
Abstract
Mitochondria play a central role in cellular energy metabolism, and their dysfunction is increasingly recognized as a critical factor in the pathogenesis of diabetes-related cardiac pathophysiology, including vulnerability to ischemic events that culminate in myocardial infarction on the one hand and ventricular arrhythmias on the other. In diabetes, hyperglycemia and altered metabolic substrates lead to excessive production of reactive oxygen species (ROS) by mitochondria, initiating a cascade of oxidative stress that damages mitochondrial DNA, proteins, and lipids. This mitochondrial injury compromises the efficiency of oxidative phosphorylation, leading to impaired ATP production. The resulting energy deficit and oxidative damage contribute to functional abnormalities in cardiac cells, placing the heart at an increased risk of electromechanical dysfunction and irreversible cell death in response to ischemic insults. While cardiac mitochondria are often considered to be relatively autonomous entities in their capacity to produce energy and ROS, their highly dynamic nature within an elaborate network of closely-coupled organelles that occupies 30–40% of the cardiomyocyte volume is fundamental to their ability to exert intricate regulation over global cardiac function. In this article, we review evidence linking the dynamic properties of the mitochondrial network to overall cardiac function and its response to injury. We then highlight select studies linking mitochondrial ultrastructural alterations driven by changes in mitochondrial fission, fusion and mitophagy in promoting cardiac ischemic injury to the diabetic heart.
Similar content being viewed by others


Introduction
Diabetes mellitus is a global public health epidemic that continues to expand in both its incidence and prevalence. Among diabetics, cardiac dysfunction, stemming from both ischemic and non-ischemic insults, is the leading cause of morbidity and mortality [1]. Non-ischemic factors comprise diabetic cardiomyopathy, a condition characterized by structural and functional changes in the heart muscle that are independent of pre-existing coronary artery disease. These changes culminate in left ventricular hypertrophy, myocardial fibrosis, and impaired myocardial relaxation, driven largely by a triad of hyperglycemia, insulin resistance, and dyslipidemia. On the other hand, ischemic factors, which this review article focuses on, involve a heightened prevalence and severity of acute ischemia-reperfusion (I/R) events, leading to myocardial infarction, ischemic heart failure and sudden cardiac death due to ventricular arrhythmias.
Central to the development and exacerbation of ischemic insults in the diabetic heart is a state of oxidative stress, which arises from an imbalance in the production and scavenging of reactive oxygen species (ROS), favoring the former. Multiple sources contribute to elevated ROS production in the diabetic heart, including advanced glycation end-products (AGEs) and their receptors (RAGE), NADPH oxidases, uncoupled nitric oxide synthases and most importantly the mitochondrial network. In diabetes, the rate of glucose oxidation is decreased while that of fatty acid oxidation is enhanced, contributing to an increase in oxygen consumption [2]. This fundamental change in substrate utilization in favor of fatty acid oxidation plays a significant role in promoting mitochondrial ROS overproduction. In animal models and human samples from diabetic subjects, mitochondria are prime sources of excess ROS [3, 4]. For example, defective mitochondrial respiration associated with substrates of complexes I, II, and IV, as well as impaired state 4 → 3 transition occurs in isolated mitochondria from cardiomyocytes of leptin receptor deficient mice [5]. This, in turn, causes a major overflow of mitochondrial ROS that is exacerbated by high glucose conditions [5]. While mitochondria are clearly a major source of ROS overproduction in cardiomyocytes, they are also a victim of their own ROS generation. This establishes a vicious mitochondria-centric cycle that culminates in global oxidative stress.
Although mitochondria are often depicted as autonomous organelles that generate ATP and ROS through their intrinsic electron transport chain machinery, they comprise highly dynamic networks whose ultrastructural organization, distribution and morphology significantly impact their function both individually and as a network. The nature of these intricate networks depends on the balance between mitochondrial dynamics proteins, namely those governing biogenesis, fission, fusion, and mitophagy. In this article, we describe: (1) the importance of mitochondrial network dynamics in amplifying ROS injury to the heart culminating in arrhythmias or myocardial infarction, (2) the factors that dictate changes in the mitochondrial network morphology from biogenesis to mitophagy and their alterations in diabetes, and (3) the regulation of these processes by upstream metabolic signaling through the master metabolic sensor, AMPK. Understanding these mechanisms is crucial for developing targeted therapeutic strategies to address cardiovascular complications in diabetic patients.
Cardiomyocyte mitochondrial network dynamics, ROS-induced ROS-release and global cardiac dysfunction
Mitochondria are central mediators of the cardiac response to oxidative stress (OS), as they can either amplify or limit ROS-induced injury through a host of ROS-sensitive mitochondrial ion channels [6,7,8]. The inner membrane anion channel (IMAC) and components of the mitochondrial permeability transition pore (mPTP) are crucial in OS-induced mitochondrial dysfunction. Both channel complexes are activated by increasing ROS levels, but they follow a hierarchical activation pattern [9, 10]. Initially, IMAC responds to moderate OS levels, and subsequently, the large conductance mPTP activates, causing irreversible mitochondrial membrane potential (ΔΨm) depolarization [10]. Indeed, both channels have been implicated in mitochondrial dysfunction through a regenerative, autocatalytic process known as ROS-induced ROS-release (RIRR) which can culminate in electrical dysfunction or cell death (Fig. 1) [11,12,13,14,15].
RIRR, a fundamental mechanism by which cardiac mitochondria respond to elevated ROS levels by stimulating their own endogenous ROS production, is an emergent property of the mitochondrial network, which depends not only on ROS-sensitive mitochondrial ion channels, but also on the ultrastructure, distribution, morphology and functional coupling of mitochondria within the cardiomyocyte [9, 16]. Once a threshold level of ROS is exceeded across a critical mass of the mitochondrial population within the cardiomyocyte, emergent network behavior in the form of IMAC-mediated synchronized metabolic oscillations or mPTP -mediated global mitochondrial collapse can arise (Fig. 1) [17]. The former involve cell-wide ΔΨm oscillations that significantly impact cardiomyocyte function and lead to inexcitability at the cellular level through cyclical activation of ATP-sensitive potassium channels [17,18,19]. At the organ level, heterogeneity in ΔΨm-driven inexcitability promotes a form of conduction failure via a mechanism termed metabolic sink (Fig. 2) [17]. Stabilization of ΔΨm using pharmacological interventions that target the mitochondrial translocator protein (TSPO) to inhibit IMAC can lead to action potential (AP) stabilization and prevention of post-ischemic arrhythmias [17]. More sustained ROS induced injury, on the other hand, has been shown to activate mPTP culminating in irreversible cellular damage and death leading to cellular necrosis and myocardial infarction, which is a known substrate for malignant ventricular arrhythmias and sudden cardiac death (Fig. 1) [20, 21].
