
Abstract
Background
The filamentous fungus Aspergillus niger is widely exploited as an important expression host for industrial production. The glucoamylase high-producing strain A. niger CICC2462 has been used as a host strain for the establishment of a secretion expression system. It expresses recombinant xylanase, mannase and asparaginase at a high level, but some high secretory background proteins in these recombinant strains still remain, such as alpha-amylase and alpha-glucosidase; lead to a low-purity of fermentation products. The aim was to construct an A. niger host strain with a low background of protein secretion.
Results
The transcription factor amyR was deleted in A. niger CICC2462, and the results from enzyme activity assays and SDS-PAGE analysis showed that the glucoamylase and amylase activities of the ∆amyR strains were significantly lower than those of the wild-type strain. High-throughput RNA-sequencing and shotgun LC–MS/MS proteomic technology analysis demonstrated that the expression of amylolytic enzymes was decreased at both the transcriptional and translational levels in the ∆amyR strain. Interestingly, the ∆amyR strain growth rate better than the wild-type strain.
Conclusions
Our findings clearly indicated that the ∆amyR strain of A. niger CICC2462 can be used as a host strain with a low background of protein secretion.
Similar content being viewed by others


Background
The filamentous fungus Aspergillus niger is one of the most important industrial filamentous species and is used extensively for the production of organic acids and industrial enzymes [1–3] and for basic genetic research. Compared with E. coli and Pichia expression systems, Aspergillus has better expression and secretion capacity [4, 5]. The GRAS (generally recognized as safe) status of A. niger makes it attractive as a host for recombinant protein expression, and A. niger is known as “a cell factory of eukaryotic protein expression [6, 7]”. The main challenge in industry using filamentous fungi is the expression of homologous and/or heterologous proteins that are functional. Strategies for improving protein production have been discussed in detail by Archer et al. in 1994 [8], including the use of strong homologous promoters, increased gene copy number and gene fusions etc. Nevalainen and Peterson [9] described the current obstacles for the production of recombinant proteins, including the mode of glycosylation and the problems related to the processing in the endoplasmic reticulum. They also proposed that exploration of metabolic pathway engineering may result in the improvement of the production of recombinant proteins.
The glucoamylase-producing strain A. niger CICC2462 has been used as a host strain for the establishment of a secretion expression system by our research team. The target gene was integrated into the glaA gene locus for high expression by homologous gene replacement. It could express recombinant xylanase, mannase and asparaginase at a high level [10–12], but some high secretory background proteins still remain, such as alpha-amylase and alpha-glucosidase, the low proportion of target proteins relative to the total protein not only restrict the continued ascension of target protein production but also lead to a low-purity of fermentation products, thus increasing the costs of target protein purification. Therefore, one possible method that could be effective to solve this problem is to regulate highly expressed genes at the transcriptional level and subsequently reduce the amount of secretory proteins in the whole expression system.
The regulation of secretory proteins in Aspergillus species has been well studied. The cis-elements SYGGRG, CGGN8(C/A)GG, GGCTAR and CCAAT have been found upstream of the major secretory protein genes amylase, cellulase and hemicellulase, which are regulated by the regulatory factors CreA/B/C, AmyR, XlnR, ClbR and the Hap complex [13, 14]. AmyR is a Zn(II)2Cys6-type transcriptional activator responsible for the induction of the amylolytic genes in Aspergillus species, and is known to bind to the CGGN8(C/A)GG sequence in various amylase promoters to activate gene transcription [15–17]. Furthermore, many details of the structure and regulatory function of the amyR gene have been elucidated [18–20].
