
Abstract
Background
Post-acute sequelae of COVID-19 (PASC), also now known as long COVID, has become a major global health and economic burden. Previously, we provided evidence that there is a significant insoluble fibrin amyloid microclot load in the circulation of individuals with long COVID, and that these microclots entrap a substantial number of inflammatory molecules, including those that might prevent clot breakdown. Scientifically, the most challenging aspect of this debilitating condition is that traditional pathology tests such as a serum CRP (C-reactive protein) may not show any significant abnormal inflammatory markers, albeit these tests measure only the soluble inflammatory molecules. Elevated, or abnormal soluble biomarkers such as IL-6, D-Dimer or fibrinogen indicate an increased risk for thrombosis or a host immune response in COVID-19. The absence of biomarkers in standard pathology tests, result in a significant amount of confusion for patients and clinicians, as patients are extremely sick or even bed-ridden but with no regular identifiable reason for their disease. Biomarkers that are currently available cannot detect the molecules present in the microclots we identified and are therefore unable to confirm their presence or the mechanisms that drive their formation.
Methods
Here we analysed the protein content of double-digested microclots of 99 long COVID patients and 29 healthy controls. The patients suffering from long COVID reported their symptoms through a questionnaire completed by themselves or their attending physician.
Results
Our long COVID cohort’s symptoms were found to be in line with global findings, where the most prevalent symptoms were constant fatigue (74%,) cognitive impairment (71%) and depression and anxiety (30%). Our most noteworthy findings were a reduced level of plasma Kallikrein compared to our controls, an increased level of platelet factor 4 (PF4) von Willebrand factor (VWF), and a marginally increased level of α-2 antiplasmin (α-2-AP). We also found a significant presence of antibodies entrapped inside these microclots.
Conclusion
Our results confirm the presence of pro-inflammatory molecules that may also contribute to a failed fibrinolysis phenomenon, which could possibly explain why individuals with long COVID suffer from chronic fatigue, dyspnoea, or cognitive impairment. In addition, significant platelet hyperactivation was noted. Hyperactivation will result in the granular content of platelets being shed into the circulation, including PF4. Overall, our results provide further evidence of both a failed fibrinolytic system in long COVID/PASC and the entrapment of many proteins whose presence might otherwise go unrecorded. These findings might have significant implications for individuals with pre-existing comorbidities, including cardiovascular disease and type 2 diabetes.
Similar content being viewed by others

Introduction
Post-acute sequelae of COVID-19 (PASC), now commonly referred to as long COVID, has quickly become a major global health and economic burden [1,2,3,4]. SARS-CoV-2 typically makes use of the cell surface receptor angiotensin-converting enzyme 2 (ACE2), to enter target cells. ACE2 receptors are expressed on the surface of lung epithelial cells, enterocytes of the small intestine and are also present in arterial and venous endothelial cells as well as arterial smooth muscle cells of multiple organs [5]. Viral replication leads to cellular damage and the release of pro-inflammatory alarmins due to cell injury and death [6]. The complement system may be activated by locally formed immune complexes which would inevitably boost the inflammatory response [7]. The activation of the complement cascade directly leads to endothelial damage and C3a and C5a formation recruits leucocytes that are responsible for a substantial local release of pro-inflammatory cytokines such as interleukin IL-1, IL-6, IL-8 and interferon-γ [8]. Fabio Ciceri and co-authors suggested the use of the term MicroCLOTS (microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome) [9]. They used this term as an atypical acute respiratory distress syndrome because of lung damage, primarily explained by dramatic alveolar endothelial damage leading to a progressive endothelial pulmonary syndrome with microvascular thrombosis [9]. In the acute phase of COVID-19, patients present with multi-organ symptoms [10] and this phase is characterized by acute clinical pathologies, including various coagulopathies that may be accompanied by hypercoagulation and platelet hyperactivation [11,12,13,14,15,16,17]. If these coagulopathies are not adequately treated during the acute phase of the disease, possible tissue hypoxia and impaired oxygen exchange may linger for months. The resulting underlying pathologies can directly perpetuate lingering long COVID symptoms that include fatigue or muscle weakness, dyspnoea, cognitive impairment, insomnia, and anxiety or depression.
We recently provided evidence, based on a relatively small cohort of patients, that individuals with long COVID present with a significant fibrin amyloid microclot load in their circulation and that these persistent microclots are resistant to fibrinolysis [1]. Microclots as described further in this paper, will refer to microscopic (fibrin-amyloid-containing) clots present in the plasma of recruited individuals and not MicroCLOTS, the syndrome as suggested by Ciceri and co-authors [9]. In our previously mentioned work, we used techniques including proteomics and fluorescence microscopy to study plasma samples from healthy individuals, individuals with type 2 diabetes mellitus (T2DM), those with acute COVID-19, and those with long COVID symptoms. Plasma samples from long COVID patients still contained large amounts of anomalous (amyloid) microclots that are resistant to fibrinolysis (compared to the plasma from controls and T2DM), even after trypsinisation. We could solubilize the microclots only after a second trypsinization step. The most noteworthy inflammatory molecules that were trapped in the solubilized microclots present in plasma of long COVID patients, versus the equivalent volume of fully digested plasma fluid of control samples, were α(2)-antiplasmin (α2AP), various fibrinogen chains, von Willebrand factor (VWF), and Serum Amyloid A (SAA). In addition to the natural resistance of protein amyloid structures to proteolysis [18] these trapped molecules could indeed result in fibrinolytic-resistant pellet microclots [1, 19].
We have now embarked on a considerably more extensive analysis of these microclot contents. We used proteomics to study blood samples from 99 long COVID patients and 29 healthy controls. There is increasing evidence that autoantibodies may be involved in the lingering symptoms of these patients [20,21,22]. Thus, in addition to molecules of interest that are related to clotting, we also focused on the presence of antibodies or autoantibodies that might be associated with or trapped inside the fibrinolysis-resistant microclots.
Materials and methods
Ethical clearance
Ethical clearance for the study was obtained from the Health Research Ethics Committee (HREC) of Stellenbosch University (South Africa) (references: B21/03/001_COVID-19, project ID: 21,911 (long COVID registry) and N19/03/043, project ID 9521 with yearly re-approval). Participants were either recruited via the long COVID registry or identified from our clinical collaborators’ practice. The experimental objectives, risks, and details were explained to volunteers and informed consent were obtained prior to blood collection. Strict compliance to ethical guidelines and principles Declaration of Helsinki, South African Guidelines for Good Clinical Practice, and Medical Research Council Ethical Guidelines for Research were kept for the duration of the study and for all research protocols.
Sample demographics and considerations
Blood was collected from 29 volunteers (9 males; 20 females; median age 52 [range 41–57]) to serve as controls. Control patients did not smoke or suffer from coagulopathies, were not pregnant and only 1 volunteer suffered from cardiovascular disease. Those suffering from hypertension or hyperlipidaemia, were well controlled on treatment and none of our controls took anticoagulation medication. Initially, we included 99 long COVID patients in the study (30 males, 69 females.) Liquid chromatography and mass spectrometry were performed on all of the samples and data collected. 66 of the long COVID samples were found to be “high responders” and 24 “low responders” (please refer to the mass spectrometry data analysis section for an in-depth explanation of the classification.) Mass spectrometry data analysis was only carried out on the high responder long COVID samples. The median age of high responders was 51 [40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60] (21 males; 45 females) and the median age of low responders 45 [31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58] (5 males; 19 females.) Healthy participants, patients or their attending physicians completed a questionnaire, either as part of the long COVID registry or when they visited their attending clinician. The questionnaire contained information regarding their age, gender, pre-existing comorbid conditions, chronic medication, information regarding the onset, diagnosis, and severity of their acute COVID-19 illness, self-reported long COVID symptoms, as well as their COVID-19 vaccination history (where available).
Blood sample collection
Either a qualified phlebotomist or medical practitioner drew blood samples. Blood samples were collected in 4.5 mL sodium citrate (3.2%), 4 ml lithium heparin (75 USP units) and 5 ml rapid serum (thrombin-based clot activator) tubes (BD Vacutainer®, 369714)), via venepuncture, adhering to the standard sterile protocol. On the day of blood collection, natriuretic peptide tests measuring levels of BNP or NT-proBNP, serum ferritin and Interleukin-6 (IL-6) levels were analysed by a pathology lab. NT-proBNP were analysed to determine that our cohort did not have underlying heart failure that might falsely suggest coagulopathies are due to long COVID. If NT-proBNP levels were not available, the attending clinician ensured that the patients had normal echocardiograms. Whole blood (WB) was centrifuged at 3000xg for 15 min at room temperature and the supernatant platelet poor plasma (PPP) samples were collected in 1.5 mL Eppendorf tubes and stored at – 80 °C.
Double trypsin digestion protocol to solubilize microclots in PPP
Stored PPP was used to determine protein concentration using nanodrop technology where a 20 × dilution was made with 10 mM ammonium bicarbonate and protein concentration determined using a nanodrop spectrophotometer (Thermo Fischer) and measuring the absorbance at 280 nm. The samples were then standardised to the lowest protein concentration and 50 µg total protein removed for digestion (25 µL). For the first trypsin digestion step, 50 µg of protein per sample was placed in a tube and 25 µL ammonium bicarbonate and 1 µg of trypsin was added to a final volume of 50 µL. The samples were incubated over night at 37 °C.