Using various experimental and computational approaches, we and others described the biophysical properties and functional consequences of RIRR at the tissue-network level and across broad myocardial regions. Zhou et al. developed a mathematical, reaction–diffusion model of RIRR that emphasized the importance of superoxide (O2−) diffusion in mediating mitochondrial dysfunction across 2-dimensional in silico networks of virtual mitochondria [22]. We experimentally tested these model predictions using a semi-quantitative approach of O2− mapping across the heart [14]. Consistent with cellular studies of RIRR, we demonstrated that exposure of intact hearts to high doses of exogenous pro-oxidants, such as H2O2, provoked two distinct ROS peaks. While the initial low amplitude peak coincided with the exogenous stressor, the secondary large amplitude peak occurred following, not during the exogenous stress, consistent with a regenerative RIRR response [14]. Functionally, hearts that exhibited the autocatalytic secondary ROS peak were prone to ventricular arrhythmias, whereas those that did not were relatively immune [14]. In a subsequent study, we investigated the relationship between the stability of the mitochondrial membrane in response to oxidative stress and the pro-arrhythmic potential of guinea pig hearts [23]. Specifically, we modulated the threshold and rate of decline of ΔΨm in response to exogenous pro-oxidant challenge using a variety of agents that affected the activities of key mitochondrial ion channels and their interaction with one another. In doing so, we uncovered functional cross-talk between the energy-dissipative mPTP and the cardioprotective mitochondrial K-ATP channels that ultimately governed the arrhythmic response of the heart to pro-oxidant challenge [23]. Once again, hearts that exhibited rapid ΔΨm decline were associated with low thresholds for sustained arrhythmias [23]. Consistent with the notion that ΔΨm stability is protective against electrical dysfunction at the organ level, Alleman et al. [24] demonstrated that exercise-mediated protection against reperfusion arrhythmias was indeed associated with and likely dependent upon proper ΔΨm polarization [24].
Strong mechanistic links between mitochondrial stability, oxidative stress and arrhythmias in the diabetic heart have been documented by multiple groups [16, 25,26,27,28,29,30,31]. For example, we found that glutathione (GSH) oxidation and therefore impaired ROS scavenging, were effective in unmasking mitochondrial depolarization and arrhythmic vulnerability of chronically hyperglycemic guinea pigs [32]. Slodzinski et al. [33] used two-photon microscopy to demonstrate that acute oxidative stress by GSH oxidation readily produces heterogeneous fluctuations in ΔΨm and ROS levels between neighboring myocytes within the intact heart [33]. Since ΔΨm oscillations can drive action potential duration oscillations [17], spatiotemporal changes in ΔΨm may indeed increase electrical heterogeneity across the heart, which we found was directly linked to pro-arrhythmic vulnerability in diabetic guinea pigs [32]. Finally, using wavelet analysis of the mitochondrial network’s response to pro-oxidant challenge, Vetter et al [26]. demonstrated that cardiomyocytes from diabetic guinea pig hearts were in a state of heightened vulnerability to cell-wide mitochondrial oscillations compared to their non-diabetic counterparts, an effect that could be rescued by insulin treatment [26]. Collectively, these studies and others emphasized the importance of the functional coupling between individual mitochondria within the cardiomyocyte in the response to injury. Such functional coupling is modulated by the ultrastructural features of the mitochondrial network which is composed of organelles that continuously fuse, divide and turnover in an effort to meet the changing energetic requirements of the cardiomyocyte and the organ as a whole [34, 35]. Indeed, remodeling of the mitochondrial network in acquired diseases such as heart failure in favor of fragmented mitochondria have been shown to inhibit the synchronization of metabolic oscillations while sensitizing individual mitochondria to permanent depolarization [36]. While not explicitly investigated, these effects would be expected to suppress post-ischemic arrhythmias at the expense of expanding the infarct size. As will be discussed next, the dynamic morphological adaptations of the mitochondrial network that regulate its function as a unit are orchestrated by fundamental cellular processes, namely mitochondrial biogenesis, fusion, fission, and mitophagy. Furthermore, these processes are remodeled in the context of common metabolic diseases and contribute to the enhanced propensity of the diabetic heart to pro-oxidant injury and post-ischemic arrhythmias, as we and others have observed [5, 25, 32].
Mitochondrial dynamics and the response of the mitochondrial Network to oxidative stress: from biogenesis to mitophagy
As mentioned previously, a host of ROS sensitive mitochondrial ion channel complexes in the cardiomyocyte activate in a hierarchal manner to produce metabolic oscillations that provoke arrhythmias on the one hand or irreversible mitochondrial depolarization leading to necrosis on the other. The amplification of ROS injury, however, is dependent not only on the intrinsic mitochondrial ion channels within individual mitochondria, but also on their network properties, namely their ultrastructural organization and morphology (Fig. 1). Indeed, cardiac mitochondria are densely packed within a 3-dimensional lattice structure adjacent to the myofilaments and T-tubules of the cardiomyocyte. Their unique network architecture and extensive subcellular organization localizes mitochondria in close spatial proximity to critical loci where: (1) energy is consumed (i.e. the myofilaments), (2) excitation-contraction coupling is initiated (i.e. the dyads), and (3) energy-dependent ion channels, exchangers and transporters are concentrated (i.e. the sarcolemma). While this network organization optimizes the efficient delivery of energy to critical microdomains within the cardiomyocyte, this same proximity also contributes to the transmission and amplification of pro-oxidant injury resulting in global cellular dysfunction and death. In what follows, we briefly introduce the mitochondrial dynamics processes of biogenesis, fission, fusion, and mitophagy, which govern the ultrastructural morphology and architecture of the mitochondrial network. For comprehensive reviews on the subject, the reader is referred to excellent reviews [37, 38].
Biogenesis
Mitochondrial biogenesis is the orchestrated process by which new mitochondria are generated to maintain or expand the existing mitochondrial pool in a given cell. Under conditions of energy supply-to-demand mismatch, mitochondrial biogenesis acts as a compensatory mechanism to restore mitochondrial capacity of the diabetic heart. Mitochondrial biogenesis requires the activation of various signaling pathways, including peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α), nuclear respiratory factors [39], and mitochondrial transcription factor A [40]. The coordinated activation of these factors causes a surge in mitochondrial mass, which bolsters the respiratory capacity and bioenergetic status of the cardiomyocyte; thereby, enhancing cardiac function in the face of increased metabolic demands.
Fission and fusion
In the native myocardium, fission and fusion events are required to maintain mitochondrial integrity and metabolic homeostasis [41]. Disruption of these opposing processes represents an early response to stress and has been observed in the onset and progression of cardiac disorders [41]. Mitochondrial fusion and fission are mediated by mechanisms that determine both the ultrastructure and function of mitochondria. Fusion describes the merger of the outer and inner membranes (OMM and IMM, respectively) of adjacent mitochondria, leading to the formation of elongated and interconnected networks with synchronized DYm, a key functional metric that fuels electron transport across the inner membrane. In addition to promoting functional coupling across the mitochondrial network, fusion also plays a crucial role in maintaining uniform mitochondrial DNA within neighboring mitochondria [42]. The process of mitochondrial fusion is coordinated by specific membrane-bound GTPases, namely mitofusins 1 & 2 (Mfn1 & Mfn2) and optic atrophy protein 1 (Opa1) [43,44,45,46]. Mitofusins form tethering connections which undergo GTP hydrolysis-driven conformational changes that merge the OMMs of neighboring mitochondria. A similar process involving Opa1 and cardiolipin mediates the fusion of the IMM; thereby completing the union of the two adjacent organelles [47].
Mitochondrial fission, on the other hand, is an evolutionarily conserved process that facilitates the division of mitochondria. Mitochondrial fission serves multiple purposes that are essential to cellular life, including the regulation of mitochondrial inheritance, the removal of damaged organelles, and the release of pro-apoptotic factors [42]. The primary mediator of mitochondrial fission is the dynamin-related protein 1 (Drp1), also known as dynamin-1-like protein. Drp1 is recruited from the cytoplasm to specific sites on the OMM [48, 49] through interactions with various Drp1 adaptor proteins, including mitochondrial fission 1 (Fis1), mitochondrial fission factor (Mff) and others [50,51,52]. Binding of Drp1 by these adaptors leads to the formation of Drp1 multimeric spirals that trigger GTP hydrolysis to induce a constricting conformational change [53]. This, in turn, facilitates the severing of the inner and outer mitochondrial membranes of the dividing organelles.
