cells trigger effector T cell DNA damage and senescence caused by metabolic competition. Nat Commun. 2018;9(1):249.
Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, Brunazzi EA, Vignali KM, Sun M, Stolz DB, et al. Treg cells promote the SREBP1-dependent metabolic fitness of tumor-promoting macrophages via repression of CD8(+) T cell-derived interferon-gamma. Immunity. 2019;51(2):381-397 e3386.
Mantovani A, Sica A, Allavena P, Garlanda C, Locati M. Tumor-associated macrophages and the related myeloid-derived suppressor cells as a paradigm of the diversity of macrophage activation. Hum Immunol. 2009;70(5):325–30.
Ricketts TD, Prieto-Dominguez N, Gowda PS, Ubil E. Mechanisms of macrophage plasticity in the tumor environment: manipulating activation state to improve outcomes. Front Immunol. 2021;12: 642285.
Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42.
Hotamisligil GS. Inflammation, metaflammation and immunometabolic disorders. Nature. 2017;542(7640):177–85.
Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, Valle LD, Trillo-Tinoco J, Maj T, Zou W, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. 2015;3(11):1236–47.
Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, Corzo A, Cho HI, Celis E, Lennox B, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880–6.
Gao F, Liu C, Guo J, Sun W, Xian L, Bai D, Liu H, Cheng Y, Li B, Cui J, et al. Radiation-driven lipid accumulation and dendritic cell dysfunction in cancer. Sci Rep. 2015;5:9613.
de Pablo MA, de Cienfuegos GA. Modulatory effects of dietary lipids on immune system functions. Immunol Cell Biol. 2000;78(1):31–9.
Gevariya N, Besancon M, Robitaille K, Picard V, Diabate L, Alesawi A, Julien P, Fradet Y, Bergeron A, Fradet V. Omega-3 fatty acids decrease prostate cancer progression associated with an anti-tumor immune response in eugonadal and castrated mice. Prostate. 2019;79(1):9–20.
Tzeng HT, Chyuan IT, Chen WY. Shaping of innate immune response by fatty acid metabolite palmitate. Cells. 2019. https://doi.org/10.3390/cells8121633.
Tan J, McKenzie C, Potamitis M, Thorburn AN, Mackay CR, Macia L. The role of short-chain fatty acids in health and disease. Adv Immunol. 2014;121:91–119.
Hansen L, Skeie G, Landberg R, Lund E, Palmqvist R, Johansson I, Dragsted LO, Egeberg R, Johnsen NF, Christensen J, et al. Intake of dietary fiber, especially from cereal foods, is associated with lower incidence of colon cancer in the HELGA cohort. Int J Cancer. 2012;131(2):469–78.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6.
Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–73.
Nakajima A, Nakatani A, Hasegawa S, Irie J, Ozawa K, Tsujimoto G, Suganami T, Itoh H, Kimura I. The short chain fatty acid receptor GPR43 regulates inflammatory signals in adipose tissue M2-type macrophages. PLoS ONE. 2017;12(7): e0179696.
Wu W, Sun M, Chen F, Cao AT, Liu H, Zhao Y, Huang X, Xiao Y, Yao S, Zhao Q, et al. Microbiota metabolite short-chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 2017;10(4):946–56.
Guaita-Esteruelas S, Guma J, Masana L, Borras J. The peritumoural adipose tissue microenvironment and cancer. The roles of fatty acid binding protein 4 and fatty acid binding protein 5. Mol Cell Endocrinol. 2018;462(Pt B):107–18.
Kobayashi S, Wannakul T, Sekino K, Takahashi Y, Kagawa Y, Miyazaki H, Umaru BA, Yang S, Yamamoto Y, Owada Y. Fatty acid-binding protein 5 limits the generation of Foxp3(+) regulatory T cells through regulating plasmacytoid dendritic cell function in the tumor microenvironment. Int J Cancer. 2021. https://doi.org/10.1002/ijc.33777.
Wen X, Zhao WH, Chen LZ, Qu W, Liu HX, Yan HY, Hou LF, Ping J. Attenuated cholesterol metabolism pathway suppresses regulatory T cell development in prenatal nicotine exposed female mice. Toxicology. 2019;428: 152309.
Kolonin MG. Bad cholesterol uptake by CD36 in T-cells cripples anti-tumor immune response. Immunometabolism. 2021. https://doi.org/10.20900/immunometab20210028.
Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity. 2021;54(7):1561-1577 e1567.
Das UN. Bioactive lipids as modulators of immune check point inhibitors. Med Hypotheses. 2020;135: 109473.
Bi J, Ichu TA, Zanca C, Yang H, Zhang W, Gu Y, Chowdhry S, Reed A, Ikegami S, Turner KM, et al. Oncogene amplification in growth factor signaling pathways renders cancers dependent on membrane lipid remodeling. Cell Metab. 2019;30(3):525-538 e528.
Gouw AM, Eberlin LS, Margulis K, Sullivan DK, Toal GG, Tong L, Zare RN, Felsher DW. Oncogene KRAS activates fatty acid synthase, resulting in specific ERK and lipid signatures associated with lung adenocarcinoma. Proc Natl Acad Sci USA. 2017;114(17):4300–5.
Zhu A, Marcus DM, Shu HKG, Shim H. Application of metabolic PET imaging in radiation oncology. Radiat Res. 2012;177(4):436–48.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (Grant No. 81801904).
Author information
Authors and Affiliations
Contributions
XL and PZ worked on the literature study, figures, and manuscript draft. JX and GL contributed to study idea, manuscript writing and editing, and clinical consultant. YL contributed to manuscript editing, and English checking. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, X., Zhang, P., Xu, J. et al. Lipid metabolism in tumor microenvironment: novel therapeutic targets. Cancer Cell Int 22, 224 (2022). https://doi.org/10.1186/s12935-022-02645-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-022-02645-4