For the second trypsin digestion step, the samples were centrifuged the next day at 16,000g for 10 min (volume in each sample were still 50 µL). Next, 42 µL of supernatant were removed carefully to not do not disturb the microclots at bottom of tube. To the remaining 8 µL, 10 µL of 250 mM Triethyamonium bicarbonate (TEAB) (ThermoFisher) containing 5 mM tris carboxyethyl phosphine (TCEP) (Sigma), was added. The samples were vortexed for 5 s followed by incubation of 10 min at 90 °C. Samples where then incubated for a further 50 min at 60 °C, followed by a cooling-down step. Cysteine residues were blocked using 10 mM (final concentration) methyl methanethiosulfonate (MMTS) (we added 1 µl into each sample). Samples were left at room temp for 30 min. Thereafter, 20 µL of 50% (Sigma) methanol was added and samples were vortexed for 5 s (to denature last intact proteins that might remain). Now 10 µL of trypsin solution in 100 mM TEAB (containing 1 µg of trypsin) (Thermo) was added to each sample followed by incubation for 18 h at 37 °C. Samples were then acidified with 5 µL trifluoroacetic acid (TFA) (Sigma) of which the final concentration of 0.5% was used to precipitate trypsin. Samples where centrifuged at 16,000g to pellet the trypsin precipitate and the supernatant removed and placed into HPLC inserts. Samples were allowed to evaporate to dryness under vacuum (roto-evaporator) followed by resuspending in 20µL loading buffer consisting of 2% acetonitrile (Burdick and Jackson) in deionized water containing 0.1% Formic acid (Sigma) spiked with Biognosys 11 indexed retention times standards (Biognosys.) The dissolved samples were loaded directly in the autosampler set to 4C.
Liquid chromatography of digested microclots
Liquid chromatography was performed on a Thermo Scientific Ultimate 3000 RSLC [23, 24] equipped with a 20 mm × 100 µm C18 trap column (Thermo Scientific) and a CSH 25cmx75µm 1.7 µm particle size C18 column (Waters) analytical column. The solvent system employed was loading: 2% acetonitrile:water; 0.1% FA; Solvent A: water; 0.1% FA and Solvent B: 100% acetonitrile, 0.15% FA. Methods were previously described in [1]. Separation was effected using a linear gradient from 2 B to 30% B over 65 min followed by 30 B to 45% B from 65 to 80 min. The sample was loaded onto a trap column at 2 µL/min before switching to the analytical column at three minutes with a flow rate of 300 nL/min. Chromatography was performed at 45 °C and the outflow delivered to the mass spectrometer through a stainless-steel nano-bore emitter.
Mass spectrometry of digested microclots
Mass spectrometry was performed using a Thermo Scientific Fusion mass spectrometer equipped with a Nanospray Flex ionization source. Data was collected in positive ion mode with spray voltage set to 1.8 kV and ion transfer capillary set to 275 °C. The mass spectrometer auto-calibration was activated using polysiloxane ions at m/z = 445.12003. MS1 scans were performed using the orbitrap detector set at 120,000 resolution over the scan range 375–1500 with AGC target at 2 E4 and maximum injection time of 50 ms. Data were acquired in profile mode. MS2 acquisitions were performed using monoisotopic precursor selection for ion with charges + 2 to + 7 with error tolerance set to ± 10 ppm. Precursor ions were excluded from fragmentation once for a period of 60 s. Precursor ions were selected for fragmentation in HCD mode using the quadrupole mass analyser with HCD energy set to 30%. Quadrupole isolation was set at m/z = 1.2. Fragment ions were detected in the Orbitrap mass analyser set to 30,000 resolution. The AGC target was set to 5E4 and the maximum injection time to 100 ms. The data was acquired in centroid mode. Samples were randomised by using a randomization schedule. Technical replicates are not feasible in an experiment with a large sample size such as this.
Mass spectrometry data analysis
The raw files generated by the mass spectrometer were imported into SearchGUI [25] and processed using the MsGF + algorithm. Database interrogation was performed against the 2019-nCOVpFASTA database [26]. Semi-tryptic cleavage with 2 missed cleavages was allowed for. Precursor mass tolerance was set to 10 ppm and fragment mass tolerance set to 0.02 Da. Deamidation (NQ), oxidation (M) and Phosho-ST were allowed as dynamic modifications and methylthio of C as a fixed modification. Data was imported into Skyline and the extracted ion chromatograms of positively identified peptides summated as indication of relative protein abundance. Spectral data were evaluated in Skyline and the proteomics sample was divided into high and low responders. Samples where 90% of all the protein intensity level read outs (or the area under the peak) were above the 3rd quartile when compared to the protein levels observed in our other samples, were classified as “high responders". If 90% of all the protein intensity level read outs were below the 1st quartile when compared to the proteins in other samples, they were classified as “low responders.” For example, if there were 100 proteins identified but 90 of those proteins had concentrations/responses that were below the 1st quartile compared to the other samples in the sample set, the patient was classified as a low responder. Low responders were excluded from further analysis. We ensured that it was not attributable to instrument performance and batch effects were also excluded. The search results were imported into Scaffold Q + for further validation (www.proteomesoftware.com) and statistical testing. The (FDR) false discovery rate was set to 1%. Normalization was selected in Scaffold Q + with a weighted spectra quantitation method and missing values were processed by the algorithm contained within the software. A Fischer’s exact test (F-test) was performed on the dataset and Benjaminin-Hochberg multiple testing correction applied. This resulted in a p-value of < 0.012. For immunoglobulin specific searches the FASTA files were obtained from uniprot (www.uniprot.org) by searching the database for human immunoglobulins and downloading the returned protein sequences. Protein parsimony for immunoglobulin searches was handled by peptide and Protein Prohpet algorithms within Scaffold. The immunoglobulins listed later in the paper (as shown in Table 3 and Table 4,) were cross referenced on THE INTERNATIONAL IMMUNOGENETICS INFORMATION SYSTEM®’s IMGT/LIGM-DB database (nucleotide sequences of IG and TR from 360 species.) The citable accession numbers of the immunoglobulins were used to determine the EMBLi reference number to search for the particular immunoglobulin on the database: https://www.imgt.org/ligmdb/ Our proteomics data will be deposited in an international repository such as PRIDE (EBI) for public access.
PPP and the detection of amyloid fibrin(ogen) protein and anomalous micro-clotting
Fluorescence microscopy of microclot formation in PPP was performed on some of our samples as described in our previous papers [17]. PPP were exposed to the fluorescent amyloid dye, Thioflavin T (ThT) (final concentration: 0.005 mM) (Sigma-Aldrich, St. Louis, MO, USA) for 30 min (protected from light) at room temperature [18, 27,28,29,30]. After incubation, 3 µL stained PPP was placed on a glass slide and covered with a coverslip. The excitation wavelength band for ThT was set at 450 to 488 nm and the emission at 499 to 529 nm and processed samples were viewed using a Zeiss Axio Observer 7 fluorescent microscope with a Plan-Apochromat 63x/1.4 Oil DIC M27 objective (Carl Zeiss Microscopy, Munich, Germany).
Platelet pathology / evaluation
Haematocrit samples of a few of our patients in the cohort were exposed to the two fluorescent markers, CD62P (PE-conjugated) (platelet surface P-selectin) (IM1759U, Beckman Coulter, Brea, CA, USA) and PAC-1 (FITC-conjugated) (340507, BD Biosciences, San Jose, CA, USA) (17). CD62P is a marker for P-selectin that is either found on the membrane of platelets or inside them. PAC-1 identifies platelets through marking the glycoprotein IIb/IIIa (gpIIb/IIIa) on the platelet membrane. To study platelet pathology, 4 µL CD62P and 4 µL PAC-1 was added to 20 µL haematocrit, followed by incubation for 30 min and protected from light at room temperature. The excitation wavelength band for PAC-1 was set at 450 to 488 nm and the emission at 499 to 529 nm and for the CD62P marker it was 540 to 570 nm and the emission 577 to 607 nm [17]. Samples were viewed using a Zeiss Axio Observer 7 fluorescent microscope with a Plan-Apochromat 63x/1.4 Oil DIC M27 objective (Carl Zeiss Microscopy, Munich, Germany).
Statistics
Statistical analysis was done using Graphpad Prism 8 (version 8.4.3). All data were subjected to Shapiro-Wilks normality tests. An unpaired T-test was performed on parametric data with the data expressed as mean ± standard deviation, whereas the Mann–Whitney U test was used on unpaired non-parametric data and the data expressed as median [Q1–Q3] (all two-tailed).
Results
Individuals suffering from long COVID in South Africa, were invited to fill in an online South African long COVID registry and they could, in addition, opt to be contacted to provide a blood sample (ethics number for registry: B21/03/001_COVID-19, project ID: 21911). Individuals that served as controls, were also recruited for a blood sample and filled in consent forms as per ethics number N19/03/043, project ID 9521.
