
Abstract
Apolipoprotein C1 (apoC1) is a small size apolipoprotein whose exact role is not totally clarified but which seems to modulate significantly the metabolism of lipoproteins. ApoC1 is involved in the metabolism of triglyceride-rich lipoproteins by inhibiting the binding of very low density lipoproteins (VLDL) to VLDL-receptor (VLDL-R), to low density lipoprotein receptor (LDL-R) and to LDL receptor related protein (LRP), by reducing the activity of lipoprotein lipase (LPL) and by stimulating VLDL production, all these effects leading to increase plasma triglycerides. ApoC1 takes also part in the metabolism of high density lipoproteins (HDL) by inhibiting Cholesterol Ester Transfer Protein (CETP). The functionality of apoC1 on CETP activity is impaired in diabetes that might account, at least in part, for the increased plasma CETP activity observed in patients with diabetes. Its different effects on lipoprotein metabolism with a possible role in the modulation of inflammation makes the net impact of apoC1 on cardiometabolic risk difficult to figure out and apoC1 might be considered as pro-atherogenic or anti-atherogenic depending on the overall metabolic context. Making the link between total plasma apoC1 levels and the risk of cardio-metabolic diseases is difficult due to the high exchangeability of this small protein whose biological effects might depend essentially on its association with VLDL or HDL. The role of apoC1 in humans is not entirely elucidated and further studies are needed to determine its precise role in lipid metabolism and its possible pleiotropic effects on inflammation and vascular wall biology. In this review, we will present data on apoC1 structure and distribution among lipoproteins, on the effects of apoC1 on VLDL metabolism and HDL metabolism and we will discuss the possible links between apoC1, atherosclerosis and diabetes.
Similar content being viewed by others

Introduction
ApoC1 is the smallest plasma apolipoprotein, composed of 57 amino-acids only. Its very simple structure, together with the analysis of sequence similarities, suggest that apoC1 reflects most closely the ancestor gene from which derived many other apolipoproteins (apo) such as apoC2, apoC3 and apoE [1]. While the involvement of the three latter in the regulation of plasma triglyceride hydrolysis and lipoprotein remnant uptake has been extensively studied and clearly established over years, apoC1 has long appeared as the “poor relative” of the family. Although its similarities with the ApoE/C2/C4 group initially oriented investigations toward a role in triglyceride-rich lipoprotein (TRL) metabolism [2], apoC1 appears to have a wider impact on different lipoprotein classes, as reflected by its association with HDL (80%) and to a lesser extent with VLDL (20%) in normolipidemic plasma [3, 4]. Accordingly, a substantial amount of studies demonstrated that, beyond its impact on VLDL and chylomicron metabolism, apoC1 plays a key role in the regulation of HDL remodelling [5,6,7,8,9,10,11]. This wide range of effects together with a possible role in the modulation of inflammation makes the net impact of apoC1 on cardiometabolic risk difficult to figure out and apoC1 might be considered as pro-atherogenic or anti-atherogenic depending on the overall metabolic context. Investigations of the functions of apoC1 in animal models can somehow be complex, since most rodent models substantially differ from humans concerning several features of lipoprotein metabolism, especially the absence of active CETP, i.e. one of the main target of apoC1 in humans [12,13,14]. Genetic studies in humans are also hampered by the fact that apoC1 gene polymorphisms are tightly linked to apolipoprotein E variants [15,16,17,18]. In addition, making the link between total plasma apoC1 levels and the risk of cardio-metabolic diseases is difficult due to the high exchangeability of this small protein whose biological effects might depend essentially on its association with VLDL or HDL. In this review we will present data on apoC1 structure and distribution among lipoproteins, on the effects of apoC1 on VLDL metabolism and HDL metabolism and we will discuss the possible links between apoC1, atherosclerosis and diabetes.
ApoC1 structure and gene
The human apoC1 gene is located on chromosome 19 (locus 19q13.2) [1, 19] within a 45-kilobase cluster encompassing also the apoE, apoC1’, apoC2 and apoC4 genes (Fig. 1). It is located 4.3 kb [20,21,22] or 5.3 kb [19] downstream from the apoE gene, in the same transcriptional orientation. A copy of the apoC1 gene, apoC1’ is located 7.5 kb downstream from the apoC1 gene (Fig. 1). The apoC1 gene is 4653 base pairs long while the apoC1’ gene is 4387 base pairs long. Both genes contain 4 exons and 3 introns (Fig. 2). No mRNA product of the apoC1’ gene can be detected in any tissue, suggesting that it is a pseudogene. The similar structure and the proximity of apoC1 with apoE, C1’, C2 and C4 genes suggest that they are derived from a common ancestor gene, with a primordial apolipoprotein gene that was most likely very similar to current apoC1 [1].

Position of apoC1 gen on chromosome 19 within a cluster encompassing the apoE, apoC1’, apoC2 and apoC4 genes. Genes are shown in green boxes, with their respective length indicated below. Black arrows indicate gene orientation. The length of noncoding regions between genes are indicated in italic. ApoC1’ pseudogene is hatched in grey. Regulatory regions are shown in brown (ME 1 and ME2) and yellow boxes (HCR1 and HCR2). ME and HCR regions promote gene expression in macrophages and hepatocyte, respectively. Transcription factors involved in the regulation of APOC1 gene are shown in ovals, with activators in green and repressors in red. Kb kilobase, ME multienhancer, HCR hepatic control region, LXR liver X receptor, PPAR peroxisome proliferator-activated receptor, Znf, Zfp zinc finger protein

Schematic representation of apoC1. A Structure of the transcribed region of the apoC1 gene. Exons are shown as boxes numbered with Roman numerals. Bold numbers are exon lengths, in nucleotides. Italicized numbers are intron lengths. The total length is indicated at the 3’-end of the gene. Beginning (ATG) and end (Stop) of translation are shown with black arrows. Untranslated and translated exonic sequences are shown in brown and green, respectively. B Primary structure of apoC1. Basic aminoacid residues are highlighted in dark blue for lysine and light blue for arginine. A lysine-rich cluster spans from residues 48 to 54. The vertical arrow represents the described possible site of truncation of apoC1 [35]. C Conformation of apoC1 with 2 α-helices according to [36] (permission validated by editor). The N-terminal helix spans from residues 7 to 29, the C-terminal helix from residue 38 to 52. The two helices are separated by an unstructured, hinge region. The conformation according to 2 or one α-helix might depend on the hydrophobicity of the environment. The blue frame delineates the region associated with the CETP-inhibitory effect. D Conformation of apoC1 with one α-helix with two possible dimeric associations according to [37] (permission validated by editor). Positively charged residues are shown in blue; negatively charged residues are shown in red. Hydrophobic clusters resulting from dimerization are visible as white/grey patches
The regulation apoC1 gene expression is under the control of regulatory elements located throughout the ApoE/C1/C1’/C2/C4 cluster. These include, among others, two copies of hepatic control regions (HCR-1 and HCR-2) and two copies of multienhancers (ME-1 and ME-2) (Fig. 1). While HCR sequences are required for the expression of apoC1 gene in hepatocytes [23], ME-1 and ME-2 play a significant role in stimulating its expression in macrophages through the binding of the Liver X Receptor (LXR) via LXR response elements [24]. On the opposite, a Peroxisome Proliferator-activated Receptor (PPAR) gamma response element located the intergenic region between apoE and apoC1 was shown to be involved in a negative effect of glitazones on apoC1 gene expression in hepatocytes [25, 26]. Finally, apoC1 gene expression was also shown to be downregulated by zinc-finger proteins Znf202 and Zfp125 which play a role in the regulation of genes involved in HDL metabolism and hepatic cholesterol /bile acid elimination on the one hand, and hepatic steatosis and VLDL secretion on the other hand [27, 28]. Importantly, all cis- and trans- regulatory elements mostly exert their effects not only on the apoC1 gene alone but on the whole ApoE/C1/C1’/C2/C4 gene cluster [23, 24, 26,27,28]. Overall, although apoC1 gene is expressed in many tissues such as lung, skin, testes, spleen, and adipose tissue, the main sites for its expression are the liver and differentiated macrophages [19, 24, 29, 30]. In addition to regulation of apoC1 gene expression at the transcriptional level, the secretion of apoC1 protein can also be modulated post-transcriptionally, with a stimulatory effect in the context of increased cholesterol pool, while cellular triglyceride content had no effect [31].
ApoC1 is synthesized with a 26-residue signal peptide, which is cleaved cotranslationally in the rough endoplasmic reticulum [32]. The mature protein encompasses 57 amino acids for a total molecular weight of 6613 Da, which makes apoC1 the smallest of all circulating apolipoproteins [33, 34] (Fig. 2). In addition, due to its high relative content in basic amino acid residues (7 lysine, 3 arginine), apoC1 is the apolipoprotein with the highest isoelectric point (pH(I) = 8.3) [33, 34]. On the opposite, apoC1 contains no histidine, tyrosine, or cysteine residues (for complete primary structure, see Fig. 2). A truncated form of apoC1, devoid of the N-terminal threonine and proline residues could also be detected in plasma and might result from the action of plasma dipeptidyl peptidase IV [35]. While initial nuclear magnetic resonance (NMR) studies indicated that apoC1 is composed of two amphipathic α-helices (residues 7–29 and 38–52) [36] more recent investigations using X-ray diffraction on apoC1 crystals suggest that the whole molecule can form a single α-helix [37] (Fig. 2). The conformation according to 2 or one α-helix might depend on the hydrophobicity of the environment [36, 37]. The amphipathic properties are important for the affinity of apoC1 for different lipoprotein classes [36, 38, 39] and might also influence its exchangeability between them [40]. Interestingly, apoC1 proteins have the ability to form dimers, or to fold their two α-helices upon one another [37, 41]. Mature apoC1 is devoid of glycosylation and, due to the absence of cysteine residues, of disulphide bonds.
Plasma levels and distribution of apoC1 between lipoproteins
The serum concentration of apoC1 is about 60 mg/L in normolipidemic subjects [3, 13, 42,43,44]. ApoC1 is mainly measured by enzyme-linked immunosorbent assay (ELISA) [13, 45, 46] but the use of mass spectrometry is becoming more and more widespread [47]. ApoC1 is mainly associated with HDL and VLDL in humans [4, 48, 49]. Methodological reports raised the possibility that small, exchangeable apolipoproteins, such as apoC1 [2, 3], might undergo shedding or redistribution between lipoprotein classes especially during extensive ultracentrifugation procedures [50]. Pioneering studies reported that up to 14% of apolipoproteins C are shed from VLDL after 2 ultracentrifugation runs [51], and the recovery of labeled apoC1 in the lipoprotein-free fraction after ultracentrifugation (less than 10% of total pool) was reported in kinetic studies [4].
In normolipidemic subjects, the majority of apoC1 is associated with HDL (80% to 95%) [3, 4]. Additionally, substantial amounts of apoC1 could also be found in chylomicrons in the post-prandial phase [52, 53].
