
Abstract
Background
Small, cysteine-rich and cationic antifungal proteins (APs) from filamentous ascomycetes, such as NFAP from Neosartorya fischeri and PAF from Penicillium chrysogenum, are promising candidates for novel drug development. A prerequisite for their application is a detailed knowledge about their structure–function relation and mode of action, which would allow protein modelling to enhance their toxicity and specificity. Technologies for structure analyses, such as electronic circular dichroism (ECD) or NMR spectroscopy, require highly purified samples and in case of NMR milligrams of uniformly 15N-/13C-isotope labelled protein. To meet these requirements, we developed a P. chrysogenum-based expression system that ensures sufficient amount and optimal purity of APs for structural and functional analyses.
Results
The APs PAF, PAF mutants and NFAP were expressed in a P. chrysogenum ∆paf mutant strain that served as perfect microbial expression factory. This strain lacks the paf-gene coding for the endogenous antifungal PAF and is resistant towards several APs from other ascomycetes. The expression of the recombinant proteins was under the regulation of the strong paf promoter, and the presence of a paf-specific pre-pro sequence warranted the secretion of processed proteins into the supernatant. The use of defined minimal medium allowed a single-step purification of the recombinant proteins. The expression system could be extended to express PAF in the related fungus Penicillium digitatum, which does not produce detectable amounts of APs, demonstrating the versatility of the approach. The molecular masses, folded structures and antifungal activity of the recombinant proteins were analysed by ESI–MS, ECD and NMR spectroscopy and growth inhibition assays.
Conclusion
This study demonstrates the implementation of a paf promoter driven expression cassettes for the production of cysteine-rich, cationic, APs in different Penicillium species. The system is a perfect tool for the generation of correctly folded proteins with high quality for structure–function analyses.
Similar content being viewed by others
Background
Antifungal proteins (APs) from filamentous ascomycetes are small in size, cysteine-rich and cationic. These pre-pro proteins are synthesized in the ribosome and processed to mature forms when secreted into the fungal culture broth [1]. Extensively studied examples are PAF from Penicillium chrysogenum [2], NFAP from Neosartorya fischeri [3–5] and AFP from Aspergillus giganteus [6]. A significant diversity of PAF related genes and proteins has been found in ascomycetes, with genomes encoding up to three different sequence-related AP groups that provide a rich source of potentially divergent antifungals [7]. These natural proteins inhibit the growth of human-, animal- and plant-pathogenic moulds [8]. No detrimental effects on plant or on mammalian cells in vitro and in vivo could be observed for PAF [9, 10] and AFP [11, 12]. These findings strongly support their applicability as new antifungal drugs or their use for the development of new antifungal strategies.
Detailed structure–function analyses, however, are indispensable for a potential improvement of activity and specificity by rational design and for any future application. To this end, high quality protein preparations in considerable amounts are required. However, the bio-molecules are mostly expressed in small quantities by the producing moulds. Recombinant techniques for over-expression of APs in heterologous systems encounter major problems: correct processing and disulphide bond formation are essential for full protein activity [13]. Gene over-expression in microbial cells is still a challenging issue. Protein production by Escherichia coli offers some advantages for its easy and cost-effective cultivation and high protein yields [14]. However, this expression system also exhibits some disadvantages: (1) codon bias when expressing eukaryotic genes; (2) endotoxin contamination of the protein preparation; (3) incorrect folding and disulphide bridge formation, and low solubility of proteins can lead to inclusion body formation, which complicates purification and makes protein refolding necessary [14, 15].
A more reliable expression system is the yeast Pichia pastoris; it copes with disulphide bond formation, glycosylation and proper protein processing and folding [14]. However, the P. pastoris expression system bears the risk of unwanted protein modifications, such as O-linked and non-covalently linked sugars. Furthermore, P. pastoris secretes high concentrations of mannan into the expression medium [16]. Such carbohydrates need to be extensively removed otherwise they are detrimental for NMR studies and limit the amount of structural information [17].
Filamentous ascomycetes, finally, have been developed for homologous and heterologous gene expression [18, 19]. P. chrysogenum has been successfully used as expression system applying inducible (xylanase xylA) or constitutive (NADP-dependent glutamate dehydrogenase gdhA) promoters [20, 21] and efforts were undertaken to define new promoters for strain engineering in Aspergillus and Penicillium spp. [22, 23].
In this study we provide a new and important example for the appropriateness of Penicillium spp. as expression systems. We present an expression cassette consisting of the strong paf gene promoter, the paf pre-pro sequence for correct protein processing and secretion and the paf gene termination signal [24]. This expression cassette was used in P. chrysogenum for the production of high yields of recombinant, cysteine-rich APs for structural and functional analyses. The cultivation of the genetically engineered P. chrysogenum strains in defined minimal medium allowed an easy, single-step chromatographic purification of the proteins from the culture broth. With this expression system we generated PAF and PAF mutants with amino acid exchanges to investigate the role of specific protein motifs in antifungal function. Moreover, we used this system for the heterologous expression of the aforementioned AP NFAP from N. fischeri, which was found to be harmless for P. chrysogenum (unpublished data). Finally, we also tested the applicability of the P. chrysogenum expression cassette in another Penicillium species: we generated PAF in the post-harvest phyto-pathogenic fungus Penicillium digitatum that does not produce detectable amounts of APs [7] and is tolerant to PAF (unpublished data). The mature APs PAF and NFAP are 55 and 57 amino acids long and contain six cysteine-residues (Fig. 1). In PAF, these cysteines form three disulphide bonds in abcabc pattern, which is essential for proper folding and full antifungal activity [13, 25, 26]. A similar folding is highly probable for NFAP [4]. Both proteins do not undergo any posttranslational modifications, except for cleavage of the pre-pro sequence and protein folding (Fig. 1).

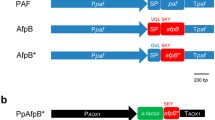
Schematic representation of the linearized expression plasmids a pSK275paf for the transformation in P. chrysogenum and b the T-DNA region of pBHt2_PAF binary vector for the A. tumefaciens mediated transformation of P. digitatum. Colour code of the expression cassette: black, paf 5′ UTR (promoter); green, paf 3′ UTR (terminator); yellow, gene of interest to be expressed (pre-pro sequence included); c ClustalW (BioEdit) alignment of the pre-pro proteins PAF[Pc] (equivalent to PAF[Pd]), PAFF31N, PAFY48Q and NFAP[Pc]. Top: PAF-specific pre-pro sequence; Bottom: mature proteins. The amino acid positions are numbered. Distinct amino acids are represented in colour code, identical amino acids are aligned in coloured dots, cysteine residues are boxed. The arrow indicates the predicted signal sequence cleavage site, the first amino acid of the mature proteins is marked with an asterisk. GOI gene of interest, hph hygromycin resistance gene cassette, ptrA pyrithiamine resistance gene cassette, ampR ampicillin resistance gene cassette, LB/RB left/right borders, F1 ori single-stranded phagemid origin; ColE1 origin, plasmid origin of replication, NotI restriction site for linearization of pSK275paf
Since not only the protein amount, but also the correct processing and folding is important [27], we paid special attention to characterize these features of recombinant PAF, PAF mutants and NFAP by using electro-spray ionization mass spectrometry (ESI–MS), electronic circular dichroism (ECD) and nuclear magnetic resonance (NMR) spectroscopy. The activity of the recombinant APs was determined in broth microdilution assays with Aspergillus niger as test organism.
