
Abstract
Background
To determine the association between circadian pathway genetic variants and the risk of prostate cancer progression.
Methods
We systematically evaluated 79 germline variants in nine circadian pathway genes in a cohort of 458 patients with localized prostate cancer as the discovery phase. We then replicated the significant findings in another cohort of 324 men with more advanced disease. The association of each variant with prostate cancer progression was evaluated by a log-rank test and Cox regression.
Results
A single nucleotide polymorphism of the neuronal PAS domain protein 2 (NPAS2) gene (rs6542993 A>T) was found to be associated with a significantly higher risk of disease progression in both localized (P = 0.001) and advanced (P = 0.039) prostate cancer cases. In silico analysis revealed decreased expression levels of NPAS2 in carriers of the T allele of rs6542993 compared with those carrying the A allele. Consistently, downregulation of NPAS2 expression was associated with more aggressive prostate cancer and poor progression-free survival (log-rank P = 0.002).
Conclusions
The NPAS2 rs6542993 polymorphism may be a promising biomarker, and may shed light on the pathways that govern prostate cancer progression.
Similar content being viewed by others
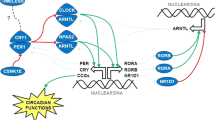
Background
The circadian rhythm is driven by an internal biological clock, which allows organisms to sustain an approximate 24-h cycle of physiological activities such as the sleep–wake cycle, basal metabolism, hormone production, and immunity [1]. The core circadian clock is mainly generated through a series of transcriptional/translational feedback loops. In the morning, clock circadian regulator (CLOCK) and neuronal PAS domain protein 2 (NPAS2) form heterodimers with aryl hydrocarbon receptor nuclear translocator like (ARNTL), bind to E-box enhancer elements, and activate the transcription of period circadian regulators (PER1, PER2, PER3) and cryptochrome circadian regulators (CRY1, CRY2) [2,3,4,5]. Late in the day, PERs and CRYs heterodimerize and activate a negative feedback loop, which directly suppresses transcriptional activity of the CLOCK/ARNTL complex. The suppression of CLOCK/ARNTL is released through the degradation of PERs and CRYs by casein kinase 1 epsilon (CSNK1E) and ubiquitin-mediated pathways [6]. After PERs and CRYs are degraded, the entire cycle repeats with a periodicity of approximately 24 h.
Shift-work, sleep deprivation, jet lag, and light exposure at night potentially cause circadian disruption, which has been linked to the risk of various diseases such as diabetes, depression, cardiovascular problems, and cancer [7,8,9,10]. In particular, the International Agency for Research on Cancer classified shift-work with circadian disruption as a probable human carcinogen (Group 2A) [11]. A few candidate gene studies have examined the associations between circadian genes and several cancers [12,13,14,15,16], including prostate cancer. The association between circadian gene variants and prostate cancer risk has been described in several case–control study in Chinese and Caucasian population [13, 17]. However, an alternative study failed finding any association of 96 variants across 12 circadian-related genes with fatal prostate cancer using three patient cohorts [18]. Therefore, this gene-disease association remains inconsistent, and very few studies have assessed the prognostic values of these genes.
Based on accumulating evidence for a relationship between the circadian rhythm and cancer, we hypothesized that genetic variants in circadian pathway genes might have impacts on the prognosis of patients with prostate cancer. To test this hypothesis, we systematically evaluated the influence of 79 circadian gene variants on disease progression in patients with localized prostate cancers and then verified the findings in another group of patients with advanced prostate cancers.
Methods
Patient population and clinical data collection
A total of 458 patients with localized prostate cancer who underwent radical prostatectomy as initial treatment and 324 patients with advanced prostate cancer who were on androgen-deprivation therapy (ADT) were included in the present study. The recruitment process and patient characteristics were described previously [19,20,21,22]. In the localized prostate cancer cohort, biochemical recurrence (BCR) was defined as two consecutive prostate-specific antigen (PSA) level increments of > 0.2 ng/mL during an interval of > 3 weeks [23, 24]. In the advanced prostate cancer cohort, disease progression was defined as a serial rise, at least two rises over 1 week apart, in PSA over the nadir [25, 26]. Initiation of secondary hormone treatment for patients with a rising PSA level was also considered as a progression event. This study was performed in accordance with the approval procedures by the Institutional Review Board of Kaohsiung Medical University Hospital, and written informed consent was obtained from all patients.