Changes in Drp1 expression alone are often insufficient to alter mitochondrial fission; instead, this process is highly regulated by post-translational modifications and their interactions with various OMM sites. These modifications can vary under different pathophysiological conditions. For example, the phosphorylation of Ser-600 and Ser-637 have been shown to be particularly important in diabetic mouse models [54,55,56,57].
Mitophagy
As mentioned above, mitochondrial fission is a critical process that serves to segregate damaged mitochondrial components thereby enhancing their accessibility for selective clearance, which then occurs through the process of mitophagy. Mitophagy, a specialized form of autophagy, recognizes damaged mitochondria, marking them for subsequent degradation. Hence, the cooperative interplay between fission and mitophagy serves as a pivotal mechanism for safeguarding the structural and functional integrity of the mitochondrial network. Alterations in mitophagy rates, either through unregulated activation or impaired function, contribute to the progression of many cardiovascular diseases [58]. At the molecular level, PTEN-induced kinase 1 (PINK1), a mitochondrial serine/threonine-protein kinase converges on damaged mitochondria, leading to the recruitment and subsequent ubiquitination of mitochondrial OMM proteins by Parkin. This, in turn, facilitates the binding of autophagy receptors.
The impact of altered mitochondrial dynamics on cardiac dysfunction specifically in the context of diabetes mellitus will be explored in more detail next [59]. We will focus on recent advances in our understanding of how alterations in mitochondrial network ultrastructure and morphology, driven by changes in mitochondrial dynamics and mitophagy-related proteins, impact myocardial function of the diabetic heart as well as its response to ischemic injury.
Mitochondrial network dysfunction in the diabetic heart
A frequently observed characteristic in tissues of hyperglycemic patients and animal models is mitochondrial deformation, marked by excessive accumulation of fragmented mitochondria [60, 61]. The importance of these ultrastructural changes is underscored by the fact that mitochondrial homeostasis and function are reflected by their morphology. Perhaps more importantly, changes in mitochondrial morphology have often been shown to cause, rather than merely reflect the increased generation of ROS in hyperglycemic conditions. Specifically, Yu et al. [60] elegantly demonstrated that mitochondrial fragmentation mediated by the fission process is a necessary component of high glucose-induced ROS overproduction and altered mitochondrial respiration in H9c2 cells. Overexpression of a dominant negative form of Drp1 prevented high glucose-induced mitochondrial fragmentation and excessive ROS production in cell lines. In another study, Drp1 silencing using small interfering RNAs in H9c2 cells exposed to oxidative stress also resulted in reduced mitochondrial fragmentation and improved insulin signaling [62]. Similarly, overexpression of Mfn2 or Opa1 were both effective in improving mitochondrial function and suppressing ROS in response of various cells to high glucose exposure [63, 64]. These lines of evidence strongly suggest that mitochondrial dynamics may be a previously unrecognized nexus in the control of ROS production in hyperglycemia-associated disorders [63].
The functional significance of mitochondrial fission during myocardial I/R injury is evident from numerous studies demonstrating a potent cardioprotective efficacy of Drp1 inhibition or gene silencing [65,66,67,68,69]. For example, in HL-1 cells, expression of a dominant negative Drp1 mutant resulted in the genesis of elongated mitochondria with reduced sensitivity to mitochondrial permeability transition [65]. Accordingly, transgenic expression of this mutant in rat hearts resulted in decreased infarct size, reduced cell death, and improved cardiac function following in vivo I/R [68]. Moreover, indirect suppression of Drp1 through the kinase Pim-1, known for its antiapoptotic and pro-proliferative properties, preserved mitochondrial phenotype in neonatal rat cardiomyocytes exposed to simulated ischemia [67]. Finally, pharmacological Drp1 inhibition using Drpitor1a preserved cardiac function during I/R, likely by inhibiting mitochondrial fission [66]. Taken together, these studies and others demonstrate the cardioprotective effect of preventing mitochondrial fission through pharmacological, regulatory and gene-based Drp1 downregulation during I/R injury. The role of Drp1 in the development of I/R injury specifically in the context of diabetes, has received limited attention thus far [60, 62, 70, 71]. A recent study in a mouse model of diabetes provided compelling evidence that increased translocation of Drp1 to mitochondria during I/R injury was associated with decreased mitochondrial size, consistent with a pro-fission response [72]. In vivo administration of MDIVI-1 to these mice inhibited Drp1 translocation to mitochondria, reduced mitochondrial fission, limited the extent of myocardial infarction and reduced levels of serum cardiac troponin and lactate dehydrogenase [72]. Similarly, in a rat model of high fat diet, treatment with MDIVI1 before, during or even after the ischemic challenge were effective in decreasing mitochondrial ROS generation, ΔΨm depolarization and arrhythmia burden [73]. Altering mitochondrial dynamics by promoting fusion via the small molecule M1 (which targets Opa1) was found to be equally effective in mitigating the detrimental effects of I/R injury in prediabetic animals as that achieved using the chemical Drp1 inhibitor MDIVI1 [74]. Interestingly, the combined treatment with both agents did not appear to exert added benefit over mono-therapy by MDIVI1 or M1 alone [75].
It is important to note, however, that proteins that regulate mitochondrial dynamics also serve important functions that are independent of fusion or fission, per se. For example, Mfn2 plays a key role in mediating functional coupling between mitochondria and the sarco(endo)plasmic reticulum (SR/ER) via physical tethering of the two organelles [76]. Hyperglycemia increases mitochondria–ER contact, alters Ca2+ levels within both compartments to promote mitochondrial ROS production, ER stress, mitochondrial dysfunction, and apoptosis. These effects of hyperglycemia are reversed by Mfn2 gene silencing, which was reported to decrease Ca2+ transfer from the ER to mitochondria; thereby ameliorating mitochondrial ROS production, oxygen consumption, mitochondrial dysfunction and cell death [77, 78]. In contrast, exaggerated mitochondrial Ca2+ uptake in models of prediabetes due to increased mitochondria-SR tethering can exacerbate adverse remodeling and increase mortality [79, 80].
Finally, mitophagy plays a major role in the regulation of cardiac function in diabetes by limiting the extent of oxidative stress through elimination of dysfunctional mitochondria [81]. Mitophagy, which is typically stimulated under conditions of nutrient deficiency, earmarks damaged mitochondria for lysosome-mediated degradation [82]. Indeed, increased mitophagy has been identified as a relatively early event that precedes overall cardiac dysfunction and likely plays a cardioprotective role in models of pre-diabetes. To that end, injection of Beclin-1 conjugated to a Tat peptide was found to be effective in ameliorating mitochondrial dysfunction, decreasing lipid accumulation, and protecting against diastolic dysfunction in mice on a high fat diet due to activation of mitophagy [83]. Finally, in chronic high fat diet models conventional mitophagy is inactivated, while an alternative form mediated by Ulk1 and Rab9-dependent processes is activated [84,85,86,87]. Table 1 summarizes some of the key studies linking changes in mitochondrial dynamics protein expression to network ultrastructure and cardiac function in diabetes that we covered in this section.