Results
Protein expression and purification
The expression cassette consisted of the paf gene (420 bp) flanked by approximately 1280 bp of the 5′-UTR and 370 bp of the 3′-UTR and was inserted into plasmid pSK275 that contains the pyrithiamine resistance gene (ptrA) for selection of positive P. chrysogenum transformants and the ampicillin resistance gene (amp) for plasmid propagation in E. coli [28] (Fig. 1a). This plasmid pSK275paf was used to over-express PAF (PAF[Pc]) and served as basis for all further constructs (Additional File 1: Fig. S1).
The plasmid pSK275paf was used as a template for PCR-based site-directed mutagenesis to mutate distinct codons of the paf gene for amino acid substitutions and the production of PAF mutants. The preferential codon usage of P. chrysogenum was taken into account for gene modification. The amino acids Phe25, Ile26, Phe31 and Tyr48 form a hydrophobic patch on the surface of PAF [13, 26]. This motif is assumed to play a major role in the interaction of this AP with the plasma membrane of target fungi [13, 26]. To address this assumption, we exchanged Phe31 and Tyr48 with the uncharged, polar residues asparagine and glutamine, respectively and created PAFF31N and PAFY48Q (Fig. 1c).
We further used the P. chrysogenum-based expression system to increase the yield and purity of the N. fischeri AP NFAP for structural analyses. To this end, the nfap cDNA sequence coding for the mature NFAP replaced the part of the paf-gene that codes for the mature PAF in the pSK275paf vector. The nfap cDNA was fused to the paf pre-pro sequence (pSK275nfap paf_signal) and NFAP was produced in P. chrysogenum under the regulation of the strong paf-promoter (NFAP[Pc]) (Fig. 1a, c; Additional file 1: Fig. S2).
The production of PAF[Pc], PAFF31N, PAFY48Q and NFAP[Pc] required the use of the P. chrysogenum ∆paf mutant as cell factory, where the paf coding sequence was deleted by replacement with the nourseothricin (nat1) resistance gene [29].
To test the applicability of the expression cassette in other Penicillium species and with different transformation techniques (i.e., Agrobacterium tumefaciens mediated transformation, ATMT), we constructed an A. tumefaciens transformation vector with the expression cassette for paf gene insertion into the P. digitatum PHI26 genome (Fig. 1b). This experiment was intended to produce the P. chrysogenum PAF in P. digitatum (PAF[Pd]).
After single spore selection of positive P. chrysogenum and P. digitatum transformants, candidate clones were tested for highest protein production in time course experiments using small-scale fermentation. One clone each with the highest production of the desired recombinant protein was selected for further characterization. The respective producer strains were named P. chrysogenum paf, P. chrysogenum paf F31N, P. chrysogenum paf Y48Q, P. chrysogenum nfap and P. digitatum paf.
In Southern blotting experiments the random integration of the transforming DNA into the fungal genomes of the selected candidate clones was proved by using a 1.3 kb DIG-labelled PCR probe partially spanning the paf gene and the paf promoter (Additional file 1: Fig. S3). In all P. chrysogenum transformants the nat1 resistant gene was still present that originated from the deletion of the paf gene in the P. chrysogenum Δpaf strain [29] (Additional file 1: Fig. S3). This correlated well with a nourseothricin-resistant phenotype of the respective transformants, in addition to the resistance for pyrithiamine acquired by the uptake of the pSK275paf-based plasmids. In P. chrysogenum, the random plasmid integration resulted in a hybridizing fragment of 2.6 kb in length (P. chrysogenum nfap strain digested with NheI/XhoI) or of 2.7 kb in length (P. chrysogenum paf/paf mutants digested with NdeI) and additional fragments of larger or smaller sizes, depending on the integration locus. The additional signals proved that single or multiple-copy random plasmid integrations in the fungal genomes had occurred (Additional file 1: Fig. S3). The analysis of genomic DNA of P. digitatum paf strain revealed the presence of at least three copies of the paf expression cassette. No signals were detected in the P. digitatum PHI26 recipient strain that lacks the paf gene (Additional file 1: Fig. S3).
The selected clones were cultivated under larger culture conditions and, after clearing the culture broth from insoluble matter, the proteins in the supernatant were purified by cation-exchange chromatography. Optimal protein production was achieved at 72–96 h of cultivation for PAF, PAF mutants and NFAP in P. chrysogenum, whereas longer cultivation times of 7–11 days were applied for high yield expression of PAF[Pd] in P. digitatum paf, due to a lower proliferation rate of this strain in minimal medium. The protein amounts varied between a few milligrams per litre up to approximately 80 mg/L (Table 1).
The purity of the protein preparations was verified with SDS-PAGE (Fig. 2) and ESI–MS analysis (Fig. 3). In the SDS-PAGE one single band was visible for each purified protein sample corresponding to a molecular weight of approximately 6 kDa. Interestingly, the slightly larger NFAP protein (6.6 kDa) showed faster migration than the other proteins (6.2 kDa). A slight deviation from the expected migration behaviour is a phenomenon often observed with small, cysteine-rich proteins [30, 31].

Analysis of purified recombinant proteins with 18% (w/v) tris–glycine SDS-PAGE. One microgram of the protein samples was loaded per lane and visualized by Coomassie blue staining. M pre-stained protein size-standard (SeeBlue Plus2, ThermoFischer Scientific, Waltham, MA, USA), 1 PAF[Pc], 2 PAFF31N, 3 PAFY48Q, 4 PAF[Pd], 5 NFAP, 6 NFAP[Pc]

ESI-MS data showing the isotopic average molecular mass (m/z) of a PAF[Pc], b PAFF31N, c PAFY48Q, d NFAP[Pc] and e PAF[Pd]. The last four amino acid residues of the pro sequence (underlined), the first four of the predicted mature proteins, and the experimentally determined N-terminal cleavage sites (arrowheads) are shown on each major MS peak
Mass spectrometry
The identity and purity of all produced proteins and the correct amino acid conversions in case of PAF mutants were determined by ESI–MS. The average molecular (mol.) masses shown in Fig. 3 fit perfectly to the calculated theoretical mol. masses shown in Table 1 where the presence of three disulphide bridges in all proteins was taken into account. The PAF[Pc] mol. mass (6242.8 Da, Fig. 3a) was identical with that of PAF produced in P. chrysogenum Q176 [13]. The mol. mass of PAFF31N (6209.8 Da) and PAFY48Q (6208.0 Da) proved the correct exchange of the Phe and Tyr residues for an Asn and Gln residue, respectively (Fig. 3b, c). The mol. mass of NFAP[Pc] (6619.1 Da, Fig. 3d) corresponded to the calculated 6620 Da (Table 1). The PAF[Pd] MS revealed one main signal of 6242.9 Da and two additional signals that correspond to PAF with variable N-terminus: one lacking the first two amino acids Ala-Lys (6043.7 Da) and one comprising three additional amino acids Gly-Val-Leu (6513.01 Da) at the N-terminus (Fig. 3e). However, this PAF[Pd] preparation produced one single peak during chromatographic purification (Additional file 1: Fig. S4) and a single band in SDS-PAGE analysis (Fig. 2). All MS data indicated that the six cysteine residues present in all proteins were oxidized and three intra-molecular disulphide bonds were formed during intracellular protein processing. Furthermore, the data excluded any further post-translational protein modifications, except for the cleavage of the pre-pro sequence.
