
Abstract
Clear cell renal cell carcinoma (ccRCC) is the most common subtype of renal cell carcinoma (RCC), and is frequently accompanied by the genetic features of von Hippel–Lindau (VHL) loss. VHL loss increases the expression of hypoxia-inducible factors (HIFs) and their targets, including epidermal growth factor (EGF), vascular endothelial growth factor (VEGF), and platelet-derived growth factor (PDGF). The primary treatment for metastatic RCC (mRCC) is molecular-targeted therapy, especially anti-angiogenic therapy. VEGF monoclonal antibodies and VEGF receptor (VEGFR) tyrosine kinase inhibitors (TKIs) are the main drugs used in anti-angiogenic therapy. However, crosstalk between VEGFR and other tyrosine kinase or downstream pathways produce resistance to TKI treatment, and the multi-target inhibitors, HIF inhibitors or combination strategies are promising strategies for mRCC. HIFs are upstream of the crosstalk between the growth factors, and these factors may regulate the expression of VEGR, EGF, PDGF and other growth factors. The frequent VHL loss in ccRCC increases HIF expression, and HIFs may be an ideal candidate to overcome the TKI resistance. The combination of HIF inhibitors and immune checkpoint inhibitors is also anticipated. Various clinical trials of programmed cell death protein 1 inhibitors are planned. The present study reviews the effects of current and potential TKIs on mRCC, with a focus on VEGF/VEGFR and other targets for mRCC therapy.
Similar content being viewed by others

Background
Renal cell carcinoma (RCC) is the most common kidney solid neoplasm, and 12 drugs are approved in US for metastatic RCC (mRCC). RCC is distinguished into three major histopathological classifications: clear cell RCC (ccRCC; 70–75%), papillary RCC (pRCC; 10–16%), and chromophobe RCC (chRCC; 5%) [1]. Approximately 60–80% of ccRCC cases exhibit the most frequent genetic feature, the loss of von Hippel–Lindau (VHL) [2, 3], which increases the expression of hypoxia-inducible factors (HIFs), their targets, and cell survival [4, 5]. HIF-2 is implicated in angiogenesis, and some ccRCCs are HIF-2 independent [6], which triggered biomarker-driven clinical trials. Biomarkers to predict outcome using targeted therapy in metastatic ccRCC exhibited some promise but further validation is needed [7,8,9,10,11]. Patients confronted with rare kidney cancers are often treated in the same manner as ccRCC patients [12]. The prognosis of mRCC is poor and the primary treatment is molecular-targeted therapy. Targeted therapy developed quickly and tyrosine kinase inhibitors (TKIs), mammalian target of rapamycin (mTOR) inhibitors and the programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) checkpoint inhibitors (such as nivolumab) are the standard target therapies for mRCC [13,14,15].
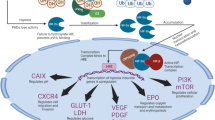
Receptor tyrosine kinases (RTKs), include epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor (VEGFR), fibroblast growth factor receptor (FGFR), platelet-derived growth factor receptor (PDGFR), and insulin-like growth factor 1 receptor (IGF-1R). Activation of tyrosine kinases (TKs) initiates multiple downstream signalling pathways, including phosphatidylinositol 3-kinase (PI3K)/AKT, Ras/Raf/MEK/ERK1/2, phospholipase C (PLC), signal transducer and activator of transcription (STAT)3 and STAT5 pathways [16, 17]. These multiple downstream signalling pathways are the basis of the crosstalk between TKs (Fig. 1).

Receptor tyrosine kinases, including EGFR, VEGFR, FGFR, PDGFR, and IGF-1R, are shown. Activation of tyrosine kinases initiates multiple downstream signalling pathways, including PI3K/AKT, MAPK, and JAK/STAT pathways and so on, which become the basis of the crosstalk between TKs
Twelve TKs (e.g., ABL2, CSF1R, and MET) are significantly upregulated in ccRCC, and 7 TKs (e.g., ERBB4, PDGFRA, ERBB2, and FGFR3) are downregulated [18]. Selective TKIs exhibited promise in the treatment of cancers driven by activated TKs. For example, TKIs for direct to Bcr-Abl, c-Kit and EGFR exhibited promise in the treatment of chronic myelogenous leukaemia, stromal tumours, and non-smallcell lung cancer (NSCLC) respectively. Numerous monoclonal antibodies directed against receptors or ligands and TKIs, such as cabozantinib [19], XMD8-87 (ACK inhibitor) [20] and axitinib [21, 22], were developed or approved (Table 1).
VEGF/VEGFR downstream pathway and VEGFR-TKI
VEGF family members in mammals consist of VEGF-A, -B, -C, -D, -E and placenta growth factor (PLGF). There are three main isoforms of VEGFR, VEGFR-1, VEGFR-2 and VEGFR-3, and VEGFR-2 plays a key role in angiogenesis [23]. VEGFR-3 is primarily expressed on lymphatic vessels, but the other VEGFR and the Tie receptor family are primarily expressed specifically in the endothelium. VEGF-A stimulates VEGFR2, which is autophosphorylated and activates various downstream signaling pathways [24]. Anti-angiogenesis, especially VEGF/VEGFR targeted theraphy, emerged as the standard of care for mRCC. Numerous VEGFR-TKIs were designed and developed (Table 2). VEGFR2-TKIs, such as sorafenib or sunitinib, are valuable treatment approaches for patients with mRCC [25]. VEGF and VEGFR polymorphisms affected outcomes in sunitinib-treated mRCC patients, especially VEGFR1 polymorphisms [26].