In the post-genomics era, various-omics technologies have been applied in filamentous fungus to generate a new approach for improving the expression system of host strains for the industrial production of proteins. Proteomic analysis is a powerful tool for high-throughput global protein expression analysis using gel-based or gel-free protein separation techniques coupled with mass spectrometry (MS/MS). Proteomic methods have been used to study the effect of different culture conditions on the secretome of A. niger [21–23]. Furthermore, recent advances in high-throughput RNA sequencing (RNA-Seq) technology have markedly reshaped the landscape of transcriptome analysis [24, 25]. Transcriptomics sequences of A. niger strains have been used to give new knowledge about the regulation of carbohydrate metabolism [26, 27]. Such knowledge could provide new strategies for strain improvement.
In this study, an amyR-deletion strain was constructed, transcriptome, secretome, amylolytic enzyme activity and growth rate were analysed and compared in the amyR deletion strain and the wild-type strain. Ultimately, a host strain with a low background of protein secretion and without growth inhibition was obtained.
Methods
Strains and culture conditions
Aspergillus niger CICC2462 was provided by an enzyme preparation company (Zhaodong Richeng Enzyme Preparation Co., Ltd.), and the pSZH-xynB plasmid vector was constructed by our laboratory. Escherichia coli DH5α, Agrobacterium tumefaciens AGLI and the pAN7-1 vector were used for DNA manipulation. A mutant strain (∆amyR) of A. niger CICC2462 was constructed in this study. The A. niger strain was grown at 30 °C in PDA medium (20 g/L glucose, 3 g/L KH2PO4, 1.5 g/L MgSO4·7H2O, and 200 g/L potato piece). Plasmid-harbouring E. coli cells were grown at 37 °C in LB medium (5 g/L yeast extract, 10 g/L peptone, and 10 g/L NaCl, pH 7.0). Agrobacterium tumefaciens was grown at 28 °C in YEB medium (1 g/L yeast extract, 5 g/L peptone, 0.493 g/L MgSO4·7H2O, 5 g/L beef extract paste, and 5 g/L sucrose, pH 6.5). Cultures where grown in shake flasks at 30 °C in industrial fermentation medium (100 g/L glucose, 20 mL/L corn steep liquor, and 20 g/L soybean powder, pH 5.5–6.0) at 260 rpm/min with 10 % of the inoculation amount.
Shotgun LC–MS/MS proteome analysis
Shotgun LC–MS/MS was performed at Shanghai GeneCore Bio-Technologies Co., Ltd. (Shanghai, China). Peptides were purified through reverse-phase high-performance liquid chromatography on a surveyor LC system (Thermo Finnigan, San Jose, CA, USA) with an autosampler. Peptides were ionized in the positive ion mode and introduced into an LTQ linear ion trap mass spectrometer equipped with a microelectrospray source for MS/MS. Protein identification was performed with Bioworks Browser 3.3 against the NCBI Uniprot A. niger database and the results were extracted from the SEQUEST file using in-house software. The SEQUEST search parameters were Delta CN (≥0.1) and Xcorr (one charge ≥1.9, two charges ≥2.2, three charges ≥3.75). Proteins with a unique peptide count ≥2 were considered accurately identified and used for bioinformatic analysis.
Construction of amyR-deletion cassettes
Plasmid vectors were constructed using a previously published enzymatic assembly method [28]. The PCR primers are listed in Table 1. The amyR gene deletion vector, pAmyRd, was constructed in three steps as follows. First, a 1.3-kb SpeI/HindIII amyR3SH fragment was amplified from A. niger CICC2462 genomic DNA with the primer pairs amyR3SH-R/amyR3SH-F, and the amyR3SH fragment was placed between the XbaI and HindIII sites located immediately in front of the hph gene on pAN7-1 to construct pAN-amyR3SH. Second, this plasmid was introduced between the XhoI and HindIII sites of pSZH-xynB to yield the plasmid pSZH-amyR3SH. Finally, a 1.2-kb XbaI/KpnI amyR5 fragment and a 1.3-kb XhoI/KpnI amyR3KX fragment were amplified from A. niger CICC2462 genomic DNA with the primer pairs amyR5-R/amyR5-F and amyR3KX-R/amyR3KX-F, respectively. These fragments were then introduced between the XbaI and XhoI sites of pSZH-amyR3SH to obtain amyR gene-deletion vector, pAmyRd. This vector was subsequently used for the transformation of A. niger CICC2462 to obtain the reporter A. niger ∆amyR strain.