Antifungal activity assay
The antifungal activity of all recombinant proteins was tested on the PAF- and NFAP-sensitive model organism A. niger and the minimal inhibitory concentration (MIC) was determined (Table 2; Additional file 1: Fig. S5). Susceptibility data demonstrated that PAF[Pc] and PAF[Pd] showed the same antifungal activity as the wild-type PAF from P. chrysogenum Q176 (MIC 1.2 μg/mL; Additional file 1: Fig. S5). The recombinant NFAP[Pc] exhibited identical activity as the wild-type NFAP from N. fischeri (MIC 1.2 μg/mL; Additional file 1: Fig. S5). In contrast, PAFF31N and PAFY48Q dramatically lost antifungal activity: for PAFF31N the MIC was 800 μg/mL, whereas for PAFY48Q no MIC could be determined at the concentrations tested (Table 2; Additional file 1: Fig. S5).
ECD spectroscopy
ECD spectroscopy is a sensitive tool for the determination of the secondary structure of proteins. Low sample volume (0.1–1 mL) and concentration (µM range) requirements make this method a sensible choice for the rapid determination of protein conformation in order to verify if an expressed, purified protein is correctly folded, or how different mutations may affect its structure and thermal stability [32]. The ECD spectrum of PAF[Pc] at 25 °C was highly similar to that reported previously for this protein [26] and other disulphide bridged, β-structured proteins [33] (Fig. 4a). The spectrum had two maxima at 195 and 229 nm, a low intensity minimum centred at 210 nm and a weak shoulder at around 200 nm. The maximum at 229 nm was mainly attributed to the presence of disulphide bridges while the low intensity minimum at 210 nm indicated β-pleated conformation. The maximum centred at around 195 nm reflected contributions from both β-pleated conformation and the spectral transitions of disulphide bridges. The spectrum of PAF[Pd] at 25 °C (Fig. 4b) was almost identical to that of PAF[Pc]. The spectra of PAFF31N and PAFY48Q at this temperature (Fig. 4c, d, respectively) were similar too, differing only in the shoulder at 200 nm, which was missing or less pronounced in the spectra of these PAF mutants. In general, ECD spectra measured at 25 °C indicated that the structure of PAF[Pd] and the two PAF mutants are highly similar to the native fold of PAF. Spectra measured at 95 °C reflected the loss of ordered secondary structure in case of all PAF proteins ([Pc], [Pd] and mutants). After heat treatment and cooling back to 25 °C, gradual structural reorganization of PAF[Pc] and PAF[Pd] took place (Fig. 4a, b). However, this reorganization was very slow and in the case of PAF[Pd] refolding was incomplete even after four weeks (Fig. 4a, b; yellow line). This observation may be attributed to the variation of the N-terminus in PAF[Pd]. In contrast to PAF[Pc] and PAF[Pd], thermal unfolding of PAF mutants was irreversible, even after four weeks (Fig. 4c, d). The unfolding curves of PAF proteins (Fig. 5a) reflected their high thermal stability, which was not affected by the amino acid exchanges in the PAF mutants or when PAF was expressed in P. digitatum. These curves also indicated that unfolding was not complete in the studied temperature range that reached 95 °C as the curves did not present the usual sigmoidal shape. Nevertheless, the loss of secondary structure was apparent on the spectra measured at high temperatures (Fig. 4). The data of the unfolding experiment did not allow the fitting of a sigmoidal function of which inflexion point would have defined the melting temperature (Tm) of the protein structure. Hence Tm of the PAF proteins could only be estimated to be around 85 °C.

ECD spectra of a PAF[Pc], b PAF[Pd], c PAFF31N, d PAFY48Q, e NFAP and f NFAP[Pc], recorded at 25 °C (black), 95 °C (red), and at 25 °C immediately (blue), 72 h (green) and 4 weeks (orange, only in a–d) after annealing

Thermal unfolding of a PAF[Pc] (black), PAF[Pd] (red), PAFF31N (green), PAFY48Q (blue) and b NFAP (black) and NFAP[Pc] (red) followed by ECD spectroscopy at 229 nm
The ECD spectrum of the native NFAP (Fig. 4e) and the recombinant NFAP[Pc] (Fig. 4f) measured at 25 °C displayed similar features as the PAF spectra presented above, which suggests that this closely related protein has similar structural features as PAF[Pc]. The main difference between the spectra of NFAP and PAF[Pc] was the intensity ratio of the two components (195 and 200 nm) of the large positive maximum to be the opposite from that observed for PAF[Pc]. This might be due to a slightly different disulphide bridge pattern and/or different relative contribution of β-sheets to the overall structure of the protein. The spectrum of NFAP measured at 95 °C indicated only partial unfolding of the protein. The band at 229 nm reflecting the presence of disulphide bridges remained present opposed to PAF[Pc] where the dominance of unordered structures was observed at this temperature. Moreover, after cooling back to 25 °C the native fold of NFAP was restored completely and immediately. The heterologous expressed NFAP[Pc] adopted the same three-dimensional structure as NFAP and behaved in the same manner when heated and then cooled (Fig. 4f). The thermal stability of NFAP[Pc] appeared to be similar to that of NFAP, although the Tm was again roughly estimated (>85 °C) (Fig. 5b).
NMR analysis
To provide a proof of principle that the P. chrysogenum expression system is suitable for the production of uniformly isotope-labelled and pure proteins for NMR analyses, PAF[Pc] was 15N-labelled or 15N-and13C-labelled (15N/13C-PAF[Pc]), purified and analysed by heteronuclear NMR experiments. These experiments provide direct information about the chemical environment of almost all atoms within a protein at atomic resolution (all hydrogens which are directly attached to a nitrogen or a carbon atom are generally resolved in the heteronuclear dimension). The lack of NMR signals from unlabelled PAF[Pc] in single or double labelled samples proved that the incorporation of stable isotopes was close to 100%. The comparison of the 15N-1H and 13C-1H heteronuclear single quantum coherence (HSQC) spectra of the 15N/13C-PAF[Pc] sample to the previously recorded NMR data of 15N-labelled or 15N/13C-labelled PAF generated in P. chrysogenum wild-type Q176 demonstrated the absolute spectral identity of these protein samples [13, 26] (Fig. 6).

Overlaid view of 15N–1H HSQC (left) and Cα-Hα region of 13C-1H HSQC (right) of 15N-labelled PAF produced in the wild-type P. chrysogenum Q176 (blue) and 15N/13C-PAF[Pc] (red). Chemical shift scale was intentionally shifted in the F1 dimension for clarity
1H homonuclear spectra of PAF[Pc], PAFF31N, and PAFY48Q were acquired using solvent water suppression scheme (Fig. 7). The excellent dispersion of the amide signals in the 1H-NMR spectra proved the correct processing and folding of PAF[Pc] and the PAF mutants. These results were consistent with the data generated by ECD spectroscopy (Fig. 6). Moreover, all the samples were found to be pure by NMR standards: signals of impurities or minor forms of the products were not observed or were negligible for the given detection limit of the 500 MHz NMR spectrometer.