Regulation of mitochondrial dynamics by upstream metabolic signaling: role of AMPK
Upstream metabolic signaling via the master metabolic sensor 5’ adenosine monophosphate-activated protein kinase (AMPK) is vital in the diabetic heart, particularly in its response to ischemic injury. Diabetes exacerbates cardiovascular complications by impairing metabolic flexibility and energy homeostasis, placing the heart at a heightened risk to ischemic injury. AMPK functions as a cellular energy sensor, activated by an increase in the AMP/ATP ratio, which is a common occurrence during ischemic stress. In addition, AMPK is activated by the upstream kinases LKB1 and CaMKKβ (Ca2+/calmodulin-dependent protein kinase β). LKBI activation of AMPK is intertwined with AMP binding to the AMPKγ-subunit. In contrast, CaMKKβ activates AMPK in response to increases in cellular Ca2+ without significant changes in AMP/ADP/ATP levels [88, 89]. The activation of AMPK by these various substrates was originally thought to be tissue specific, with the primary activators in the heart being AMP, ADP, and LKB1. More recent data have revealed the importance of CaMKK2 in the reduction of oxidative stress and inflammation via the AMPK-AKT-GSK-3beta/Nrf2 pathway in response to myocardial I/R injury [90].
In addition to cellular stressors such as low nutrients or prolonged exercise, AMPK is also activated by various pharmacologic agents (see full review Mihaylova & Shaw, 2011) [89]. Most relevant to this review is the activation of AMPK by metformin, the most widely prescribed Type 2 diabetes drug. Metformin activates AMPK in a LKB1 dependent manner. Indeed, Shaw et al. have shown that deletion of LKB1 in the adult mouse liver leads to nearly complete loss of AMPK activity and that metformin requires hepatic LKB1 to lower blood glucose levels [91]. Metformin, however, is an AMPK agonist that also blocks mitochondrial complex I in many cell types and in doing so elicits numerous pleiotropic effects that impact cardiac function. These include a reducion of glucose output from the liver, a decrease in glycation end products and ROS production in the endothelium, and altered regulation of glucose and lipid metabolism in cardiomyocytes [92].
Upon activation, AMPK restores energy balance by stimulating catabolic processes such as glucose uptake, enhancing glycolysis, and promoting fatty acid oxidation. Additionally, AMPK activation initiates protective autophagy and mitophagy processes that remove damaged cellular components and dysfunctional mitochondria, thus preserving cellular integrity. By regulating these adaptive responses, AMPK not only mitigates the detrimental effects of ischemic injury in the diabetic heart but also improves overall cardiac function and survival. In what follows, we highlight the intricate control of mitochondrial biogenesis, fission, fusion and mitophagy by AMPK signaling in the context of diabetes.
AMPK regulation of mitochondrial biogenesis
AMPK plays a critical role in the regulation of mitochondrial biogenesis in skeletal muscle [39, 93, 94] and likely in heart [95, 96]. Upon activation, AMPK enhances the activity of PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha), a key transcriptional coactivator that drives the expression of genes involved in mitochondrial biogenesis [97]. In turn, PGC-1α activates several nuclear transcription factors, including NRF-1 (nuclear respiratory factor 1) and NRF-2, which promote the expression of Tfam (mitochondrial transcription factor A, also known as mtTFA), leading to the transcription of mitochondrial DNA-encoded and nuclear-encoded mitochondrial genes [39, 40, 98]. In addition to transcriptional activation through NRFs, PGC-1α is also the co-activator of the PPARs family, thyroid hormone, glucocorticoid, oestrogen, and ERRs (oestrogen-related receptors) [40, 99]. Thus, the activation of PGC-1α via AMPK leads to the regulation of energy homeostasis via the synthesis of new mitochondrial proteins and the formation of new mitochondria, thereby enhancing the oxidative capacity of cardiac cells.
In addition to its role in activating PGC-1α, AMPK influences mitochondrial biogenesis through other signaling pathways. Whereas PGC-1α has been found to activate mitochondrial biogenesis in response to low temperature and prolonged exercise, activation of SIRT1 (sirtuin 1) occurs in response to fasting through a nutrient-signaling response [40, 97, 100, 101]. Indeed, AMPK directly phosphorylates and activates SIRT1, a deacetylase that further activates PGC-1α [100]. This, in turn, amplifies the mitochondrial biogenesis response. Finally, AMPK regulates the expression of mitochondrial genes by directly phosphorylating and activating transcription factors that are crucial for mitochondrial DNA replication and transcription [39, 40, 98].
The regulation of mitochondrial biogenesis by AMPK is particularly important in the context of cardiac diseases, such as heart failure and ischemic heart disease, where mitochondrial dysfunction is a key pathological feature and energy demand often exceeds supply. In this context, AMPK activation can support cellular survival by expanding the mitochondrial pool to increase ATP synthesis. Moreover, AMPK deficiency has been reported to result in a significant decrease in PGC-1α driven mitochondrial biogenesis in aged hearts that are prone to ROS production and concomitant contractile dysfunction [102]. Both genetic knockout of PGC-1α and pharmacologic competition for its coactivator, PPARα/γ, lead to inhibition of the PGC-1α/SIRT1 pathway, lower mitochondrial abundance, and decreased cardiac function [95, 103]. Therefore, targeting AMPK signaling to enhance mitochondrial biogenesis represents a promising therapeutic strategy to improve cardiac function and resilience in various heart diseases and conditions, such as aging [41].
AMPK regulation of mitochondrial fission and fusion
Multiple lines of evidence have highlighted the robust regulation of mitochondrial fission in the heart by AMPK signaling [70, 104,105,106]. AMPK has multiple phosphorylation targets leading to mitochondrial fission and fusion, including Mff, ARMC10 (Armadillo repeat-containing protein 10 ), and MTFR1L (mitochondrial fission regulator 1-like protein). Pharmacological activation of AMPK directly promotes mitochondrial fission by phosphorylating serine-155 and serine-173 on Mff [107], which is involved in the recruitment of Drp1 to the mitochondrial membrane [108]. In fact, Drp1 localizes to mitochondria only when the AMPK phosphorylation sites on Mff are intact. Another key substrate of AMPK phosphorylation is the serine-45 site on ARMC10 [109]. ARMC10 localizes to the OMM and interacts with Mff and Drp1 to modulate mitochondrial fission. Lastly, AMPK has been shown to phosphorylate MTFR1L, and in doing so, to promote mitochondrial fragmentation in response to stress [110]. In this regard, depletion of AMPK was found to abolish mitochondrial fission induced by inhibitors of mitochondrial complexes I or III [106]. Yet, the regulation of mitochondrial fission by AMPK is multi-factorial and complex as a growing body of evidence has shown that increases in AMPK signaling inhibit pathological fission through direct phosphorylation of Drp1 at Ser-637, and in doing so to protect against cardiac dysfunction in a variety of settings.
As mentioned previously, AMPK mediates mitochondrial fission through direct targeting of Mff and MTFR1L. In the context of diabetes, however, activation of AMPK-mediated cardioprotective signaling has been shown to inhibit rather than promote pathological mitochondrial fission by altering Drp1 phosphorylation at Ser-637 [70]. In fact, there is a growing body of evidence suggesting the crucial involvement of the so-called AMPK-Drp1 axis in the cardioprotective effects of various interventions in type 2 diabetes mellitus. For instance, Zhou et al. [70] demonstrated that the sodium/glucose cotransporter 2 inhibitor Empagliflozin improved myocardial function, preserved microvascular barrier integrity, sustained endothelial nitric oxide synthase phosphorylation, and enhanced endothelium-dependent relaxation through AMPK-dependent inhibition of mitochondrial fission [70]. Together these data show the importance of AMPK in the regulation of mitochondrial dynamics and function in the diabetic heart.