Agrobacterium tumefaciens-mediated transformation of Aspergillus niger
The following steps were taking throughout the procedure: a pAmyRd Agrobacterium transformant colony which was obtained by the freeze-thawing method in YEB liquid medium (containing 50 mg/L rifampicin and 100 mg/L kanamycin) and incubated at 28 °C with 200 rpm rotary shaking for 24 h. The pAmyRd Agrobacterium was inoculated at a proportion of 1:10 into fresh YEB liquid medium (containing 50 mg/L rifampicin and 100 mg/L kanamycin) for secondary activation until the OD 600 reached 0.4–0.6. 80 μL of the A. niger CICC2462 mycelium suspension and 80 μL of the pAmyRd Agrobacterium suspension were coated on a covering cellophane PDA solid medium (containing 200 μmol/L acetosyringone). The cellophane was transferred to a new PDA solid medium (containing 200 mmol/L cefotaxime sodium and 200 mmol/L hygromycin B) after being co-cultivated at 28 °C for 2 days and removed it after cultivation at 30 °C for 1 day. Cultivation for 6–8 days until growth of the A.niger colonies were detected. The resistant A.niger colony was transfered to PDA liquid medium (containing 200 mmol/L hygromycin B) for secondary screening. A. niger genomic DNA was extracted for identification. The primer pairs amyR5-R and amyR3KX-F were used for PCR identification.
Enzyme and protein assays
Glucoamylase activity was assayed using the DNS method. Protein fermentation supernatant samples were mixed with an equal volume of 2× protein loading buffer and boiled for 10 min. The samples were subjected to SDS-PAGE using 4 % stacking gels and 12 % resolving gels in a mini-vertical electrophoresis system (Bio-Rad Laboratories, USA). The gels were stained with CBB-R250.
RNA isolation, cDNA synthesis and quantitative RT-PCR
Total RNA extraction, cDNA synthesis and quantitative real-time PCR were performed. An RNA isolation reagent was used for the total RNA extraction, and the PrimeScript RT Reagent Kit with gDNA Eraser was used for cDNA synthesis according to the manufacturer’s protocols. Quantitative real-time PCR was performed on a Stratagene Mx3000P instrument (USA) using SYBR Premix Ex Taq. Quantitative RT-PCR analysis of each gene was performed in triplicate with the primers listed in Table 1. Each amplification reaction was conducted in a total reaction volume of 20 μL. The thermal cycling protocol was as follows: initial denaturation for 2 min at 95 °C followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The fluorescence signal was measured at the end of each extension step at 95 °C. The transcript number of the actin gene from the same sample was used as an internal quantitative standard.
Transcriptome sequencing and annotation
Transcriptome sequencing of the A. niger CICC2462 and ∆amyR strains were performed with an Illumina HiSeq™2000 system. Reads were mapped to the reference genome of A. niger CBS513.88. The possible functions of all unigenes and the differentially expressed unigenes were determined with the Gene Ontology (GO) classification system [29]. The Gene Ontology terms for each A. niger unigene were obtained with the software Blast2GO using the default parameters. GO enrichment analyses were performed with the DAVID database. Blast2GO was also used for the GO functional enrichment analysis of certain genes by performing Fisher’s exact test with a robust false discovery rate (FDR) correction to obtain an adjusted P value between the gene and the entire annotation.