In hyperlipidemic individuals, several groups reported increased plasma levels of apoC1, especially in the context of hypertriglyceridemia or mixed hyperlipidemia, with values reaching 200 mg/L in some studies [3, 43, 45]. However, this phenotype was not always observed [13]. Hypertriglyceridemia or mixed hyperlipidemia, were also associated with alterations in the distribution of apoC1 between lipoproteins, i.e. an enrichment in VLDL at the expense of HDL. Indeed, 49% of apoC1 is associated with VLDL in subjects with hypertriglyceridemia [3] and 43% in subjects with combined hyperlipidemia (high level of triglycerides and LDL cholesterol), whereas the relative amounts of apoC1 in HDL decreased (respectively 51% and 57%) [3]. This relative enrichment of VLDL during hypertriglyceridemia has been proposed to result from 1) increased hepatic apoC1 production coupled with enhanced VLDL secretion and/or 2) redistribution from HDL to VLDL in the circulation [3] since apoC1 has been demonstrated to be a highly exchangeable protein [4, 40].
Role of apoC1 in the VLDL and chylomicron metabolism
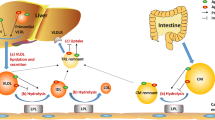
Pioneering in vitro and ex vivo studies demonstrated that apoC1 impairs the clearance of triglyceride-rich lipoproteins (TRLs). It was shown, in vitro, in isolated-perfused rat liver models, that adding human apoC1 to chylomicrons [54] or triglycerides (TG) emulsions [55] led to the inhibition of their hepatic uptake. It was then shown that apoC1 inhibited the apoE-dependent binding of β-VLDL to LRP [56, 57]. It was suggested that this inhibiting action of ApoC1 on the binding of lipoproteins to LRP was related to the displacement of apoE from the lipoproteins since apoC1 peptides were able to displace significant quantities of apoE from β-VLDL and to inhibit the binding of β-VLDL to LRP [58]. In addition, apoC1 could impair the apoE-mediated binding of VLDL to the LDL-R, either by masking the apoE, or by changing the conformation of apoE [59, 60]. Altogether, these studies led to the conclusion that apoC1 inhibited hepatic uptake of TRLs via the impaired binding of these lipoproteins either to LDL-R or to LRP by impairing their interaction via apoE.
The inhibitory effect of apoC1 on TRL clearance could be confirmed in vivo in different transgenic mouse models overexpressing human apoC1. All of these transgenic mice displayed an increase in their plasma triglyceride levels and to a lesser extent an increase in their plasma levels of total cholesterol [61,62,63]. One study showed that the increase in plasma triglyceride levels occurred in a dose-dependent manner according to the expression of the apoC1 transgene [64]. This hypertriglyceridemic phenotype was related to a considerable increase in VLDL levels [63, 65]. It was then shown in VLDL turnover studies that the clearance of VLDL from the circulation was less efficient in apoC1 transgenic mice as compared with control mice. Hepatic VLDL production and extrahepatic lipolysis was not different between mice overexpressing human apoC1 and control mice [65]. These data suggest that the hypertriglyceridemia in apoC1 transgenic mice results primarily from impaired hepatic VLDL particle uptake, in line with the in vitro observations. However, the inhibition of VLDL uptake in mice overexpressing human apoC1 was also observed in the context of LDL-R deficiency, suggesting that apoC1 inhibition of VLDL uptake involves another pathway than LDL-R, probably LRP [65]. In addition, it was shown that the overexpression of the VLDL-R in apoC1 transgenic mice had no effect on the hyperlipidemia [66]. This suggests that apoC1 also inhibits the binding of lipoproteins to the VLDL-R, which was also supported by in vitro studies [66].
Inhibition of the apoE-mediated hepatic uptake of VLDL is not the sole mechanism accounting for the hypertriglyceridemic effect of apoC1 overexpression since apoC1 transgenic mice also develop hypertriglyceridemia in an apoE-deficient context [8, 67]. Reduced lipoprotein lipase (LPL) activity might account for this effect since it was shown in vitro that apoC1 could inhibit LPL [68]. This inhibitory effect of apoC1 on LPL activity was confirmed in vivo in transgenic mice models [67] and was shown to be independent from apoC3 (physiological inhibitor of LPL) and VLDL-R (known modulator of LPL) [69]. The role of apoC1 in inhibiting LPL activity was further confirmed in humans in 28 subjects with kidney failure and on haemodialysis [70]. In this population, the dialysis procedure led to apoC1 depletion in VLDL, making them better substrates for LPL ex vivo. Accordingly, the apoC1 content of VLDL was shown to correlate negatively with LPL activity in 81 aged volunteers [71]. Finally, one study conducted in mice deficient for apoE and expressing apoC1 at physiological levels found that apoC1 increased the production rate of hepatic VLDL-TG and VLDL-apoB [64]. This observation is consistent with increased production rate of apoC1 in hypertriglyceridemic individuals [3], although the causal link between both phenomena was not investigated in this clinical study.
Overall, apoC1 affects VLDL metabolism (Fig. 3) as shown by the marked increase in plasma TG levels and the lesser increase in plasma levels of total cholesterol observed in transgenic mice expressing human apoC1. Many data suggest that apoC1 could directly or indirectly inhibit LPL activity, leading to a decrease on the clearance of VLDL. Interestingly, these effects are very similar to the widely documented impact of another small apolipoprotein which is present in high amounts in TRL, apoC3, on TG levels. This might suggest that both proteins could have overlapping functions (see Table 1). In addition, this lipid phenotype could be explained by the inhibitory effect of apoC1 on hepatic receptors LDL-R and LRP, which leads to a decrease in the hepatic uptake of VLDL. However, it should be borne in mind that LDL-R and LRP- mediated uptake is far from being the main pathway for VLDL clearance in humans. In addition, these effects on hepatic receptors have been shown only in animal models expressing apoC1 at supra-physiological levels. Furthermore, one study in a knock-out mouse model on a high-fat, high-cholesterol diet showed that apoC1 deficiency also led to decreased fractional catabolic rate of VLDL [72] while a recent study using transgenic rabbits expressing human apoC1 failed to observe an increase in plasma TG levels on a western diet [49]. These latter data further demonstrate that the impact of apoC1 on TRL metabolism is more complex than that of apoC3 which was consistently shown to exert hypertriglyceridemic effects [73]. Additional kinetic studies in human populations presenting with various clinical conditions are required to get a comprehensive view of the mechanisms involved in triglyceride homeostasis which are modulated by apoC1 in our species.

Influence of apoC1 on lipoprotein metabolism. ApoC1 acts on the metabolism of triglyceride-rich lipoproteins, leading to an increase in plasma TG levels by promoting TRL secretion by and inhibiting TG hydrolysis as well as TRL remnant clearance. ApoC1 also acts on the metabolism of HDL by favouring HDL maturation and by limiting the net loss of cholesteryl esters toward VLDL and LDL. (1) ApoC1 inhibits LPL activity. (2) ApoC1 inhibits the binding of triglyceride-rich lipoproteins to LDL-R. (3) ApoC1 inhibits the binding of triglyceride-rich lipoproteins to LRP. (4) ApoC1 inhibits the binding of triglyceride-rich lipoproteins to VLDL-R. (5) ApoC1 stimulates the hepatic production of VLDL. (6) ApoC1 is the physiological inhibitor of CETP. (7) ApoC1 stimulates LCAT activity. (8) ApoC1 inhibits SR-BI. ABCA1 ATP-binding cassette A1, ABCG1 ATP-binding cassette G1, CETP cholesteryl ester transfer protein, HDL high density lipoprotein, HL hepatic lipase, IDL intermediate density lipoprotein, LCAT lecithin-cholesterol acyltransferase, LDL low density lipoprotein, LDL-R LDL receptor, LPL lipoprotein lipase, LRP LDLR related protein, PLTP phospholipid transfer protein, VLDL very low density lipoprotein, VLDL-R VLDL receptor
Role of ApoC1 in HDL metabolism
In vitro studies have shown that apoC1 could play a role in HDL metabolism by modulating several plasma proteins and cellular receptors involved in the remodelling of these lipoproteins. Indeed, apoC1 partially activates lecithin-cholesterol acyltransferase (LCAT) [5, 6] which is involved in the maturation of HDL into spherical particles rich in esterified [74, 75]. This effect of apoC1 is opposite to that of apoC3 which inhibits LCAT activity (Table 1) [76]. Conversely, apoC1 is able to inhibit phospholipase A2 in vitro [7] which is responsible for increased HDL catabolism. However, none of these observations could be confirmed in vivo.
ApoC1 inhibits also hepatic lipase [77] which mediates triglyceride hydrolysis of cholesterol-rich lipoproteins, including HDL. Conde-Knape et al. [8] confirmed the modulating effect of apoC1 on hepatic lipase in vitro but only when apoC1 was located on HDL, and not on the surface of VLDL. In addition, as apoC2 and apoC3 were shown to inhibit hepatic uptake of cholesterol esters of HDL by the scavenger receptor B1 (SR-B1) [78], the effect of apoC1 on SR-B1 was also studied in vitro and in vivo in mice expressing high levels of apoC1 or in mice lacking apoC1. It was shown that apoC1 substantially diminished the uptake of HDL-bound cholesterol esters via SR-B1 [9].
Overall, these various effects of apoC1 on HDL metabolism (Fig. 3) suggest that the presence of apoC1 would lead to an increase in HDL-cholesterol levels, possibly by reduction of HDL catabolism. This could be confirmed in genetically engineered mice, with significantly lower HDL-C in apoC1 knock-out mice [9, 11] and increased HDL-C in mice overexpressing apoC1 [9, 10]. Once again, these effects should be confirmed in humans but clinical studies are lacking in this respect. Furthermore, studying the impact of apoC1 on HDL metabolism in mice suffers a major drawback since this species, unlike humans, is devoid of CETP which plays a major role in HDL metabolism by promoting the exchange of neutral lipids between HDL and triglyceride-rich lipoproteins. Importantly, apoC1 plays a major role in CETP activity, as discussed below.
ApoC1, a physiological regulator of CETP
CETP is a lipoprotein-associated glycoprotein that enables the exchange of cholesterol esters (CE) and TG between HDL and TRLs [79, 80]. In vitro studies [81], observations in CETP transgenic mouse models [82] and clinical studies with genetic variants of CETP [83, 84] or treated with CETP inhibitors [85] consistently demonstrated that CETP promotes the net cholesterol enrichment of pro-atherogenic apoB-containing lipoproteins (VLDL and LDL) in parallel with a decrease in the cholesterol content of atheroprotective HDL. This pejorative effect of CETP on plasma lipoprotein profile raised the hypothesis that CETP might represent a relevant target to prevent cardiovascular diseases [86]. However, the pharmacological inhibitors of CETP that were tested so far in large clinical trials led to modest cardiovascular protection despite dramatic reductions in CETP activity [87].