AMPK regulation of mitophagy
As with mitochondrial biogenesis and dynamics, AMPK is also a major regulator of mitophagy. AMPK activates Unc-51-like Kinase 1 (ULK1), a key initiator of mitophagy, directly by phosphorylation as well as indirectly through suppression of mTOR signaling. AMPK-dependent activation of ULK1 promotes autophagosome formation around dysfunctional mitochondria and has also been suggested to activate Pink1/Parkin dependent mitophagy [111, 112]. Of note, an alternative mitophagy process mediated by a Parkin-independent, but Ulk1-Rab9 dependent, mechanism has also been documented in various settings by the Sadoshima group [84,85,86]. Finally, mitochondria-localized pools of AMPK (so-called mitoAMPK) are activated by local energetic stress to induce mitophagy, at least in skeletal muscle; thereby, acting as local sensors that help maintain quality control [113]. AMPK is the functional connection between energy sensing and mitochondrial homeostasis given its role in the biogenesis of new mitochondria via activation of PGC-1α and its regulation of mitophagy through a ULK1-dependent mechanism.
Additionally, AMPK enhances mitophagy at the transcriptional level by regulating the expression of genes involved in autophagic and lysosomal pathways. It upregulates proteins such as BNIP3 (BCL2/adenovirus E1B 19 kDa interacting protein 3) and NIX (NIP3-like protein X), which are essential for the recognition and targeting of damaged mitochondria for autophagic degradation. Furthermore, AMPK influences the activity of TFEB (transcription factor EB), a master regulator of lysosomal biogenesis and function. By promoting the nuclear translocation and activity of TFEB, AMPK ensures an adequate supply of lysosomal enzymes and an efficient degradation capacity, facilitating the clearance of damaged mitochondria.
Finally, AMPK also exerts strong regulation over mitochondrial function independently of its influence on network dynamics. Indeed, cardiac mitochondria isolated from mice expressing a kinase dead mutant of AMPK exhibit reduced oxidative capacity, increased H2O2 production and decreased resistance to mitochondrial permeability transition pore opening [114]. These intrinsic mitochondrial deficits correlate with increased rates of necrosis during reperfusion after coronary occlusion [114]. In this context, it is important to note that AMPK signaling in diabetes appears to be salutary in nature both in terms of its effects on individual mitochondria and the network as a whole.
Key unresolved questions
Despite the robust body of work linking mitochondrial dynamics to the cardiovascular pathophysiology of I/R injury, it is worth noting that contradictory evidence does exist in the literature that suggests a more nuanced role of mitochondrial fission and fusion in the regulation of I/R injury as highlighted in Table 2. For example, cardiac-specific Drp1 knockout mice exhibit greater mitochondrial dysfunction, left ventricular remodeling, and premature death than their control counterparts [115]. This strongly suggests that a basal level of myocardial Drp1 expression is likely required for maintenance of physiological mitochondrial function. Mfn2 deletion, which increases the rate of mitochondrial fission, was also reported in one study to paradoxically prevent rather than promote cell death and to reduce rather than expand the size of the infarct [116]. Silencing of Mfn2 in H9c2 cells or Mfn1 in cardiomyocytes also elicited an unexpected protection against ROS-induced apoptosis [117, 118]. Furthermore, overexpression of Opa1 did not confer protection against apoptosis induced by simulated ischemia in H9c2 cells [119]. These seemingly paradoxical findings emphasize the need for further examination to fully understand the precise role of mitochondrial dynamics in the context of I/R injury in various settings. These studies also indicate that therapeutic treatments to manipulate mitochondrial dynamics processes might need to be restricted to acute interventions because chronic alterations in the balance between fusion and fission can potentially disrupt adaptive mechanisms that are required for long-term mitochondrial and cardiac homeostasis. It is also likely that achieving cardioprotection against I/R might require a more nuanced orchestrated activation of multiple mitochondrial dynamics proteins, rather than more dramatic manipulation of single targets that may disrupt mitochondrial feedback processes. In this regard, AMPK activation through its salutary regulation of mitochondrial network dynamics from biogenesis to mitophagy represents an attractive target. Nonetheless, acute pharmacological inhibition or transient gene silencing of Drp1 at the time of injury represent therapeutic strategies that warrant further development and testing. Indeed, the mitochondrial division inhibitors MDIVI-1, Drpitor1a and P110, which act on Drp1 through distinct mechanisms, have all shown promising effects in reducing myocyte apoptosis in the context of ischemic injury, both in vitro and in vivo [120]. Moreover, customizing the therapeutic approach to specific molecular targets (Mfn2, Opa1, Drp1 or others) is likely to be required for achieving an optimal therapeutic effect. In addition, therapeutic strategies will also have to avoid potentially deleterious non-canonical processes, such as interfering with mitophagy in the case with Drp1 [121] or mitochondria/SR tethering in the case with Mfn2 [122, 123].
Conclusions
Diabetes is associated with a very high burden of coronary artery disease leading to myocardial infarction (MI) and ventricular arrhythmias [124]. Central to the pathophysiology of the diabetic heart and its susceptibility to acute and chronic ischemic injuries is the mitochondrial network which amplifies ROS injury to produce metabolic oscillations underlying arrhythmias or irreversible depolarization leading to cell death. The delicate interplay of mitochondrial biogenesis, fusion, fission and mitophagy is crucial for adapting to the physiological and pathological demands of the cardiomyocyte as a whole. Ongoing investigations in our laboratories are focused on understanding the regulation of cardiac function and arrhythmogenesis by upstream metabolic signaling that controls mitochondrial biogenesis, dynamics and mitophagy. It is our hope that these studies will uncover new therapeutic approaches for treating cardiac dysfunction in the context of metabolic diseases by improving mitochondrial function.