Results
amyR gene deletion decreases the amount of secretory proteins in A. niger CICC2462
Previous studies have shown that amylolytic-related proteins are regulated by the amyR transcriptional factor [15, 16]. To decrease the total quantity of proteins secreted in A. niger CICC2462, a deletion vector (pAmyRd) was transformed into it using the Agrobacterium tumefaciens-mediated method. Through PCR analysis, we found that 11 out of 14 transformants were positive recombinant transformants (primers of amyR5-R and amyR3KX-F), resulting in a transformation efficiency of 78.5 %, and three transformants of the ∆amyR strain (amyR3, amyR6, and amyR12) were further characterized.
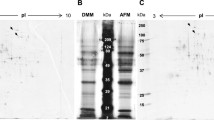
The enzyme activities of the fermentation supernatant of the reference strain A. niger CICC2462 and the ∆amyR strain were measured. The enzyme activities reached a peak after 7 days of culture. The data showed that the amylolytic enzyme activities of the ∆amyR strain was barely detected and they were 144- to 157-fold lower than that of the A. niger CICC2462 strain (Fig. 1a). SDS-PAGE analysis demonstrated that the protein bands corresponding to glucoamylase, acid-stable amylase A and alpha-amylase A were absent in the ∆amyR strains (Fig. 1b). These results suggest that the expression of glaA, asAA and amyA in A. niger CICC2462 depends on the presence of the amyR gene. The amount of total protein was assayed and compared in the supernatant after growth of the two strains through a BCA protein assay kit. The protein concentration of A.niger CICC2462 and amyR12 strain was 7.70 and 0.47 g/L, respectively. The date showed that the ∆amyR strain was 16.4-fold lower than that of the A.niger CICC2462 strain (Fig. 1c).

a Activities of amylolytic enzymes from A. niger CICC2462 and ∆amyR (amyR3, amyR6 and amyR12) strains grown on fermentation medium. b Separation of identified amylolytic enzyme proteins from the fermentation supernatant of A. niger CICC2462 and ∆amyR (amyR3, amyR6 and amyR12) strains by SDS-PAGE. 1, 2 and 3 represent glucoamylase, acid-stable alpha-amylase and alpha-amylase, respectively. An volume of 10 μL of each sample was separated on a 12 % resolving gel. c The protein concentration of A. niger CICC2462 and amyR12 strains
Furthermore, samples from the fermentation supernatant used for enzyme activity analysis were also analyzed at the proteomic level using shotgun LC–MS/MS directly. The main metabolic enzymes are listed in Tables 2 and 3. By comparing the major identified proteins with the A. niger CICC2462 proteomics, as observed from the number of peptides from the two strains, the overall quantity of proteins decreased. The proteins presenting the greatest reductions were identified as glucoamylase and amylase. Proteases also decreased, whereas alpha-galactosidase B and beta-mannosidase A increased, xylanase presented both decreased and increased.
Transcriptome analysis of A. niger strains
RNA-Seq data analysis
A total of 49,072,138 and 48,989,116 high-quality reads were obtained from two strains (A. niger CICC2462 and amyR12) by RNA-Seq, respectively. We specifically observed 11,699 and 11,646 expressed genes from these two strains (see Table 4). A total of 1956 differentially expressed unigenes (DEUs) between A. niger CICC2462 and amyR12 were detected, and these included 1331 and 635 unigenes that were up- and down-regulated in amyR12, respectively. Of the total reads, 89.19 and 87.93 % matched to unique genomic locations, respectively, whereas 54.31 and 53.36 % matched to unique gene locations, respectively. To better analyse the genes that presented marked differences, we downsized the scope of the analysis and took into account those with FPKM (Fragments per kilobase of transcript per million fragments mapped) >100 and an absolute value of the Log2 ratio (amyR12/CICC2462) >1.0 and <−1.0. The numbers of genes were then markedly decreased to less than 350. For CICC2462-VS-amyR12, 290 unigenes presented marked expression differences, and 243 and 47 unigenes were up- and down-regulated in amyR12, respectively.