Amide and aromatic signal region of the 1H-NMR spectra of PAF[Pc], PAFF31N and PAFY48Q. Sharp signals and dispersion indicate the folded state of the analysed proteins
Discussion
There is a strong interest in investigating the potential of natural molecules for the development of new antifungal strategies. Filamentous ascomycetes are valuable and promising sources for various APs most of which await identification and further characterization [7]. This requires the generation of considerable protein amounts for analyses. The expression of APs however, varies significantly under laboratory conditions and might be low or repressed in some cases.
To overcome these restrictions, we introduced in this study a new expression system for the recombinant production of small cysteine-rich APs that are generally difficult to obtain in a soluble and correctly folded conformation when expressed in heterologous systems [34]. Correct disulphide bond pattern and protein folding, however, are essential for full antifungal activity [13]. Furthermore, we could show that the cultivation of two different Penicillium spp. in defined minimal medium for protein expression allowed single-step purification and provided high yields of proteins without unwanted carbohydrate impurities or posttranslational protein modifications for structural analyses using ECD spectroscopy and NMR techniques with 15N- or 15N/13C-labelled proteins.
Optimal AP production in P. chrysogenum under the control of the paf promoter required the use of the ∆paf mutant to avoid co-expression of the wild-type PAF. In contrast, the PAF-related AP of P. digitatum (AfpB) could not be detected so far even under constitutive expression conditions of the respective afpB gene [7] and therefore, the wild-type P. digitatum PHI26 was applied as recipient strain in this study. Here we could show that the promoter, the pre-pro and terminator sequences of the P. chrysogenum paf gene work efficiently also in a heterologous system, e.g. to produce PAF in P. digitatum with high yields. The additional MS signals detected in PAF[Pd] may indicate potential problems of P. digitatum proteases with the recognition of the pro sequence during protein maturation. However, the processing of the signal sequence and the disulphide bond formation seems to work properly as high yields of correctly folded and bioactive PAF[Pd] were recovered from the supernatant. The biological function of the pro sequence in APs is less understood, but a chaperone-like role for proper protein folding is suggested [1]. Naturally occurring variations of the N-terminus have been observed in related, non-recombinant APs, such as the A. giganteus AFP [35]. However, our data demonstrate that subtle differences in the processing of the pro sequence do not affect the antifungal activity of PAF[Pd].
The N. fischeri AP NFAP slightly differs from PAF in predicted structure, antifungal spectrum and mechanistic function [3–5]. Despite the knowledge of the transcriptional regulation elements, the bulk production of pure NFAP for structural investigations has not been achieved yet in the native producer N. fischeri NRRL181 where the average NFAP yield was not more than 1 mg/L [4]. It was previously proved that P. pastoris KM71H produces high amounts (average yield 6 mg/L) of folded and active recombinant NFAP [4], but the carbohydrate impurities as a consequence of the secretion system severely disturbed the structural analyses. In this study, we adopted the P. chrysogenum ∆paf expression system to produce this protein. Two different pSK275paf-based expression vectors were constructed for the production of NFAP in P. chrysogenum ∆paf. The attempt to put the NFAP encoding cDNA with the nfap specific pre-pro sequence (pSK275nfap) under the control of the strong paf promoter did not succeed in raising the NFAP protein yield (unpublished data). Instead, the average NFAP[Pc] yield of the pSK257nfap paf_signal transformant was approximately 3 mg/L, three-fold higher than in the natural producer N. fischeri NRRL181. This amount was sufficient for the structural and functional experiments in this study and isotope-labelling of NFAP[Pc] is in progress to resolve the three dimensional structure by NMR in the near future.
Furthermore, our data indicate that the gene copy number and the protein yield did not directly correlate. However, the NFAP[Pc] yield did not reach that of the other recombinant proteins. This could be a consequence of the genomic integration site and/or the high copy-number of the plasmid pSK257nfap paf_signal in the Penicillium genome. High gene dosage was shown to possibly hamper the transcription or translation machinery of microbial cell factories [36, 37]. However, protein-specific determinants, such as the signal sequence, may also modulate heterologous protein production, processing and secretion in the Penicillium sp. cell factories [38]. To elucidate the molecular background that regulates these processes further investigations are currently in progress.
Since we are interested to unravel the structure–function relation of PAF for a better understanding of its antifungal mode of action, we took advantage of the P. chrysogenum ∆paf cell factory to express two PAF mutants PAFF31N and PAFY48Q with exchanged hydrophobic residues (Phe, Tyr) at position 31 and 48, respectively (Fig. 8).

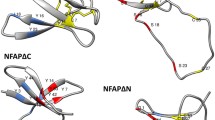
Cartoon representation of the PAF solution structure. Purple arrows show β-sheets, lines in light red indicate the loops, yellow lines highlight the disulphide bridges between the corresponding cysteines. The residues Phe31 and Tyr48 are indicated
The Phe31 participates in the most extended hydrophobic region of PAF with low primary sequence similarity to related APs [7, 13, 38], whereas Tyr48 represents a highly conserved residue that forms, together with two other tyrosines (Tyr3 and Tyr50), a well-defined aromatic region in PAF [13]. Both amino acids reside in loop regions of PAF, Phe31 in loop 3 and Tyr48 in loop 4, and are surface and solvent exposed. These loop regions show increased mobility and structural heterogeneity, which points towards a role as possible interaction sites with fungal target molecules [13, 26]. The replacement of these two residues by the neutral residues Asn and Gln resulted in a dramatic loss of activity and refolding capability (Fig. 4), underlining the important role of the postulated hydrophobic and aromatic patch in PAF for full antifungal function. Importantly, the data acquired by ECD spectroscopy and NMR analyses proved that the loss of activity could not be assigned to unfolding or misfolding of the protein mutants. The presence of β-sheets characteristic for the secondary structures of PAF[Pc] and PAF mutants PAFY48Q and PAFF31N was proven by the well dispersed 1H and/or 1H/15N HSQC NMR spectra, represented by the fingerprint amide spectral regions (Figs. 6, 7). The abcabc type disulphide pattern was unambiguously verified for PAF (pdb code: 2mhv, RCSB:10362) [25, 26] and corresponded to the pattern suggested for the A. giganteus AFP (abcdabcd) [38]. Cysteines that are oxidized and form intramolecular disulphide bonds elicit cysteine β 13CH2 signals that are well above 35 ppm. In case of PAF[Pc], the signals reached 37–39 ppm (NMR data of PAF can be found at BMRB code: 19657). Therefore, the abcabc pattern is highly probable for PAF[Pc] and the PAF mutants. If the disulphide bonds were intentionally broken by heat and/or reducing agents, the scrambled protein aggregates would result in much less dispersed NMR signals typical for unstructured proteins. Accordingly, the disappearance of the intense positive band at 229 nm from ECD spectra recorded at elevated temperatures could be attributed to either the disruption or conformational change of disulphide bridges during this measurement [39]. It has been reported that UV excitation of aromatic residues may result in electron or H ejection, which can reduce nearby disulphide groups [40]. Both the PAF and NFAP sequence contains aromatic amino acid residues adjacent to cysteines. Such UV light-induced reduction may occur at elevated rate at high temperatures (95 °C). Disruption of disulphide bridges and formation of possible new disulphide bonds in random oligomers at the experimental conditions applied could explain the apparent differences between the ECD spectra regarding the unfolding of PAF, NFAP and especially of PAF mutants.