Beside pharmacological compounds, the presence of an endogenous inhibitor of CETP in plasma has long been suspected. In vitro studies showed that CETP has a greater affinity for HDL than for LDL [88] and it could be demonstrated that, while LDL dose-dependently increases CETP activity, HDL exert an inhibitory effect at high doses [89]. These data suggest that a CETP inhibitory factor is associated with HDL. Although some studies found in vitro that apoA1 and apoA2 played a role in this inhibitory effect of HDL, others found a neutral or even an activating effect of apoAs on cholesteryl esters [90,91,92,93,94,95,96]. In vitro experiments comparing the modulatory effect of different purified apolipoproteins showed that apoC1 had the strongest inhibitory effect on CETP activity [97,98,99]. In comparison, apoC3 does not inhibit CETP in vitro [97]. These observations were confirmed in an exhaustive screening of HDL apolipoproteins allowed us to demonstrate that apoC1 was the main contributor to the inhibition of CETP when present as a component of HDL [95]. Furthermore, isolated apoC1 was found to decrease CETP activity in a concentration-dependant manner [95] while the effect of other apolipoproteins on CETP varied depending on the dose used [92]. The mechanisms involved in the inhibitory effect of apoC1 on CETP were investigated in vitro. We could show that the presence of apoC1 decreases the electronegativity of HDL, leading to a dissociation of HDL-CETP complexes [100]. This observation confirms the key role of surface potential in the interaction of CETP with lipoproteins [101, 102]. More into details, our group could demonstrate that this alteration of negative surface charge of HDL could be attributed to the presence of a positively charged, lysine-rich cluster in the C-terminal α helix of human apoC1, and blocking these amino-acid residues through chemically induced acetylation was associated with a significant decrease in the inhibitory effect of apoC1 on CETP [100]. Interestingly, rabbit apoC1, which unlike human or mouse apoC1 does not modify the electrostatic charge of HDL, is unable to inhibit CETP [12]. These studies thus showed that the electrostatic charge of apoC1 plays a major role in its ability to inhibit CETP.
The physiological relevance of CETP inhibition by apoC1 was first confirmed in vivo in transgenic mice expressing human CETP (CETP-Tg) crossed either with apoC1-knocked out mice (apoC1-KO mice), or with mice overexpressing human apoC1 (apoC1-Tg mice). In CETP-Tg/apoC1-KO mice, we observed that specific CETP activity was dramatically increased when compared with CETP-Tg mice. This was accompanied with significant decreases in the CE content and in the CE-to-TG ratio of HDL, in line with enhanced CETP-mediated neutral lipid exchanges between HDL and apoB-containing lipoproteins in the absence of apoC1 [103]. Ex vivo, the HDL of CETP-Tg/apoC1-KO mice interacted more readily with purified CETP than did HDL of CETP-Tg mice, even though these HDL only differed by their apoC1 content [103]. On the opposite, CETP-Tg-apoC1-Tg mice presented a twofold decrease in specific CETP activity compared with CETP-Tg mice [10]. More recently, we developed transgenic rabbits expressing human apoC1 which also helped to demonstrate that the presence of human apoC1 at physiological levels could rescue the lack of inhibitory potential of rabbit apoC1 against CETP, leading once again to an increase in plasma HDL-cholesterol [49].
The inhibitory effect of apoC1 on CETP activity was also confirmed in humans. Indeed, we observed significant negative correlations between plasma levels of apoC1 and CETP specific activity in three cohorts of normolipidemic subjects [12,13,14]. Although the correlation was less marked in patients with coronary artery disease (CAD) and treated with statins, apoC1 concentration was also positively correlated with the HDL to LDL-cholesterol ratio, suggesting once again that apoC1 might have a positive impact on plasma cholesterol profile through CETP inhibition [13]. However, this negative correlation between apoC1 levels and CETP activity was lost in CAD patients with hypertriglyceridemia or combined hyperlipidemia [13].
The presence of high amounts of TRLs might explain the loss of inhibitory potential of apoC1 in these dyslipidemic patients. Firstly, the abundance of triglyceride-rich lipoproteins, which are preferential acceptors of cholesterol esters from HDL [104, 105], might further stimulate the CETP-mediated cholesterol ester transport and thus overcome the inhibitory effect of apoC1 on CETP. Secondly, as mentioned above (see “Plasma levels and distribution of apoC1 between lipoproteins” Section), apoC1 is a highly exchangeable protein [4, 40] and the accumulation of VLDL in the bloodstream may lead to its preferential association with this lipoprotein class at the expense of HDL [3]. Importantly, we could show that apoC1 does not inhibit CETP when bound to VLDL [70]. Therefore, the decrease in the relative amounts of functional apoC1 present in the HDL might also explain the loss of negative correlation between apoC1 and CETP activity in subjects with high triglyceride levels. The fact that plasma triglyceride levels correlated positively with apoC1 bound to VLDL (i.e. apoC1 that cannot inhibit CETP), in a sub-group of healthy subjects reinforces the idea that the inhibitory of apoC1 is impaired when TRL accumulate [13].
ApoC1 in diabetes
Plasma apoC1 levels are higher in type 1 and type 2 diabetic patients than in control subjects and we observed a positive correlation with plasma TG [14, 106, 107]. In the context of diabetes, apoC1 would thus appear as positively associated with some cardiometabolic risk factors. However, the causal relationship between high apoC1 levels and diabetes is questionable. In vivo studies in transgenic mice suggest that apoC1 overexpression protects against insulin resistance through reduced fat storage despite elevated plasma TG [108]. On the opposite, apoC3 appears as pro-diabetic by triggering pancreatic beta-cell apoptosis and by promoting insulin resistance via Toll-Like Receptor 2 (TLR2) activation in transgenic mouse models (Table 1) [109, 110]. In men with metabolic syndrome, elevated apoC1 levels were also associated with higher TG levels but lower visceral fat mass [111]. In a study conducted in a small cohort of subjects of American Indian or Mexican ancestry, a structural polymorphism of apoC1, the T45S variant, which is associated with a higher propensity of the protein to undergo N-terminal truncation, was found to be associated with higher prevalence of diabetes secondarily to the onset of obesity [112]. In a 5-year prospective study, although plasma levels of apoC1 correlated positively with a high risk of T2D, this link was lost after adjusting for TG levels [113]. The only genetic study which investigated a most common apoC1 gene polymorphism (rs11568822) in relation with diabetes failed to find a correlation [114]. These observations suggest that apoC1 itself is protective or neutral regarding diabetes risk, and that high apoC1 levels in diabetic subjects might rather be in relation with altered TG metabolism, i.e. increased production and/or lower catabolism of TRLs. Although apoC1, through is involvement in TRL clearance, might influence fat content in different tissues [11, 115], we observed an association between apoC1 plasma levels neither with adipose tissue areas or distribution (visceral or subcutaneous) nor with steatosis in patients with type 1 or type 2 diabetes, as the correlation between apoC1 and liver fat content disappeared after adjustment for triglyceride-level [106, 107]. High apoC1 levels in diabetic patients thus appear to be a consequence rather than a cause of impaired TG metabolism and fat storage in diabetic patients, but the precise mechanisms involved remain poorly understood. The elevation of plasma TG levels in most diabetic patients might explain only in part this phenomenon since our study in type 1 diabetics showed significantly elevated apoC1 despite TG levels similar to that of control subjects [107].
A loss of the negative correlation between apoC1 and CETP activity was observed in patients with type 1 or type 2 diabetes [14], similarly to what was reported CAD patients with hyperlipidemia. More strikingly, we also reported that CETP activity in both type 1 and type 2 diabetics was higher than that in controls (in agreement with the literature [116,117,118,119,120] despite higher apoC1 levels [14]. This reinforces the notion that apoC1 is dysfunctional in this population. Furthermore, CETP activity was significantly higher in type 2 diabetic patients than in type 1 diabetic patients [14]. This difference can be explained in part by TG levels, which directly stimulate CETP activity [104, 105] and which were 13% and 170% higher in type 1 and type 2 diabetic patients, respectively, than in controls. However, in a subgroup of type 1 diabetics with normal TG levels (below 1.7 mmol/L) CETP activity was still significantly higher than that in controls although TG levels in these 2 groups were not statistically different. In addition, in these type 1 diabetic patients with normal TG levels, there was no correlation between CETP activity and triglyceride levels. Therefore, the greater ability of CETP to transfer cholesterol esters in type 1 diabetic patients is probably related to mechanisms other than hypertriglyceridemia. Hyperglycaemia, which correlated with CETP activity in type 1 diabetic patients, arise as a possible explanation. It is proposed that glycation phenomena plays a key role in the loss of the inhibitory potential of apoC1 in diabetic patients. The presence of apoC1 with glycated lysine residues was reported in hyperglycemic diabetic subjects [121]. In vitro glycation of apoC1 was shown to modify its electrostatic properties, leading to a decrease in its ability to inhibit CETP activity [14]. Accordingly, we could confirm in vivo that the electrostatic properties of apoC1 were modified in type 1 diabetic patients [14]. These results are in agreement with the in vitro studies in which acetylation of lysine residues of apoC1 was accompanied by a reduction in its electropositive charge and by a weakening of its ability to inhibit CETP-induced cholesterol ester transport [100].
A meta-analysis reported an association between a polymorphism of apoC1 (rs4420638) and the risk of developing diabetic nephropathy [122]. In mice overexpressing apoC1, albuminuria and glomerulosclerosis with increased numbers of glomerular M1 macrophages were observed while no renal abnormalities were reported in wild-type mice [123]. In the clinical part of the same study, apoC1 was detected only in the kidneys from autopsied diabetic subjects and not in control subjects and the amount of apoC1 in glomeruli was significantly higher in patients with diabetic nephropathy than in those without diabetic nephropathy [123]. The authors suggest that apoC1 might play a local role in the development of diabetic nephropathy, probably by increasing macrophage activation in renal tissue.
Overall, diabetic states not only lead to elevated apoC1 levels, but also to the generation of structurally modified and dysfunctional apoC1. Beyond the use of apoC1 level as a new marker of diabetes severity, the restoration of apoC1 functionality might help to counteract the deleterious effects of increased CETP activity on plasma lipoprotein profile in this population at high cardiovascular risk.
ApoC1 and atherosclerosis
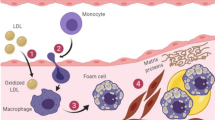
Atherosclerosis is a complex, multifactorial disease which involves disturbed lipoprotein metabolism, inflammation, and alterations in the function of numerous cell types present in the vascular wall including endothelial cells, smooth muscle cells (VSMCs) or myeloid cells such as macrophages. ApoC1 is involved not only in many aspects of lipoprotein metabolism, but also in the regulation of inflammatory response and in the biology of some cell components of vascular wall (Fig. 4), making its net impact on atherosclerosis difficult to anticipate at first glance.