Important down-regulated genes in amyR12
As expected, the transcriptome data showed that the transcription levels of amylolytic enzyme, glucoamylase, glucosidase and amylase were markedly decreased in amyR12 (see Table 5). This finding is consistent with the above-presented results, which showed that the number of high secretory protein was significantly decreased. Along with a decrease in the total secreted protein, the level of proteases were also reduced. Moreover, some of the sugar transporter genes were also down-regulated. Similar changes have been found after the deletion of xlnR in A. niger [26]. However, the regulatory mechanism of sugar transporter remains unclear. We analysed the amyR-binding motif of the above-mentioned down-regulated genes by Blast searches. By screening the amyR-binding sequence in the promoter regions (−1 to −1000 with the putative translational start site as +1) of the genes, we found that the amylolytic enzyme genes contained at least one putative amyR-binding site in their promoter regions. The protease genes without the amyR-binding sequence were possible indirectly regulated by amyR, and some sugar transport genes have amyR-binding sequences (see Table 6).
To validate the reliability of the transcriptome analysis, quantitative real-time PCR analysis was performed on three differentially expressed genes, which were selected for their high expression levels (one transcription factor, amyR, and two highly expressed genes, glaA and amyA). The transcript abundance patterns of A. niger CICC2462 and amyR12 were compared with the RNA-Seq data. The results showed that for these three genes, real-time PCR revealed the same expression patterns as those detected in the RNA-Seq data (Fig. 2). The transcription levels of the glaA and amyA genes in the ∆amyR strain decreased to very low levels compared with the levels detected in the wild-type strain.

Quantitative real-time PCR of some representative transcripts that were differentially expressed in A. niger CICC2462 and ∆amyR (amyR3, amyR6 and amyR12) strains grown on fermentation medium. The selected genes were: glucoamylase gene glaA, amylase gene amyA and transcriptional activator amyR
Influence of the deletion of amyR on protease activity
As mentioned above, the down-regulated genes in transcriptome data showed the transcription situation of protease genes. To analyze the mutant strain in more detail, protease activity analysis was carried out from these two strains (Fig. 3), compared with the wild-type strain, the protease activity was two- to tenfold lower in the amyR deletion strain.

Activities of protease activities from A. niger CICC2462 and ∆amyR strains grown on fermentation medium
Up-regulation of genes in amyR12
Most of the oxidative phosphorylation and tricarboxylic acid cycle genes were up-regulated in amyR12 (see Table 7). These include some genes relevant to oxidative phosphorylation, cytochrome C oxidase classes with heme-copper terminal oxidase activity (GO:0015002), ATP synthase classes and cytochrome C reductase classes with hydrogen ion transmembrane transporter activity (GO:0015078), NADH-ubiquinone oxidoreductase classes with NADH dehydrogenase (quinone) activity (GO:0050136), and succinate dehydrogenase (ubiquinone) class with succinate dehydrogenase activity (GO:0000104). These genes are mainly involved in the tricarboxylic acid cycle and include malate dehydrogenase (An07g02160), fumarate hydratase (An12g07850), succinate dehydrogenase (ubiquinone) flavoprotein subunit (An02g12770), and isocitrate dehydrogenase (NAD) subunit 2 (An08g05580). The transcriptome data showed that most of these genes related to these two central metabolic pathways are constitutively expressed. Normal cellular metabolism and the generation and utilization of energy were unaffected by deletion of the amyR gene in this organisms. In addition, ribosomal proteins (60S or 40S) with structural molecular activity (GO:0005198) that are mainly involved in the process of gene expression (GO:0010467) are concentrated in the up-regulated genes. Many unnamed protein products and hypothetical protein genes related to cytochrome and ribosomal proteins were found, although most have not been annotated, and some of these are available for novel gene prediction.