Conclusion
We developed an expression cassette for homologous and heterologous gene expression under the strong paf-promoter for its application in P. chrysogenum and P. digitatum using different transformation techniques. This system was used to successfully increase the yield of correctly folded, small, cysteine-rich and cationic APs. Single-step purification from the Penicillium spp. culture broth of defined minimal nutrient composition ensured high protein quality and uniform isotope-labelling suitable for structural analyses employing NMR technology and ECD spectroscopy. NMR-based techniques provide information about the structure and dynamics of macromolecules. Since even the most recent NMR spectrometers are relatively insensitive, e.g. compared to MS, a protein concentration of 0.1–1 mM in a 250–500 µL sample volume is generally needed. Efficient protein production is especially important when an expensive 13C-labelled carbon source is utilized. Therefore, any improvement in protein yield and purity makes the research more cost-effective and supports approaches to investigate protein function, applying for example alanine scan methodology [41, 42]. The availability of restriction enzyme free cloning strategies, such as the seamless cloning technique [43], allows a broad application of our expression cassette using the strong paf promoter/terminator. Penicillium sp. represents an ideal microbial cell factory for the expression of a multitude of genes, originating also from non-fungal species [20], as protein processing and maturation is similar to higher eukaryotes [44, 45]. Taking into account the recent approaches for “humanizing” the glycosylation profile (as one of the most common post-translational modifications) in filamentous fungi [27], the fungal expression system presented in this study could be further refined in the future for the expression of human therapeutic proteins. This system is exceptionally promising for studying structure–function relations supporting the rational design of biotechnological interesting proteins and peptides.
Methods
Strains and growth conditions
Plasmids and vectors were propagated in E. coli DH5α, grown in LB medium [1% tryptone, 0.5% yeast extract, 1% NaCl (w/v)], supplemented with 100 μg/mL ampicillin. Fungal strains used in this study are listed in Table 3. For transformation, P. chrysogenum ∆paf [29] shaking cultures were grown in complete medium (CM; Additional file 1: Table S1) for 36 h at 25 °C. Conidia were harvested with spore buffer (0.9% NaCl (w/v), 0.01% Tween 80 (v/v)). For protein production, 2 × 108 conidia of P. chrysogenum or P. digitatum were inoculated per 200 mL PcMM or 450 mL PdMM, respectively (Additional file 1: Table S1) and cultivated at 25 °C under continuous shaking (200 rpm). Isotopic 15N- and/or 15N/13C-labelling of recombinant proteins for NMR analysis was performed by replacing the nitrogen/carbon source by 0.3% Na15NO3 and 1% 13C-glucose, respectively (Euriso-Top, Saarbrücken, Germany) in PcMM. As a PAF- and NFAP-sensitive test organism A. niger was cultivated in 20-fold diluted potato dextrose broth (0.05 × PDB, Sigma-Aldrich, St Louis, MO, USA) for growth inhibition assays. A. niger conidia were harvested from cultures grown at 37 °C on CM agar (Additional file 1: Table S1). Single use working solutions of conidia were prepared by adding 10% glycerol (v/v) to conidia suspensions, freezing them in liquid nitrogen and storing at –80 °C.
Vector constructions
All PCR reactions were performed with Q5 High Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA). From the plasmid pSK275 [28], pSK275paf was created by inserting the paf gene (420 bp) and approximately 1280 bp of the 5′-UTR and 370 bp of the 3′-UTR (Fig. 1; Additional file 1: Fig. S1). To generate PAF protein mutants, site-directed mutagenesis was applied to pSK275paf, using specific mutation primers that included mismatch nucleotides encoding for the amino acid to be exchanged (Additional file 1: Table S2). First, two PCR reactions were carried out with the primer pairs M13 and the respective forward (fw) mutation primer or opaf10 and the respective reverse (rev) mutation primer. In the next step, the generated fragments were combined in a PCR with the primers T7var and opaf9. The received mutated paf gene was cloned into pSK275paf at its NotI/NruI restriction sites. Thus the plasmids for protein expression were named pSK257paf F31N and pSK257paf Y48Q.
The expression cassette for NFAP production was constructed by preparing a PCR fragment from plasmid pBSK(-)nfap [4] containing the cDNA encoding the mature NFAP. This was fused to the paf pre-pro sequence and flanked by parts of the paf 5′-UTR and 3′-UTR. The DNA fragment was cloned into the BspMI and NotI digested pSK275paf vector, exchanging the paf-coding sequence (pSK275nfap paf_signal). A detailed description of the cloning strategy is given in the Additional file 1: Fig. S2.
To generate the P. digitatum paf strain, the paf expression cassette was obtained from the pSK275paf vector by PCR amplification, using the primers OJM483 and OJM484 to introduce restriction sites XmaI and XbaI, respectively (Additional file 1: Table S2). This cassette was first subcloned into the pGEM®-T Easy vector system (Promega, Fitchburg, WI, USA), from where it was excised with XmaI and XbaI and inserted into the appropriately digested binary vector pBHt2 (pBHt2_PAF), containing the hygromycin (hph) resistance selection marker [46].
In all cases, the correct vector assembly and nucleotide sequences were verified with Sanger sequencing.
Fungal transformation and confirmation of gene integration
For the production of PAF, PAF mutants and NFAP in P. chrysogenum the paf-deletion strain P. chrysogenum ∆paf [29] served as a recipient for the plasmids pSK257paf, pSK257paf F31N, pSK257paf Y48Q, pSK257nfap paf_signal. P. chrysogenum ∆paf was grown as described above and transformation was carried out according to [47, 48], using 10 µg of NotI linearized plasmids per transformation. Transformants were single spored three times on PcMM agar plates supplemented with 0.3–0.6 µg/mL pyrithiamine hydrobromide (Sigma-Aldrich, St Louis, MO, USA) to obtain genetically homogeneous strains.
For the production of PAF protein in P. digitatum, the binary vector pBHt2_PAF was transformed into A. tumefaciens AGL-1, and used to transform the parental CECT 20796 strain by ATMT as previously described [49, 50] with modifications as indicated in [51].
To confirm gene integration Southern blotting was performed. A DIG-labelled probe (1.3 kb) was PCR amplified using the paf-specific oligonucleotides intron1paf (within the paf-coding region) and opaf13 (within the paf 5′-UTR). Genomic DNA was extracted according to [52] and restriction enzyme digested. A detailed description is given in Additional file 1.