Single nucleotide polymorphism (SNP) selection and genotyping
TagSNPs were chosen from nine circadian-related genes (ARNTL, CLOCK, CRY1, CRY2, CSNK1E, NPAS2, PER1, PER2, and PER3) using the Tagger pairwise method [27] based on an r2 value of 0.8 or higher and a minor allele frequency of at least 0.2 in the HapMap Chinese Han population [28]. Ultimately, a total of 96 SNPs were selected for genotyping. Genomic DNA was extracted from peripheral blood samples using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, USA). Genotyping was performed as described previously [29] using Agena Bioscience iPLEX matrix-assisted laser desorption/ionization time-of-flight mass-spectrometry technology at the National Center for Genome Medicine, Taiwan. Any SNP that failed at assay design (N = 7), deviated from Hardy–Weinberg equilibrium (P < 0.01, N = 5), or fell below a genotyping call rate of 0.85 (N = 3) was removed, leaving 79 SNPs for further analysis. The average genotype call rate was 99.2%, and the concordance rate was 100% among 10 duplicated samples.
Bioinformatics analysis
We annotated the regulatory potential of the region adjoining the tagSNPs using HaploReg v4.1 [30] and ENCODE [31]. The GENe Expression VARiation (Genevar) database was used to identify potential SNP-gene expression quantitative trait loci (eQTL) associations within a locus [32]. The prognostic effect of NPAS2 on prostate cancer was analysed using datasets from Memorial Sloan Kettering Cancer Center (MSKCC) Prostate Oncogenome Project [33].
Statistical analysis
Kaplan–Meier analysis with the log-rank test was first used to assess the association of time to disease progression with each tagSNP under dominant, recessive, and additive models of inheritance. Since many tagSNPs were analysed, we conducted bootstrap resampling [34] to internally validate the significance of the tagSNPs by performing 1000 bootstrap runs. The risk of disease progression was estimated using the hazard ratios (HRs) and 95% confidence intervals (CIs) obtained by multivariate Cox regression adjusting for age, PSA level at diagnosis, pathologic Gleason score, and pathologic stage in the localized prostate cancer cohort [35], or adjusting for age, PSA level at ADT initiation, biopsy Gleason score, clinical stage, PSA nadir, and treatment modality in the advanced prostate cancer cohort [36]. Nonparametric analysis of variance followed by post hoc multiple comparison tests were applied to compare the level of NPAS2 expression with clinical characteristics of the patients. All statistical analyses were performed using Statistical Package for the Social Sciences (SPSS) software version 19.0.0 (IBM, Armonk, NY, USA), and a two-sided P value of < 0.05 was considered statistically significant.
Results
The clinical characteristics of the study groups are presented in Table 1. The median age of the patients in the localized prostate cancer cohort was 66 years. Over a median follow-up time of 54 months, 184 (40.2%) of the patients experienced a disease relapse. The median age of patients in the advanced prostate cancer cohort was 72 years, and 296 (91.4%) patients progressed to castration-resistant prostate cancer during the median follow-up of 93 months.
In the localized prostate cancer cohort, we screened 79 SNPs within nine circadian-related genes. NPAS2 rs6542993 was significantly associated with BCR (P = 0.039, Table 2 and Fig. 1a), while no significant association was detected for the other selected variants (Additional file 1: Table S1). The prognostic value of rs6542993 remained significant with 1000 bootstrap resampling (P = 0.042, Table 2), and in multivariate analysis after adjusting for age, PSA level at diagnosis, pathologic Gleason score, and stage (HR 1.50, 95% CI 1.18–1.91, P = 0.001; Table 2), providing support for the validity of this result. Interestingly, NPAS2 rs6542993 was also found to be associated with an increased risk of progressive disease (HR 1.32, 95% CI 1.01–1.71, P = 0.032; Table 3 and Fig. 1b) after adjusting for known clinicopathological variables that are associated with advanced prostate cancer, confirming NPAS2 rs6542993 as a biomarker for prostate cancer progression.

Impact of the SNP NPAS2 rs6542993 on prostate cancer progression. Kaplan–Meier estimates of a biochemical recurrence (BCR)-free survival in localized prostate cancer patients who received radical prostatectomy, and b progression-free survival in patients with advanced prostate cancer who received androgen-deprivation therapy (ADT) according to NPAS2 rs6542993 genotypes
We annotated all correlated variants within the linkage disequilibrium (LD) block (r2 ≥ 0.9) of rs6542993 with respect to regulatory elements, and predicted functional motifs catalogued in ENCODE and HaploReg (Fig. 2). All SNPs coincided with promoter or enhancer elements: five SNPs are located in DNase hypersensitivity domains and are predicted to alter transcription factor-binding motifs, suggesting that these SNPs could influence gene expression. Genevar analysis using HapMap data to investigate the association of these SNPs with NPAS2 expression demonstrated that rs6542993 and linked SNPs coincide with a probable eQTL and are the best candidates to influence NPAS2 expression. The risk allele T of rs6542993 was associated with a decreased NPAS2 expression level in the HapMap Han Chinese in Beijing (CHB) population (empirical P = 0.005, Fig. 3).