The demographics, serum ferritin and IL-6 levels performed on the day of blood collection, self-reported symptom analysis and comorbidities before suffering from acute COVID-19 of our long COVID patients and controls are shown in Table 1 below. Our results follow trends reported on previously [34, 35]. Interestingly, previously it has been reported that misfolded protein aggregates behave as infectious agents [36]. Of interest is that 24% of the participants in our current long COVID cohort, reported to have hypertension and 19% hyperlipidaemia, prior to contracting COVID-19. 24% of our control patients suffered from hypertension, 24% from hyperlipidaemia and 3% (1 volunteer) from cardiovascular disease. The most prevalent long COVID symptoms of the current cohort were noted as constant fatigue (74%) and cognitive impairment [reported by the patients as “brain fog”, forgetfulness, or poor concentration (71%.)] We wish to emphasize the fact that our control patients did not suffer from long COVID and were asymptomatic. The severity of acute COVID-19 disease in our patients suffering from long COVID varied from mild or moderate to severe requiring oxygen or ventilation. We analysed our long COVID cohort and divided them into groups of severity as per the WHO clinical progression scale [31]. Patients with mild disease included asymptomatic patients as well as patients with clinical symptoms but were not hospitalized; patients with moderate disease were hospitalized and included patients that didn’t receive oxygen or received oxygen via face mask or nasal prongs and patients with severe disease were hospitalized and received high flow oxygen or were mechanically ventilated. 71% had mild disease, 5% moderate disease, 17% had severe disease and 7% were unknown (patients that we lost to follow up.) 21% of the long COVID cohort were hospitalized, 72% did not require admission and 7% were unknown. The duration of symptoms from diagnosis to the taking of blood samples from the long COVID cohort was 221 ± 99 days.
We evaluated serum ferritin and IL-6 levels of our high and low responder cohorts and excluded other samples from the analysis. There were no significant differences between long COVID patients and controls in their serum ferritin concentrations. The IL-6 concentrations between high responder long COVID patients and healthy controls were significantly different (see Table 1). We excluded individuals with high Pro-BNP values, as that might point to underlying heart conditions and would influence our results. Some of the most noteworthy molecules we found were VWF, plasma Kallikrein and Platelet Basic Protein or Platelet Factor 4 (PF4.) Fig. 1 depicts stripe plots comparing the unique peptide counts of VWF, plasma Kallikrein and PF4 between patients suffering from hypertension and hyperlipidaemia in our long COVID and control population. This demonstrates that there is no correlation between patients suffering from hypertension or hyperlipidaemia and that it does not serve to differentiate between the groups.

Stripe plots comparing the unique peptide counts of VWF, plasma Kallikrein and PF4 between patients suffering from hypertension and hyperlipidaemia in our long COVID and control population. This demonstrates that there is no correlation between patients suffering from hypertension or hyperlipidaemia and that it doesn’t serve to differentiate between the groups. (Created with BioRender.com.)
We present our proteomics results as fold changes in levels of proteins. Changes are shown as fold changes for the most significant proteins for pair-wise comparisons. The most noteworthy (i.e. differentially observed) molecules of the digested microclots from the long COVID patients, versus the equivalent volume of solubilized plasma from healthy controls are listed in Table 2 below. As described in Methods, we divided the sample into high and low responders and here we present the data from only the high responders (N = 66). Low responders were excluded from further data analysis. We also show the fold changes of immunoglobulins discovered in Table 3. The quantity of immunoglobulins, expressed as a percentage, present in 63 of our 66 long COVID high responder samples and not found in the control group, is shown in Table 4. Unfortunately, 3 of the long COVID samples were not included in the data analysis of the immunoglobulins shown in Table 4. We only show the relevant immunoglobulins/molecules that were present in 13% or more of the samples (13% was an arbitrarily chosen minimum cut-off).
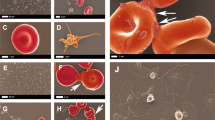
Figure 2 shows activated platelets and fibrin amyloid microclots present in representative blood samples from the current cohort. This was in line with the findings of our previous papers [1, 17, 34, 35].

Fluorescence microscopy showing platelets and microclots in individuals with long COVID and control samples. The first 2 columns show platelets in the haematocrit and the last column shows microclots in PPP. PPP were exposed to ThT, a fluorescent amyloid dye (final concentration: 0.005 mM) (Sigma-Aldrich, St. Louis, MO, USA) for 30 min at room temperature and protected from light. The same scoring system can be utilized to evaluate and interpret the fibrinaloid microclot load severity in PPP as published in our previous work [17]. We are aware that this isn’t a perfect system to aid as a quantitative score for qualitative data. Stage 1 would represent a minimal fibrinaloid microclot load as seen in healthy/control PPP, whereas Stage 4 represents a severe fibrinaloid microclot load. The fluorescent micrographs of representative samples of our long COVID cohort and control patients of haematocrit samples are stained with PAC-1 (green fluorescence) and CD62P-PE (purple fluorescence). The white areas represent areas where the two markers overlap, and the images were taken at 63 × magnification. The scoring system as described by Laubscher and Lourens et al. can be used to evaluate platelet activation and clumping in these fluorescence micrographs. In Stage 1 platelets are minimally activated and are seen as small and round with few pseudopodia (representative of healthy/control platelets). Severe platelet activation, or Stage 4 activation, is characterized by large, egg-shaped morphology with aggregations and is indicative of hyperactivated platelets. Similarly, platelet clumping can also be assessed with no clumping seen in healthy/control samples, which is classified as Stage 1 and severe clumping of platelets seen in Stage 4. Therefore, in our long COVID cohort, the fibrinaloid microclot load severity would fit with a severity of stage 2 to 3, and in some instances even a stage 4 (severe microclot load). Mild to moderate platelet activation (stage 2 to 3), as well as clumping are seen in micrographs D, E, G and H
Discussion
The current proteomics analysis of the content of the fibrin amyloid (plasma protein) microclots we found in our samples, identified increased or decreased levels of inflammatory molecules related to cellular function, coagulation, and lipid metabolism, as well as antibodies trapped inside these microclots. The fluorescent marker used was ThT (demarcated as the green signal in smears of the PPP as seen in Fig. 2). Small areas of protein misfolding will always be present in individuals, for example patients with type II diabetes mellitus, which has been implicated as a protein misfolding disease [36]. However, healthy individuals will also have minor plasma protein misfolding. Long COVID, as a disease, is still so new involves numerous organ systems and affects individuals with numerous comorbidities. In the current paper, we wish to shed more light on the levels of the molecules we found contained within these microclots, albeit increased or decreased and provide researchers and clinicians with a snapshot of the molecules present in patients suffering from long COVID. These molecules may contribute to the symptomatology of patients with long COVID, be involved in the pathophysiology thereof or potentially contribute to autoimmunity in these patients. Hopefully, as researchers piece together the disease pathophysiology, some of the molecules noted in this paper, will give some answers to unravel this very complex condition.
Many regulatory health bodies still do not recognize long COVID (PASC) as a separate disease entity and refer to it under the broad terminology of “COVID”. There is still a lot to learn about long COVID pathophysiology, but our understanding of the disease is represented in Fig. 3, illustrating the optimal time for intervention and disease progression. From the literature, we do know that many individuals that develop long COVID, are those that might suffer from comorbidities, including hypertension, dyslipidaemia, cardiovascular disease, and previous viral infection(s) [37,38,39,40]. Predisposing risk factors or co-morbidities that may also lead to a poor prognosis of acute COVID-19 are well known and include cardiovascular disease, diabetes, arterial hypertension, obesity [12, 13, 41,42,43,44,45], as well as cancer [46].

A representation of our understanding of disease progression and pathophysiology from acute COVID-19 to long COVID. A Depending on the severity of acute COVID-19 disease, dysregulation with increased levels of the following biomarkers have been found, namely P-selectin [47], fibrinogen, D-dimer and VWF [11]. Abnormal clotting and hypercoagulability are seen early in acute COVID-19 disease with the development of thrombocytopaenia and a risk of bleeding as disease severity progresses. Therefore, the optimal time for intervention is early on in acute COVID-19 disease to address the presence of abnormal clotting. B Progression to long COVID disease with patients presenting with debilitating symptoms such as “brainfog,” dyspnoea, chest pain and chronic fatigue and the presence of microclots that may block capillaries with limited passage of red blood cells resulting in reduced O2 and CO2 exchange. Created with BioRender.com
Individuals with previous reports of autoimmune diseases may also be more prone to develop long COVID. We also know that symptoms that develop during acute COVID infection can become chronic or persistent. We have recently discussed reasons for these persistent symptoms and suggested that it can particularly be ascribed to abnormal blood clotting, a dysfunction in clot lysis, and an entrapment of large numbers of inflammatory molecules inside of fibrinolytic-resistant microclots [35]. These microclots, together with large-scale platelet dysfunction and systemic endothelialitis, may possibly result in generalized cellular hypoxia [17, 35]. Cellular hypoxia, resulting from blockages of the capillaries by microclots might explain most of the persistent (albeit multifarious) symptoms that are noted in long COVID patients. When microclots block capillaries, the passage of red blood cells are limited, resulting in a reduced O2 and CO2 exchange [35].
We have previously noted that soluble fibrinogen in blood can clot into an anomalous ‘amyloid’ form of fibrin (fibrinaloids), that is comparatively resistant to fibrinolysis [35]. We have also shown that the reason for failed fibrinolysis, might be due to the presence of various molecules entrapped inside the clots, that might prevent clot breakdown [1, 11, 17]. This failed fibrinolysis phenomenon can explain why individuals with long COVID have extensive insoluble fibrinaloid microclots that can persist in circulation, albeit some are trapped in places that are not observed in a standard blood draw. An important consideration is that current pathology tests measure only the soluble inflammatory molecules in blood and standard pathology blood tests such as a full blood count or CRP (C-reactive protein) appear to be normal in these patients. However, if inflammatory molecules are entrapped inside (insoluble) microclots in circulation, the pathology tests will indeed show that the levels of inflammatory molecules are within normal pathology laboratory concentration ranges. From literature we know that IL-6, D-Dimer and fibrinogen levels are elevated in patients with COVID-19, indicating activation of coagulation pathways and a hypercoagulable state or thrombotic risk [48, 49]. The absence of biomarkers in standard pathology tests, result in a significant amount of confusion for patients and clinicians, as patients are extremely sick or even bed-ridden but with no regular identifiable reason for their disease. Biomarkers that are currently available cannot detect the molecules present in the microclots we identified and are therefore unable to confirm their presence or the mechanisms that drive their formation. Further studies are therefore necessary to develop a method or biomarker that would be able to detect and confirm these microclots in patients with long COVID and therefore the appropriate treatment thereof. Our current study, investigating the composition of these microclots, pave the way for future studies to detect their presence.