Mechanisms of TKI resistance
TKIs treatments are associated with dynamic changes in relevant biomarkers, including other protein TKs [27]. For example, VEGFR-TKI treatment significantly reduced vessel density (CD31) and phospho-S6K, PD-L1, and FOXP3 expression and significantly increased Ki-67, cytoplasmic FGF-2 and MET receptor expression in vessels [27]. However, long-lasting efficacy is seldom achieved, and evasive resistance eventually occurs under anti-angiogenic TKI therapy [28]. A recent study suggested that long non-coding RNAs (lncRNAs) play a significant role in TKI resistance in RCC. lncRNA-SRLR may be resistant to sorafenib and serve as a predictive biomarker for sorafenib tolerance via directly binding to NF-κB and the promotion of IL-6 transcription, which leads to STAT3 activation [29].
Exosomes also play a key role in resistance to drug theraphy. Stromal cells orchestrate an intricate crosstalk with tumour cells via utilization of exosomes to expand therapy resistance and reinitiate tumour growth [30]. lncARSR may promote and disseminate sunitinib resistance via competitively binding to miR-34/miR-449 to facilitate AXL and c-MET expression, incorporating into exosomes and transmitting to sensitive in RCC cells [31]. MiR-21 and miR-126 are targets of lncRNAs, and these molecules may be probable prognostic markers and therapeutic targets in RCC [32].
Various multi-targeted TKIs were developed because resistance to TKI treatment is inevitable. Sorafenib is a multi-targeted TKI that significantly improved clinical outcomes of mRCC patients [33]. However, no significant differences between sorafenib and sunitinib were observed in the treatment of advanced renal cancer in Chinese patients [34]. Axitinib, bevacizumab, and pazopanib are also anti-angiogenic drugs that approved for use in mRCC. Interleukin (IL)-8 expression is elevated, during sunitinib resistance, which suggests that IL-8 is also an key contributor and a potential therapeutic target to reverse sunitinib resistance in ccRCC [35]. Patients with high concentrations of IL-8, osteopontin and HGF exhibited shorter progression-free survival (PFS) compared to patients with low IL-8 concentrations using pazopanib as a treatment drug [36]. Cabozantinib is an important new standard-of-care treatment option for patients with advanced RCC who previously received anti-angiogenic therapy [37, 38]. VEGF is the downstream target of the HIF signal, and drugs that inhibit HIF-2 are in various stages of clinical testing [5]. The targeting of angiogenesis and hypoxia pathways may provide a resolution for the anti-angiogenesis resistance [28]. The HIF2α antagonist PT2385 is a novel therapeutics for RCC, and it exhibited cogent preclinical efficacy and improved tolerability [39]. Table 3 shows the factors, genes, proteins and other molecules (e.g., P-gp, MRP, and GSTs) involved in TKI resistance.
Side effects of TKIs
The development of TKIs is revolutionary progress, but TKIs exhibit side effects, including cardiovascular side effects, especially hypertension and congestive heart failure, and continual clinical monitoring should be emphasized in the use of new TKI agents [40, 41]. Cardiac damage from TKIs (sorafenib and sunitinib) treatment is a largely underrated phenomenon, but it is manageable with careful cardiovascular monitoring and cardiac treatment at the first signs of myocardial damage [42]. Another TKIs, cabozantinib, also exhibited a manageable adverse events profile in patients with advanced RCC [37]. Sunitinib increases buccodental toxicity compared to chemotherapy [43].
TKIs and immunotherapy
Immunotherapy enjoyed tremendous development recently in the form of immune checkpoint inhibition and vaccines [44]. VEGF-A/VEGFR-2 is also related to with tumour escape. VEGF-A directly triggers Treg proliferation, and VEGF-A/VEGFR-2 blockade inhibits this effect. Therefore, anti-VEGF-A therapies may also exert immunological effects [45]. A combination of immunotherapy treatment is also in process [46]. The combination of an IL-6 inhibitors (tocilizumab) and TKIs (sorafenib) may be a novel therapeutic approach for RCC [47]. Anti-VEGF (bevacizumab) in combination with an anti-PD-L1 (atezolizumab) improved antigen-specific T cell migration in mRCC [48]. More drug combination experiments will be performed with the design and development of less toxic novel immune checkpoint inhibitors and TKIs.
VEGFR-TKIs and other signalling pathways
Molecular crosstalk between VEGFR and other TKs or downstream pathways, such as EGFR, c-Met, FGFR, PDGFR, IGF-1R, c-Kit and PI3K/AKT/mTOR, may have great therapeutic and resistance implications [23, 49]. The crosstalk between these factors contributes to TKI resistance, but multi-targets or combination drugs may exhibit good synergy. Therefore, various multi-target inhibitors were examined or in process and some of these are listed below.
The mTOR pathway and its inhibitors
The mTOR/Raptor complex (mTORC1) is a key molecule in the PI3K/AKT/mTOR signalling pathway, and its activation increases protein synthesis and cell survival via direct phosphorylation of its effectors. Inhibitors of mTOR, such as everolimus and temsirolimus, are approved for the treatment of mRCC. Administration of everolimus alone or with lenvatinib is one of the most effective options for mRCC [50]. However, the inhibition of mTORC1 produces a loss of negative feedback loops, which upregulates the downstream effectors of the PI3K/AKT/mTOR pathway and activates of HIFs (an activator of angiogenesis) [51].