a ENCODE data for the linkage disequilibrium (LD) block containing NPAS2 rs6542993. H3K4Me1, H3K4Me3, and H3K27Ac tracks show the levels of enrichment of the mono-methylation of lysine 4, tri-methylation of lysine 4, and acetylation of lysine 27 of the H3 histone protein, respectively, across the genome as determined by chromatin immunoprecipitation sequencing (ChIP-seq) assays. These marks are thought to be associated with enhancer and promoter regions. Chromatin State Segmentation track displays chromatin state segmentations by integrating the ChIP-seq data using a Hidden Markov Model for HepG2 hepatocellular carcinoma cells, HMEC normal mammary epithelial cells, and NHLF normal lung fibroblast cells. The chromatin state regions predicted for promoters and enhancers are highlighted. The DNase clusters track shows DNase hypersensitivity areas. The Tnx factor track shows regions of transcription factor binding to DNA as assayed by ChIP-seq experiments. b Regulatory annotation of variants within the LD block containing NPAS2 rs6542993. In the LD block with the tagSNP rs6542993, there are strong enrichments of promoter and enhancer marks among the several different cell types tested. In addition, multiple regulatory motifs are predicted to be affected by the linked variants

Expression quantitative trait loci (eQTL) analyses targeting the NPAS2 locus with data of the HapMap CHB population. a eQTL SNPs are visualized as a regional plot (2 Mb), where a dotted line represents the P value threshold (0.01). b Best candidate eQTL SNPs after running 10,000 permutations. rho, Spearman’s rank correlation coefficient; P, P value; Pemp, adjusted P value after running 10,000 permutations. Note that NPAS2 is located between coordinates 100,803,039 and 100,979,719 according to the NCBI36/Ensembl 50 assembly
We further evaluated the association of NPAS2 expression with prostate cancer outcome using the MSKCC Prostate Oncogenome Project data. Consistently, the NPAS2 gene expression level was significantly lower in cases of more aggressive forms of prostate cancer (P ≤ 0.003, Fig. 4a, b), and a low expression level of NPAS2 was associated with poor BCR-free survival in patients with prostate cancer (P = 0.002, Fig. 4c).

Negative correlation of NPAS2 mRNA expression with prostate cancer progression. The associations between NPAS2 expression and prostate cancer aggressiveness were analysed using data of the MSKCC Prostate Oncogenome Project. More advanced prostate cancers, pathologic stage T3/T4 vs. T2 (a), and pathologic Gleason score ≥ 7 vs. < 7 (b) were associated with significantly lower mRNA expression levels of NPAS2. c Kaplan–Meier curves of biochemical recurrence (BCR)-free survival according to the expression of NPAS2. Patients were dichotomized based on the median expression level of NPAS2
Discussion
We conducted a two-stage study to investigate the effects of common germline genetic variants in the circadian pathway on the prognosis of prostate cancer patients. Among the 79 variants assessed in nine genes, NPAS2 rs6542993 was identified as an independent prognostic factor for prostate cancer progression across both cohorts. In addition, bioinformatics analysis provided further evidence that rs6542993 is an eQTL that affects the expression of NPAS2, and down-regulation of NPAS2 expression was correlated with a shorter progression-free survival of prostate cancer patients. Overall, these findings highlight the importance of NPAS2 in prostate cancer progression.