Influence of the deletion of amyR on mycelium morphology
Up-regulation of the oxidative phosphorylation and tricarboxylic acid cycle genes is a good indication of increase in growth rate. The A. niger CICC2462 and amyR12 strains were grown in PDA medium and the shake flask fermentation medium with glucose as the carbon source. Compared with the reference strain, amyR12 on PDA medium did not differ in size and colour for the first few days (Fig. 4a), but a significant difference was observed after 8 days incubation in that the colony of the amyR12 strain is larger (Fig. 4b). In the shake flask fermentation medium, the mycelium of amyR12 were thicker than those of the wild-type strain, and the dry weight of the amyR12 strain increased higher growth rate than the wild type (Fig. 4c).

a Growth of A. niger CICC2462 and ∆amyR strains on PDA medium. b Growth of A. niger CICC2462 and ∆amyR strains on PDA medium after 8 days. c Biomass weights of A. niger CICC2462 and ∆amyR grown in fermentation medium
Future perspectives
The research and development of filamentous fungus expression systems has become a hotspot in the field of recombinant protein expression throughout the world. Previous studies have demonstrated that Aspergillus has great economic potential as an expression host for the secretion of proteins with industrial applications [4, 30, 31]. Aspergillus niger and A. oryzae are the most commonly used Aspergillus species for industrial production, and the host is crucial for the efficient expression of proteins [32]. The exhibited morphology influences not only the culture broth viscosity but also the productivity, substrate diffusion and downstream processing [33]. Aspergillus niger CICC2462 was selected since it is used in the industrial production of glucoamylase. This strain is a morphological mutant strain of A. niger that does not produce spores, has short mycelia, thick hyphae, which results in a low-viscosity fermentation broth, is a strong enzyme producer, has low protease activity, is osmotolerant, and is suitable for high-density submerged liquid fermentation. As described previously, glucose generally represses the expression of amylolytic genes in creA-dependent manner [34]. To determine the function of the creA gene, transcriptome data (CICC2462-FPKM = 117.34, amyR12-FPKM = 146.9 and log2 Ratio = 0.3242) and quantitative real-time PCR analysis (Fig. 5) of creA gene were performed in the two strains. Transcription is not different comparing A. niger CICC2462 and the amyR12 strains. Moreover, amylolytic enzyme activity in the creA deletion strain was detected in the presence of glucose as the carbon source. The enzyme activity of A. niger CICC2462 strain and creA deletion strain was 48966.0960 and 49121.0520 U/mL, respectively. The results show that there is only a small difference. The activity of the creA gene product is probably weaker in A. niger CICC2462. In the shake flask fermentation experiment, glucose still remain at a concentration of 3.8 % (A. niger CICC2462) and 4.5 % (∆amyR) in the medium after 4 days. There was no significant difference in consumption of glucose comparing the wild type and amyR deleted strains. The A.niger CICC2462 strain can also secrete a large number of enzyme proteins into the growth medium in the presence of a high concentration of glucose which was used as the sole carbon source. The regulation of the secretome is obviously different from other strains, indicating the great research value of this strain.

Quantitative real-time PCR of creA gene that was differentially expressed in A. niger CICC2462 and amyR12 strains grown on fermentation medium
amyR has been studied in detail in several Aspergillus species in relation to amylolytic enzyme genes. The enzyme activity, SDS–PAGE and -omics analyses performed in our study provide clear indications for a key regulation role of amyR, because we observed strongly decreased levels of glucoamylase, amylase and glucosidase in our amyR-deletion strain compared with the wild type strain. We observed a reduction in protease secretion, possibly because the production of highly secreted proteins (glucoamylase, amylase and glucosidase) in amyR12 was decreased to a very low level. Considering the morphology, the amyR12 strain without growth inhibition compared to the wild type strain, and the dry weight of the amyR12 strain increased higher growth rate than the wild type. This result may be due to the reduced of highly expressed proteins in the liquid fermentation medium, thus nutrients are essentially used for mycelium growth.