A combination of agents targeting the multiple pathways of angiogenesis, including HIF, VEGFR, PI3K and mTORC1/2, will likely be a beneficial choice. Lenvatinib plus everolimus and lenvatinib alone improved PFS in patients with mRCC who progressed after administration of one previous VEGF-targeted therapy [52]. The combination of bevacizumab and temsirolimus in patients previously treated with VEGFR-TKI is possible, but with dose reductions and treatment discontinuations [53]. The antidiabetic drug metformin blocks cell growth via TORC1 inhibition, and the combination of metformin and VEGF-TKI may be effective [54]. Combined treatment with everolimus and a Toll-like receptor 9 agonist immune modulatory oligonucleotide effectively interfered with tumour growth and angiogenesis in VHL wild-type and mutant models of RCC [55].
VEGFR and other tyrosine kinase pathway or TKIs
EGF/EGFR pathway and TKIs
EGF family ligands include EGF, heparin-binding EGF-like growth factor (HB-EGF), transforming growth factor alpha (TGFα), amphiregulin, epiregulin, epigen, β-cellulin, and neuregulin 2β (NRG2β) [56]. The human EGFR family consists of EGFR (ERBB1), HER2 (ERBB2), HER3 (ERBB3), and HER4 (ERBB4) [57]. Activation of HER2 and EGFR activates intracellular pathways, such as RAS/RAF/MEK/ERK, PI3K/AKT/TOR, Src kinases, and STAT transcription factors. The EGFR gene is upregulated in ccRCC [58], and the HIF may activate the TGF-α/EGFR pathway to promote the growth of VHL(−/−) RCC cells [59].
Three generations of EGFR-TKIs were developed. The first generation of EGFR-TKIs such as erlotinib or gefitinib, exhibit resistance after several months of treatment in patients with EGFR-activating mutations, especially in NSCLC patients [60]. The EGFR T790M mutation confers resistance to gefitinib via blockade of drug binding [61]. Therefore, the second generation of EGFR-TKIs such as afatinib and dacomitinib, were developed. However, the expression of FGFR1 and its ligand FGF2 is enhanced in afatinib-resistant cancer cells, which provide an escape mechanism for cell survival [62]. Second-generation drugs inhibit EGFR T790M, but these agents also inhibit wild-type EGFR. Therefore the dose-limiting toxicities from wild-type EGFR inhibition prevent the administration of doses that are sufficient to fully suppress T790M. The third generation of EGFR-TKIs, such as osimertinib, were developed to overcome these limitations [63].
Compensatory TK signalling is observed in EGFR-TKI therapy, and KRAS, anaplastic lymphoma kinase (ALK), c-MET and BRAF mutations are also associated with poor responses to anti-EGFR therapy in some cancers. Adaptation to TKI treatment also reactivates ERK signalling in TK-driven malignancies [64]. A novel dual inhibitor of EGFR and c-MET, TC-N19, was investigated as a potential new-generation TKI inhibitor to treat resistance to current TKI-targeting therapies [65]. Resistance to EGFR-targeted agents may also be related to increased VEGF levels. Vandetanib, is an inhibitor of EGFR, VEGFR and RET TK that exhibited therapeutic efficacy, and it received FDA approval for the treatment of advanced medullary thyroid carcinoma [66].
AEE788 is another potent inhibitor of EGFR and VEGFR TKs at the isolated enzyme level and in cellular systems [67], AEE788 profoundly reduce RCC cells growth in vitro [68]. However, the VEGF/VEGFR signal is the primary target because the universal VHL loss in ccRCC, and the mTORC1, MET and IL–8, but not the EGFR or PI3K pathways are secondary targets based on the available clinical and preclinical studies in mRCC [4].
PDGF/PDGFR and TKIs
The PDGF family consists of PDGF-A to -D polypeptide homodimers and the PDGF-AB heterodimer, and these ligands can bind to PDGFR-α and -β tyrosine kinase receptors [23]. The HIF signalling pathway regulates the target genes VEGF, EGF, TGF-β, and PDGF. High expression of PDGFR-β and α-smooth muscle actin (α-SMA) and low vessel density were significantly associated with short survival in RCC [69]. Other PTK pathways, such as PDGF/PDGFR and FGF/FGFR pathways, provide underlying escape mechanisms from anti-VEGF/VEGFR therapy that may promote resumption of tumor growth [23]. Multiple inhibitors, such as sunitinib, pazopanib, axitinib, tivozanib, linifanib, telatinib and motesanib, that inhibit VEGFR and PDGFR TKs have been used [70].
HGF/c-MET and TKIs
Met and its ligand, hepatocyte growth factor (HGF), play significant roles in multiple oncogenic cellular processes, including regulation of cell proliferation, invasion, angiogenesis and alternative pathways to the VEGF [49]. MET mutations are frequently found in Papillary RCC (pRCC) [71]. PD-L1 and PD-L2 in ccRCC is associated with adverse features of c-MET and VEGF expression, respectively [72].
The role for Met in resistance to other RTK-targeted therapies is associated with crosstalk between Met and other receptors, such as EGFR, HER2 and VEGFR [49]. Combination targeting of the VEGF and c-MET pathways in a ccRCC model exhibited a better anti-tumour effect than single agent administration [73]. Cabozantinib is a TKI inhibitor of VEGFR, c-MET and other TKs that exhibited significant clinical benefit in PFS and objective response rate over the standard-of-care sunitinib as first-line therapy in patients with intermediate- or poor-risk mRCC [74].