Pro-atherogenic and atheroprotective effects of apoC1.The biochemical and cellular effects of apoC1 were classified as dependent or not on the presence of CETP. The experimental conditions (in vitro experiments, animal models) or the clinical context (for studies in human subjects) in which the effects were observed are shown on the first two rows. LPS lipopolysaccharide, VCSMs vascular smooth muscle cells, TRL, triglycerides rich lipoproteins, LCAT lecithin cholesterol acyl transferase, PON-1 paraoxonase-1, CETP cholesteryl ester transfer protein, CVD cardiovascular disease
Several observations indicate that apoC1 is expressed by different cell types involved in the development of atherosclerotic plaque. The expression of the apoC1 gene was compared in the atheroma lesions of 10 subjects who underwent carotid or femoral endarterectomy and in healthy arteries harvested post mortem [124]. mRNA levels of apoC1 were higher in the carotid and femoral atherosclerotic lesions than in healthy arteries. Macrophages are involved in the initiation and the development of atherosclerotic lesions in vivo [125]. ApoC1 is highly expressed in macrophages, where its expression is tightly regulated by LXR which is deeply involved in the maintenance of cellular lipid homeostasis [24]. It was shown in vitro that human apoC1 increased cholesterol efflux from macrophages thanks to its α-helix conformation [126, 127], and could prevent cholesterol accumulation in macrophages exposed to modified LDL [128]. However, when bone marrow transplants from apoC1-KO or wild-type mice to apoE-KO mice were conducted to assess the specific effect of macrophage-derived apoC1 on the development of atherosclerosis, no difference was found for plasma either lipid levels or atherosclerotic lesions [128]. This indicates that apoC1 in macrophages is not likely to play a major role in the development of atherosclerotic plaques in vivo, despite its promoting effect on cholesterol efflux demonstrated in vitro. Beside macrophages, some studies reported a role of apoC1 in VSMC biology. It could be shown that apoC1 can induce apoptosis in cultured VSMCs via a mechanism involving neutral sphingomyelinase-ceramide pathway [129], and that apoC1 co-localized with markers of apoptosis (ceramide, caspase-3) in neo-intimal dissections in rabbit models of atherosclerosis [130]. These results suggest that at the local level, apoC1, like apoC3 (Table 1), could be involved in complications of the atherosclerotic process.
Inflammation plays a prominent role from initiation processes to the development of complications of atherosclerosis, as illustrated by the success of anti-inflammatory therapies to reduce major adverse cardiovascular events in high-risk patients [131, 132]. Inflammatory stimuli involved in atherosclerosis are very diverse and include, among others, oxidized lipoproteins and modified lipids but also damage- and pathogen- associated molecular patterns (DAMPS and PAMPS, respectively) [133]. The pro-apoptotic effect of apoC1 on VSMCs mentioned above can contribute to the generation of DAMPs from dying cells. Next to DAMPs, bacterial lipopolysaccharides (LPS, endotoxins) are major PAMPs triggering innate immune response. The presence of bacterial LPS in the circulation has long been known to aggravate atherosclerosis [134, 135]. In addition, LPS could also be found in atherosclerotic plaques from endarterectomy samples collected from patients with high cardiovascular risk, but not in healthy arterial wall [136,137,138]. ApoC1 was shown to interact with circulating LPS. Indeed, apoC1 can directly bind LPS via its α-helices and facilitates LPS presentation to innate immune receptors at the surface of macrophages, thereby enhancing inflammatory response [139]. Although this effect might be protective against infection and sepsis [140, 141], the presence of apoC1 was shown to enhance atherosclerotic lesion size and inflammatory status of the plaque in a genetically engineered mice where atherosclerosis was induced by repeated LPS injections [142]. ApoC3 was also reported to exert pro-inflammatory effects on components of the vascular wall, but via distinct mechanisms (Table 1).
The impact of apoC1 on atherosclerosis via its effect on plasma lipid metabolism was investigated in mice and rabbits. In two years old apoE-deficient mice expressing or not endogenous apoC1 [128], apoC1 expression was accompanied by a 70% increase in triglycerides levels and a 30% increase in cholesterol levels which were essentially related to an increase in triglycerides and cholesterol in the VLDL. Atherosclerotic lesions in mice expressing apoC1 were 87% greater than in the other mice. The macrophage content of the atherosclerotic lesions was not different between the 2 populations of mice. Data from this study suggest that systemic apoC1 aggravates atherosclerosis, probably via the accumulation of TRLs, since apoC1 specifically derived from macrophages had no effect on the pathology [128]. However, studying the lipid mediated effects of apoC1 on atherosclerosis in mice is hampered by the absence of CETP in this species, which leads to overlooking the possible impact of apoC1 on CETP-mediated remodelling of HDL as occurs in humans. ApoC1 and its effect on atherosclerosis were studied in rabbits by our group more recently. Rabbits indeed appear to be a better model than mice for the study of atherosclerotic effects of apoC1, because (1) rabbits fed with cholesterol-rich diet spontaneously develop atherosclerotic lesions, as humans but not as mice [143] (2) plasma lipid profile of rabbits is closer to humans than the one of mice and (3) rabbits, like humans, express endogenous, active CETP [144]. In this study, wild type rabbits were compared with transgenic rabbits expressing human apoC1 (HuApoC1-Tg rabbits), in terms of atherosclerosis, plasma lipid and lipoprotein profiles [49]. After 8 weeks of cholesterol-rich diet, all rabbits experienced a massive increase of plasma cholesterol levels in apoB-containing lipoproteins, without any significant difference in hypercholesterolemia between the two groups of rabbits. Of note, plasma TG levels did not differ either between both groups while HDL-cholesterol was higher in HuApoC1-Tg rabbits than in wild-type animals. This increase was associated with an impairment of HDL to serve as substrates for CETP in HuApoC1 rabbits. Human apoC1 prevented extension of aortic atherosclerotic lesions in rabbits fed with cholesterol-rich diet while, on the opposite, CETP activity was found to be correlated positively with the extent of atherosclerosis. This protective effect of human apoC1 was not due to decreased inflammatory status or increased cholesterol efflux from macrophages but was associated with a decrease in markers of lipid oxidation in plasma. This study suggests that, when CETP is expressed as in humans, moderate elevation of plasma apoC1 level leads to increased levels of atheroprotective HDL. This atheroprotective mechanism is a direct consequence of CETP inhibition and is mostly operative in the absence of concomitant, potentially deleterious changes in TRL metabolism. From this respect, apoC1 drastically differs from apoC3 which, in addition to its hypertriglyceridemic effect, has a pejorative impact on HDL functions such as cholesterol efflux and protection against apoptosis or oxidation (Table 1).
In humans, the net impact of apoC1 on atherosclerosis was assessed in several genetic studies. The most common polymorphism at the apoC1 gene (HpaI, rs11568822) consists in a 4 base pair insertion/deletion located in the proximal promoter and resulting in a high and low gene expression in vitro, respectively [145]. The insertion variant was found to be associated with metabolically unhealthy phenotypes in obese people or in people with normal weight [146] as well as with an elevation of triglyceride levels [114, 147] thereby consolidating the possible impact of high apoC1 levels on TRL metabolism in humans. However, none of these studies could find an association between this polymorphism and coronary artery disease or myocardial infarction. In a prospective study in more than 2767 subjects, the analysis of additional polymorphisms at the apoC1 gene evidenced a slight effect on coronary heart disease [15]. Additional polymorphisms located in the ApoE-apoC1 locus were also identified as possibly linked with altered plasma lipid levels or atherogenic risk by different bioinformatics approaches including genome wide association studies (GWAS) or protein network analysis [16, 148, 149]. However, these studies underlie the fact that these apoC1 polymorphisms are in strong linkage disequilibrium with apoE variants which could explain the majority of cardiovascular disease (CVD) risk [18], thus questioning the real impact of apoC1 polymorphisms per se on atherosclerosis [15,16,17].
The possible association between circulating apoC1 and cardiovascular risk was also explored in humans. A first study was conducted in patients with metabolic syndrome and demonstrated that high plasma levels of apoC1 were associated with increased carotid wall thickness only in the subgroup presenting with systemic inflammation (hsCRP > 3 mg/L) [150]. A second group set up a prospective study in patients with heart failure and measured plasma apoC1 using LC-MRM-MS at inclusion before a 3 year follow-up. Higher levels of plasma apoC1 were found in patients who died from cardiovascular diseases when compared to those who did not. This difference was observed whereas triglycerides, total cholesterol, HDL cholesterol and LDL cholesterol levels after three years of follow-up were similar between the two groups [47]. These pejorative correlations between plasma apoC1 and atherosclerosis or cardiovascular death were observed in contexts that are critical for atherosclerosis and cardiovascular events, i.e. metabolic syndrome and inflammation on the one hand and heart failure on the other hand. Interestingly, the presence of structurally altered forms of apoC1 was reported after MALDI-TOF analyses on serum from patients with CAD. One study showed indeed that CAD patient displayed a truncated form of apoC1 which appeared to be more susceptible for oxidative modifications [151]. Accordingly, another group also observed the presence of oxidized apoC1 in CAD patients but not in healthy control subjects [152]. Whether these structural modifications contribute to, or are a consequence of atherosclerosis is not known but, in a similar manner to what was observed with glycation phenomena in diabetic subjects [14], they might be associated with alterations of apoC1 functionality. In line with this latter hypothesis, it could be shown that the negative correlation between apoC1 and CETP activity was less marked in CAD patients than in healthy subjects [13].
Postprandial elevation of triglycerides is frequently observed in normolipidemic coronary heart disease patients. Some postprandial disturbances of lipid metabolism may thus serve as early markers of cardiovascular risk. It was shown that the lipid and apolipoprotein composition of VLDL was modified in the postprandial period. In normolipidemic male subjects with CAD, it was shown that the apoC1 content of VLDL was 50 to 100% higher than that in control subjects in the postprandial period [52]. The number and the composition of VLDL and of chylomicron remnants in the postprandial period were further studied in thirty 50-year-old normolipidemic men with no cardiovascular history [153]. The subjects with early signs of carotid atherosclerosis (increased intima-media thickness, IMT) did not show greater postprandial response, but they presented twofold increased levels of apoC1 in the chylomicron remnants and in VLDL when compared with the subjects with normal intima-media thickness [153]. Furthermore, the apoC1 content of postprandial TRL was independently correlated with IMT in a population of middle aged men which had all an apoE3/E3 genotype [154]. However, other studies evidenced a higher enrichment of apoC1 in VLDL in patients with carotid atherosclerosis compared to controls [155] and a positive correlation between apoC1 enrichment of VLDL and carotid plaque area [71] for VLDL analysed in the fasting state, but not in the postprandial phase. Although the relative contribution of remnants of the postprandial phase and fasting VLDL is still unclear, these data indicate that apoC1 content of TRLs could be an early marker of cardiovascular risk. These observations further support tight connection between apoC1 and TRLs although the causative relationship between apoC1 enrichment of TRL and elevated TG levels still needs to be explored. Abnormalities of apoC1 distribution during hypertriglyceridemic states, with preferential association with TRLs [71], could once again be linked with alterations of apoC1 functionality, redirecting apoC1 function towards inhibition of TG catabolism, thereby increasing CVD risk [156] at the expense of its inhibitory effect against CETP when present in the HDL [157]. Interestingly, it was shown in a prospective family-based study that plasma levels of apoC1 were positively associated with large HDL at birth and in infants, while it was correlated with TG levels, but not HDL, in adults [158]. It suggests that a progressive shift of apoC1 location and function occurs along life with an increase of the relative importance of its inhibitory effect on TG metabolism along the build-up of cardiometabolic risk.
Conclusion
Data available for apoC1 principally concern in vitro studies and transgenic animal models. Strikingly, it appears that the balance between protective and deleterious effects of apoC1 mostly rely on the presence of CETP in the experimental models (Fig. 4), as illustrated, among others by opposite effects human apoC1 on atherosclerosis between mice [64] and rabbits [49]. Beyond animals, the very limited amount of studies conducted in humans might also explain why the precise role of apoC1 on lipid metabolism and cardiovascular risk remains unclear. On the one side, several data indicate that apoC1 seems to be pro-atherogenic by increasing TG level which may be explained by the inhibition of binding to LDL-R, to LRP and to VLDL-R, the inhibition of LPL or the stimulation of VLDL production. On the other side, apoC1 associated with HDL seems to be anti-atherogenic in HDL metabolism by inhibiting CETP, when VLDL-bound apoC1 is unable to inhibit CETP. In addition, apoC1 loses its ability to inhibit CETP in a context of hyperlipidemia and diabetes.