Our findings that microclots contain and entrap numerous inflammatory molecules, can immediately explain the reasons why traditional pathology tests such as a CRP or full blood count do not find many of the insoluble inflammatory molecules that are and may directly be involved in the disease presentation, pathophysiology and progression [1, 11, 17, 35].
Fibrin-amyloid microclots represent a novel and potentially important target for understanding their inflammatory content and also a target for the treatment of long COVID and related disorders [35]. If we look closely at the entrapped microclot molecules shown in Table 2 that might be directly relevant for cellular function, coagulation, and lipid metabolism we note both increased and decreased levels of molecules when we compare fold changes between healthy individuals and those with long COVID (see Table 2).
Molecules regulating coagulation
We found increased levels of molecules that may modulate coagulation (see Fig. 4 for the areas in the clotting cascade where these molecules will have physiological activity). The most significant ones were VWF and PF4. VWF is a well-known molecule in vascular (endothelial) injury, platelet-platelet adhesion and is generally involved in hypercoagulation, if found to be increased [11, 50] which could contribute to coagulopathies seen in patients with long COVID. PF4 is a small cytokine belonging to the CXC chemokine family that is also known as chemokine (C-X-C motif) ligand 4 (CXCL4). Platelets store PF4, and it is released from the platelet during activation [51]. Heparin can bind to PF4 and promotes PF4 aggregation, resulting in the formation of PF4/heparin complexes, that have antigenic properties [51]. Recently, it was reported that COVID-19 patients might present with reactivity in PF4/heparin antigen tests without the presence of platelet-activating antibodies [52]. PF4 and VWF may also form complexes; recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation [53]. Recently, in a smaller cohort of samples, we found that α-2AP was increased in long COVID samples [1]. In the current analysis, it was marginally increased. Of significant importance, is that platelets are significantly hyperactivated in long COVID patients. This is noted in both microscopy (as shown in Fig. 2) and the proteomics analysis. Also, it is well-known that individuals with long COVID struggle with anxiety [54, 55]. This was also seen in our long COVID cohort, where 30% of the participants reported anxiety as a symptom (see Table 1). Platelets are well-known for their storage of serotonin (5-HT) [56] and PF4 and serotonin are stored in α- and δ-granules [57]. Circulating immune complexes may also activate platelets via receptor-receptor binding followed by the release of serotonin from platelet granules [57]. Hyperactivated platelets may shed serotonin (and other molecules stored inside platelets), resulting in depletion of serotonin and other molecules stored inside of platelets. The serotonin transporter (SERT or 5HTT) (https://www.ncbi.nlm.nih.gov/books/NBK53687/) is expressed on the plasma membrane of several bodily tissues, for example blood platelets [58, 59], the brain, lungs, bone, gastrointestinal tract as well as the cardiovascular system and transports the neurotransmitter serotonin (5-hydroxytryptamine) across the cell membrane. Prior research suggested that the transport of serotonin (5-HT) via the SERT as well as the storage, metabolism and release mechanisms thereof are similar in platelets and serotonergic neurons. Serotonin depletion may be a significant contributor to the depressed mood or anxiety noted in individuals suffering from long COVID.

The coagulation pathway to demonstrate the areas of action of the molecules involved in coagulation in our long COVID cohort compared to controls. Activation is illustrated by a line and arrow and inhibition by a line ending in a circular end. The Uniprot ID numbers of our molecules of interest were entered on the David Bioinformatics website and converted to an Entrez_Gene_ID gene list. The generated list was used to search the DAVID Bioinformatics website for molecular pathways where these molecules could potentially play a role. Here we demonstrate the coagulation cascade found on the KEGG pathway database. https://david.ncifcrf.gov/ and https://www.genome.jp/kegg/pathway.html. Diagram created with BioRender.com
Plasma kallikrein (KLKB1) levels were found to be decreased in the current long COVID cohort, and is known to activate factor XII, and it also belongs to a subgroup of trypsin-like serine proteases, enzymes capable of cleaving peptide bonds in proteins [60]. It is also involved in the bradykinin pathway, that is seen as an inflammatory product of the coagulation system [61]. Both neutrophil and mast-cell activation are functionally linked to the bradykinin pathways [61], and mast cell activation is a known feature of PASC, consistent with the known benefits of antihistamine treatment [62] KLKB1 also digests plasminogen to plasmin and participates in surface-dependent activation of blood coagulation, fibrinolysis, and inflammation. Under physiological conditions, plasma kallikrein serves as a cardioprotective enzyme, but when it is increased in circulation, it is involved in cardiovascular disease [63]. A decrease in the level of this molecule may prevent adequate plasminogen activation and may directly impact downstream on clot breakdown and therefore fibrinolysis (see Fig. 5.)

A Progression from acute COVID-19 to B long COVID and the persistence of microclots and a general hypoxic state, accompanied by increased circulating antibodies, proteins related to cellular function and liver protein dysfunction (C)
Molecules regulating cellular function
Molecules that modulate cellular function that we found to be increased, included galectin-3-binding protein, Thrombospondin-1, Alpha-1-acid glycoprotein-2 as well as Inter-alpha-trypsin inhibitor heavy chain H1 and 2 (ITIH1 and ITIH2). Galectin-3 expression is known to be induced in viral infections and by a multitude of molecules that either mimic or are characteristic of ongoing inflammation and microbial infection, such as IFN-α, IFN-β, IFN-γ, TNF-α, poly(I:C), dsRNA, and dsDNA [64]. In addition, it is commonly overexpressed in most types of cancers [65] and is an important player in cancer development, progression, and metastasis via its interactions with galactoside-terminated glycans [66]. Furthermore, it interacts with the cell-surface glycoprotein CD146 (MCAM, MUC18) and induces secretion of metastasis-promoting cytokines from vascular endothelial cells [66]. Recently, it was also identified as a novel predictor of venous thromboembolism in systemic lupus erythematosus [67].
Thrombospondin-1 was also found to be increased in our cohort of long COVID participants. It is an adhesive glycoprotein that is released from platelet alpha-granules in response to thrombin stimulation and is also a transient component of the extracellular matrix in developing and repairing tissues [68]. Recently it was suggested that it might regulate haemostasis in vivo through modulation of platelet cAMP signalling at sites of vascular injury [69], and that it could mark a prothrombotic state and an underlying inflammatory process [70]. It is also increased in the tumour microenvironment [64] and might be involved in antiphospholipid syndrome [70], a condition well known to be associated with thrombosis.
Alpha-1-acid glycoprotein-2, which we also found to be elevated, is known to modulate the immune system during the acute-phase reaction. ITIH1 and ITIH2 were found to be elevated in our long COVID population. These molecules belong to the inter-α-trypsin inhibitors. Previously, it was reported that ITIH1 and ITIH2 were more abundant in a COVID-19 survivor group [71]. ITIH1 has previously been found to be elevated in patients with osteoarthritis [72]. ITIH1 is synthesized by chondrocytes and binds to hyaluronic acid and also to the extracellular matrix [73]. ITIH2 has been associated with demyelinating diseases [74].
We found decreased levels of a few molecules, namely Long palate, lung and nasal epithelium carcinoma-associated protein 1 (LPLC1_HUMAN,) Lactotransferrin, Adiponectin and Alpha-1-acid glycoprotein 1 (A1AG1_HUMAN) that might point to a type of immunosuppression. Our research group also suggested that Lactoferrin may be beneficial for preventing adverse effects of acute COVID-19, mainly due to its antiviral properties [75]. Previously, it was found that some critically ill COVID-19 patients could not be discharged from the ICU even though they exhibited negative viral tests [76]. Zhou and co-workers in 2021 suggested that in such patients, adaptive immunosuppression might be present, and ascribed it to a dysregulated host response to severe infection, similar to the immunosuppression that exists in patients with sepsis [76]. In long COVID, there has also been reports of immune suppression [54].
Of importance may be LPLC1, which may play an important role in innate immunity, as its function is related to binding of LPS, as a LPS-binding protein (https://www.uniprot.org/uniprot/C3TTP7). If LPS is not bound effectively, it may have significant implications for the response to LPS in circulation. This could possibly indicate an altered immune response to infectious pathogens. Related to LPS binding, is Lactotransferrin also known as lactoferrin (LF,) that was also decreased in our long COVID cohort. LF has important immunological properties and is both antibacterial and antiviral. In particular, there is evidence that it can bind to at least some of the receptors used by coronaviruses and thereby block their entry [75]. LF makes use of Heparan Sulfate Proteoglycans (HSPGs) on the surface of epithelial cells of the stomach to facilitate entry into the bloodstream via endocytosis [77, 78]. Coronaviruses bind to host cells by first attaching to HSPGs utilizing them as preliminary docking sites to assist with viral entry and LF is able to interfere with some of the receptors used by coronaviruses. LF can inhibit the entry of viral particles into host cells by blocking cellular receptors or via direct attachment to viral particles. In COVID-19 infection, LF might be able to competitively bind receptors as well as HSPGs and therefore prevent viral entry and the subsequent cytokine storm [75] but this still requires further investigation. Our finding of decreased levels of lactoferrin possibly indicates suppression of the host immune response in patients with long COVID.