The risk variant rs6542993 is located in the intronic region of NPAS2, displaying histone marks with transcriptional enhancer activity. In addition to rs6542993, multiple SNPs in high LD were also identified as significant eQTLs for NPAS2. In fact, rs10172146, a SNP in high LD (r2 = 0.9) with the risk allele, showed a strong association with NPAS2 expression. The risk allele rs6542993 T was associated with reduced levels of NPAS2 expression. Only a few studies have examined the role of circadian gene variants on prostate cancer risk, with some suggestive general associations reported [13, 17, 18]. NPAS2 has consistently emerged as one of the top genes with the greatest contribution to the observed statistical association between the circadian pathway and risk of prostate cancer in two meta-analyses of data from genome-wide association studies [37, 38]. NPAS2 is suggested to be a putative tumour suppressor playing an important role in transcriptional suppression of the protooncogene c-Myc [39], DNA damage response [40], and cell cycle control by regulating diverse downstream genes [41]. In addition, NPAS2 has been identified as a prognostic biomarker in breast and colorectal cancers [42, 43], in which reduced NPAS2 expression was associated with decreased disease-free and overall survival rates in patients with breast cancer [43], in line with our present results in patients with prostate cancer. Silencing NPAS2 expression promoted the proliferation, invasion, and wound healing abilities of colorectal cancer cells [42], indicating a crucial role of NPAS2 in tumour growth and metastasis. We found no association between NPAS2 rs6542993 polymorphism and prostate cancer risk when comparing our patient population to 1500 healthy controls from the Taiwan Biobank (data not shown). The NPAS2 gene expression levels also did not vary significantly when comparing prostate cancers and normal tissues using the MSKCC Prostate Oncogenome Project data. These data suggested that NPAS2 might play a role in prostate cancer progression but not in prostate cancer initiation. However, the functional roles of NPAS2 in prostate cancer progression remain largely unclear and need further investigation.
The modest sample size of both cohorts included in this study did not allow for optimal statistical power to detect associations; therefore, the observed P values could not reach a more stringent significance level to avoid multiple comparisons. However, the association between NPAS2 rs6542993 and prostate cancer progression was replicated across two independent and different types of cohorts, which largely reduces the chance of false-positive findings. In addition, functional analyses support the role of NPAS2 rs6542993 in patient prognosis. Since the tagSNPs evaluated in this study were selected based on haplotype diversities, the linked causal variants remain to be determined. Since circadian genes are expressed in a circadian manner, the timing of specimen collection and the measurements of genetic variants and gene expression should be consistent in the same population. Another potential limitation of the study is that our findings may not be generalized to other ethnic groups since the majority of patients in both cohorts were Taiwanese. However, this may also be a benefit of our study in reducing the effects of population heterogeneity to more clearly detect an association. Nevertheless, further independent, larger, and inter-ethnic studies, as well as functional experiments are needed to validate our findings.
Conclusions
This is the first attempt to systematically evaluate the effect of genetic variants in the circadian pathway on prostate cancer progression. NPAS2 rs6542993 was consistently associated with disease progression after adjusting for clinical confounders in two independent prostate cancer cohorts. This SNP might affect prostate cancer progression by reducing the expression level of NPAS2. Thus, our findings might help to improve understanding of the interaction of circadian dysfunction and prostate cancer progression, and can provide a promising biomarker toward realizing the personalized management of patients with prostate cancer.
References
Merbitz-Zahradnik T, Wolf E. How is the inner circadian clock controlled by interactive clock proteins?: Structural analysis of clock proteins elucidates their physiological role. FEBS Lett. 2015;589:1516–29.
Chen R, Schirmer A, Lee Y, Lee H, Kumar V, Yoo SH, et al. Rhythmic PER abundance defines a critical nodal point for negative feedback within the circadian clock mechanism. Mol Cell. 2009;36:417–30.
Gekakis N, Staknis D, Nguyen HB, Davis FC, Wilsbacher LD, King DP, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science. 1998;280:1564–9.
Honma S, Ikeda M, Abe H, Tanahashi Y, Namihira M, Honma K, et al. Circadian oscillation of BMAL1, a partner of a mammalian clock gene Clock, in rat suprachiasmatic nucleus. Biochem Biophys Res Commun. 1998;250:83–7.
Thresher RJ, Vitaterna MH, Miyamoto Y, Kazantsev A, Hsu DS, Petit C, et al. Role of mouse cryptochrome blue-light photoreceptor in circadian photoresponses. Science. 1998;282:1490–4.
Lee HM, Chen R, Kim H, Etchegaray JP, Weaver DR, Lee C. The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci USA. 2011;108:16451–6.
Altman BJ. Cancer clocks out for lunch: disruption of circadian rhythm and metabolic oscillation in cancer. Front Cell Dev Biol. 2016;4:62.
Blakeman V, Williams JL, Meng QJ, Streuli CH. Circadian clocks and breast cancer. Breast Cancer Res. 2016;18:89.
Karatsoreos IN. Links between circadian rhythms and psychiatric disease. Front Behav Neurosci. 2014;8:162.