We can hypothesize that apoC1 may have different functions according to its distribution between lipoproteins (with a pro-atherogenic effect when associated with VLDL and an anti-atherogenic effect when associated with HDL) and that qualitative changes of apoC1 occurring in clinical situations as diabetes and atherosclerosis may also modify its properties and expression pattern. In this aspect, apoC1 differs from another small, highly exchangeable apolipoprotein, i.e. apoC3. Indeed, although both proteins impair plasma TG clearance when bound to TRLs, their impact on HDL functionality is opposite: apoC3 exerts a negative effect and apoC1 a positive effect on the atheroprotective functions of HDL, including its inhibitory effect on CETP (see Table 1). Overall, while apoC1 harbors either beneficial or deleterious effects on lipoprotein metabolism and cardiometabolic diseases, apoC3 appears in most cases as a risk factor. Accordingly, the strong positive correlations between apoC3 levels or genetic variants with cardiovascular risk [159] led to the development of multiple anti-apoC3 therapeutic strategies [160]. In contrast, using apoC1 as a therapeutic target might be more complex.
Despite the possible beneficial use of apoC1 as a CETP inhibitor against atherosclerosis, strategies aiming at inducing very high plasma levels of apoC1 would not be relevant because of the possible hypertriglyceridemic effect [10]. Importantly however, it must be borne in mind that beneficial effects of apoC1 on atherosclerosis might be reached on the long term even when keeping its plasma levels within a physiological range as suggested by our studies in transgenic rabbits [46]. Alternatively, it is tempting to speculate that therapeutic interventions that could prevent the sequestration of apoC1 in TRLs or its structural modifications (truncation, oxidation, glycation) could improve its inhibitory function against CETP and restore, maintain or magnify its anti-atherogenic function.
Other studies are still necessary, particularly in humans, to determine the precise role of apoC1 lipoprotein metabolism but also for its possible pleiotropic effects on inflammation and vascular wall biology, in order to get a comprehensive view of its potential impact in the development of atherosclerosis.
Availability of data and materials
Not applicable.
Abbreviations
- ApoC1:
-
Apolipoprotein C1
- CAD:
-
Coronary artery disease
- CE:
-
Cholesteryl ester
- CETP:
-
Cholesterol Ester Transfer Protein
- CVD:
-
Cardiovascular disease
- DAMPs:
-
Damage- associated molecular patterns
- HDL:
-
High density lipoprotein
- HCR-1/HCR-2:
-
Hepatic control regions-1 and -2
- IMT:
-
Increased intima-media thickness
- LCAT:
-
Lecithin-cholesterol acyltransferase
- LDL:
-
Low density lipoprotein
- LDL-R:
-
Low density lipoprotein receptor
- LRP:
-
LDL receptor related protein
- LPL:
-
Lipoprotein lipase
- LPS:
-
Lipopolysaccharide
- LXR:
-
Liver X receptor
- ME-1 and ME-2:
-
Multienhancers 1 and 2
- PAMPs:
-
Pathogen-associated molecular patterns
- SR-B1:
-
Scavenger receptor B1
- TG:
-
Triglycerides
- TRL:
-
Triglyceride-rich lipoprotein
- VLDL:
-
Very low density lipoprotein
- VSMCs:
-
Vascular smooth muscle cells
References
Li WH, Tanimura M, Luo CC, Datta S, Chan L. The apolipoprotein multigene family: biosynthesis, structure, structure-function relationships, and evolution. J Lipid Res. 1988;29(3):245–71.
Shachter NS. Apolipoproteins C-I and C-III as important modulators of lipoprotein metabolism. Curr Opin Lipidol. 2001;12(3):297–304.
Cohn JS, Tremblay M, Batal R, Jacques H, Veilleux L, Rodriguez C, et al. Plasma kinetics of VLDL and HDL apoC-I in normolipidemic and hypertriglyceridemic subjects. J Lipid Res. 2002;43(10):1680–7.
Malmendier CL, Lontie JF, Grutman GA, Delcroix C. Metabolism of apolipoprotein C-I in normolipoproteinemic human subjects. Atherosclerosis. 1986;62(2):167–72.
Jonas A, Sweeny SA, Herbert PN. Discoidal complexes of A and C apolipoproteins with lipids and their reactions with lecithin: cholesterol acyltransferase. J Biol Chem. 1984;259(10):6369–75.
Soutar AK, Garner CW, Baker HN, Sparrow JT, Jackson RL, Gotto AM, et al. Effect of the human plasma apolipoproteins and phosphatidylcholine acyl donor on the activity of lecithin: cholesterol acyltransferase. Biochemistry. 1975;14(14):3057–64.
Poensgen J. Apolipoprotein C-1 inhibits the hydrolysis by phospholipase A2 of phospholipids in liposomes and cell membranes. Biochim Biophys Acta. 1990;1042(2):188–92.
Conde-Knape K, Bensadoun A, Sobel JH, Cohn JS, Shachter NS. Overexpression of apoC-I in apoE-null mice: severe hypertriglyceridemia due to inhibition of hepatic lipase. J Lipid Res. 2002;43(12):2136–45.
de Haan W, Out R, Berbée JFP, van der Hoogt CC, van Dijk KW, van Berkel TJC, et al. Apolipoprotein CI inhibits scavenger receptor BI and increases plasma HDL levels in vivo. Biochem Biophys Res Commun. 2008;377(4):1294–8.
Gautier T, Masson D, Jong MC, de Pais Barros JP, Duverneuil L, Le Guern N, et al. Apolipoprotein CI overexpression is not a relevant strategy to block cholesteryl ester transfer protein (CETP) activity in CETP transgenic mice. Biochem J. 2005;385(Pt 1):189–95.
Gautier T, Tietge UJF, Boverhof R, Perton FG, Le Guern N, Masson D, et al. Hepatic lipid accumulation in apolipoprotein C-I-deficient mice is potentiated by cholesteryl ester transfer protein. J Lipid Res. 2007;48(1):30–40.
de Barros JPP, Boualam A, Gautier T, Dumont L, Vergès B, Masson D, et al. Apolipoprotein CI is a physiological regulator of cholesteryl ester transfer protein activity in human plasma but not in rabbit plasma. J Lipid Res. 2009;50(9):1842–51.
Pillois X, Gautier T, Bouillet B, de Pais Barros JP, Jeannin A, Vergès B, et al. Constitutive inhibition of plasma CETP by apolipoprotein C1 is blunted in dyslipidemic patients with coronary artery disease. J Lipid Res. 2012;53(6):1200–9.
Bouillet B, Gautier T, Blache D, de Pais Barros JP, Duvillard L, Petit JM, et al. Glycation of apolipoprotein C1 impairs its CETP inhibitory property: pathophysiological relevance in patients with type 1 and type 2 diabetes. Diabetes Care. 2014;37(4):1148–56.
Ken-Dror G, Talmud PJ, Humphries SE, Drenos F. APOE/C1/C4/C2 gene cluster genotypes, haplotypes and lipid levels in prospective coronary heart disease risk among UK healthy men. Mol Med. 2010;16(9–10):389–99.
CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45(1):25–33.
Geisel MH, Coassin S, Heßler N, Bauer M, Eisele L, Erbel R, et al. Update of the effect estimates for common variants associated with carotid intima media thickness within four independent samples: the Bonn IMT Family Study, the Heinz Nixdorf Recall Study, the SAPHIR Study and the Bruneck Study. Atherosclerosis. 2016;249:83–7.
Hubácek JA, Pitha J, Adámková V, Skodová Z, Lánská V, Poledne R. Apolipoprotein E and apolipoprotein CI polymorphisms in the Czech population: almost complete linkage disequilibrium of the less frequent alleles of both polymorphisms. Physiol Res. 2003;52(2):195–200.
Lauer SJ, Walker D, Elshourbagy NA, Reardon CA, Levy-Wilson B, Taylor JM. Two copies of the human apolipoprotein C-I gene are linked closely to the apolipoprotein E gene. J Biol Chem. 1988;263(15):7277–86.
Myklebost O, Rogne S. The gene for human apolipoprotein CI is located 4.3 kilobases away from the apolipoprotein E gene on chromosome 19. Hum Genet. 1986;73(4):286–9.
Smit M, van der Kooij-Meijs E, Frants RR, Havekes L, Klasen EC. Apolipoprotein gene cluster on chromosome 19. Definite localization of the APOC2 gene and the polymorphic Hpa I site associated with type III hyperlipoproteinemia. Hum Genet. 1988;78(1):90–3.
Davison PJ, Norton P, Wallis SC, Gill L, Cook M, Williamson R, et al. There are two gene sequences for human apolipoprotein CI (apo CI) on chromosome 19, one of which is 4 kb from the gene for apo E. Biochem Biophys Res Commun. 1986;136(3):876–84.
Kim E, Xie S, Yeh SD, Lee YF, Collins LL, Hu YC, et al. Disruption of TR4 orphan nuclear receptor reduces the expression of liver apolipoprotein E/C-I/C-II gene cluster. J Biol Chem. 2003;278(47):46919–26.
Mak PA, Laffitte BA, Desrumaux C, Joseph SB, Curtiss LK, Mangelsdorf DJ, et al. Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors alpha and beta. J Biol Chem. 2002;277(35):31900–8.
Dahabreh DF, Medh JD. Activation of peroxisome proliferator activated receptor-gamma results in an atheroprotective apolipoprotein profile in HepG2 cells. Adv Biol Chem. 2012;2(3):218–25.
Subramanian S, Gottschalk WK, Kim SY, Roses AD, Chiba-Falek O. The effects of PPARγ on the regulation of the TOMM40-APOE-C1 genes cluster. Biochim Biophys Acta Mol Basis Dis. 2017;1863(3):810–6.
Vrins CLJ, Out R, van Santbrink P, van der Zee A, Mahmoudi T, Groenendijk M, et al. Znf202 affects high density lipoprotein cholesterol levels and promotes hepatosteatosis in hyperlipidemic mice. PLoS ONE. 2013;8(2):e57492.
Fernandes GW, Bocco BMLC, Fonseca TL, McAninch EA, Jo S, Lartey LJ, et al. The Foxo1-inducible transcriptional repressor Zfp125 causes hepatic steatosis and hypercholesterolemia. Cell Rep. 2018;22(2):523–34.
Wassef H, Bernier L, Davignon J, Cohn JS. Synthesis and secretion of apoC-I and apoE during maturation of human SW872 liposarcoma cells. J Nutr. 2004;134(11):2935–41.
Cyr Y, Wassef H, Bissonnette S, Lamantia V, Davignon J, Faraj M. WAT apoC-I secretion: role in delayed chylomicron clearance in vivo and ex vivo in WAT in obese subjects. J Lipid Res. 2016;57(6):1074–85.