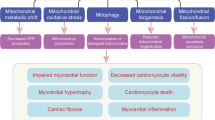
Schematic illustrating the functional and physical features of mitochondrial network dynamics. The processes of biogenesis, fusion, fission and mitophagy dictate the morphology, density and ultrastructure of the mitochondrial network within the cardiomyocyte, which in turn affect the functional coupling/synchronization of individual mitochondria. An increase in mitochondrial production can lead to rapid amplification of ROS across the mitochondrial network via activation of ROS-sensitive mitochondrial channels. A hierarchal activation pattern of ROS sensitive channels causes early activation of TSPO/IMAC leading to reversible mitochondrial membrane potential oscillations that drive electrophysiological instability via cyclical activation of sarcolemmal ATP-sensitive K channels. This promotes conduction abnormalities and arrhythmias. On the other hand, higher levels of oxidative stress can activate the mPTP to cause irreversible mitochondrial depolarization leading to cell necrosis and myocardial infarction, a major risk factor for malignant arrhythmias and sudden cardiac death

Schematic illustrating the multi-scale process by which individual mitochondria can lead to organ level arrythmias. Diabetes causes an increase in ROS production and impairment in ROS scavenging. ROS levels are amplified through an autocatalytic feed-forward process of ROS-induced ROS-release which culminates in metabolic oscillations and rapid mitochondrial uncoupling. This causes activation of ATP-sensitive potassium channels resulting in membrane inexcitability at the cellular level and a phenomenon termed metabolic sink that leads to conduction block at the tissue level. Heterogeneous formation of these metabolic sinks results in ventricular arrhythmias
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- AGE:
-
Advanced glycation end products
- AMPK:
-
5' adenosine monophosphate-activated protein kinase
- AP:
-
Action potential
- ARMC10:
-
Armadillo repeat-containing protein 10
- BNIP3:
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- CaMKKβ:
-
Ca2+/calmodulin–dependent protein kinase β
- Drp1:
-
Dynamin-related protein 1
- eNOS:
-
Endothelial nitric oxide synthase
- ETC:
-
Electron transport chain
- Fis1:
-
Mitochondrial fission 1
- GSH:
-
Glutathione
- IMM:
-
Inner mitochondrial membrane
- I/R:
-
Ischemia-reperfusion
- JNK:
-
c-Jun N-terminal kinas
- Mdivi-1:
-
Mitochondrial division inhibitor 1
- mDNA:
-
Mitochondrial DNA
- Mff:
-
Mitochondrial fission factor
- Mfn:
-
Mitofusin
- mPTP:
-
Mitochondrial permeability transition pore
- MTFR1L:
-
Mitochondrial fission regulator 1-like protein
- nDNA:
-
Nuclear DNA
- NRF:
-
Nuclear respiratory factors
- OMM:
-
Outer mitochondrial membrane
- Opa1:
-
Optic atrophy protein 1
- OS:
-
Oxidative stress
- PGC-1α:
-
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PINK1:
-
PTEN-induced kinase 1
- RIRR:
-
ROS-induced ROS-release
- ROS:
-
Reactive oxygen species
- Sirt:
-
Sirtuin
- TFAM:
-
Mitochondrial transcription factor A
- TSPO:
-
Mitochondrial translocator protein
- UCP:
-
Mitochondrial uncoupling proteins
- ULK1:
-
Unc-51-like kinase 1
- ΔΨm:
-
Mitochondrial membrane potential
References
Matheus AS, Tannus LR, Cobas RA, Palma CC, Negrato CA, Gomes MB. Impact of diabetes on cardiovascular disease: an update. Int J Hypertens. 2013;2013:653789.
Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. 2010;88:229–40.
Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20.
Shah MS, Brownlee M. Molecular and Cellular mechanisms of Cardiovascular disorders in Diabetes. Circ Res. 2016;118:1808–29.
Tocchetti CG, Caceres V, Stanley BA, Xie C, Shi S, Watson WH, O’Rourke B, Spadari-Bratfisch RC, Cortassa S, Akar FG, Paolocci N, Aon MA. GSH or palmitate preserves mitochondrial energetic/redox balance, preventing mechanical dysfunction in metabolically challenged myocytes/hearts from type 2 diabetic mice. Diabetes. 2012;61:3094–105.
Akar FG, O’Rourke B. Mitochondria are sources of metabolic sink and arrhythmias. Pharmacol Ther. 2011;131:287–94.
O’Rourke B, Cortassa S, Akar F, Aon M. Mitochondrial ion channels in cardiac function and dysfunction. Novartis Found Symp. 2007;287:140–51. discussion 152– 146.
O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49.
Aon MA, Cortassa S, Akar FG, O’Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta. 2006;1762:232–40.
Aon MA, Cortassa S, Maack C, O’Rourke B. Sequential opening of mitochondrial ion channels as a function of glutathione redox thiol status. J Biol Chem. 2007;282:21889–900.
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–14.
Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial ROS-induced ROS release: an update and review. Biochim Biophys Acta. 2006;1757:509–17.
Akar FG. Mitochondrial targets for arrhythmia suppression: is there a role for pharmacological intervention? J Interv Card Electrophysiol. 2013;37:249–58.
Biary N, Xie C, Kauffman J, Akar FG. Biophysical properties and functional consequences of reactive oxygen species (ROS)-induced ROS release in intact myocardium. J Physiol. 2011;589:5167–79.
Yang L, Korge P, Weiss JN, Qu Z. Mitochondrial oscillations and waves in cardiac myocytes: insights from computational models. Biophys J. 2010;98:1428–38.
Ilkan Z, Akar FG. The mitochondrial translocator protein and the emerging link between oxidative stress and arrhythmias in the Diabetic Heart. Front Physiol. 2018;9:1518.
Akar FG, Aon MA, Tomaselli GF, O’Rourke B. The mitochondrial origin of postischemic arrhythmias. J Clin Investig. 2005;115:3527–35.
Aon MA, Cortassa S, Marban E, O’Rourke B. Synchronized whole cell oscillations in mitochondrial metabolism triggered by a local release of reactive oxygen species in cardiac myocytes. J Biol Chem. 2003;278:44735–44.
Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248–58.
Assaly R, de Tassigny A, Paradis S, Jacquin S, Berdeaux A, Morin D. Oxidative stress, mitochondrial permeability transition pore opening and cell death during hypoxia-reoxygenation in adult cardiomyocytes. Eur J Pharmacol. 2012;675:6–14.
Morciano G, Pedriali G, Bonora M, et al. A naturally occurring mutation in ATP synthase subunit c is associated with increased damage following hypoxia/reoxygenation in STEMI patients. Cell Rep. 2021;35:108983.
Zhou L, Aon MA, Almas T, Cortassa S, Winslow RL, O’Rourke B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput Biol. 2010;6:e1000657.
Xie C, Kauffman J, Akar FG. Functional crosstalk between the mitochondrial PTP and KATP channels determine arrhythmic vulnerability to oxidative stress. Front Physiol. 2014;5:264.
Alleman RJ, Tsang AM, Ryan TE, Patteson DJ, McClung JM, Spangenburg EE, Shaikh SR, Neufer PD, Brown DA. Exercise-induced protection against reperfusion arrhythmia involves stabilization of mitochondrial energetics. Am J Physiol Heart Circ Physiol. 2016;310:H1360–1370.
Xie C, Hu J, Motloch LJ, Karam BS, Akar FG. The classically cardioprotective Agent Diazoxide elicits arrhythmias in type 2 diabetes Mellitus. J Am Coll Cardiol. 2015;66:1144–56.
Vetter L, Cortassa S, O’Rourke B, Armoundas AA, Bedja D, Jende JME, Bendszus M, Paolocci N, Sollot SJ, Aon MA, Kurz FT. Diabetes increases the vulnerability of the cardiac mitochondrial network to Criticality. Front Physiol. 2020;11:175.
Gong M, Yuan M, Meng L, et al. Wenxin Keli regulates mitochondrial oxidative stress and homeostasis and improves atrial remodeling in Diabetic rats. Oxid Med Cell Longev. 2020;2020:2468031.
Shao Q, Meng L, Lee S, Tse G, Gong M, Zhang Z, Zhao J, Zhao Y, Li G, Liu T. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019;18:165.
Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol. 2018;17:144.
Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP. Inhibition of NAPDH oxidase 2 (NOX2) prevents oxidative stress and mitochondrial abnormalities caused by Saturated Fat in Cardiomyocytes. PLoS ONE. 2016;11:e0145750.
Zhang X, Zhang Z, Zhao Y, Jiang N, Qiu J, Yang Y, Li J, Liang X, Wang X, Tse G, Li G, Liu T. Alogliptin, a Dipeptidyl Peptidase-4 inhibitor, alleviates atrial remodeling and improves mitochondrial function and Biogenesis in Diabetic rabbits. J Am Heart Assoc. 2017. https://doi.org/10.1161/JAHA.117.005945.