Mukherji A, Kobiita A, Damara M, Misra N, Meziane H, Champy MF, et al. Shifting eating to the circadian rest phase misaligns the peripheral clocks with the master SCN clock and leads to a metabolic syndrome. Proc Natl Acad Sci USA. 2015;112:E6691–8.
Straif K, Baan R, Grosse Y, Secretan B, El Ghissassi F, Bouvard V, et al. Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007;8:1065–6.
Alexander M, Burch JB, Steck SE, Chen CF, Hurley TG, Cavicchia P, et al. Case-control study of the PERIOD3 clock gene length polymorphism and colorectal adenoma formation. Oncol Rep. 2015;33:935–41.
Chu LW, Zhu Y, Yu K, Zheng T, Yu H, Zhang Y, et al. Variants in circadian genes and prostate cancer risk: a population-based study in China. Prostate Cancer Prostatic Dis. 2008;11:342–8.
Couto P, Miranda D, Vieira R, Vilhena A, De Marco L, Bastos-Rodrigues L. Association between CLOCK, PER3 and CCRN4L with nonsmall cell lung cancer in Brazilian patients. Mol Med Rep. 2014;10:435–40.
Jim HS, Lin HY, Tyrer JP, Lawrenson K, Dennis J, Chornokur G, et al. Common genetic variation in circadian rhythm genes and risk of epithelial ovarian cancer (EOC). J Genet Genome Res. 2015;2:2.
Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y, et al. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008;107:421–5.
Zhu Y, Stevens RG, Hoffman AE, Fitzgerald LM, Kwon EM, Ostrander EA, et al. Testing the circadian gene hypothesis in prostate cancer: a population-based case–control study. Cancer Res. 2009;69:9315–22.
Markt SC, Valdimarsdottir UA, Shui IM, Sigurdardottir LG, Rider JR, Tamimi RM, et al. Circadian clock genes and risk of fatal prostate cancer. Cancer Causes Control. 2015;26:25–33.
Bao BY, Pao JB, Huang CN, Pu YS, Chang TY, Lan YH, et al. Significant associations of prostate cancer susceptibility variants with survival in patients treated with androgen-deprivation therapy. Int J Cancer. 2012;130:876–84.
Huang CN, Huang SP, Pao JB, Chang TY, Lan YH, Lu TL, et al. Genetic polymorphisms in androgen receptor-binding sites predict survival in prostate cancer patients receiving androgen-deprivation therapy. Ann Oncol. 2012;23:707–13.
Huang SP, Ting WC, Chen LM, Huang LC, Liu CC, Chen CW, et al. Association analysis of Wnt pathway genes on prostate-specific antigen recurrence after radical prostatectomy. Ann Surg Oncol. 2010;17:312–22.
Yu CC, Lin VC, Huang CY, Liu CC, Wang JS, Wu TT, et al. Prognostic significance of cyclin D1 polymorphisms on prostate-specific antigen recurrence after radical prostatectomy. Ann Surg Oncol. 2013;20(Suppl 3):S492–9.
Freedland SJ, Sutter ME, Dorey F, Aronson WJ. Defining the ideal cutpoint for determining PSA recurrence after radical prostatectomy. Prostate-specific antigen. Urology. 2003;61:365–9.
Huang SP, Huang LC, Ting WC, Chen LM, Chang TY, Lu TL, et al. Prognostic significance of prostate cancer susceptibility variants on prostate-specific antigen recurrence after radical prostatectomy. Cancer Epidemiol Biomarkers Prev. 2009;18:3068–74.
Bao BY, Pao JB, Huang CN, Pu YS, Chang TY, Lan YH, et al. Polymorphisms inside microRNAs and microRNA target sites predict clinical outcomes in prostate cancer patients receiving androgen-deprivation therapy. Clin Cancer Res. 2011;17:928–36.
Ross RW, Oh WK, Xie W, Pomerantz M, Nakabayashi M, Sartor O, et al. Inherited variation in the androgen pathway is associated with the efficacy of androgen-deprivation therapy in men with prostate cancer. J Clin Oncol. 2008;26:842–7.
de Bakker PI, Yelensky R, Pe’er I, Gabriel SB, Daly MJ, Altshuler D. Efficiency and power in genetic association studies. Nat Genet. 2005;37:1217–23.
International HapMap C, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61.
Huang CN, Huang SP, Pao JB, Hour TC, Chang TY, Lan YH, et al. Genetic polymorphisms in oestrogen receptor-binding sites affect clinical outcomes in patients with prostate cancer receiving androgen-deprivation therapy. J Intern Med. 2012;271:499–509.
Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016;44:D877–81.
Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63.
Yang TP, Beazley C, Montgomery SB, Dimas AS, Gutierrez-Arcelus M, Stranger BE, et al. Genevar: a database and Java application for the analysis and visualization of SNP-gene associations in eQTL studies. Bioinformatics. 2010;26:2474–6.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22.
Henderson AR. The bootstrap: a technique for data-driven statistics. Using computer-intensive analyses to explore experimental data. Clin Chim Acta. 2005;359:1–26.
Huang SP, Levesque E, Guillemette C, Yu CC, Huang CY, Lin VC, et al. Genetic variants in microRNAs and microRNA target sites predict biochemical recurrence after radical prostatectomy in localized prostate cancer. Int J Cancer. 2014;135:2661–7.
Huang SP, Lin VC, Lee YC, Yu CC, Huang CY, Chang TY, et al. Genetic variants in nuclear factor-kappa B binding sites are associated with clinical outcomes in prostate cancer patients. Eur J Cancer. 2013;49:3729–37.
Gu F, Zhang H, Hyland PL, Berndt S, Gapstur SM, Wheeler W, et al. Inherited variation in circadian rhythm genes and risks of prostate cancer and three other cancer sites in combined cancer consortia. Int J Cancer. 2017;141:1794–802.
Mocellin S, Tropea S, Benna C, Rossi CR. Circadian pathway genetic variation and cancer risk: evidence from genome-wide association studies. BMC Med. 2018;16:20.
Fu L, Pelicano H, Liu J, Huang P, Lee C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell. 2002;111:41–50.
Hoffman AE, Zheng T, Ba Y, Zhu Y. The circadian gene NPAS2, a putative tumor suppressor, is involved in DNA damage response. Mol Cancer Res. 2008;6:1461–8.
Yi CH, Zheng T, Leaderer D, Hoffman A, Zhu Y. Cancer-related transcriptional targets of the circadian gene NPAS2 identified by genome-wide ChIP-on-chip analysis. Cancer Lett. 2009;284:149–56.
Xue X, Liu F, Han Y, Li P, Yuan B, Wang X, et al. Silencing NPAS2 promotes cell growth and invasion in DLD-1 cells and correlated with poor prognosis of colorectal cancer. Biochem Biophys Res Commun. 2014;450:1058–62.
Yi C, Mu L, de la Longrais IA, Sochirca O, Arisio R, Yu H, et al. The circadian gene NPAS2 is a novel prognostic biomarker for breast cancer. Breast Cancer Res Treat. 2010;120:663–9.
Authors’ contributions
C-CY, VCL, C-YH, I-LL, S-PH, and B-YB conceived and designed the experiments. L-CC, C-YC, Y-JC, T-YC, and T-LL performed the experiments and analyzed the data. All authors read and approved the final manuscript.
Acknowledgements
We thank Chao-Shih Chen for data analysis, and the National Center for Genome Medicine, Ministry of Science and Technology of Taiwan, for technical support. The results published here are based in part on data generated by the HapMap, HaploReg, and MSKCC Prostate Oncogenome projects.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The data of the study are available from the corresponding author on reasonable request.
Consent for publication
All authors approved publication of the manuscript.
Ethics approval and consent to participate
This study was performed in accordance with the approval protocols by the Institutional Review Board of Kaohsiung Medical University Hospital, and written informed consent was obtained from all participants before the questionnaire interview and specimen collection.
Funding
This work was supported by the Ministry of Science and Technology of Taiwan (Grant Nos: 103-2314-B-037-060, 104-2314-B-650-006, 104-2314-B-037-052-MY3, 105-2314-B-650-003-MY3, 106-2314-B-039-018, and 107-2320-B-039-010), the Kaohsiung Medical University Hospital (Grant No: KMUH106-6R57), the E-Da Hospital (Grant Nos: EDPJ104059, EDPJ105054, EDPJ106038, and EDAHP104053), and the China Medical University (Grant Nos: CMU106-S-24 and CMU107-S-42). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
Additional file 1: Table S1.
Genotyped SNPs and the P values of their association with biochemical recurrence after radical prostatectomy.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yu, CC., Chen, LC., Chiou, CY. et al. Genetic variants in the circadian rhythm pathway as indicators of prostate cancer progression. Cancer Cell Int 19, 87 (2019). https://doi.org/10.1186/s12935-019-0811-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-019-0811-4