Bouchard C, Dubuc G, Davignon J, Bernier L, Cohn JS. Post-transcriptional regulation of apoC-I synthesis and secretion in human HepG2 cells. Atherosclerosis. 2005;178(2):257–64.
Knott TJ, Robertson ME, Priestley LM, Urdea M, Wallis S, Scott J. Characterisation of mRNAs encoding the precursor for human apolipoprotein CI. Nucleic Acids Res. 1984;12(9):3909–15.
Jackson RL, Sparrow JT, Baker HN, Morrisett JD, Taunton OD, Gotto AM Jr. The primary structure of apolopoprotein-serine. J Biol Chem. 1974;249(16):5308–13.
Shulman RS, Herbert PN, Wehrly K, Fredrickson DS. Thf complete amino acid sequence of C-I (apoLp-Ser), an apolipoprotein from human very low density lipoproteins. J Biol Chem. 1975;250(1):182–90.
Bondarenko PV, Cockrill SL, Watkins LK, Cruzado ID, Macfarlane RD. Mass spectral study of polymorphism of the apolipoproteins of very low density lipoprotein. J Lipid Res. 1999;40(3):543–55.
Rozek A, Sparrow JT, Weisgraber KH, Cushley RJ. Conformation of human apolipoprotein C-I in a lipid-mimetic environment determined by CD and NMR spectroscopy. Biochemistry. 1999;38(44):14475–84.
McPherson A, Larson SB. The structure of human apolipoprotein C-1 in four different crystal forms. J Lipid Res. 2019;60(2):400–11.
Segrest JP, Jackson RL, Morrisett JD, Gotto AM. A molecular theory of lipid-protein interactions in the plasma lipoproteins. FEBS Lett. 1974;38(3):247–58.
Segrest JP, Feldmann RJ. Amphipathic helixes and plasma lipoproteins: a computer study. Biopolymers. 1977;16(9):2053–65.
Meyers NL, Wang L, Gursky O, Small DM. Changes in helical content or net charge of apolipoprotein C-I alter its affinity for lipid/water interfaces. J Lipid Res. 2013;54(7):1927–38.
Cushley RJ, Okon M. NMR studies of lipoprotein structure. Annu Rev Biophys Biomol Struct. 2002;31:177–206.
Carlson LA, Holmquist L. Concentrations of apolipoproteins B, C-I, C-II, C-III and E in sera from normal men and their relation to serum lipoprotein levels. Clin Chim Acta. 1982;124(2):163–78.
Curry MD, McConathy WJ, Fesmire JD, Alaupovic P. Quantitative determination of apolipoproteins C-I and C-II in human plasma by separate electroimmunoassays. Clin Chem. 1981;27(4):543–8.
Fuior EV, Gafencu AV. Apolipoprotein C1: its pleiotropic effects in lipid metabolism and beyond. Int J Mol Sci. 2019;20(23):E5939.
Riesen WF, Sturzenegger E. Enzyme-linked immunosorbent assay for apolipoprotein C-I. J Clin Chem Clin Biochem. 1986;24(10):723–7.
Cohn JS, Tremblay M, Boulet L, Jacques H, Davignon J, Roy M, et al. Plasma concentration and lipoprotein distribution of ApoC-I is dependent on ApoE genotype rather than the Hpa I ApoC-I promoter polymorphism. Atherosclerosis. 2003;169(1):63–70.
Lemesle G, Chouraki V, de Groote P, Turkieh A, Beseme O, Drobecq H, et al. Apolipoprotein proteomic profiling for the prediction of cardiovascular death in patients with heart failure. Proteomics Clin Appl. 2020;14(6):e2000035.
Fredrickson DS, Lux SE, Herbert PN. The apolipoproteins. Adv Exp Med Biol. 1972;26:25–56.
Gautier T, Deckert V, Aires V, Le Guern N, Proukhnitzky L, Patoli D, et al. Human apolipoprotein C1 transgenesis reduces atherogenesis in hypercholesterolemic rabbits. Atherosclerosis. 2021;320:10–8.
Sattler W, Mohr D, Stocker R. Rapid isolation of lipoproteins and assessment of their peroxidation by high-performance liquid chromatography postcolumn chemiluminescence. Methods Enzymol. 1994;233:469–89.
Herbert PN, Forte TM, Shulman RS, La Piana MJ, Gong EL, Levy RI, et al. Structural and compositional changes attending the ultracentrifugation of very low density lipoproteins. Prep Biochem. 1975;5(2):93–129.
Björkegren J, Boquist S, Samnegârd A, Lundman P, Tornvall P, Ericsson CG, et al. Accumulation of apolipoprotein C-I-rich and cholesterol-rich VLDL remnants during exaggerated postprandial triglyceridemia in normolipidemic patients with coronary artery disease. Circulation. 2000;101(3):227–30.
Wassef H, Salem H, Bissonnette S, Baass A, Dufour R, Davignon J, et al. White adipose tissue apolipoprotein C-I secretion in relation to delayed plasma clearance of dietary fat in humans. Arterioscler Thromb Vasc Biol. 2012;32(11):2785–93.
Windler E, Havel RJ. Inhibitory effects of C apolipoproteins from rats and humans on the uptake of triglyceride-rich lipoproteins and their remnants by the perfused rat liver. J Lipid Res. 1985;26(5):556–65.
Quarfordt SH, Michalopoulos G, Schirmer B. The effect of human C apolipoproteins on the in vitro hepatic metabolism of triglyceride emulsions in the rat. J Biol Chem. 1982;257(24):14642–7.
Kowal RC, Herz J, Weisgraber KH, Mahley RW, Brown MS, Goldstein JL. Opposing effects of apolipoproteins E and C on lipoprotein binding to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265(18):10771–9.
Weisgraber KH, Mahley RW, Kowal RC, Herz J, Goldstein JL, Brown MS. Apolipoprotein C-I modulates the interaction of apolipoprotein E with beta-migrating very low density lipoproteins (beta-VLDL) and inhibits binding of beta-VLDL to low density lipoprotein receptor-related protein. J Biol Chem. 1990;265(36):22453–9.
Swaney JB, Weisgraber KH. Effect of apolipoprotein C-I peptides on the apolipoprotein E content and receptor-binding properties of beta-migrating very low density lipoproteins. J Lipid Res. 1994;35(1):134–42.
Sehayek E, Eisenberg S. Mechanisms of inhibition by apolipoprotein C of apolipoprotein E-dependent cellular metabolism of human triglyceride-rich lipoproteins through the low density lipoprotein receptor pathway. J Biol Chem. 1991;266(27):18259–67.
Clavey V, Lestavel-Delattre S, Copin C, Bard JM, Fruchart JC. Modulation of lipoprotein B binding to the LDL receptor by exogenous lipids and apolipoproteins CI, CII, CIII, and E. Arterioscler Thromb Vasc Biol. 1995;15(7):963–71.
Simonet WS, Bucay N, Pitas RE, Lauer SJ, Taylor JM. Multiple tissue-specific elements control the apolipoprotein E/C-I gene locus in transgenic mice. J Biol Chem. 1991;266(14):8651–4.
Shachter NS, Ebara T, Ramakrishnan R, Steiner G, Breslow JL, Ginsberg HN, et al. Combined hyperlipidemia in transgenic mice overexpressing human apolipoprotein Cl. J Clin Invest. 1996;98(3):846–55.
Jong MC, Gijbels MJ, Dahlmans VE, Gorp PJ, Koopman SJ, Ponec M, et al. Hyperlipidemia and cutaneous abnormalities in transgenic mice overexpressing human apolipoprotein C1. J Clin Invest. 1998;101(1):145–52.
Westerterp M, de Haan W, Berbée JFP, Havekes LM, Rensen PCN. Endogenous apoC-I increases hyperlipidemia in apoE-knockout mice by stimulating VLDL production and inhibiting LPL. J Lipid Res. 2006;47(6):1203–11.
Jong MC, Dahlmans VE, van Gorp PJ, van Dijk KW, Breuer ML, Hofker MH, et al. In the absence of the low density lipoprotein receptor, human apolipoprotein C1 overexpression in transgenic mice inhibits the hepatic uptake of very low density lipoproteins via a receptor-associated protein-sensitive pathway. J Clin Invest. 1996;98(10):2259–67.
Jong MC, van Dijk KW, Dahlmans VE, Van der Boom H, Kobayashi K, Oka K, et al. Reversal of hyperlipidaemia in apolipoprotein C1 transgenic mice by adenovirus-mediated gene delivery of the low-density-lipoprotein receptor, but not by the very-low-density-lipoprotein receptor. Biochem J. 1999;338(Pt 2):281–7.
Berbée JFP, van der Hoogt CC, Sundararaman D, Havekes LM, Rensen PCN. Severe hypertriglyceridemia in human APOC1 transgenic mice is caused by apoC-I-induced inhibition of LPL. J Lipid Res. 2005;46(2):297–306.
Havel RJ, Fielding CJ, Olivecrona T, Shore VG, Fielding PE, Egelrud T. Cofactor activity of protein components of human very low density lipoproteins in the hydrolysis of triglycerides by lipoproteins lipase from different sources. Biochemistry. 1973;12(9):1828–33.
van der Hoogt CC, Berbée JFP, Espirito Santo SMS, Gerritsen G, Krom YD, van der Zee A, et al. Apolipoprotein CI causes hypertriglyceridemia independent of the very-low-density lipoprotein receptor and apolipoprotein CIII in mice. Biochim Biophys Acta. 2006;1761(2):213–20.
Dautin G, Soltani Z, Ducloux D, Gautier T, de Pais Barros JP, Gambert P, et al. Hemodialysis reduces plasma apolipoprotein C-I concentration making VLDL a better substrate for lipoprotein lipase. Kidney Int. 2007;72(7):871–8.
Hansen JB, Fernández JA, Notø ATW, Deguchi H, Björkegren J, Mathiesen EB. The apolipoprotein C-I content of very-low-density lipoproteins is associated with fasting triglycerides, postprandial lipemia, and carotid atherosclerosis. J Lipids. 2011;2011:271062.
Jong MC, van Ree JH, Dahlmans VE, Frants RR, Hofker MH, Havekes LM. Reduced very-low-density lipoprotein fractional catabolic rate in apolipoprotein C1-deficient mice. Biochem J. 1997;321(Pt 2):445–50.
Zheng C. Updates on apolipoprotein CIII: fulfilling promise as a therapeutic target for hypertriglyceridemia and cardiovascular disease. Curr Opin Lipidol. 2014;25(1):35–9.
Forte T, Norum KR, Glomset JA, Nichols AV. Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency: structure of low and high density lipoproteins as revealed by elctron microscopy. J Clin Invest. 1971;50(5):1141–8.
Vaisman BL, Neufeld EB, Freeman LA, Gordon SM, Sampson ML, Pryor M, et al. LCAT enzyme replacement therapy reduces LpX and improves kidney function in a mouse model of familial LCAT deficiency. J Pharmacol Exp Ther. 2019;368(3):423–34.
Nishida HI, Nakanishi T, Yen EA, Arai H, Yen FT, Nishida T. Nature of the enhancement of lecithin-cholesterol acyltransferase reaction by various apolipoproteins. J Biol Chem. 1986;261(26):12028–35.