IGF system pathway
The insulin-like growth factor (IGF) system is comprised of multiple growth factor receptors, including IGF-1R, insulin receptor (IR)-A and -B [75]. IR is primarily expressed in adipose tissue, the muscle and liver in adult tissues, and IGF-1R is expressed in most human tissues [76]. As a TK receptor for IGF-1 and IGF-2, IGF-1R plays a key role in proliferation, malignant transformation, anti-apoptosis and metastasis. IGF-1R expression in RCC is associated with poor long-term patient survival [77]. The risk of death for patients with IGF-1R overexpression increases 70% compared to ccRCC patients with tumours without IGF-1R expression [78].
IGF-1 co-culture with cells facilitates angiogenesis via the PI3K/Akt signalling pathway [79]. IGF-1R also exhibits crosstalk with the VEGF/VEGFR, EGF/EGFR pathway [80, 81]. IGF-1R also confers resistance to EGFR or VEGFR family targeted therapies [75]. A bi-functional antibody-receptor domain fusion protein that targeted IGF-IR and VEGF for degradation, bi-AbCap, exhibited superior inhibition of tumour growth in RCC, colon cancer, and pancreatic cancer compared to a combination of anti-IGF-IR and anti-VEGF therapies [82].
Other TKIs
Table 1 shows other RTKs, such as FGFR (FGFR1-4), tyrosine receptor kinase (Trk), ephrin receptor, ALK and Src. FGF/FGFR regulates normal and tumour cells growth, differentiation and angiogenesis, and the complex interaction and crosstalk between tumour angiogenic factors, such as FGF2 and PDGFR, promoted tumour growth and metastasis [83].
TrkA and TrkB are neurotrophin receptors. TrkB activation or overexpression could promotes proliferation, survival, angiogenesis, anoikis-resistance and metastasis in tumours. Brain-derived neurotrophic factor binds to TrkB and p75NTR and induces cell survival and migration via p75NTR, which is independent of TrkB activation [84], which indicates a resistance mechanism of TKIs for TrkB. TrkB silencing improved the anticancer efficiency of sorafenib in anoikis-resistant ACHN (a renal cancer cell line derived from metastatic site) RCC cells via inactivation of PI3K/Akt and MEK/ERK pathways [85].
Many other non-receptor TKs exist and numerous TKIs are under investigation.
Conclusions
System treatment using multi-target drugs, immune checkpoint inhibitors or drugs combinations may be a promising approach to RCC therapy in the future because of the emergence of drug resistance to VEGFR-TKI, which may facilitate tumour invasiveness and metastasis. Three new second-line treatments received FDA approval in the last year for use after anti-angiogenic therapy: nivolumab, cabozantinib, and the combination of the TKI lenvatinib and everolimus (the mTOR inhibitor) [86]. Nivolumab is an immune checkpoint inhibitor, and cabozantinib is a multi-target TKI. The potential synergistic activity of antiangiogenic agents and TKIs or immunotherapy in NSCLC was summarized previously [87].
Combinations of VEGFR-TKIs, immune checkpoint inhibitors, and HIF inhibitors may provide good options to overcome drug resistance. Increasing evidence supports a primary role for the HIF-2α subunit in ccRCC over HIF-1α. Due to the frequently VHL loss in ccRCC, which causes increased expression of HIFs, targeting HIFs may be a promising strategy for ccRCC because of frequent VHL loss, which increases HIF expression. HIFs are upstream of the crosstalk between growth factors and regulate the expression of VEGR, EGF, PDGF and other growth factors, which supports targeting HIFs to overcome TKI resistance. Recent preclinical and clinical data on ccRCC indicate that PT2385 and PT2399 effectively inhibit cancer cell growth, proliferation, and tumour angiogenesis characteristic [88, 89]. The combination of HIF and immune checkpoint inhibitors is also anticipated, and various clinical trials for PD-1 inhibitors are planned.
Abbreviations
- RCC:
-
renal cell carcinoma
- ccRCC:
-
clear cell renal cell carcinoma
- mRCC:
-
metastatic renal cell carcinoma
- VHL:
-
von Hippel–Lindau
- HIFs:
-
hypoxia-inducible factors
- EGF:
-
epidermal growth factor
- VEGF:
-
vascular endothelial growth factor
- PDGF:
-
platelet-derived growth factor
- TKIs:
-
tyrosine kinase inhibitors
- mTOR:
-
mammalian target of rapamycin
- PD-1:
-
programmed cell death protein 1
- PD-L1:
-
programmed death ligand 1
- EGFR:
-
epidermal growth factor receptor
- VEGFR:
-
vascular endothelial growth factor receptor
- FGFR:
-
fibroblast growth factor receptor
- PDGFR:
-
platelet-derived growth factor receptor
- IGF-1R:
-
insulin-like growth factor 1 receptor
- TK:
-
tyrosine kinase
- RTK:
-
receptor tyrosine kinase
- PI3K:
-
phosphatidylinositol 3-kinase
- PLC:
-
phospholipase C
- STAT:
-
the signal transducer and activator of transcription
- NSCLC:
-
non-smallcell lung cancer
- lncRNA:
-
long non-coding RNA
- IL:
-
interleukin
- mTORC1:
-
mTOR/raptor complex
- PFS:
-
progression-free survival
- HB-EGF:
-
heparin-binding EGF-like growth factor
- TGFα:
-
transforming growth factor alpha
- α-SMA:
-
α-smooth muscle actin
- EMT:
-
epithelial to mesenchymal transformation
- BIM:
-
Bcl-2 interacting mediator of cell death
- P-gp:
-
P-glycoprotein
- MDR1:
-
multidrug resistance protein 1
- GSTs:
-
glutathione S-transferase
- MRP:
-
multidrug resistance-associated protein
- CSF-1:
-
colony stimulating factor 1
- FLT3:
-
Fms-like tyrosine kinase 3
- BDNF:
-
brain-derived neurotrophic factor
- GDNF:
-
glial cell line-derived neurotrophic factor
- NGF:
-
nerve growth factor
- NRG2β:
-
neuregulin 2β
- Trk:
-
tyrosine receptor kinase
- HGF:
-
hepatocyte growth factor
- ALK:
-
anaplastic lymphoma kinase
References
Shuch B, Amin A, Armstrong AJ, Eble JN, Ficarra V, Lopez-Beltran A, Martignoni G, Rini BI, Kutikov A. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. Eur Urol. 2015;67(1):85–97.
Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499(7456):43–9.
Schodel J, Grampp S, Maher ER, Moch H, Ratcliffe PJ, Russo P, Mole DR. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur Urol. 2016;69(4):646–57.
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009.
Shen C, Kaelin WJ. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23(1):18–25.
Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, Homayoun F, Ma Y, Patel N, Yell P, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature. 2016;539(7627):112–7.
Stewart GD, O’Mahony FC, Laird A, Rashid S, Martin SA, Eory L, Lubbock AL, Nanda J, O’Donnell M, Mackay A, et al. Carbonic anhydrase 9 expression increases with vascular endothelial growth factor-targeted therapy and is predictive of outcome in metastatic clear cell renal cancer. Eur Urol. 2014;66(5):956–63.
Xu J, Pham CG, Albanese SK, Dong Y, Oyama T, Lee CH, Rodrik-Outmezguine V, Yao Z, Han S, Chen D, et al. Mechanistically distinct cancer-associated mTOR activation clusters predict sensitivity to rapamycin. J Clin Investig. 2016;126(9):3526–40.
Voss MH, Chen D, Marker M, Hakimi AA, Lee CH, Hsieh JJ, Knox JJ, Voi M, Motzer RJ. Circulating biomarkers and outcome from a randomised phase II trial of sunitinib vs everolimus for patients with metastatic renal cell carcinoma. Br J Cancer. 2016;114(6):642–9.
Voss MH, Hakimi AA, Pham CG, Brannon AR, Chen YB, Cunha LF, Akin O, Liu H, Takeda S, Scott SN, et al. Tumor genetic analyses of patients with metastatic renal cell carcinoma and extended benefit from mTOR inhibitor therapy. Clin Cancer Res. 2014;20(7):1955–64.
Funakoshi T, Lee CH, Hsieh JJ. A systematic review of predictive and prognostic biomarkers for VEGF-targeted therapy in renal cell carcinoma. Cancer Treat Rev. 2014;40(4):533–47.
Giles RH, Choueiri TK, Heng DY, Albiges L, Hsieh JJ, Linehan WM, Pal S, Maskens D, Paseman B, Jonasch E, et al. Recommendations for the management of rare kidney cancers. Eur Urol. 2017;72(6):974–83.
Escudier B, Lougheed JC, Albiges L. Cabozantinib for the treatment of renal cell carcinoma. Expert Opin Pharmacother. 2016;17(18):2499–504.
Hsieh JJ, Chen D, Wang PI, Marker M, Redzematovic A, Chen YB, Selcuklu SD, Weinhold N, Bouvier N, Huberman KH, et al. Genomic biomarkers of a randomized trial comparing first-line everolimus and sunitinib in patients with metastatic renal cell carcinoma. Eur Urol. 2017;71(3):405–14.
Powles T, Albiges L, Staehler M, Bensalah K, Dabestani S, Giles RH, Hofmann F, Hora M, Kuczyk MA, Lam TB, et al. Updated european association of urology guidelines recommendations for the treatment of first-line metastatic clear cell renal cancer. Eur Urol. 2018:73(3):311–5.
Mitsudomi T, Yatabe Y. Epidermal growth factor receptor in relation to tumor development: EGFR gene and cancer. FEBS J. 2010;277(2):301–8.
Roskoski RJ. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74.
Behbahani TE, Thierse C, Baumann C, Holl D, Bastian PJ, von Ruecker A, Muller SC, Ellinger J, Hauser S. Tyrosine kinase expression profile in clear cell renal cell carcinoma. World J Urol. 2012;30(4):559–65.
Yakes FM, Chen J, Tan J, Yamaguchi K, Shi Y, Yu P, Qian F, Chu F, Bentzien F, Cancilla B, et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol Cancer Ther. 2011;10(12):2298–308.
Maxson JE, Abel ML, Wang J, Deng X, Reckel S, Luty SB, Sun H, Gorenstein J, Hughes SB, Bottomly D, et al. Identification and characterization of tyrosine kinase nonreceptor 2 mutations in leukemia through Integration of kinase inhibitor screening and genomic analysis. Cancer Res. 2016;76(1):127–38.
Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14(22):7272–83.
Pemovska T, Johnson E, Kontro M, Repasky GA, Chen J, Wells P, Cronin CN, McTigue M, Kallioniemi O, Porkka K, et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature. 2015;519(7541):102–5.
Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist. 2015;20(6):660–73.
Kowanetz M, Ferrara N. Vascular endothelial growth factor signaling pathways: therapeutic perspective. Clin Cancer Res. 2006;12(17):5018–22.
Zhang L, Wang X, Bullock AJ, Callea M, Shah H, Song J, Moreno K, Visentin B, Deutschman D, Alsop DC, et al. Anti-S1P antibody as a novel therapeutic strategy for VEGFR TKI-resistant renal cancer. Clin Cancer Res. 2015;21(8):1925–34.