Furthermore, promoter is a crucial element for controlling protein production and can largely regulate gene expression at the transcriptional level. The commonly used PglaA and PamyA promoters reduced their function in the ∆amyR strain. In general, homologous strong promoters appear to result in higher expression yields compared with heterologous promoters [31, 35]. As such, we need to determine the strong promoters in the ∆amyR strain. We primarily chose genes with a FPKM that was relatively high from the ∆amyR transcriptome data (see Table 8). The design and functional verification of promoters are a future line of research.
Conclusion
In this study, the regulatory effect of amyR on amylolytic enzymes in the expression system is apparent; specifically, the transcription and translation levels of glucoamylase and amylase were obviously decreased in the expression system. We now have a host strain with a low background of protein secretion and without growth inhibition, our work lays a foundation for continued improvements this expression system.
References
Pariza MW, Cook M. Determining the safety of enzymes used in animal feed. Regul Toxicol Pharmacol. 2010;56:332–42.
Karaffa L, Kubicek CP. Aspergillus niger citric acid accumulation: do we understand this well working black box? Appl Microbiol Biotechnol. 2003;61:189–96.
Goldberg I, Rokem JS, Pines O. Organic acids: old metabolites, new themes. J Chem Technol Biotechnol. 2006;81:1601–11.
Ward OP. Production of recombinant proteins by filamentous fungi. Biotechnol Adv. 2012;30:1119–39.
Fleißner A, Dersch P. Expression and export: recombinant protein production systems for Aspergillus. Appl Microbiol Biotechnol. 2010;87:1255–70.
Schröder M. Engineering eukaryotic protein factories. Biotechnol Lett. 2008;30:187–96.
Punt PJ, Biezen N, Conesa A, Albers A, Mangnus J, Hondel C. Filamentous fungi as cell factories for heterologous protein production. Trends Biotsechnol. 2002;20:200–6.
Archer DB, Jeenes DJ, Mackenzie DA. Strategies for improving heterologous protein production from filamentous fungi. Antonie Van Leeuwenhoek. 1994;65:245–50.
Nevalainen H, Peterson R. Making recombinant proteins in filamentous fungi- are we expecting too much? Front Microbiol. 2014;5:75.
Li J, Zhang H. Construction of food-grade xylanase engineering strain of Aspergillus niger. J Northeast Agric Univ. 2013;44:7–13.
Wei SZ, Zhang H, Li J. Homologous expression of asparaginase gene in Aspergillus niger. J Food Sci Biotechnol. 2015;34:554–9.
Zhang YY, Zhang H, Li J. Construction of β-mannanase engineering strain of Aspergillus niger. Biotechnology. 2013;23:31–6.
Amore A, Giacobbe S, Faraco V. Regulation of cellulase and hemicellulase gene expression in fungi. Curr Genom. 2013;14:230–49.
Tsukagoshi N, Kobayashi T, Kato M. Regulation of the amylolytic and (hemi-) cellulolytic genes in Aspergilli. J General Appl Microbiol. 1999;47:1–19.
Petersen KL, Lehmbeck J, Christensen T. A new transcriptional activator for amylase genes in Aspergillus. Mol General Genet MGG. 1999;262:668–76.
Gomi K, Minetoki T, Ozeki K, Kumagai C, Akeno T. Molecular cloning and characterization of a transcriptional activator gene, amyR, involved in the amylolytic gene expression in Aspergillus oryzae. Biosci Biotechnol Biochem. 2000;64:816–27.
Nakamura T, Makita T, Maeda Y, Tanoue N, Kato M, Kobayashi T. Expression profile of amylolytic genes in Aspergillus nidulans. Biosci Biotechnol Biochem. 2006;70:2363–70.
Watanabe J, Tanaka H, Mogi Y, Yamazaki T, Suzuki K, Watanabe T, Yamada O, Akita O. Loss of Aspergillus oryzae amyR function indirectly affects hemicellulolytic and cellulolytic enzyme production. J Biosci Bioeng. 2011;111:408–13.