Kinnunen PK, Ehnolm C. Effect of serum and C-apoproteins from very low density lipoproteins on human postheparin plasma hepatic lipase. FEBS Lett. 1976;65(3):354–7.
Huard K, Bourgeois P, Rhainds D, Falstrault L, Cohn JS, Brissette L. Apolipoproteins C-II and C-III inhibit selective uptake of low- and high-density lipoprotein cholesteryl esters in HepG2 cells. Int J Biochem Cell Biol. 2005;37(6):1308–18.
Chajek T, Fielding CJ. Isolation and characterization of a human serum cholesteryl ester transfer protein. Proc Natl Acad Sci USA. 1978;75(7):3445–9.
Barter PJ, Lally JI. The activity of an esterified cholesterol transferring factor in human and rat serum. Biochim Biophys Acta. 1978;531(2):233–6.
Barter PJ, Hopkins GJ, Calvert GD. Transfers and exchanges of esterified cholesterol between plasma lipoproteins. Biochem J. 1982;208(1):1–7.
Agellon LB, Walsh A, Hayek T, Moulin P, Jiang XC, Shelanski SA, et al. Reduced high density lipoprotein cholesterol in human cholesteryl ester transfer protein transgenic mice. J Biol Chem. 1991;266(17):10796–801.
Inazu A, Brown ML, Hesler CB, Agellon LB, Koizumi J, Takata K, et al. Increased high-density lipoprotein levels caused by a common cholesteryl-ester transfer protein gene mutation. N Engl J Med. 1990;323(18):1234–8.
Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NGD, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69(7):823–36.
Clark RW, Sutfin TA, Ruggeri RB, Willauer AT, Sugarman ED, Magnus-Aryitey G, et al. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24(3):490–7.
Barter PJ, Brewer HB, Chapman MJ, Hennekens CH, Rader DJ, Tall AR. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23(2):160–7.
Armitage J, Holmes MV, Preiss D. Cholesteryl ester transfer protein inhibition for preventing cardiovascular events: JACC review topic of the week. J Am Coll Cardiol. 2019;73(4):477–87.
Barter PJ, Jones ME. Kinetic studies of the transfer of esterified cholesterol between human plasma low and high density lipoproteins. J Lipid Res. 1980;21(2):238–49.
Ihm J, Quinn DM, Busch SJ, Chataing B, Harmony JA. Kinetics of plasma protein-catalyzed exchange of phosphatidylcholine and cholesteryl ester between plasma lipoproteins. J Lipid Res. 1982;23(9):1328–41.
Moulin P, Cheung MC, Bruce C, Zhong S, Cocke T, Richardson H, et al. Gender effects on the distribution of the cholesteryl ester transfer protein in apolipoprotein A-I-defined lipoprotein subpopulations. J Lipid Res. 1994;35(5):793–802.
Sparks DL, Pritchard PH. Transfer of cholesteryl ester into high density lipoprotein by cholesteryl ester transfer protein: effect of HDL lipid and apoprotein content. J Lipid Res. 1989;30(10):1491–8.
Guyard-Dangremont V, Lagrost L, Gambert P, Lallemant C. Competitive enzyme-linked immunosorbent assay of the human cholesteryl ester transfer protein (CETP). Clin Chim Acta. 1994;231(2):147–60.
Lagrost L, Perségol L, Lallemant C, Gambert P. Influence of apolipoprotein composition of high density lipoprotein particles on cholesteryl ester transfer protein activity. Particles containing various proportions of apolipoproteins AI and AII. J Biol Chem. 1994;269(5):3189–97.
Nishikawa O, Yokoyama S, Okabe H, Yamamoto A. Enhancement of non-polar lipid transfer reaction through stabilization of substrate lipid particles with apolipoproteins. J Biochem. 1988;103(1):188–94.
Gautier T, Masson D, de Barros JP, Athias A, Gambert P, Aunis D, et al. Human apolipoprotein C-I accounts for the ability of plasma high density lipoproteins to inhibit the cholesteryl ester transfer protein activity. J Biol Chem. 2000;275(48):37504–9.
Nishida HI, Kato H, Nishida T. Affinity of lipid transfer protein for lipid and lipoprotein particles as influenced by lecithin-cholesterol acyltransferase. J Biol Chem. 1990;265(9):4876–83.
Sparks D, Frohlich JJ, Pritchard PH. Lipid transfer proteins, hypertriglyceridemia, and reduced high-density lipoprotein cholesterol. Am Heart J. 1991;122(2):601–7.
Kushwaha RS, Hasan SQ, McGill HC, Getz GS, Dunham RG, Kanda P. Characterization of cholesteryl ester transfer protein inhibitor from plasma of baboons (Papio sp.). J Lipid Res. 1993;34(8):1285–97.
Connolly DT, Krul ES, Heuvelman D, Glenn KC. Inhibition of cholesteryl ester transfer protein by apolipoproteins, lipopolysaccharides, and cholesteryl sulfate. Biochim Biophys Acta. 1996;1304(2):145–60.
Dumont L, Gautier T, de Barros JPP, Laplanche H, Blache D, Ducoroy P, et al. Molecular mechanism of the blockade of plasma cholesteryl ester transfer protein by its physiological inhibitor apolipoprotein CI. J Biol Chem. 2005;280(45):38108–16.
Nishida HI, Arai H, Nishida T. Cholesterol ester transfer mediated by lipid transfer protein as influenced by changes in the charge characteristics of plasma lipoproteins. J Biol Chem. 1993;268(22):16352–60.
Masson D, Athias A, Lagrost L. Evidence for electronegativity of plasma high density lipoprotein-3 as one major determinant of human cholesteryl ester transfer protein activity. J Lipid Res. 1996;37(7):1579–90.
Gautier T, Masson D, Jong MC, Duverneuil L, Le Guern N, Deckert V, et al. Apolipoprotein CI deficiency markedly augments plasma lipoprotein changes mediated by human cholesteryl ester transfer protein (CETP) in CETP transgenic/ApoCI-knocked out mice. J Biol Chem. 2002;277(35):31354–63.
Tato F, Vega GL, Grundy SM. Determinants of plasma HDL-cholesterol in hypertriglyceridemic patients. Role of cholesterol-ester transfer protein and lecithin cholesteryl acyl transferase. Arterioscler Thromb Vasc Biol. 1997;17(1):56–63.
de Vries R, Perton FG, Dallinga-Thie GM, van Roon AM, Wolffenbuttel BHR, van Tol A, et al. Plasma cholesteryl ester transfer is a determinant of intima-media thickness in type 2 diabetic and nondiabetic subjects: role of CETP and triglycerides. Diabetes. 2005;54(12):3554–9.
Bouillet B, Gautier T, Aho LS, Duvillard L, Petit JM, Lagrost L, et al. Plasma apolipoprotein C1 concentration is associated with plasma triglyceride concentration, but not visceral fat, in patients with type 2 diabetes. Diabetes Metab. 2016;42(4):263–6.
Bouillet B, Gautier T, Rouland A, Duvillard L, Petit JM, Lagrost L, et al. Plasma apolipoprotein C1 concentration is associated with plasma triglyceride concentration but not with visceral fat and liver fat content in people with type 1 diabetes. Acta Diabetol. 2019;56(10):1155–7.
Jong MC, Voshol PJ, Muurling M, Dahlmans VE, Romijn JA, Pijl H, et al. Protection from obesity and insulin resistance in mice overexpressing human apolipoprotein C1. Diabetes. 2001;50(12):2779–85.
Åvall K, Ali Y, Leibiger IB, Leibiger B, Moede T, Paschen M, et al. Apolipoprotein CIII links islet insulin resistance to β-cell failure in diabetes. Proc Natl Acad Sci USA. 2015;112(20):E2611-2619.
Botteri G, Montori M, Gumà A, Pizarro J, Cedó L, Escolà-Gil JC, et al. VLDL and apolipoprotein CIII induce ER stress and inflammation and attenuate insulin signalling via Toll-like receptor 2 in mouse skeletal muscle cells. Diabetologia. 2017;60(11):2262–73.
van der Ham RLM, Alizadeh Dehnavi R, Berbée JFP, Putter H, de Roos A, Romijn JA, et al. Plasma apolipoprotein CI and CIII levels are associated with increased plasma triglyceride levels and decreased fat mass in men with the metabolic syndrome. Diabetes Care. 2009;32(1):184–6.
Kasthuri RS, McMillan KR, Flood-Urdangarin C, Harvey SB, Wilson-Grady JT, Nelsestuen GL. Correlation of a T45S variant of apolipoprotein C1 with elevated BMI in persons of American Indian and Mexican ancestries. Int J Obes. 2007;31(8):1334–6.
Croyal M, Wargny M, Chemello K, Chevalier C, Blanchard V, Bigot-Corbel E, et al. Plasma apolipoprotein concentrations and incident diabetes in subjects with prediabetes. Cardiovasc Diabetol. 2022;21(1):21.
Ozuynuk AS, Erkan AF, Ekici B, Erginel-Unaltuna N, Coban N. Cholesterol-related gene variants are associated with diabetes in coronary artery disease patients. Mol Biol Rep. 2021;48(5):3945–54.
Jong MC, Rensen PC, Dahlmans VE, van der Boom H, van Berkel TJ, Havekes LM. Apolipoprotein C-III deficiency accelerates triglyceride hydrolysis by lipoprotein lipase in wild-type and apoE knockout mice. J Lipid Res. 2001;42(10):1578–85.
Colhoun HM, Scheek LM, Rubens MB, Van Gent T, Underwood SR, Fuller JH, et al. Lipid transfer protein activities in type 1 diabetic patients without renal failure and nondiabetic control subjects and their association with coronary artery calcification. Diabetes. 2001;50(3):652–9.
Dullaart RP, Groener JE, Dikkeschei BD, Erkelens DW, Doorenbos H. Elevated cholesteryl ester transfer protein activity in IDDM men who smoke. Possible factor for unfavorable lipoprotein profile. Diabetes Care. 1991;14(4):338–41.
Dullaart RP, Groener JE, Dikkeschei LD, Erkelens DW, Doorenbos H. Increased cholesterylester transfer activity in complicated type 1 (insulin-dependent) diabetes mellitus–its relationship with serum lipids. Diabetologia. 1989;32(1):14–9.
Bagdade JD, Lane JT, Subbaiah PV, Otto ME, Ritter MC. Accelerated cholesteryl ester transfer in noninsulin-dependent diabetes mellitus. Atherosclerosis. 1993;104(1–2):69–77.
Karpe F. Postprandial lipid metabolism in relation to coronary heart disease. Proc Nutr Soc. 1997;56(2):671–8.
Curtiss LK, Witztum JL. Plasma apolipoproteins AI, AII, B, CI, and E are glucosylated in hyperglycemic diabetic subjects. Diabetes. 1985;34(5):452–61.
Mooyaart AL, Valk EJJ, van Es LA, Bruijn JA, de Heer E, Freedman BI, et al. Genetic associations in diabetic nephropathy: a meta-analysis. Diabetologia. 2011;54(3):544–53.
Bus P, Pierneef L, Bor R, Wolterbeek R, van Es LA, Rensen PC, et al. Apolipoprotein C-I plays a role in the pathogenesis of glomerulosclerosis. J Pathol. 2017;241(5):589–99.