Protein expression and purification
PAF, PAF mutants and NFAP were purified from the supernatants of P. chrysogenum and P. digitatum. Shaking cultures of P. chrysogenum were first cleared from mycelia. The cell-free supernatant was ultra-filtered (Ultracell 30 kDa, Millipore, Billerica, MA, USA) and applied to a CM-Sepharose (Fast Flow, GE Healthcare Life Sciences, Little Chalfont, UK) column, equilibrated in phosphate buffer (10 mM NaPO4, 25 mM NaCl, 0.15 mM EDTA, pH 6.6). The P. digitatum cell-free supernatant was dialyzed (2 K MWCO, Sigma-Aldrich, St Louis, MO, USA) against the phosphate buffer before applying to an AKTA Purifier system equipped with a 6 mL RESOURCE™ S column (GE Healthcare Life Sciences, Little Chalfont, UK), equilibrated in phosphate buffer. In all cases, the proteins were eluted applying 0.1-0.5 M NaCl. The protein containing fractions were pooled and dialyzed (3.5 K MWCO, ThermoFisher Scientific, Waltham, MA, USA) against ultra-pure ddH2O and filter sterilized (0.22 µm, Millex-GV, PVDF, Millipore, Billerica, MA, USA). Protein concentrations were determined spectrophotometrically (A280) considering the respective molar extinction coefficients and the purity was checked by SDS-PAGE using Coomassie blue staining.
Mass spectrometry
The mass of the purified proteins was determined by ESI–MS on a CESI 8000 (AB Sciex, Framingham, MA, USA) coupled to a Q Exactive (ThermoFisher Scientific, Waltham, MA, USA) at the Protein Micro-Analysis Facility (Medical University of Innsbruck). PAF protein samples were directly diluted in 100 mM acetic acid and analysed (180 nL/min flow rate). NFAP samples were ZipTip (EMD, Millipore, Billerica, MA, USA) enriched, dissolved in 100 mM acetic acid and analysed by capillary electrophoresis (CE)-ESI–MS (20 kV separation voltage, 10 psi pressure). Protein masses were determined by deconvolution using the integrated Xcalibur pXtract software (ThermoFisher Scientific, Waltham, MA, USA).
ECD spectroscopy
ECD spectroscopic measurements were performed in the 195–260 nm wavelength range (far-UV) to determine the secondary structure and examine the structural stability of the recombinant proteins. Protein samples were dissolved in pure ddH2O at approximately 0.1 mg/mL concentration and measured in a 0.1 cm path-length quartz cuvette using a Jasco J-815 spectropolarimeter (JASCO, Tokyo, Japan) at a scan speed of 100 nm/s. In brief, first ECD spectrum of the sample was recorded at 25 °C, then the temperature was increased gradually up to 95 °C at a rate of 1 °C/min using a Peltier thermoelectronic controller (TE Technology, Traverse City, MI, USA) while ellipticity data was recorded as a function of temperature at three wavelengths, appointed by the extrema of the spectrum measured at 25 °C. The system was allowed to equilibrate for 1 min at each temperature point before measurements were taken. The resulting melting curves were used to estimate the Tm of the protein structures. Then, an ECD spectrum in the 195–260 nm wavelength range was recorded at 95 °C, the final temperature point of the unfolding experiments. The sample was left to cool and the full spectrum was measured again 1 min after the temperature reached 25 °C. Spectrum acquisition was repeated 72 h later and when the observed changes made it necessary. The presented spectra are accumulations of 10 scans, from which the similarly recorded spectrum of ddH2O was subtracted. Ellipticity data were given in mdeg units.
NMR analysis
All NMR experiments were performed with a Bruker Avance II 500 spectrometer equipped with TXI z-gradient probe head (Bruker, Rheinstetten, Germany). Typically, 2-10 mg lyophilized protein was dissolved in 275 μL buffer (10 mM Na2HPO4/NaH2PO4, pH 6.0, 5% D2O (v/v), 0.04% NaN3 (w/v), 40 mM NaCl) and filled into a Shigemi NMR tube (Shigemi, Allison Park, PA, USA). 1H-NMR experiments were recorded with the watergate-5 sequence [53] for water suppression and 64 transients were collected in every case at 297 K. Sensitivity enhanced HSQC experiment was performed for 1H-15N and 1H-13C correlation [54] that allowed to obtain spectra even at natural abundance. In these heteronuclear experiments 1024 × 128 and 2048 × 350 points were acquired, respectively.
Antifungal activity assay
For testing the antifungal activity of the recombinant produced proteins, A. niger was used as a sensitive test organism. Susceptibility tests were carried out in 96-well, flat-bottom microtitre plates (Nunclon Delta, Thermo Scientific, Waltham, MA, USA) as described before [55]. Briefly, 5 × 103 conidia/mL were incubated in 0.05 × PDB with increasing concentrations of APs (0–800 µg/mL) in a total volume of 200 µL. The plates were incubated at 30 °C for 48 h and the growth was monitored microscopically using an inverted microscope (Leica DM IL Led, Leica Microsystems, Vienna, Austria) equipped with an AxioCam MR digital camera (Zeiss, Jena, Germany) for imaging. The images were processed with AxioVision software (Zeiss, Jena, Germany). The MIC was defined as the minimal protein concentration that inhibited fungal growth by ≥90%. Experiments were prepared in triplicates and performed at least three times.
Statistical analysis
Statistical analysis was performed using Microsoft Excel 2010 (Microsoft Corp.).
Abbreviations
- AFP:
-
Aspergillus giganteus antifungal protein
- AP:
-
antifungal protein
- ATMT:
-
Agrobacterium tumefaciens mediated transformation
- CM:
-
complete medium
- DIG:
-
digoxigenin
- ECD:
-
electronic circular dichroism
- ESI-MS:
-
electro-spray ionization mass spectrometry
- HSQC:
-
heteronuclear single quantum coherence
- MIC:
-
minimal inhibitory concentration
- MS:
-
mass spectrometry
- MWCO:
-
molecular weight cut-off
- NFAP:
-
Neosartorya fischeri strain NRRL181 antifungal protein
- NFAP[Pc]:
-
recombinant NFAP produced in Penicillium chrysogenum
- NMR:
-
nuclear magnetic resonance
- PAF:
-
Penicillium chrysogenum strain Q176 antifungal protein
- PAF[Pc]:
-
recombinant PAF produced in Penicillium chrysogenum
- PAF[Pd]:
-
recombinant PAF produced in Penicillium digitatum
- PcMM:
-
Penicillium chrysogenum minimal medium
- PdMM:
-
Penicillium digitatum minimal medium
- SDS-PAGE:
-
sodium dodecyl sulphate polyacrylamide gel electrophoresis
- UTR:
-
untranslated region
References
Marx F, Salvenmoser W, Kaiserer L, Graessle S, Weiler-Görz R, Zadra I, Oberparleiter C. Proper folding of the antifungal protein PAF is required for optimal activity. Res Microbiol. 2005;156:35–46.
Marx F, Binder U, Leiter É, Pócsi I. The Penicillium chrysogenum antifungal protein PAF, a promising tool for the development of new antifungal therapies and fungal cell biology studies. Cell Mol Life Sci. 2008;65:445–54.
Kovács L, Virágh M, Takó M, Papp T, Vágvölgyi C, Galgóczy L. Isolation and characterization of Neosartorya fischeri antifungal protein (NFAP). Peptides. 2011;32:1724–31.