Murakoshi Y, Makita T, Kato M, Kobayashi T. Comparison and characterization of ɑ-amylase inducers in Aspergillus nidulans based on nuclear localization of AmyR. Appl Microbiol Biotechnol. 2012;94:1629–35.
VanKuyk PA, Benen JAE, Wösten HAB, Visser J, de Vries RP. A broader role for AmyR in Aspergillus niger: regulation of the utilisation of d-glucose or d-galactose containing oligo- and polysaccharides. Appl Genet Mol Biotechnol. 2012;93:285–93.
Braaksma M, Martens-Uzunova ES, Punt PJ, Schaap PJ. An inventory of the Aspergillus niger secretome by combining in silico predictions with shotgun proteomics data. BMC Genom. 2010;11:2–11.
Lu X, Sun J, Nimtz M, Wissing J, Zeng AP, Rinas U. The intra- and extracellular proteome of Aspergillus niger growing on defined medium with xylose or maltose as carbon substrate. Microb Cell Fact. 2010;9:23.
de Oliveira JMPF, van Passel MWJ, Schaap PJ, de Graaff LH. Shotgun proteomics of Aspergillus niger microsomes upon D-xylose induction. Appl Environ Microbiol. 2010;76:4421–9.
Wang B, Guo GW, Wang C, Lin Y, Wang XN. Survey of the transcriptome of Aspergillus oryzae via massively parallel mRNA sequencing. Nucleic Acids Res. 2010;38:5075–87.
Chang PK, Scharfenstein LL, Mack B, Yu JJ, Ehrlich KC. Transcriptomic profiles of Aspergillus flavus CA42, a strain that produces small sclerotia, by decanal treatment and after recovery. Fungal Genet Biol. 2014;68:39–47.
de Souza WR, Maitan-Alfenas GP, de Gouvêa PF, Brown NA, Savoldi M, Goldman GH. The influence of Aspergillus niger transcription factors AraR and XlnR in the gene expression during growth in D-xylose, L-arabinose and steam-exploded sugarcane bagasse. Fungal Genet Biol. 2013;60:29–45.
Jacobs DI, Olsthoorn MMA, Maillet I, Sagt CMJ. Effective lead selection for improved protein production in Aspergillus niger based on integrated genomics. Fungal Genet Biol. 2009;46:141–52.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO:a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Zoglowek M, Lübeck PS, Ahring BK, Lübeck M. Heterologous expression of cellobiohydrolases in filamentous fungi -An update on the current challenges, achievements and perspectives. Process Biochem. 2015;50:211–20.
Nevalainen KMH, Te’o VSJ, Bergquist PL. Heterologous protein expression in filamentous fungi. Trends Biotechnol. 2005;9:468–74.
Meyer V. Genetic engineering of filamentous fungi-progress, obstacles and future trends. Biotechnol Adv. 2008;26:177–85.
Wucherpfennig T, Kiep KA, Driouch H, Wittmann R, Krull R. Chapter 4-morphology and rheology in filamentous cultivations. Adv Appl Microbiol. 2010;72:89–136.
Felenbok B, Kelly JM. Regulation of carbon metabolism in mycelial fungi. Biochem Mol Biol. 1996;3:369–80.
Radzio R, Kück U. Synthesis of biotechnologically relevant heterologous proteins in filamentous fungi. Process Biochem. 1997;32:529–39.
Authors’ contributions
HZ carried out the molecular genetic studies and drafted the manuscript. JL and YZ participated in the design of the study and performed the statistical analysis. SW and XXZ participated in the sequence alignment. WJ and FPS helped assemble the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by Grants from the Special Scientific Research Fund of Grain Public Welfare Profession of China (Project Number 201313005).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, H., Wang, S., Zhang, X.x. et al. The amyR-deletion strain of Aspergillus niger CICC2462 is a suitable host strain to express secreted protein with a low background. Microb Cell Fact 15, 68 (2016). https://doi.org/10.1186/s12934-016-0463-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0463-1