Adiponectin has anti-inflammatory and anti-apoptotic properties that has shown to protect the vasculature, heart, lung, and colon [79], and was also reduced in our long COVID population. Pro-inflammatory and anti-inflammatory cytokines or adipokines namely adiponectin, leptin and resistin may be released from adipose tissue [80]. Adiponectin exists in two isoforms namely HMW (high molecular weight) and LMW (low molecular weight) adiponectin and is an important regulator of cytokine immune responses. These cytokine responses are isoform specific. IL-6 secretion by human monocytes and human monocytic leukemia cell line cells are stimulated by HMW adiponectin and does not inhibit LPS-induced IL-6 secretion. On the other hand, LMW adiponectin promotes IL-10 secretion and inhibits LPS-mediated IL-6 release. For example, in chronic hepatitis C infection these cytokines play an important role in the inflammatory and immune response of affected individuals. This could explain the different outcomes seen in hepatitis C infection. Some of the metabolic and inflammatory effects of adiponectin are regulated through the mitogen-activated protein kinase (MAPK) signalling pathway [80]. The MAPK pathway forms a crucial part in the regulation of IFN-α and INF-γ signalling in viral infections such as hepatitis C. The reduced level of adiponectin in our long COVID cohort also potentially points to an altered immune response in infected patients.
Alpha-1-acid glycoprotein 1 is an acute-phase protein [81] and functions as a transport protein in the blood stream, and it was found to be reduced in our long COVID participants. It is known to regulate inflammation, including binding of pathogens and modulating white blood cells’ activity throughout the entire leukocyte attacking sequence [82]. This molecule is also known to modulate the immune system during the acute-phase reaction and decreased levels are related to the mortality of septic patients [83]. The decreased levels seen in the current cohort, might possibly indicate a dysregulated or abnormal immune or inflammatory response. It may therefore also be a predictor of morbidity and mortality in patients with COVID-19. If we look at the physiological processes that are regulated by the aforementioned molecules, the increased or reduced levels of these molecules will have an effect on cellular function and the potentially dysregulated immune response in these patients. They also have immune-related or cellular remodelling properties and may impact on vascular function.
Molecules regulating lipid metabolism and regulation
We also found interesting apolipoproteins, known to play a role in lipid metabolism and regulation. Apolipoprotein C-II (a component of very low density lipoproteins and chylomicrons) was found to be increased while Apolipoprotein A-II (the second most abundant protein of the high density lipoprotein particles) was reduced in our cohort. Previous studies support the crucial role that lipids and lipid metabolism play in SARS CoV-2 infection [84,85,86]. Shen et al. performed proteomic and metabolomic profiling on the serum of 46 COVID-19 patients compared to 53 controls [84]. They found the downregulation of over 100 lipids, including sphingolipids, glycerophospholipids and fatty acids which correlated with disease severity in patients with COVID-19. Lipids, including sphingolipids and glycerophospholipids form an essential part of biomembranes. Specialized membrane domains exist in the plasma membrane and sphingolipids are an example of this. Sphingolipids are also involved in the regulation of signal transduction, immune activation pathways and inflammatory responses [87]. The downregulation of lipids in the sera of patients suffering from COVID-19 points to liver damage and changes in bilirubin and bile acids supports this. Song et al. studied the lipidome and metabolome of 50 patients with different severities (mild, moderate, and severe) of SARS CoV2 infection compared to 26 healthy controls by utilizing targeted and untargeted mass spectrometry of plasma samples that were collected [85]. They noted reductions of major classes of plasma glycerophospholipids, phosphatidic acids (PAs), phosphatidylinositols (PIs), and phosphatidylcholines (PCs) and increased concentrations of lysophospholipids namely lysophosphatidic acids (LPAs,) lysophosphatidylinositols (LPIs,) and lysophosphatidylcholines (LPCs.) Another finding was that the plasma lipidome of patients with COVID19 had similarities to monosialodihexosyl ganglioside (GM3)-enriched exosomes where levels of sphingomyelins (SMs) and GM3s are elevated, and diacylglycerols (DAGs) decreased. Increasing levels of GM3s were seen with progressive disease severity indicating that GM3-rich exosomes also play a role in the pathophysiology of COVID19.
From the literature it is evident that fluctuations in DGs (diglycerides,) FFAs (free fatty acids), and TGs (triglycerides) occur in pathological conditions. For example, increased lipolysis of adipose tissue is seen in Ebola virus disease where TGs are converted to FFAs and DGs, with increased recycling of fatty acids back into TGs [88, 89]. Bruzzone et al. found elevated levels of serum TG, TG-VLDL (VLDL, very low-density lipoproteins), TG-IDL (IDL, intermediate density lipoproteins), TG-LDL (LDL, low-density lipoproteins) and TG-HDL (HDL, high density lipoproteins) in patients with COVID-19 in contrast to a decreased total cholesterol (TC) concentration. They propose that serum accumulation of TG and TG-VLDL may indicate decreased oxidation of acetyl-CoA in the mitochondria of the liver. Ketone bodies are produced by the liver from fatty acid oxidation-derived acetyl-CoA [90]. This pathogenic redistribution of the lipoprotein particle size and composition increases the cardiovascular risk for patients with COVID-19 infection.
Literature supports the role that apolipoproteins and steroid hormones have to play in SARS CoV-2 infection. Shen and co-workers in 2020 [84] found decreased levels of apolipoproteins APOA1, APOA2, APOH, APOL1, APOD, and APOM in the serum of patients following SARS CoV2 infection. It has also previously been reported that APOA1 levels decreased when patients progressed from mild to severe disease [91]. These apolipoproteins are involved in the functioning of macrophages [92]. Arachidonic acid (AA) is an essential fatty acid. Prostaglandins, leukotrienes and thromboxanes are pro-inflammatory metabolites of AA and eicosapentaenoic acid (EPA). Lipoxins, resolvins, protectins and maresins are derivates of AA, EPA and docosahexaenoic acid (DHA) and are able to suppress the inflammatory response, enhance phagocytosis of macrophages as well as other immune cells and assist with microbial clearance [92, 93]. Therefore, lipids such as glycerophospholipids and fatty acids play a pivotal role in inflammation and the host immune response against viral infections.
In summary, lipid metabolism plays an integral role in COVID-19 disease, resulting in an imbalance between immune-regulating lipids, pro-inflammatory lipids and lipid mediators during the host immune response. It is evident that our findings of increased levels of Apolipoprotein C-II and reduced levels of Apolipoprotein A-II also supports this dysregulation of lipid metabolism in patients suffering from COVID-19 (see Fig. 5).
Immunoglobulins of interest
Table 3 shows selected immunoglobulins involved in the immune response and that might act as antibodies or autoantibodies. As can be noted in Table 3, there are numerous molecules with no data available on their function. However, we do list these molecules here, as it may be of value for immunologists to note the presence of these potentially important antibodies. Of specific interest was the presence of Immunoglobulin kappa chain V-III region POM (plasma membrane), that is known as rheumatoid factor (RF) and that may act as an autoantibody [33]. Another RF factor that was found to be elevated was Immunoglobulin kappa chain V-III region CLL.
Of significance also, was that there were numerous antibodies only present in 63 of our long COVID high responder samples and we present those in Table 4 that were present in 13% or more (arbitrary chosen cut-off) of the cohort. IG c1641_light_IGKV1-6_IGKJ4 (Fragment), IGL c3450_light_IGKV1-39_IGKJ1 (Fragment) and IGH c2558_heavy_IGHV3-20_IGHD2-2_IGHJ2 (Fragment) were found to be present in 27%, 24% and 22% of the 63 samples respectively. These antibodies were cross referenced on THE INTERNATIONAL IMMUNOGENETICS INFORMATION SYSTEM®’s IMGT/LIGM-DB database, but no data are available on these antibodies. These antibodies may have significance in the immune response following acute COVID-19 illness or driving auto-immunity in some long COVID participants.
Another important consideration is that patients with long-term persistent microclots, may also progress to develop auto-immune diseases. We suggest that this phenomenon might directly be due to autoantibodies that develop against antibodies or other molecules that are entrapped inside the fibrinaloid microclots. It appears that the long-term sequelae of many viral infections [94, 95], including long COVID [96, 97] may be attributed to autoimmunity. These microclots contain many different proteins that may present new epitopes if a conformational change has taken place. These epitopes may be recognized as ‘non-self’ eliciting an immune response with the possible development of autoantibodies. Additionally, oxidative stress induces the nitration of proteins leading to the production of autoantibodies. Autoantibodies formed against actin lead to muscle weakness [98]. In ME/CFS (Myalgic Encephalomyelitis/Chronic Fatigue Syndrome), autoimmunity is well described [94]; and might also play an important role in long COVID because some autoantibodies share elements of epitope with the SARS-CoV-2 virus [99]. It has also been suggested that some of the viral sequences are amyloidogenic [100, 101]. Viral persistence may play a key role in patients suffering from long COVID [102], and therefore driving an ongoing immune response or the development of autoimmunity (see Fig. 5).
Pascolini et al. studied 33 consecutive patients with COVID-19 of whom 94% had interstitial pneumonia and compared them to 25 age- and sex-matched controls with fever and/or pneumonia of an etiology other than COVID-19 to evaluate for autoimmune serological markers [103]. 45% of patients with COVID-19 tested positive for at least one autoantibody and they found that 33% tested positive for antinuclear antibodies (ANAs), 24% tested positive for anti-cardiolipin antibodies (immunoglobulin (Ig)G and/or IgM) and 9% tested positive for anti-β2-glycoprotein antibodies (IgG and/or IgM.) Antineutrophil cytoplasmic antibodies (ANCA) were not present in any of their patients. A higher mortality rate was seen amongst patients with autoantibodies compared to those without.