Virágh M, Vörös D, Kele Z, Kovács L, Fizil Á, Lakatos G, Maróti G, Batta G, Vágvölgyi C, Galgóczy L. Production of a defensin-like antifungal protein NFAP from Neosartorya fischeri in Pichia pastoris and its antifungal activity against filamentous fungal isolates from human infections. Protein Expr Purif. 2014;94:79–84.
Virágh M, Marton A, Vizler C, Tóth L, Vágvölgyi C, Marx F, Galgóczy L. Insight into the antifungal mechanism of Neosartorya fischeri antifungal protein. Protein Cell. 2015;6:518–28.
Meyer V. A small protein that fights fungi: AFP as a new promising antifungal agent of biotechnological value. Appl Microbiol Biotechnol. 2008;78:17–28.
Garrigues S, Gandía M, Marcos JF. Occurrence and function of fungal antifungal proteins: a case study of the citrus postharvest pathogen Penicillium digitatum. Appl Microbiol Biotechnol. 2016;100:2243–56.
Hegedüs N, Marx F. Antifungal proteins: more than antimicrobials? Fungal Biol Rev. 2013;26:132–45.
Palicz Z, Jenes Á, Gáll T, Miszti-Blasius K, Kollár S, Kovács I, Emri M, Márián T, Leiter É, Pócsi I, Csősz E, Kalló G, Hegedűs C, Virág L, Csernoch L, Szentesi P. In vivo application of a small molecular weight antifungal protein of Penicillium chrysogenum (PAF). Toxicol Appl Pharmacol. 2013;269:8–16.
Szappanos H, Szigeti GP, Pál B, Rusznák Z, Szűcs G, Rajnavölgyi É, Balla J, Balla G, Nagy E, Leiter É, Pócsi I, Marx F, Csernoch L. The Penicillium chrysogenum-derived antifungal peptide shows no toxic effects on mammalian cells in the intended therapeutic concentration. Naunyn-Schmiedebergs Arch Pharmacol. 2005;371:122–32.
Szappanos H, Szigeti GP, Pál B, Rusznák Z, Szűcs G, Rajnavölgyi É, Balla J, Balla G, Nagy E, Leiter É, Pócsi I, Hagen S, Meyer V, Csernoch L. The antifungal protein AFP secreted by Aspergillus giganteus does not cause detrimental effects on certain mammalian cells. Peptides. 2006;27:1717–25.
Theis T, Marx F, Salvenmoser W, Stahl U, Meyer V. New insights into the target site and mode of action of the antifungal protein of Aspergillus giganteus. Res Microbiol. 2005;156:47–56.
Batta G, Barna T, Gáspári Z, Sándor S, Kövér KE, Binder U, Sarg B, Kaiserer L, Chhillar AK, Eigentler A, Leiter E, Hegedüs N, Pócsi I, Lindner H, Marx F. Functional aspects of the solution structure and dynamics of PAF—a highly-stable antifungal protein from Penicillium chrysogenum. FEBS J. 2009;276:2875–90.
Demain AL, Vaishnav P. Production of recombinant proteins by microbes and higher organisms. Biotechnol Adv. 2009;27:297–306.
Rosano GL, Ceccarelli EA. Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol. 2014;5:1–17.
Palczewska M, Batta G, Groves P. Concanavalin A-agarose removes mannan impurities from an extracellularly expressed Pichia pastoris recombinant protein. Cell Mol Biol Lett. 2003;8:783–92.
O’Leary JM, Radcliffe CM, Willis AC, Dwek RA, Rudd PM, Downing AK. Identification and removal of O-linked and non-covalently linked sugars from recombinant protein produced using Pichia pastoris. Protein Expr Purif. 2004;38:217–27.
Su X, Schmitz G, Zhang M, Mackie RI, Cann IKO. Heterologous gene expression in filamentous fungi. Adv Appl Microbiol. 2012;81:1–61.
Nevalainen KMH, Te’o VSJ, Bergquist PL. Heterologous protein expression in filamentous fungi. Trends Biotechnol. 2005;23:468–74.
Graessle S, Haas H, Friedlin E, Kürnsteiner H, Stöffler G, Redl B. Regulated system for heterologous gene expression in Penicillium chrysogenum. Appl Environ Microbiol. 1997;63:753–6.
Díez B, Mellado E, Rodríguez M, Bernasconi E, Barredo JL. The NADP-dependent glutamate dehydrogenase gene from Penicillium chrysogenum and the construction of expression vectors for filamentous fungi. Appl Microbiol Biotechnol. 1999;52:196–207.
Blumhoff M, Steiger MG, Marx H, Mattanovich D, Sauer M. Six novel constitutive promoters for metabolic engineering of Aspergillus niger. Appl Microbiol Biotechnol. 2013;97:259–67.
Polli F, Meijrink B, Bovenberg RAL, Driessen AJM. New promoters for strain engineering of Penicillium chrysogenum. Fungal Genet Biol. 2016;89:62–71.
Marx F, Haas H, Reindl M, Stöffler G, Lottspeich F, Redl B. Cloning, structural organization and regulation of expression of the Penicillium chrysogenum paf gene encoding an abundantly secreted protein with antifungal activity. Gene. 1995;167:167–71.
Váradi G, Tóth GK, Kele Z, Galgóczy L, Fizil Á, Batta G. Synthesis of PAF, an antifungal protein from P. chrysogenum, by native chemical ligation: native disulfide pattern and fold obtained upon oxidative refolding. Chem A Eur J. 2013;19:12684–92.
Fizil Á, Gáspári Z, Barna T, Marx F, Batta G. “Invisible” conformers of an antifungal disulfide protein revealed by constrained cold and heat unfolding, CEST-NMR experiments, and molecular dynamics calculations. Chem A Eur J. 2015;21:5136–44.
Nevalainen H, Peterson R. Making recombinant proteins in filamentous fungi- are we expecting too much? Front Microbiol. 2014;5:1–10.
Krappmann S, Jung N, Medic B, Busch S, Prade RA, Braus GH. The Aspergillus nidulans F-box protein GrrA links SCF activity to meiosis. Mol Microbiol. 2006;61:76–88.
Hegedüs N, Sigl C, Zadra I, Pócsi I, Marx F. The paf gene product modulates asexual development in Penicillium chrysogenum. J Basic Microbiol. 2011;51:253–62.
Seibold M, Wolschann P, Bodevin S, Olsen O. Properties of the bubble protein, a defensin and an abundant component of a fungal exudate. Peptides. 2011;32:1989–95.
Rath A, Glibowicka M, Nadeau VG, Chen G, Deber CM. Detergent binding explains anomalous SDS-PAGE migration of membrane proteins. Proc Natl Acad Sci. 2009;106:1760–5.
Greenfield NJ. Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc. 2007;1:2876–90.
Lees JG, Miles AJ, Wien F, Wallace BA. A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics. 2006;22:1955–62.
Kiedzierska A, Czepczynska H, Smietana K, Otlewski J. Expression, purification and crystallization of cysteine-rich human protein muskelin in Escherichia coli. Protein Expr Purif. 2008;60:82–8.