Miao C, Cao J, Wang Y, Liu B, Wang Z. Effects of VEGF and VEGFR polymorphisms on the outcome of patients with metastatic renal cell carcinoma treated with sunitinib: a systematic review and meta-analysis. Oncotarget. 2017;8(40):68854–62.
Sharpe K, Stewart GD, Mackay A, Van Neste C, Rofe C, Berney D, Kayani I, Bex A, Wan E, O’Mahony FC, et al. The effect of VEGF-targeted therapy on biomarker expression in sequential tissue from patients with metastatic clear cell renal cancer. Clin Cancer Res. 2013;19(24):6924–34.
Lai XM, Liu SY, Tsai YT, Sun GH, Chang SY, Huang SM, Cha TL. HAF mediates the evasive resistance of anti-angiogenesis TKI through disrupting HIF-1alpha and HIF-2alpha balance in renal cell carcinoma. Oncotarget. 2017;8(30):49713–24.
Xu Z, Yang F, Wei D, Liu B, Chen C, Bao Y, Wu Z, Wu D, Tan H, Li J, et al. Long noncoding RNA-SRLR elicits intrinsic sorafenib resistance via evoking IL-6/STAT3 axis in renal cell carcinoma. Oncogene. 2017;36(14):1965–77.
Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, Azzam DJ, Twyman-Saint VC, Wiemann BZ, Ishwaran H, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. 2014;159(3):499–513.
Qu L, Ding J, Chen C, Wu ZJ, Liu B, Gao Y, Chen W, Liu F, Sun W, Li XF, et al. Exosome-transmitted lncARSR promotes sunitinib resistance in renal cancer by acting as a competing endogenous RNA. Cancer Cell. 2016;29(5):653–68.
Gu L, Li H, Chen L, Ma X, Gao Y, Li X, Zhang Y, Fan Y, Zhang X. MicroRNAs as prognostic molecular signatures in renal cell carcinoma: a systematic review and meta-analysis. Oncotarget. 2015;6(32):32545–60.
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356(2):125–34.
Sheng X, Chi Z, Cui C, Si L, Li S, Tang B, Mao L, Lian B, Wang X, Yan X, et al. Efficacy and safety of sorafenib versus sunitinib as first-line treatment in patients with metastatic renal cell carcinoma: largest single-center retrospective analysis. Oncotarget. 2016;7(19):27044–54.
Huang D, Ding Y, Zhou M, Rini BI, Petillo D, Qian CN, Kahnoski R, Futreal PA, Furge KA, Teh BT. Interleukin-8 mediates resistance to antiangiogenic agent sunitinib in renal cell carcinoma. Cancer Res. 2010;70(3):1063–71.
Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, Figlin RA, Hutson TE, Sternberg CN, Amado RG, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13(8):827–37.
Al-Salama ZT, Keating GM. Cabozantinib: a review in advanced renal cell carcinoma. Drugs. 2016;76(18):1771–8.
Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016;17(7):917–27.
Wallace EM, Rizzi JP, Han G, Wehn PM, Cao Z, Du X, Cheng T, Czerwinski RM, Dixon DD, Goggin BS, et al. A small-molecule antagonist of HIF2alpha is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 2016;76(18):5491–500.
Brown SA, Nhola L, Herrmann J. Cardiovascular toxicities of small molecule tyrosine kinase inhibitors: an opportunity for systems-based approaches. Clin Pharmacol Ther. 2017;101(1):65–80.
Santoni M, Guerra F, Conti A, Lucarelli A, Rinaldi S, Belvederesi L, Capucci A, Berardi R. Incidence and risk of cardiotoxicity in cancer patients treated with targeted therapies. Cancer Treat Rev. 2017;59:123–31.
Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008;26(32):5204–12.
Gilabert M, Provansal M, Cappiello M, Walz Y, Salem N, Tarpin C, Brunelle S, Thomassin J, Gravis G. Buccodental side effects of sunitinib in patients with metastatic renal cell carcinoma. Br J Cancer. 2013;109(7):1750–4.
Liu KG, Gupta S, Goel S. Immunotherapy: incorporation in the evolving paradigm of renal cancer management and future prospects. Oncotarget. 2017;8(10):17313–27.
Terme M, Pernot S, Marcheteau E, Sandoval F, Benhamouda N, Colussi O, Dubreuil O, Carpentier AF, Tartour E, Taieb J. VEGFA-VEGFR pathway blockade inhibits tumor-induced regulatory T-cell proliferation in colorectal cancer. Cancer Res. 2013;73(2):539–49.
Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, Voss MH, Sharma P, Pal SK, Razak A, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 Study. J Clin Oncol. 2017. https://doi.org/10.1200/JCO.2016.72.1985.
Ishibashi K, Haber T, Breuksch I, Gebhard S, Sugino T, Kubo H, Hata J, Koguchi T, Yabe M, Kataoka M, et al. Overriding TKI resistance of renal cell carcinoma by combination therapy with IL-6 receptor blockade. Oncotarget. 2017;8(33):55230–45.
Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, Hernandez G, Mier J, He X, Hodi FS, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun. 2016;7:12624.
Maroun CR, Rowlands T. The Met receptor tyrosine kinase: a key player in oncogenesis and drug resistance. Pharmacol Ther. 2014;142(3):316–38.
Ghidini M, Petrelli F, Ghidini A, Tomasello G, Hahne JC, Passalacqua R, Barni S. Clinical development of mTor inhibitors for renal cancer. Expert Opin Investig Drugs. 2017;26(11):1–9.
Figlin RA, Kaufmann I, Brechbiel J. Targeting PI3K and mTORC2 in metastatic renal cell carcinoma: new strategies for overcoming resistance to VEGFR and mTORC1 inhibitors. Int J Cancer. 2013;133(4):788–96.
Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, Mellado B, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015;16(15):1473–82.
Mahoney KM, Jacobus S, Bhatt RS, Song J, Carvo I, Cheng SC, Simpson M, Fay AP, Puzanov I, Michaelson MD, et al. Phase 2 study of bevacizumab and temsirolimus after VEGFR TKI in metastatic renal cell carcinoma. Clin Genitourin Cancer. 2016;14(4):304–13.
Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012;2(4):344–55.
Damiano V, Rosa R, Formisano L, Nappi L, Gelardi T, Marciano R, Cozzolino I, Troncone G, Agrawal S, Veneziani BM, et al. Toll-like receptor 9 agonist IMO cooperates with everolimus in renal cell carcinoma by interfering with tumour growth and angiogenesis. Br J Cancer. 2013;108(8):1616–23.
Faria J, de Andrade C, Goes AM, Rodrigues MA, Gomes DA. Effects of different ligands on epidermal growth factor receptor (EGFR) nuclear translocation. Biochem Biophys Res Commun. 2016;478(1):39–45.
Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25(3):282–303.
Matusan-Ilijas K, Damante G, Fabbro D, Dordevic G, Hadzisejdic I, Grahovac M, Avirovic M, Grahovac B, Jonjic N, Lucin K. EGFR expression is linked to osteopontin and Nf-kappaB signaling in clear cell renal cell carcinoma. Clin Transl Oncol. 2013;15(1):65–71.
Gunaratnam L, Morley M, Franovic A, de Paulsen N, Mekhail K, Parolin DA, Nakamura E, Lorimer IA, Lee S. Hypoxia inducible factor activates the transforming growth factor-alpha/epidermal growth factor receptor growth stimulatory pathway in VHL(−/−) renal cell carcinoma cells. J Biol Chem. 2003;278(45):44966–74.
van der Wekken AJ, Kuiper JL, Saber A, Terpstra MM, Wei J, Hiltermann T, Thunnissen E, Heideman D, Timens W, Schuuring E, et al. Overall survival in EGFR mutated non-small-cell lung cancer patients treated with afatinib after EGFR TKI and resistant mechanisms upon disease progression. PLoS ONE. 2017;12(8):e182885.
Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, Possemato A, Nardone J, Innocenti G, Wetzel R, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci USA. 2008;105(2):692–7.
Azuma K, Kawahara A, Sonoda K, Nakashima K, Tashiro K, Watari K, Izumi H, Kage M, Kuwano M, Ono M, et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget. 2014;5(15):5908–19.
Mazza V, Cappuzzo F. Treating EGFR mutation resistance in non-small cell lung cancer—role of osimertinib. Appl Clin Genet. 2017;10:49–56.
Bruner JK, Ma HS, Li L, Qin A, Rudek MA, Jones RJ, Levis MJ, Pratz KW, Pratilas CA, Small D. Adaptation to TKI treatment reactivates ERK signaling in tyrosine kinase-driven leukemias and other malignancies. Cancer Res. 2017;77(20):5554–63.
Wu DW, Chen TC, Huang HS, Lee H. TC-N19, a novel dual inhibitor of EGFR and cMET, efficiently overcomes EGFR-TKI resistance in non-small-cell lung cancer cells. Cell Death Dis. 2016;7(6):e2290.
Wells SJ, Robinson BG, Gagel RF, Dralle H, Fagin JA, Santoro M, Baudin E, Elisei R, Jarzab B, Vasselli JR, et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2012;30(2):134–41.
Traxler P, Allegrini PR, Brandt R, Brueggen J, Cozens R, Fabbro D, Grosios K, Lane HA, McSheehy P, Mestan J, et al. AEE788: a dual family epidermal growth factor receptor/ErbB2 and vascular endothelial growth factor receptor tyrosine kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2004;64(14):4931–41.
Juengel E, Engler J, Mickuckyte A, Jones J, Hudak L, Jonas D, Blaheta RA. Effects of combined valproic acid and the epidermal growth factor/vascular endothelial growth factor receptor tyrosine kinase inhibitor AEE788 on renal cell carcinoma cell lines in vitro. BJU Int. 2010;105(4):549–57.
Frodin M, Mezheyeuski A, Corvigno S, Harmenberg U, Sandstrom P, Egevad L, Johansson M, Ostman A. Perivascular PDGFR-beta is an independent marker for prognosis in renal cell carcinoma. Br J Cancer. 2017;116(2):195–201.
Roskoski RJ. Vascular endothelial growth factor (VEGF) and VEGF receptor inhibitors in the treatment of renal cell carcinomas. Pharmacol Res. 2017;120:116–32.
Albiges L, Guegan J, Le Formal A, Verkarre V, Rioux-Leclercq N, Sibony M, Bernhard JC, Camparo P, Merabet Z, Molinie V, et al. MET is a potential target across all papillary renal cell carcinomas: result from a large molecular study of pRCC with CGH array and matching gene expression array. Clin Cancer Res. 2014;20(13):3411–21.
Shin SJ, Jeon YK, Kim PJ, Cho YM, Koh J, Chung DH, Go H. Clinicopathologic analysis of PD-L1 and PD-L2 expression in renal cell carcinoma: association with oncogenic proteins status. Ann Surg Oncol. 2016;23(2):694–702.
Ciamporcero E, Miles KM, Adelaiye R, Ramakrishnan S, Shen L, Ku S, Pizzimenti S, Sennino B, Barrera G, Pili R. Combination strategy targeting VEGF and HGF/c-met in human renal cell carcinoma models. Mol Cancer Ther. 2015;14(1):101–10.