Chang and coworkers searched for autoantibodies in hospitalized patients with COVID-19 and found that approximately 50% of hospitalized patients developed autoantibodies against one or more antigens in the array they tested for with 25% of patients being ANA positive [104]. They also discovered antibodies that are associated with relatively rare connective tissue diseases and have been predicted to be pathogenic. A large number of anti-cytokine antibodies (ACA) were identified as well as antibodies recognizing non-structural SARS-CoV-2 proteins which correlate with autoantibodies. A few of the antibodies they discovered include Anti-C1q, which is a SLE autoantigen, anti-β2GP1 (anti-beta 2 glycoprotein 1) a thrombogenic antibody, anti-BPI (anti-bactericidal permeability inducing protein) and anti-ACE2 antibodies.
Conversely, a study performed by Klein et al. suggest that autoantibodies have a limited role to play in the pathogenesis of long COVID [105]. Their findings point to the involvement of persistent viral antigen, reactivation of latent herpesviruses, and chronic inflammation in long COVID pathophysiology. There have been previous reports of persistent viral antigen in intestinal biopsies of patients recovering from COVID-19 infection and antigen persistence may therefore contribute to the host immune response in patients with long COVID [106, 107].
Conclusion
The present proteomics study provides further evidence of the importance of not only looking at the soluble inflammatory molecules present in blood samples of individuals who suffer from long COVID. Insoluble inflammatory molecules and immunoglobulins may be of significant importance in not only causing the persistent long COVID symptoms, but also eventually leading to these individuals developing auto-immune diseases. An important consideration is to resolve these microclots, address platelet hyperactivation and wide-spread vascular inflammation preferably in the acute phase of the disease. In that way auto-immunity and the resulting severe and debilitating symptoms due to these pathologies, might be prevented. In addition, if the microclots are resolved early in the disease, general hypoxia that may lead to irreversible tissue damage, could possibly be prevented. Such pathologies may have significant implications if patients already have co-morbidities such as type II diabetes or cardiovascular disease. There is therefore an urgent need for randomized controlled clinical trials to target the prevention or resolution of these microclots in circulation and reversal of the underlying associated endotheliopathy.
Availability of data and materials
The datasets generated as well as figure micrographs analyzed during the current study are available on request. The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
References
Pretorius E, Vlok M, Venter C, Bezuidenhout JA, Laubscher GJ, Steenkamp J, Kell DB. Persistent clotting protein pathology in long COVID/post-acute sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. 2021;20(1):172.
Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of 1 biological factors that may contribute to persistent symptoms. Front Microbiol. 2021. https://doi.org/10.3389/fmicb.2021.698169.
Deer RR, Rock MA, Vasilevsky N, Carmody L, Rando H, Anzalone AJ, Basson MD, Bennett TD, Bergquist T, Boudreau EA, et al. Characterizing long COVID: deep phenotype of a complex condition. EBioMedicine. 2021;74: 103722.
Davis HE, Assaf GS, McCorkell L, Wei H, Low RJ, Re’em Y, Redfield S, Austin JP, Akrami A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine. 2021;38: 101019.
Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7.
Yang D, Han Z, Oppenheim JJ. Alarmins and immunity. Immunol Rev. 2017;280(1):41–56.
Giani M, Seminati D, Lucchini A, Foti G, Pagni F. Exuberant plasmocytosis in bronchoalveolar lavage specimen of the first patient requiring extracorporeal membrane oxygenation for SARS-CoV-2 in Europe. J Thorac Oncol. 2020;15(5):e65–6.
Monteleone G, Sarzi-Puttini PC, Ardizzone S. Preventing COVID-19-induced pneumonia with anticytokine therapy. Lancet Rheumatol. 2020;2(5):e255–6.
Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A, Peccatori J, D’Angelo A, De Cobelli F, Rovere-Querini P, et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis. Crit Care Resusc. 2020;22(2):95–7.
Higgins V, Sohaei D, Diamandis EP, Prassas I. COVID-19: from an acute to chronic disease? Potential long-term health consequences. Crit Rev Clin Lab Sci. 2021;58(5):297–310.
Grobler C, Maphumulo SC, Grobbelaar LM, Bredenkamp JC, Laubscher GJ, Lourens PJ, Steenkamp J, Kell DB, Pretorius E. Covid-19: the rollercoaster of fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-selectin and their interactions with endothelial cells, platelets and erythrocytes. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21145168.
Pretorius E, Venter C, Laubscher GJ, Lourens PJ, Steenkamp J, Kell DB. Prevalence of readily detected amyloid blood clots in “unclotted” Type 2 Diabetes Mellitus and COVID-19 plasma: a preliminary report. Cardiovasc Diabetol. 2020;19(1):193.
Venter C, Bezuidenhout JA, Laubscher GJ, Lourens PJ, Steenkamp J, Kell DB, Pretorius E. Erythrocyte, platelet, serum ferritin, and P-selectin pathophysiology implicated in severe hypercoagulation and vascular complications in COVID-19. Int J Mol Sci. 2020. https://doi.org/10.3390/ijms21218234.
Görlinger K, Levy JH. COVID-19–associated coagulopathy: less fibrinolysis can be more harmful! Anesthesiology. 2021;134(3):366–9.
Libby P, Lüscher T. COVID-19 is, in the end, an endothelial disease. Eur Heart J. 2020;41(32):3038–44.
Wool GD, Miller JL. The impact of COVID-19 disease on platelets and coagulation. Pathobiology. 2021;88(1):15–27.
Laubscher GJ, Lourens PJ, Venter C, Kell DB, Pretorius E. TEG®, microclot and platelet mapping for guiding early management of severe COVID-19 coagulopathy. J Clin Med. 2021. https://doi.org/10.3390/jcm10225381.
Kell DB, Pretorius E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog Biophys Mol Biol. 2017;123:16–41.
Willyard C. Could tiny blood clots cause long COVID’s puzzling symptoms? Nature. 2022;608(7924):662–4.
Wallukat G, Hohberger B, Wenzel K, Fürst J, Schulze-Rothe S, Wallukat A, Hönicke AS, Müller J. Functional autoantibodies against G-protein coupled receptors in patients with persistent long-COVID-19 symptoms. J Transl Autoimmun. 2021;4: 100100.
NewsCAP: Autoantibody reactivity implicated in 'long' COVID-19. Am J Nurs 2021, 121(3):17.
Bertin D, Kaphan E, Weber S, Babacci B, Arcani R, Faucher B, Ménard A, Brodovitch A, Mege JL, Bardin N. Persistent IgG anticardiolipin autoantibodies are associated with post-COVID syndrome. Int J Infect Dis. 2021;113:23–5.
Barsnes H, Vaudel M. SearchGUI: a highly adaptable common interface for proteomics search and de novo engines. J Proteome Res. 2018;17(7):2552–5.
Rigden DJ, Fernández XM. The 27th annual nucleic acids research database issue and molecular biology database collection. Nucleic Acids Res. 2020;48(D1):D1-d8.
Page MJ, Thomson GJA, Nunes JM, Engelbrecht AM, Nell TA, de Villiers WJS, de Beer MC, Engelbrecht L, Kell DB, Pretorius E. Serum amyloid A binds to fibrin(ogen), promoting fibrin amyloid formation. Sci Rep. 2019;9(1):3102.
Pretorius E, Mbotwe S, Kell DB. Lipopolysaccharide-binding protein (LBP) reverses the amyloid state of fibrin seen in plasma of type 2 diabetics with cardiovascular co-morbidities. Sci Rep. 2017;7(1):9680.
Pretorius E, Page MJ, Engelbrecht L, Ellis GC, Kell DB. Substantial fibrin amyloidogenesis in type 2 diabetes assessed using amyloid-selective fluorescent stains. Cardiovasc Diabetol. 2017;16(1):141.
Pretorius E, Mbotwe S, Bester J, Robinson CJ, Kell DB. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J R Soc Interface. 2016. https://doi.org/10.1098/rsif.2016.0539.
WHO. A minimal common outcome measure set for COVID-19 clinical research. Lancet Infect Dis. 2020; 20(8):e192–e197.
Stamatopoulos K, Belessi C, Hadzidimitriou A, Smilevska T, Kalagiakou E, Hatzi K, Stavroyianni N, Athanasiadou A, Tsompanakou A, Papadaki T, et al. Immunoglobulin light chain repertoire in chronic lymphocytic leukemia. Blood. 2005;106(10):3575–83.
Kobayashi R, Rassenti LZ, Meisenholder G, Carson DA, Kipps TJ. Autoantigen inhibits apoptosis of a human B cell leukemia that produces pathogenic rheumatoid factor. J Immunol. 1993;151(12):7273–83.
Pretorius E, Venter C, Laubscher GJ, Kotze MJ, Oladejo SO, Watson LR, Rajaratnam K, Watson BW, Kell DB. Prevalence of symptoms, comorbidities, fibrin amyloid microclots and platelet pathology in individuals with Long COVID/Post-Acute Sequelae of COVID-19 (PASC). Cardiovasc Diabetol. 2022;21(1):148. https://doi.org/10.1186/s12933-022-01579-5. PMID: 35933347; PMCID: PMC9356426.
Kell DB, Laubscher GJ, Pretorius E. A central role for amyloid fibrin microclots in long COVID/PASC: origins and therapeutic implications. Biochem J. 2022;479(4):537–59.