Martínez-Ruiz A, Martínez del Pozo A, Lacadena J, Mancheño JM, Oñaderra M, Gavilanes JG. Characterization of a natural larger form of the antifungal protein (AFP) from Aspergillus giganteus. Biochim Biophys Acta. 1997;1340:81–7.
Meyer P. Repeat-induced gene silencing: common mechanisms in plants and fungi. Biol Chem Hoppe Seyler. 1996;377:87–95.
Braumann I, van den Berg M, Kempken F. Repeat induced point mutation in two asexual fungi, Aspergillus niger and Penicillium chrysogenum. Curr Genet. 2008;53:287–97.
Campos-Olivas R, Bruix M, Santoro J, Lacadena J, del Pozo AM, Gavilanes JG, Rico M. NMR solution structure of the antifungal protein from Aspergillus giganteus: evidence for cysteine pairing isomerism. Biochemistry. 1995;34:3009–21.
Hider RC, Kupryszewski G, Rekowski P, Lammek B. Origin of the positive 225–230 nm circular dichroism band in proteins. Its application to conformational analysis. Biophys Chem. 1988;31:45–51.
Neves-Petersen MT, Gryczynski Z, Lakowicz J, Fojan P, Pedersen S, Petersen E, Petersen SB. High probability of disrupting a disulphide bridge mediated by an endogenous excited tryptophan residue. Protein Sci. 2009;11:588–600.
Lee SY, Pullen L, Virgil DJ, Castañeda CA, Abeykoon D, Bolon DNA, Fushman D. Alanine scan of core positions in ubiquitin reveals links between dynamics, stability, and function. J Mol Biol. 2014;426:1377–89.
Zamora-Carreras H, Strandberg E, Mühlhäuser P, Bürck J, Wadhwani P, Jiménez MÁ, Bruix M, Ulrich AS. Alanine scan and 2H NMR analysis of the membrane-active peptide BP100 point to a distinct carpet mechanism of action. Biochim Biophys Acta Biomembr. 2016;1858:1328–38.
Benoit RM, Ostermeier C, Geiser M, Li JSZ, Widmer H, Auer M. Seamless insert-plasmid assembly at high efficiency and low cost. PLoS ONE. 2016;11:e0153158.
Jami M-S, Barreiro C, García-Estrada C, Martín J-F. Proteome analysis of the penicillin producer Penicillium chrysogenum. Mol Cell Proteomics. 2010;9:1182–98.
Conesa A, Punt PJ, van Luijk N, van den Hondel CA. The secretion pathway in filamentous fungi: a biotechnological view. Fungal Genet Biol. 2001;33:155–71.
Mullins ED, Chen X, Romaine P, Raina R, Geiser DM, Kang S. Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology. 2001;91:173–80.
Cantoral JM, Diez B, Barredo JL, Alvarez E, Martin JF. High-frequency transformation of Penicillium chrysogenum. Nat Biotechnol. 1987;5:494–7.
Kolar M, Punt PJ, van den Hondel CAMJJ, Schwab H. Transformation of Penicillium chrysogenum using dominant selection markers and expression of an Escherichia coli lacZ fusion gene. Gene. 1988;62:127–34.
Khang CH, Park SY, Hee-Sool R, Lee Y-H, Kang S. Filamentous fungi (Magnaporthe grisea and Fusarium oxysporum). In: Wang K, editor. Agrobacterium Protocols. 2nd ed. New York city: Humana Press; 2006. p. 403–20.
Michielse CB, Hooykaas PJJ, van den Hondel CAMJJ, Ram AFJ. Agrobacterium-mediated transformation of the filamentous fungus Aspergillus awamori. Nat Protoc. 2008;3:1671–8.
Harries E, Gandía M, Carmona L, Marcos JF. The Penicillium digitatum protein O-mannosyltransferase Pmt2 is required for cell wall integrity, conidiogenesis, virulence and sensitivity to the antifungal peptide PAF26. Mol Plant Pathol. 2015;16:748–61.
Zadra I, Abt B, Parson W, Haas H. xylP promoter-based expression system and its use for antisense downregulation of the Penicillium chrysogenum nitrogen regulator NRE. Appl Environ Microbiol. 2000;66:4810–6.
Liu M, Mao X, Ye C, Huang H, Nicholson JK, Lindon JC. Improved WATERGATE pulse sequences for solvent suppression in NMR spectroscopy. J Magn Reson. 1998;132:125–9.
Palmer AG III, Cavanagh J, Wright PE, Rance M. Sensitivity improvement in proton-detected two-dimensional heteronuclear correlation NMR spectroscopy. J Magn Reson. 1991;93:151–70.
Kaiserer L, Oberparleiter C, Weiler-Görz R, Burgstaller W, Leiter E, Marx F. Characterization of the Penicillium chrysogenum antifungal protein PAF. Arch Microbiol. 2003;180:204–10.
Authors’ contributions
FM coordinated the study and prepared the first draft of the manuscript. FM, LG and JFM designed the experiments. LG produced NFAP proteins in P. chrysogenum. SG and PM produced PAF in P. digitatum. CS, NH and AH produced PAF and PAF protein mutants in P. chrysogenum, carried out activity assays and Southern blotting. AB performed ECD measurements and GB and AF carried out NMR experiments. All authors read, revised and approved the final manuscript.
Acknowledgements
We thank Doris Bratschun-Khan, Laura Burtscher, Julia Brindlinger, Stefan Vogt and Adrián Aranda for technical assistance.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its Additional file 1 and are available in the following repositories: Genbank: U22944.2 (paf gene) and AM983570.1 (nfap gene). The data of the PAF solution structure were deposited at: BMRB ID code 16087, 19658; RCSB ID code 100954, 10362; PDB ID code 2kcn, 2mhv.
Funding
This study was supported by the Austrian Science Fund (FWF P25894-B20 to FM, FWF I1644-B20 to FM), the Hungarian Science Fund (OTKA ANN 110821 to GB), and the Spanish Ministry of Economy (BIO2015-68790-C2-1-R MINECO/FEDER to PM and JFM). LG holds a Lise Meitner fellowship from the Austrian Science Fund FWF (M1776-B20). SG holds a FPU doctoral fellowship (MECD FPU13/04584) from the Spanish Ministry of Education. The research of AB was supported by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Additional files
12934_2016_586_MOESM1_ESM.docx
Additional file 1. Table S1. Composition of media used in this study. Table S2. Oligonucleotides used in this study. Figure S1. Nucleotide sequence of plasmid pSK275paf containing the paf expression cassette. Figure S2. Cloning strategy of pSK275nfap paf_signal. Figure S3. Southern blot analysis of genomic integration of the P. chrysogenum expression cassettes in the Penicillium spp. Figure S4. Chromatogram of the purification of PAF[Pd]. Figure S5. Microscopic images of growth inhibition assays with A. niger exposed to PAF, PAF[Pc], PAFF31N, PAFY48Q, PAF[Pd], NFAP and NFAP[Pc].
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sonderegger, C., Galgóczy, L., Garrigues, S. et al. A Penicillium chrysogenum-based expression system for the production of small, cysteine-rich antifungal proteins for structural and functional analyses. Microb Cell Fact 15, 192 (2016). https://doi.org/10.1186/s12934-016-0586-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0586-4