Eo HS, Kim DI. Apolipoprotein C1 and apolipoprotein E are differentially expressed in atheroma of the carotid and femoral artery. J Surg Res. 2008;144(1):132–7.
Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352(16):1685–95.
Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG, et al. Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun. 2001;280(3):818–23.
Smith LE, Segrest JP, Davidson WS. Helical domains that mediate lipid solubilization and ABCA1-specific cholesterol efflux in apolipoproteins C-I and A-II. J Lipid Res. 2013;54(7):1939–48.
Westerterp M, Van Eck M, de Haan W, Offerman EH, Van Berkel TJC, Havekes LM, et al. Apolipoprotein CI aggravates atherosclerosis development in ApoE-knockout mice despite mediating cholesterol efflux from macrophages. Atherosclerosis. 2007;195(1):e9-16.
Kolmakova A, Kwiterovich P, Virgil D, Alaupovic P, Knight-Gibson C, Martin SF, et al. Apolipoprotein C-I induces apoptosis in human aortic smooth muscle cells via recruiting neutral sphingomyelinase. Arterioscler Thromb Vasc Biol. 2004;24(2):264–9.
Steen H, Kolmakova A, Stuber M, Rodriguez ER, Gao F, Chatterjee S, et al. MRI visualized neo-intimal dissection and co-localization of novel apoptotic markers apolipoprotein C-1, ceramide and caspase-3 in a Watanabe hyperlipidemic rabbit model. Atherosclerosis. 2007;191(1):82–9.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119–31.
Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. 2020;383(19):1838–47.
Soehnlein O, Libby P. Targeting inflammation in atherosclerosis—from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20(8):589–610.
Lehr HA, Sagban TA, Ihling C, Zähringer U, Hungerer KD, Blumrich M, et al. Immunopathogenesis of atherosclerosis: endotoxin accelerates atherosclerosis in rabbits on hypercholesterolemic diet. Circulation. 2001;104(8):914–20.
Ostos MA, Recalde D, Zakin MM, Scott-Algara D. Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 2002;519(1–3):23–9.
Danesh J, Collins R, Peto R. Chronic infections and coronary heart disease: is there a link? Lancet. 1997;350(9075):430–6.
Rangé H, Labreuche J, Louedec L, Rondeau P, Planesse C, Sebbag U, et al. Periodontal bacteria in human carotid atherothrombosis as a potential trigger for neutrophil activation. Atherosclerosis. 2014;236(2):448–55.
Carnevale R, Nocella C, Petrozza V, Cammisotto V, Pacini L, Sorrentino V, et al. Localization of lipopolysaccharide from Escherichia Coli into human atherosclerotic plaque. Sci Rep. 2018;8(1):3598.
Berbée JFP, Coomans CP, Westerterp M, Romijn JA, Havekes LM, Rensen PCN. Apolipoprotein CI enhances the biological response to LPS via the CD14/TLR4 pathway by LPS-binding elements in both its N- and C-terminal helix. J Lipid Res. 2010;51(7):1943–52.
Berbée JFP, van der Hoogt CC, Kleemann R, Schippers EF, Kitchens RL, van Dissel JT, et al. Apolipoprotein CI stimulates the response to lipopolysaccharide and reduces mortality in gram-negative sepsis. FASEB J. 2006;20(12):2162–4.
Berbée JFP, Mooijaart SP, de Craen AJM, Havekes LM, van Heemst D, Rensen PCN, et al. Plasma apolipoprotein CI protects against mortality from infection in old age. J Gerontol A Biol Sci Med Sci. 2008;63(2):122–6.
Westerterp M, Berbée JFP, Pires NMM, van Mierlo GJD, Kleemann R, Romijn JA, et al. Apolipoprotein C-I is crucially involved in lipopolysaccharide-induced atherosclerosis development in apolipoprotein E-knockout mice. Circulation. 2007;116(19):2173–81.
Taylor JM, Fan J. Transgenic rabbit models for the study of atherosclerosis. Front Biosci. 1997;2:d298-308.
Guyard-Dangremont V, Desrumaux C, Gambert P, Lallemant C, Lagrost L. Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility. Comp Biochem Physiol B Biochem Mol Biol. 1998;120(3):517–25.
Xu Y, Berglund L, Ramakrishnan R, Mayeux R, Ngai C, Holleran S, et al. A common Hpa I RFLP of apolipoprotein C-I increases gene transcription and exhibits an ethnically distinct pattern of linkage disequilibrium with the alleles of apolipoprotein E. J Lipid Res. 1999;40(1):50–8.
Park JM, Park DH, Song Y, Kim JO, Choi JE, Kwon YJ, et al. Understanding the genetic architecture of the metabolically unhealthy normal weight and metabolically healthy obese phenotypes in a Korean population. Sci Rep. 2021;11(1):2279.
Olsson B, Gigante B, Mehlig K, Bergsten A, Leander K, de Faire U, et al. Apolipoprotein C-I genotype and serum levels of triglycerides, C-reactive protein and coronary heart disease. Metab Clin Exp. 2010;59(12):1736–41.
Mao C, Howard TD, Sullivan D, Fu Z, Yu G, Parker SJ, et al. Bioinformatic analysis of coronary disease associated SNPs and genes to identify proteins potentially involved in the pathogenesis of atherosclerosis. J Proteom Genom Res. 2017;2(1):1–12.
Battram T, Hoskins L, Hughes DA, Kettunen J, Ring SM, Smith GD, et al. Coronary artery disease, genetic risk and the metabolome in young individuals. Wellcome Open Res. 2018;3:114.
van der Ham RLM, Dehnavi RA, van den Berg GA, Putter H, de Roos A, Berbée JFP, et al. Apolipoprotein CI levels are associated with atherosclerosis in men with the metabolic syndrome and systemic inflammation. Atherosclerosis. 2009;203(2):355–7.
Moore D, McNeal C, Macfarlane R. Isoforms of apolipoprotein C-I associated with individuals with coronary artery disease. Biochem Biophys Res Commun. 2011;404(4):1034–8.
Chang CT, Liao HY, Chang CM, Chen CY, Chen CH, Yang CY, et al. Oxidized ApoC1 on MALDI-TOF and glycated-ApoA1 band on gradient gel as potential diagnostic tools for atherosclerotic vascular disease. Clin Chim Acta. 2013;420:69–75.
Björkegren J, Silveira A, Boquist S, Tang R, Karpe F, Bond MG, et al. Postprandial enrichment of remnant lipoproteins with apoC-I in healthy normolipidemic men with early asymptomatic atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22(9):1470–4.
Hamsten A, Silveira A, Boquist S, Tang R, Bond MG, de Faire U, et al. The apolipoprotein CI content of triglyceride-rich lipoproteins independently predicts early atherosclerosis in healthy middle-aged men. J Am Coll Cardiol. 2005;45(7):1013–7.
Notø ATW, Mathiesen EB, Brox J, Björkegren J, Hansen JB. The ApoC-I content of VLDL particles is associated with plaque size in persons with carotid atherosclerosis. Lipids. 2008;43(7):673–9.
Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. 2019;40(2):537–57.
Gautier T, Masson D, Lagrost L. The potential of cholesteryl ester transfer protein as a therapeutic target. Expert Opin Ther Targets. 2016;20(1):47–59.
Kwiterovich PO, Virgil DG, Chu AY, Khouzami VA, Alaupovic P, Otvos JD. Interrelationships between the concentration and size of the largest high-density lipoprotein subfraction and apolipoprotein C-I in infants at birth and follow-up at 2–3 months of age and their parents. J Clin Lipidol. 2013;7(1):29–37.
de la Parra Soto LG, Gutiérrez-Uribe JA, Sharma A, Ramírez-Jiménez AK. Is Apo-CIII the new cardiovascular target? An analysis of its current clinical and dietetic therapies. Nutr Metab Cardiovasc Dis. 2022;32(2):295–308.
Tokgözoğlu L, Libby P. The dawn of a new era of targeted lipid-lowering therapies. Eur Heart J. 2022;43(34):3198–208.
Dib I, Khalil A, Chouaib R, El-Makhour Y, Noureddine H. Apolipoprotein C-III and cardiovascular diseases: when genetics meet molecular pathologies. Mol Biol Rep. 2021;48(1):875–86.
Mehta A, Shapiro MD. Apolipoproteins in vascular biology and atherosclerotic disease. Nat Rev Cardiol. 2022;19(3):168–79.
Gangabadage CS, Zdunek J, Tessari M, Nilsson S, Olivecrona G, Wijmenga SS. Structure and dynamics of human apolipoprotein CIII. J Biol Chem. 2008;283(25):17416–27.
Ooi EMM, Barrett PHR, Chan DC, Watts GF. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci. 2008;114(10):611–24.
Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, et al. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Lipid Res. 2010;51(1):150–61.
Riwanto M, Rohrer L, Roschitzki B, Besler C, Mocharla P, Mueller M, et al. Altered activation of endothelial anti- and proapoptotic pathways by high-density lipoprotein from patients with coronary artery disease: role of high-density lipoprotein-proteome remodeling. Circulation. 2013;127(8):891–904.
Zvintzou E, Lhomme M, Chasapi S, Filou S, Theodoropoulos V, Xapapadaki E, et al. Pleiotropic effects of apolipoprotein C3 on HDL functionality and adipose tissue metabolic activity. J Lipid Res. 2017;58(9):1869–83.
Chan HC, Ke LY, Lu HT, Weng SF, Chan HC, Law SH, et al. An increased plasma level of ApoCIII-rich electronegative high-density lipoprotein may contribute to cognitive impairment in Alzheimer’s disease. Biomedicines. 2020;8(12):E542.
Yingchun H, Yahong M, Jiangping W, Xiaokui H, Xiaohong Z. Increased inflammation, endoplasmic reticulum stress and oxidative stress in endothelial and macrophage cells exacerbate atherosclerosis in ApoCIII transgenic mice. Lipids Health Dis. 2018;17(1):220.
Zewinger S, Reiser J, Jankowski V, Alansary D, Hahm E, Triem S, et al. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat Immunol. 2020;21(1):30–41.
Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. Apolipoprotein CIII induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114(7):681–7.
Li H, Han Y, Qi R, Wang Y, Zhang X, Yu M, et al. Aggravated restenosis and atherogenesis in ApoCIII transgenic mice but lack of protection in ApoCIII knockouts: the effect of authentic triglyceride-rich lipoproteins with and without ApoCIII. Cardiovasc Res. 2015;107(4):579–89.
Olin-Lewis K, Krauss RM, La Belle M, Blanche PJ, Barrett PHR, Wight TN, et al. ApoC-III content of apoB-containing lipoproteins is associated with binding to the vascular proteoglycan biglycan. J Lipid Res. 2002;43(11):1969–77.
Funding
This review received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
AR, TG and BB conducted a review of the literature and contributed to conception, design and the first draft of the review. DM, LL, BV, TG and BB critically revised the initial manuscript. All authors read and approved the final manuscript. BB is the guarantor of the study.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rouland, A., Masson, D., Lagrost, L. et al. Role of apolipoprotein C1 in lipoprotein metabolism, atherosclerosis and diabetes: a systematic review. Cardiovasc Diabetol 21, 272 (2022). https://doi.org/10.1186/s12933-022-01703-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-022-01703-5