Mukherjee A, Morales-Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015;21(7):439–49.
Sudre CH, Murray B, Varsavsky T, Graham MS, Penfold RS, Bowyer RC, Pujol JC, Klaser K, Antonelli M, Canas LS, et al. Attributes and predictors of long COVID. Nat Med. 2021;27(4):626–31.
Fernández-de-Las-Peñas C, Guijarro C, Torres-Macho J, Velasco-Arribas M, Plaza-Canteli S, Hernández-Barrera V, Arias-Navalón JA. Diabetes and the risk of long-term post-COVID symptoms. Diabetes. 2021;70(12):2917–21.
Khunti K, Davies MJ, Kosiborod MN, Nauck MA. Long COVID—metabolic risk factors and novel therapeutic management. Nat Rev Endocrinol. 2021;17(7):379–80.
Su Y, Yuan D, Chen DG, Ng RH, Wang K, Choi J, Li S, Hong S, Zhang R, Xie J, et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell. 2022;185(5):881-895.e820.
Finer N, Garnett SP, Bruun JM. COVID-19 and obesity. Clin Obes. 2020;10(3): e12365.
Yates T, Razieh C, Zaccardi F, Davies MJ, Khunti K. Obesity and risk of COVID-19: analysis of UK biobank. Prim Care Diabetes. 2020;14(5):566–7.
Sattar N, Ho FK, Gill JM, Ghouri N, Gray SR, Celis-Morales CA, Katikireddi SV, Berry C, Pell JP, McMurray JJ, et al. BMI and future risk for COVID-19 infection and death across sex, age and ethnicity: preliminary findings from UK biobank. Diabetes Metab Syndr. 2020;14(5):1149–51.
Gerotziafas GT, Catalano M, Colgan MP, Pecsvarady Z, Wautrecht JC, Fazeli B, Olinic DM, Farkas K, Elalamy I, Falanga A, et al. Guidance for the management of patients with vascular disease or cardiovascular risk factors and COVID-19: position paper from VAS-European Independent Foundation in Angiology/Vascular Medicine. Thromb Haemost. 2020;120(12):1597–628.
Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, Bi Z, Zhao Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin Res Cardiol. 2020;109(5):531–8.
Malek AE, Raad II, Jabbour E. Cancer and COVID-19. Lancet. 2020;396(10257):1066–7.
Neri T, Nieri D, Celi A. P-selectin blockade in COVID-19-related ARDS. Am J Physiol Lung Cell Mol Physiol. 2020;318(6):L1237-l1238.
Coomes EA, Haghbayan H. Interleukin-6 in COVID-19: a systematic review and meta-analysis. Rev Med Virol. 2020;30(6):1–9.
Rostami M, Mansouritorghabeh H. D-dimer level in COVID-19 infection: a systematic review. Expert Rev Hematol. 2020;13(11):1265–75.
Lancellotti S, Sacco M, Basso M, De Cristofaro R. Mechanochemistry of von Willebrand factor. Biomol Concepts. 2019;10(1):194–208.
Nevzorova TA, Mordakhanova ER, Daminova AG, Ponomareva AA, Andrianova IA, Le Minh G, Rauova L, Litvinov RI, Weisel JW. Platelet factor 4-containing immune complexes induce platelet activation followed by calpain-dependent platelet death. Cell Death Discov. 2019;5:106.
Brodard J, Kremer Hovinga JA, Fontana P, Studt JD, Gruel Y, Greinacher A. COVID-19 patients often show high-titer non-platelet-activating anti-PF4/heparin IgG antibodies. J Thromb Haemost. 2021;19(5):1294–8.
Johnston I, Sarkar A, Hayes V, Koma GT, Arepally GM, Chen J, Chung DW, López JA, Cines DB, Rauova L, et al. Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood. 2020;135(15):1270–80.
Oronsky B, Larson C, Hammond TC, Oronsky A, Kesari S, Lybeck M, Reid TR. A review of persistent post-COVID syndrome (PPCS). Clin Rev Allergy Immunol. 2021. https://doi.org/10.1007/s12016-021-08848-3.
Proal AD, VanElzakker MB. Long COVID or post-acute sequelae of COVID-19 (PASC): an overview of biological factors that may contribute to persistent symptoms. Front Microbiol. 2021;12: 698169.
Lee J, Kang Y, Chang J, Song J, Kim BK. Determination of serotonin concentration in single human platelets through single-entity electrochemistry. ACS Sens. 2020;5(7):1943–8.
Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, Levesque T, Becker Y, Tessandier N, Melki I, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A. 2018;115(7):E1550-e1559.
Keyes SR, Rudnick G. Coupling of transmembrane proton gradients to platelet serotonin transport. J Biol Chem. 1982;257(3):1172–6.
Lesch KP, Wolozin BL, Murphy DL, Reiderer P. Primary structure of the human platelet serotonin uptake site: identity with the brain serotonin transporter. J Neurochem. 1993;60(6):2319–22.
Xie Z, Li Z, Shao Y, Liao C. Discovery and development of plasma kallikrein inhibitors for multiple diseases. Eur J Med Chem. 2020;190: 112137.
Hofman Z, de Maat S, Hack CE, Maas C. Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol. 2016;51(2):152–61.
Weinstock LB, Brook JB, Walters AS, Goris A, Afrin LB, Molderings GJ. Mast cell activation symptoms are prevalent in long-COVID. Int J Infect Dis. 2021;112:217–26.
Kolte D, Shariat-Madar Z. Plasma Kallikrein inhibitors in cardiovascular disease: an innovative therapeutic approach. Cardiol Rev. 2016;24(3):99–109.
Loimaranta V, Hepojoki J, Laaksoaho O, Pulliainen AT. Galectin-3-binding protein: a multitask glycoprotein with innate immunity functions in viral and bacterial infections. J Leukoc Biol. 2018;104(4):777–86.
Sindrewicz P, Yates EA, Turnbull JE, Lian LY, Yu LG. Interaction with the heparin-derived binding inhibitors destabilizes galectin-3 protein structure. Biochem Biophys Res Commun. 2020;523(2):336–41.
Colomb F, Wang W, Simpson D, Zafar M, Beynon R, Rhodes JM, Yu LG. Galectin-3 interacts with the cell-surface glycoprotein CD146 (MCAM, MUC18) and induces secretion of metastasis-promoting cytokines from vascular endothelial cells. J Biol Chem. 2017;292(20):8381–9.
Peretz ASR, Rasmussen NS, Jacobsen S, Sjöwall C, Nielsen CT. Galectin-3-binding protein is a novel predictor of venous thromboembolism in systemic lupus erythematosus. Clin Exp Rheumatol. 2021;39(6):1360–8.
Adams JC. Thrombospondin-1. Int J Biochem Cell Biol. 1997;29(6):861–5.
Aburima A, Berger M, Spurgeon BEJ, Webb BA, Wraith KS, Febbraio M, Poole AW, Naseem KM. Thrombospondin-1 promotes hemostasis through modulation of cAMP signaling in blood platelets. Blood. 2021;137(5):678–89.
Patsouras M, Tsiki E, Karagianni P, Vlachoyiannopoulos PG. The role of thrombospondin-1 in the pathogenesis of antiphospholipid syndrome. J Autoimmun. 2020;115: 102527.
Völlmy F, van den Toorn H, Zenezini Chiozzi R, Zucchetti O, Papi A, Volta CA, Marracino L, Vieceli Dalla Sega F, Fortini F, Demichev V, et al. A serum proteome signature to predict mortality in severe COVID-19 patients. Life Sci Alliance. 2021;4(9):e202101099. https://doi.org/10.26508/lsa.202101099.
Lourido L, Ayoglu B, Fernández-Tajes J, Oreiro N, Henjes F, Hellström C, Schwenk JM, Ruiz-Romero C, Nilsson P, Blanco FJ. Discovery of circulating proteins associated to knee radiographic osteoarthritis. Sci Rep. 2017;7(1):137.
Yoshihara Y, Plaas A, Osborn B, Margulis A, Nelson F, Stewart M, Rugg MS, Milner CM, Day AJ, Nemoto K, et al. Superficial zone chondrocytes in normal and osteoarthritic human articular cartilages synthesize novel truncated forms of inter-alpha-trypsin inhibitor heavy chains which are attached to a chondroitin sulfate proteoglycan other than bikunin. Osteoarthr Cartil. 2008;16(11):1343–55.
Solmaz I, Kocak E, Kaplan O, Celebier M, Anlar B. Analysis of plasma protein biomarkers in childhood onset multiple sclerosis. J Neuroimmunol. 2020;348: 577359.
Kell DB, Heyden EL, Pretorius E. the biology of lactoferrin, an iron-binding protein that can help defend against viruses and bacteria. Front Immunol. 2020;11:1221.
Zhou Y, Liao X, Song X, He M, Xiao F, Jin X, Xie X, Zhang Z, Wang B, Zhou C, et al. Severe adaptive immune suppression may be why patients with severe COVID-19 cannot be discharged from the ICU even after negative viral tests. Front Immunol. 2021;12: 755579.
Harada E, Itoh Y, Sitizyo K, Takeuchi T, Araki Y, Kitagawa H. Characteristic transport of lactoferrin from the intestinal lumen into the bile via the blood in piglets. Comp Biochem Physiol A Mol Integr Physiol. 1999;124(3):321–7.
Matsuzaki T, Nakamura M, Nogita T, Sato A. Cellular uptake and release of intact lactoferrin and its derivatives in an intestinal enterocyte model of Caco-2 Cells. Biol Pharm Bull. 2019;42(6):989–95.