Choueiri TK, Halabi S, Sanford BL, Hahn O, Michaelson MD, Walsh MK, Feldman DR, Olencki T, Picus J, Small EJ, et al. Cabozantinib versus sunitinib as initial targeted therapy for patients with metastatic renal cell carcinoma of poor or intermediate risk: the alliance A031203 CABOSUN trial. J Clin Oncol. 2017;35(6):591–7.
Liefers-Visser J, Meijering R, Reyners A, van der Zee A, de Jong S. IGF system targeted therapy: therapeutic opportunities for ovarian cancer. Cancer Treat Rev. 2017;60:90–9.
Solarek W, Czarnecka AM, Escudier B, Bielecka ZF, Lian F, Szczylik C. Insulin and IGFs in renal cancer risk and progression. Endocr Relat Cancer. 2015;22(5):R253–64.
Cardillo TM, Trisal P, Arrojo R, Goldenberg DM, Chang CH. Targeting both IGF-1R and mTOR synergistically inhibits growth of renal cell carcinoma in vitro. BMC Cancer. 2013;13:170.
Tracz AF, Szczylik C, Porta C, Czarnecka AM. Insulin-like growth factor-1 signaling in renal cell carcinoma. BMC Cancer. 2016;16:453.
Lin S, Zhang Q, Shao X, Zhang T, Xue C, Shi S, Zhao D, Lin Y. IGF-1 promotes angiogenesis in endothelial cells/adipose-derived stem cells co-culture system with activation of PI3K/Akt signal pathway. Cell Prolif. 2017. https://doi.org/10.1111/cpr.12390.
Vazquez-Martin A, Cufi S, Oliveras-Ferraros C, Torres-Garcia VZ, Corominas-Faja B, Cuyas E, Bonavia R, Visa J, Martin-Castillo B, Barrajon-Catalan E, et al. IGF-1R/epithelial-to-mesenchymal transition (EMT) crosstalk suppresses the erlotinib-sensitizing effect of EGFR exon 19 deletion mutations. Sci Rep. 2013;3:2560.
Rapraeger AC, Ell BJ, Roy M, Li X, Morrison OR, Thomas GM, Beauvais DM. Vascular endothelial-cadherin stimulates syndecan-1-coupled insulin-like growth factor-1 receptor and cross-talk between alphaVbeta3 integrin and vascular endothelial growth factor receptor 2 at the onset of endothelial cell dissemination during angiogenesis. FEBS J. 2013;280(10):2194–206.
Shen Y, Zeng L, Novosyadlyy R, Forest A, Zhu A, Korytko A, Zhang H, Eastman SW, Topper M, Hindi S, et al. A bi-functional antibody-receptor domain fusion protein simultaneously targeting IGF-IR and VEGF for degradation. MABS-AUSTIN. 2015;7(5):931–45.
Cao Y, Cao R, Hedlund EM. R regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J Mol Med (Berl). 2008;86(7):785–9.
De la Cruz-Morcillo MA, Berger J, Sanchez-Prieto R, Saada S, Naves T, Guillaudeau A, Perraud A, Sindou P, Lacroix A, Descazeaud A, et al. p75 neurotrophin receptor and pro-BDNF promote cell survival and migration in clear cell renal cell carcinoma. Oncotarget. 2016;7(23):34480–97.
Zhang P, Xing Z, Li X, Song Y, Zhao J, Xiao Y, Xing Y. Tyrosine receptor kinase B silencing inhibits anoikisresistance and improves anticancer efficiency of sorafenib in human renal cancer cells. Int J Oncol. 2016;48(4):1417–25.
Tannir NM, Schwab G, Grunwald V. Cabozantinib: an Active Novel Multikinase Inhibitor in Renal Cell Carcinoma. Curr Oncol Rep. 2017;19(2):14.
Wang J, Chen J, Guo Y, Wang B, Chu H. Strategies targeting angiogenesis in advanced non-small cell lung cancer. Oncotarget. 2017;8(32):53854–72.
Martinez-Saez O, Gajate BP, Alonso-Gordoa T, Molina-Cerrillo J, Grande E. Targeting HIF-2 alpha in clear cell renal cell carcinoma: a promising therapeutic strategy. Crit Rev Oncol Hematol. 2017;111:117–23.
Cho H, Kaelin WG. Targeting HIF2 in clear cell renal cell carcinoma. Cold Spring Harb Symp Quant Biol. 2016;81:113–21.
Prior C, Perez-Gracia JL, Garcia-Donas J, Rodriguez-Antona C, Guruceaga E, Esteban E, Suarez C, Castellano D, Del AA, Lozano MD, et al. Identification of tissue microRNAs predictive of sunitinib activity in patients with metastatic renal cell carcinoma. PLoS ONE. 2014;9(1):e86263.
Authors’ contributions
WW, GZ and YL designed the research. YL and ZZ drafted the manuscript. YL, WW and TZ critically revised the manuscript. XD, XL and DC discussed and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The materials supporting the conclusions of this review are included in the article.
Consent for publication
This is not applicable for this review.
Ethics approval and consent to participate
Not applicable.
Funding
This work was sustained in part by the National Natural Science Foundation of China (contract No. 81402430).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lai, Y., Zhao, Z., Zeng, T. et al. Crosstalk between VEGFR and other receptor tyrosine kinases for TKI therapy of metastatic renal cell carcinoma. Cancer Cell Int 18, 31 (2018). https://doi.org/10.1186/s12935-018-0530-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-018-0530-2