Xie C, Biary N, Tocchetti CG, Aon MA, Paolocci N, Kauffman J, Akar FG. Glutathione oxidation unmasks proarrhythmic vulnerability of chronically hyperglycemic guinea pigs. Am J Physiol Heart Circ Physiol. 2013;304:H916–926.
Slodzinski MK, Aon MA, O’Rourke B. Glutathione oxidation as a trigger of mitochondrial depolarization and oscillation in intact hearts. J Mol Cell Cardiol. 2008;45:650–60.
Aon MA. From isolated to networked: a paradigmatic shift in mitochondrial physiology. Front Physio. 2010;1:20.
Aon MA, Cortassa S, Akar FG, Brown DA, Zhou L, O’Rourke B. From mitochondrial dynamics to arrhythmias. Int J Biochem Cell Biol. 2009;41:1940–8.
Goh KY, Qu J, Hong H, Liu T, Dell’Italia LJ, Wu Y, O’Rourke B, Zhou L. Impaired mitochondrial network excitability in failing guinea-pig cardiomyocytes. Cardiovasc Res. 2016;109:79–89.
Morciano G, Boncompagni C, Ramaccini D, Pedriali G, Bouhamida E, Tremoli E, Giorgi C, Pinton P. Comprehensive Analysis of Mitochondrial Dynamics Alterations in Heart diseases. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms24043414I.
Morciano G, Vitto VAM, Bouhamida E, Giorgi C, Pinton P. Mitochondrial bioenergetics and dynamism in the failing heart. Life (Basel). 2021. https://doi.org/10.3390/life11050436.
Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281:E1340–1346.
Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem. 2010;47:69–84.
Forte M, Schirone L, Ameri P, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. 2021;178:2060–76.
Westermann B. Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–84.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200.
Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A. 2004;101:15927–32.
Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed in vitro. Science. 2004;305:1747–52.
Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell. 2009;20:3525–32.
Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem. 2018;62:341–60.
Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science. 2011;334:358–62.
Lewis SC, Uchiyama LF, Nunnari J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science. 2016;353:aaf5549.
Gandre-Babbe S, van der Bliek AM. The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell. 2008;19:2402–12.
James DI, Parone PA, Mattenberger Y, Martinou JC. hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem. 2003;278:36373–9.
Palmer CS, Osellame LD, Laine D, Koutsopoulos OS, Frazier AE, Ryan MT. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011;12:565–73.
van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol. 2013. https://doi.org/10.1101/cshperspect.a011072.
Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab. 2012;15:186–200.
Wikstrom JD, Israeli T, Bachar-Wikstrom E, Swisa A, Ariav Y, Waiss M, Kaganovich D, Dor Y, Cerasi E, Leibowitz G. AMPK regulates ER morphology and function in stressed pancreatic β-cells via phosphorylation of DRP1. Mol Endocrinol. 2013;27:1706–23.
Li A, Zhang S, Li J, Liu K, Huang F, Liu B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol Cell Endocrinol. 2016;434:36–47.
Li W, Ji L, Tian J, Tang W, Shan X, Zhao P, Chen H, Zhang C, Xu M, Lu R, Guo W. Ophiopogonin D alleviates diabetic myocardial injuries by regulating mitochondrial dynamics. J Ethnopharmacol. 2021;271:113853.
Rocca C, Soda T, De Francesco EM, Fiorillo M, Moccia F, Viglietto G, Angelone T, Amodio N. Mitochondrial dysfunction at the crossroad of cardiovascular diseases and cancer. J Transl Med. 2023;21:635.
AS G, D M, VL R, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–245.
Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–8.
Vanhorebeek I, De Vos R, Mesotten D, Wouters PJ, De Wolf-Peeters C, Van den Berghe G. Protection of hepatocyte mitochondrial ultrastructure and function by strict blood glucose control with insulin in critically ill patients. Lancet. 2005;365:53–9.
Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H, Satoh H, Funaki M, Hayashi H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res. 2014;323:314–25.
Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res. 2008;79:341–51.
Makino A, Suarez J, Gawlowski T, Han W, Wang H, Scott BT, Dillmann WH. Regulation of mitochondrial morphology and function by O-GlcNAcylation in neonatal cardiac myocytes. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1296–1302.
Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010;121:2012–22.
Wu D, Dasgupta A, Chen KH, et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020;34:1447–64.
Din S, Mason M, Volkers M, Johnson B, Cottage CT, Wang Z, Joyo AY, Quijada P, Erhardt P, Magnuson NS, Konstandin MH, Sussman MA. Pim-1 preserves mitochondrial morphology by inhibiting dynamin-related protein 1 translocation. Proc Natl Acad Sci U S A. 2013;110:5969–74.
Zepeda R, Kuzmicic J, Parra V, Troncoso R, Pennanen C, Riquelme JA, Pedrozo Z, Chiong M, Sanchez G, Lavandero S. Drp1 loss-of-function reduces cardiomyocyte oxygen dependence protecting the heart from ischemia-reperfusion injury. J Cardiovasc Pharmacol. 2014;63:477–87.
Bouche L, Kamel R, Tamareille S, et al. DRP1 haploinsufficiency attenuates cardiac ischemia/reperfusion injuries. PLoS ONE. 2021;16:e0248554.
Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2018;15:335–46.
Yu J, Maimaitili Y, Xie P, Wu JJ, Wang J, Yang YN, Ma HP, Zheng H. High glucose concentration abrogates sevoflurane post-conditioning cardioprotection by advancing mitochondrial fission but dynamin-related protein 1 inhibitor restores these effects. Acta Physiol (Oxf). 2017;220:83–98.
Ding M, Dong Q, Liu Z, Liu Z, Qu Y, Li X, Huo C, Jia X, Fu F, Wang X. Inhibition of dynamin-related protein 1 protects against myocardial ischemia-reperfusion injury in diabetic mice. Cardiovasc Diabetol. 2017;16:19.
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Pharmacological inhibition of mitochondrial fission attenuates cardiac ischemia-reperfusion injury in pre-diabetic rats. Biochem Pharmacol. 2020;182:114295.
Maneechote C, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Chronic pharmacological modulation of mitochondrial dynamics alleviates Prediabetes-Induced Myocardial Ischemia-Reperfusion Injury by preventing mitochondrial dysfunction and programmed apoptosis. Cardiovasc Drugs Ther. 2023;37:89–105.
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Modulating mitochondrial dynamics attenuates cardiac ischemia-reperfusion injury in prediabetic rats. Acta Pharmacol Sin. 2022;43:26–38.
Dorn GW 2nd, Song M, Walsh K. Functional implications of mitofusin 2-mediated mitochondrial-SR tethering. J Mol Cell Cardiol. 2015;78:123–8.
Hernandez-Resendiz S, Prunier F, Girao H, Dorn G, Hausenloy DJ, Action E-CC. Targeting mitochondrial fusion and fission proteins for cardioprotection. J Cell Mol Med. 2020;24:6571–85.
Ong SB, Hausenloy DJ. Mitochondrial Dynamics as a therapeutic target for treating Cardiac diseases. Handb Exp Pharmacol. 2017;240:251–79.
Tow BD, Deb A, Neupane S, Patel SM, Reed M, Loper AB, Eliseev RA, Knollmann BC, Gyorke S, Liu B. SR-Mitochondria Crosstalk shapes ca Signalling to Impact Pathophenotype in Disease models marked by dysregulated intracellular ca release. Cardiovasc Res. 2022;118:2819–32.
Federico M, Zavala M, Vico T, Lopez S, Portiansky E, Alvarez S, Abrille MCV, Palomeque J. CaMKII activation in early diabetic hearts induces altered sarcoplasmic reticulum-mitochondria signaling. Sci Rep. 2021;11:20025.