Fang H, Judd RL. Adiponectin regulation and function. Compr Physiol. 2018;8(3):1031–63.
Palmer C, Hampartzoumian T, Lloyd A, Zekry A. A novel role for adiponectin in regulating the immune responses in chronic hepatitis C virus infection. Hepatology. 2008;48(2):374–84.
Hochepied T, Berger FG, Baumann H, Libert C. Alpha(1)-acid glycoprotein: an acute phase protein with inflammatory and immunomodulating properties. Cytokine Growth Factor Rev. 2003;14(1):25–34.
Ceciliani F, Lecchi C. The immune functions of α(1) acid glycoprotein. Curr Protein Pept Sci. 2019;20(6):505–24.
Barroso-Sousa R, Lobo RR, Mendonça PR, Memória RR, Spiller F, Cunha FQ, Pazin-Filho A. Decreased levels of alpha-1-acid glycoprotein are related to the mortality of septic patients in the emergency department. Clinics (Sao Paulo). 2013;68(8):1134–9.
Shen B, Yi X, Sun Y, Bi X, Du J, Zhang C, Quan S, Zhang F, Sun R, Qian L, et al. Proteomic and metabolomic characterization of COVID-19 patient sera. Cell. 2020;182(1):59-72.e15.
Song JW, Lam SM, Fan X, Cao WJ, Wang SY, Tian H, Chua GH, Zhang C, Meng FP, Xu Z, et al. Omics-driven systems interrogation of metabolic dysregulation in COVID-19 pathogenesis. Cell Metab. 2020;32(2):188-202.e185.
Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, Hudson KE, Zimring JC, Hansen KC, Hod EA, Spitalnik SL, D'Alessandro A. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight. 2020;5(14):e140327. https://doi.org/10.1172/jci.insight.140327. PMID: 32559180; PMCID: PMC7453907.
Hannun YA, Obeid LM. Sphingolipids and their metabolism in physiology and disease. Nat Rev Mol Cell Biol. 2018;19(3):175–91.
Wu D, Shu T, Yang X, Song JX, Zhang M, Yao C, Liu W, Huang M, Yu Y, Yang Q, et al. Plasma metabolomic and lipidomic alterations associated with COVID-19. Natl Sci Rev. 2020;7(7):1157–68.
Kyle JE, Burnum-Johnson KE, Wendler JP, Eisfeld AJ, Halfmann PJ, Watanabe T, Sahr F, Smith RD, Kawaoka Y, Waters KM, et al. Plasma lipidome reveals critical illness and recovery from human Ebola virus disease. Proc Natl Acad Sci U S A. 2019;116(9):3919–28.
Bruzzone C, Bizkarguenaga M, Gil-Redondo R, Diercks T, Arana E, Garcíade Vicuña A, Seco M, Bosch A, Palazón A, San Juan I, et al. SARS-CoV-2 infection dysregulates the metabolomic and lipidomic profiles of serum. iScience. 2020;23(10): 101645.
Nie S, Zhao X, Zhao K, Zhang Z, Zhang Z, Zhang Z: Metabolic disturbances and inflammatory dysfunction predict severity of coronavirus disease 2019 (COVID-19): a retrospective study. medRxiv. 2020:2020.2003.2024.20042283.
Casari I, Manfredi M, Metharom P, Falasca M. Dissecting lipid metabolism alterations in SARS-CoV-2. Prog Lipid Res. 2021;82: 101092.
Das UN. Can bioactive lipids inactivate coronavirus (COVID-19)? Arch Med Res. 2020;51(3):282–6.
Kell DB, Pretorius E. The potential role of ischaemia-reperfusion injury in chronic, relapsing diseases such as rheumatoid arthritis, long COVID, and ME/CFS: evidence, mechanisms, and therapeutic implications. Biochem J. 2022;479(16):1653–708.
Smatti MK, Cyprian FS, Nasrallah GK, Al Thani AA, Almishal RO, Yassine HM. Viruses and autoimmunity: a review on the potential interaction and molecular mechanisms. Viruses. 2019. https://doi.org/10.3390/v11080762.
Rojas M, Rodríguez Y, Acosta-Ampudia Y, Monsalve DM, Zhu C, Li QZ, Ramírez-Santana C, Anaya JM. Autoimmunity is a hallmark of post-COVID syndrome. J Transl Med. 2022;20(1):129.
Winchester N, Calabrese C, Calabrese LH. The intersection of COVID-19 and autoimmunity: what is our current understanding? Pathog Immun. 2021;6(1):31–54.
Steinz MM, Persson M, Aresh B, Olsson K, Cheng AJ, Ahlstrand E, Lilja M, Lundberg TR, Rullman E, Möller KÄ, Sandor K, Ajeganova S, Yamada T, Beard N, Karlsson BC, Tavi P, Kenne E, Svensson CI, Rassier DE, Karlsson R, Friedman R, Gustafsson T, Lanner JT. Oxidative hotspots on actin promote skeletal muscle weakness in rheumatoid arthritis. JCI Insight. 2019;5(9):e126347. https://doi.org/10.1172/jci.insight.126347. PMID: 30920392; PMCID: PMC6538353.
Liu Y, Ebinger JE, Mostafa R, Budde P, Gajewski J, Walker B, Joung S, Wu M, Bräutigam M, Hesping F, et al. Paradoxical sex-specific patterns of autoantibody response to SARS-CoV-2 infection. J Transl Med. 2021;19(1):524.
Charnley M, Islam S, Bindra GK, Engwirda J, Ratcliffe J, Zhou J, Mezzenga R, Hulett MD, Han K, Berryman JT, et al. Neurotoxic amyloidogenic peptides in the proteome of SARS-COV2: potential implications for neurological symptoms in COVID-19. Nat Commun. 2022;13(1):3387.
Nyström S, Hammarström P. Amyloidogenesis of SARS-CoV-2 spike protein. J Am Chem Soc. 2022;144(20):8945–50.
Natarajan A, Zlitni S, Brooks EF, Vance SE, Dahlen A, Hedlin H, Park RM, Han A, Schmidtke DT, Verma R, et al. Gastrointestinal symptoms and fecal shedding of SARS-CoV-2 RNA suggest prolonged gastrointestinal infection. Med (N Y). 2022;3(6):371-387.e379.
Pascolini S, Vannini A, Deleonardi G, Ciordinik M, Sensoli A, Carletti I, Veronesi L, Ricci C, Pronesti A, Mazzanti L, et al. COVID-19 and immunological dysregulation: can autoantibodies be useful? Clin Transl Sci. 2021;14(2):502–8.
Chang SE, Feng A, Meng W, Apostolidis SA, Mack E, Artandi M, Barman L, Bennett K, Chakraborty S, Chang I, et al. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat Commun. 2021;12(1):5417.
Klein J, Wood J, Jaycox J, Lu P, Dhodapkar RM, Gehlhausen JR, Tabachnikova A, Tabacof L, Malik AA, Kamath K et al. Distinguishing features of long COVID identified through immune profiling. medRxiv. 2022:2022.2008.2009.22278592.
Chertow D, Stein, S., Ramelli, S. et al.: SARS-CoV-2 infection and persistence throughout the human body and brain. ResearchSquare 2021. https://doi.org/10.21203/rs.3.rs-1139035/v1.
Cheung CCL, Goh D, Lim X, Tien TZ, Lim JCT, Lee JN, Tan B, Tay ZEA, Wan WY, Chen EX, et al. Residual SARS-CoV-2 viral antigens detected in GI and hepatic tissues from five recovered patients with COVID-19. Gut. 2022;71(1):226–9.
Acknowledgements
We would like to thank the patients and their families who participated in this study.
Funding
All authors thank the SA long COVID Charitable trust for their generous donation to perform proteomics and pathology analysis. DBK thanks the Novo Nordisk Foundation for financial support (grant NNF20CC0035580). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AK: data analysis, patient and control identification; MV: proteomics analysis and proteomics data curation; CV: Platelet analysis and technical assistance; JL: Clinical support and patient sample identification; ST: sample curation; DBK: co-corresponding author, editing of paper; AP: editing of paper; EP: Sample analysis, edited data analysis, study leader, funding, co-corresponding author. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethical clearance for the study was obtained from the Health Research Ethics Committee (HREC) of Stellenbosch University (South Africa) (references: B21/03/001_COVID-19, project ID: 21911 and N19/03/043, project ID 9521). The experimental objectives, risks, and details were explained to volunteers and informed consent were obtained prior to blood collection. Strict compliance to ethical guidelines and principles Declaration of Helsinki, South African Guidelines for Good Clinical Practice, and Medical Research Council Ethical Guidelines for Research were kept for the duration of the study and for all research protocols. This laboratory study was carried out in strict adherence to the International Declaration of Helsinki, South African Guidelines for Good Clinical Practice and the South African Medical Research Council (SAMRC), Ethical Guidelines for research. Consent was obtained from all participant. Patients or the public WERE NOT involved in the design, or conduct, or reporting, or dissemination plans of our research.
Consent for publication
All authors approved submission of the paper.
Competing interests
EP is the managing director of BioCODE Technologies.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kruger, A., Vlok, M., Turner, S. et al. Proteomics of fibrin amyloid microclots in long COVID/post-acute sequelae of COVID-19 (PASC) shows many entrapped pro-inflammatory molecules that may also contribute to a failed fibrinolytic system. Cardiovasc Diabetol 21, 190 (2022). https://doi.org/10.1186/s12933-022-01623-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-022-01623-4