Ketenci M, Zablocki D, Sadoshima J. Mitochondrial Quality Control mechanisms during Diabetic Cardiomyopathy. JMA J. 2022;5:407–15.
Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Mitophagy is essential for maintaining cardiac function during high Fat Diet-Induced Diabetic Cardiomyopathy. Circ Res. 2019;124:1360–71.
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin Sci (Lond). 2019;133:497–513.
Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J. Alternative Mitophagy protects the heart against obesity-Associated Cardiomyopathy. Circ Res. 2021;129:1105–21.
Saito T, Nah J, Oka SI, et al. An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Investig. 2019;129:802–19.
Sadoshima J. Alternative mitophagy is a major form of mitophagy in the chronically stressed heart. Autophagy. 2022;18:2252–3.
Ikeda Y, Shirakabe A, Brady C, Zablocki D, Ohishi M, Sadoshima J. Molecular mechanisms mediating mitochondrial dynamics and mitophagy and their functional roles in the cardiovascular system. J Mol Cell Cardiol. 2015;78:116–22.
Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016;48:e224.
Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–23.
Li C, Hao J, Qiu H, Xin H. CaMKK2 alleviates myocardial ischemia/reperfusion injury by inhibiting oxidative stress and inflammation via the action on the AMPK-AKT-GSK-3β/Nrf2 signaling cascade. Inflamm Res. 2023;72:1409–25.
Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–6.
Dutta S, Shah RB, Singhal S, Dutta SB, Bansal S, Sinha S, Haque M, Metformin. A review of potential mechanism and therapeutic utility beyond diabetes. Drug Des Devel Ther. 2023;17:1907–32.
Reznick RM, Zong H, Li J, et al. Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab. 2007;5:151–6.
Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99:15983–7.
Xin T, Lu C. SirT3 activates AMPK-related mitochondrial biogenesis and ameliorates sepsis-induced myocardial injury. Aging. 2020;12:16224–37.
Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl). 2010;88:993–1001.
Irrcher I, Adhihetty PJ, Sheehan T, Joseph AM, Hood DA. PPARgamma coactivator-1alpha expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am J Physiol Cell Physiol. 2003;284:C1669–1677.
Virbasius JV, Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: a potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91:1309–13.
Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–17.
Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8.
Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. Faseb j. 2002;16:1879–86.
Turdi S, Fan X, Li J, Zhao J, Huff AF, Du M, Ren J. AMP-activated protein kinase deficiency exacerbates aging-induced myocardial contractile dysfunction. Aging Cell. 2010;9:592–606.
Kalliora C, Kyriazis ID, Oka SI, et al. Dual peroxisome-proliferator-activated-receptor-α/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.129556.
Liu J, Yan W, Zhao X, Jia Q, Wang J, Zhang H, Liu C, He K, Sun Z. Sirt3 attenuates post-infarction cardiac injury via inhibiting mitochondrial fission and normalization of AMPK-Drp1 pathways. Cell Signal. 2019;53:1–13.
Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, Zou MH. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of Drp1-Mediated mitochondrial fission. Diabetes. 2017;66:193–205.
Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Loson OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, Shaw RJ. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–81.
Ducommun S, Deak M, Sumpton D, Ford RJ, Nunez Galindo A, Kussmann M, Viollet B, Steinberg GR, Foretz M, Dayon L, Morrice NA, Sakamoto K. Motif affinity and mass spectrometry proteomic approach for the discovery of cellular AMPK targets: identification of mitochondrial fission factor as a new AMPK substrate. Cell Signal. 2015;27:978–88.
Otera H, Wang C, Cleland MM, Setoguchi K, Yokota S, Youle RJ, Mihara K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J Cell Biol. 2010;191:1141–58.
Chen Z, Lei C, Wang C, Li N, Srivastava M, Tang M, Zhang H, Choi JM, Jung SY, Qin J, Chen J. Global phosphoproteomic analysis reveals ARMC10 as an AMPK substrate that regulates mitochondrial dynamics. Nat Commun. 2019;10:104.
Tilokani L, Russell FM, Hamilton S, et al. AMPK-dependent phosphorylation of MTFR1L regulates mitochondrial morphology. Sci Adv. 2022;8:eabo7956.
Hung CM, Lombardo PS, Malik N, Brun SN, Hellberg K, Van Nostrand JL, Garcia D, Baumgart J, Diffenderfer K, Asara JM, Shaw RJ. AMPK/ULK1-mediated phosphorylation of parkin ACT domain mediates an early step in mitophagy. Sci Adv. 2021. https://doi.org/10.1126/sciadv.abg4544.
Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–61.
Drake JC, Wilson RJ, Laker RC, et al. Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci U S A; 2021. https://doi.org/10.1073/pnas.2025932118.
Zaha VG, Qi D, Su KN, Palmeri M, Lee HY, Hu X, Wu X, Shulman GI, Rabinovitch PS, Russell RR 3rd, Young LH. AMPK is critical for mitochondrial function during reperfusion after myocardial ischemia. J Mol Cell Cardiol. 2016;91:104–13.
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–78.
Papanicolaou KN, Khairallah RJ, Ngoh GA, Chikando A, Luptak I, O’Shea KM, Riley DD, Lugus JJ, Colucci WS, Lederer WJ, Stanley WC, Walsh K. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol Cell Biol. 2011;31:1309–28.
Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF Jr., Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ROS-induced mitochondrial dysfunction and cell death. Am J Physiol Heart Circ Physiol. 2012;302:H167–179.
Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen KH, Xiao RP. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J Biol Chem. 2007;282:23354–61.
Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res. 2009;84:91–9.
Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Differential temporal inhibition of mitochondrial fission by Mdivi-1 exerts effective cardioprotection in cardiac ischemia/reperfusion injury. Clin Sci (Lond). 2018;132:1669–83.
Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation. 2016;133:1249–63.
Li J, Qi X, Ramos KS, Lanters E, Keijer J, de Groot N, Brundel B, Zhang D. Disruption of Sarcoplasmic Reticulum-mitochondrial contacts underlies contractile dysfunction in experimental and human atrial fibrillation: a key role of Mitofusin 2. J Am Heart Assoc. 2022;11:e024478.
Ren L, Gopireddy RR, Perkins G, et al. Disruption of mitochondria-sarcoplasmic reticulum microdomain connectomics contributes to sinus node dysfunction in heart failure. Proc Natl Acad Sci U S A. 2022;119:e2206708119.
Milazzo V, Cosentino N, Genovese S, Campodonico J, Mazza M, De Metrio M, Marenzi G. Diabetes Mellitus and Acute Myocardial infarction: impact on short and long-term mortality. Adv Exp Med Biol. 2021;1307:153–69.
Funding
Supported by grants from the National Institutes of Health 1R01HL149344, 1R01HL148008, 1R21HL165147.
Author information
Authors and Affiliations
Contributions
M.R. wrote the initial draft of the paper. M.G. wrote and edited sections of the paper. Z.T. wrote and edited sections of the paper. J.N.E. wrote and edited sections of the paper. M.R, wrote and edited sections of the paper. L.Y. edited the paper. F.G.A. wrote and edited the paper.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rudokas, M.W., McKay, M., Toksoy, Z. et al. Mitochondrial network remodeling of the diabetic heart: implications to ischemia related cardiac dysfunction. Cardiovasc Diabetol 23, 261 (2024). https://doi.org/10.1186/s12933-024-02357-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-024-02357-1