
Abstract
Background
We have previously shown that many chronic, inflammatory diseases are accompanied, and possibly partly caused or exacerbated, by various coagulopathies, manifested as anomalous clots in the form of ‘dense matted deposits’. More recently, we have shown that these clots can be amyloid in nature, and that the plasma of healthy controls can be induced to form such clots by the addition of tiny amounts of bacterial lipopolysaccharide or lipoteichoic acid. Type 2 diabetes (T2D) is also accompanied by raised levels of LPS.
Methods
We use superresolution and confocal microscopies to investigate the amyloid nature of clots from healthy and T2D individuals.
Results
We show here, with the established stain thioflavin T and the novel stains Amytracker™ 480 and 680, that the clotting of plasma from type 2 diabetics is also amyloid in nature, and that this may be prevented by the addition of suitable concentrations of LPS-binding protein.
Conclusion
This implies strongly that there is indeed a microbial component to the development of type 2 diabetes, and suggests that LBP might be used as treatment for it and its sequelae.
Similar content being viewed by others

Introduction
Inflammation is characterised by dysregulated circulating pro-inflammatory molecules, and such molecules have a pathological effect on the haematological system. This is true not only of the immune cells, but also of erythrocytes (RBCs), platelets and plasma proteins such as fibrin(ogen). During inflammation, RBCs experience structural and biochemical changes, which include membrane changes that may be visible as agglutination, eryptosis or microparticle formation [1,2,3,4,5,6,7]. In addition, the structure of the fibrin(ogen) protein changes, and this results in anomalous clot formation when fibrinogen is hydrolysed by thrombin [8,9,10,11]. Haematological pathology therefore both reflects and is reflected by a pro-coagulant state, which is a hallmark both of inflammation and of the concomitant dysregulated profile of circulating pro-inflammatory molecules [8].
Type 2 diabetes (T2D) is an inflammatory condition that is also characterised by various dysregulated cytokines and other molecules that are immunomodulatory and of pathophysiological importance [12, 13]. This condition is accompanied by many cardiovascular complications, including a thrombotic propensity; however, the inflammatory stimulus is often unknown. Importantly, T2D plasma has aberrant fibrin(ogen) packaging, and this is associated with amyloid fibril formation, as demonstrated by the amyloid-selective stain thioflavin T (ThT) (see [14, 15]) that binds to anomalous clots prepared by adding thrombin to diabetic plasma.
We have recently highlighted the possible and potent role of the circulating bacterial-derived inflammagens lipopolysaccharide (LPS) and lipoteichoic acids (LTAs) on anomalous blood hypercoagulation. LPSs derive from the membranes of Gram-negative bacteria [16], while LTAs originate from membranes of Gram-positives [17]. Importantly, the literature does support the increased circulating LPS in T2D [18,19,20,21,22,23,24,25].
LPS and LTA added to healthy plasma cause amyloidogenic changes in fibrin(ogen), demonstrated by thioflavin T (ThT) binding as well as binding of luminescent conjugated oligothiophene dyes (LCOs), marketed under the trade name Amytracker™, that also stain classical amyloid structures [11]. Various fluorescent markers have been shown to illuminate amyloids (e.g. [26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42]). This includes the luminescent LCO markers commercialised as Amytracker™ 480 and 680 (based on the published molecules HS163 and HS169 [43,44,45]), developed by Nilsson and colleagues [43, 44, 46,47,48,49,50,51,52].
Here we use these stains to provide evidence for a link between T2D as an inflammatory disease and the presence of amyloid proteins. We also assess the effect of different concentrations of LPS-binding protein and the antioxidant l-(+)ergothioneine (see, e.g. [53, 54]) on T2D plasma, and on purified fibrinogen after addition of LPS, to determine if these can decrease amyloid formation and the extent to which this could be shown with the two LCO dyes and ThT (see Fig. 1; infographic of workflow).

Infographic of workflow and study design
Materials and methods
Sample population
The control sample consisted of 17 age- and gender-matched (to the T2D sample) healthy individuals and an additional 17 young healthy individuals. Inflammation is known to increase with age [55], so we first sought to determine if there were differences between the two control groups (young versus old), to define if age plays a role in our experimental design. This said, it is well-known that the haematological system is only modestly affected by ageing [56]. Exclusion criteria for the healthy samples were: known (chronic and acute) inflammatory conditions such as asthma, human immunodeficiency virus (HIV) or tuberculosis; auto-immune conditions; risk factors associated with metabolic syndrome; smoking; and, if female, being on contraceptive or hormone replacement treatment. This population did not take any anti-inflammatory medication. Based on these exclusion criteria we classified these control donors as ostensibly healthy. We included individuals in our control group that had a range of BMI values, and for some purposes we grouped them as individuals with a BMI of either < 25 (normal BMI) or ≥ 25 (overweight). The distribution of normal versus overweight healthy individuals were: age controlled healthy individuals: < 24.9: 47%; > 25: 53% and for the younger healthy individuals: < 24.9: 88%; > 25: 12%. Although we included overweight, seemingly healthy individuals in our control group, we recognise that it is well-known that several key inflammatory markers have been consistently associated with obesity, which suggests that a persistent, low-grade, inflammatory response is a potentially modifiable risk factor [57]. Therefore, we acknowledge that overweight apparently healthy individuals may be more prone to low-grade systemic inflammation and that this may impact on their coagulation health.
Our T2D sample consisted of 33 individuals with BMI > 25 (overweight). T2D individuals were voluntarily recruited from a Diabetic Clinic in Somerset West, South Africa. Auto-immune diseases were set as exclusion criteria. Diabetic individuals were diagnosed by an Endocrinologist per the Society for Endocrinology, Metabolism and Diabetes of South Africa Type 2 Diabetes Committee (SEMSDA) guidelines [58]. These guidelines follow the American Diabetes Association (ADA) criteria to define type 2 diabetes. Demographic data including age, gender, as well as clinical information including haemoglobin A1c (HbA1c) levels and medication used by the T2D patients were obtained. Inclusion criteria consisted of both male and female participants, aged 19 and older, with a diagnosis of T2D for more than 3 months prior to screening and without any signs of infection. Smoking and either contraceptive or hormone replacement treatment were exclusion criteria. Whole blood (WB) of all the participants was obtained in citrate tubes. A platelet poor plasma (PPP) isolate was prepared by centrifuging WB for 15 min at 3000g.
Fluorescent markers and inflammagen binding agents
We aimed to determine if the hypercoagulable clot structure that we have previously noted in T2D and confirmed by staining T2D PPP with thioflavin T (ThT), was indeed amyloid in nature [59]. We have also previously shown that both the LCO dyes Amytracker™ 480 and 680 bind to amyloid areas in clots of healthy PPP exposed to known fibrin–amyloidogenic molecules (viz., iron, lipopolysaccharide (LPS) from gram negative bacteria, and two lipoteichoic acids (LTA1 from Staphylococcus aureus (Sigma, L2515) and LTA2 from Streptococcus pyogenes [Sigma, L3140)] [11]). It was thus of interest to assess if these amyloid markers and ThT will bind to amyloid proteins in clots of T2D in a similar pattern as in the healthy PPP incubated with amyloidogenic molecules that induced amyloidogenic areas (as previously shown in [11]). In addition, we investigated if we could induce a lowered LCO binding in T2D plasma after the addition of LPS-binding protein (LBP) (Abcam, AB119721) and/or an antioxidant compound, ergothioneine (Sigma E7521), to the PPP of T2D (previously we confirmed a reduced binding of ThT in T2D plasma after the addition of LBP [10]). Here, we analysed all diabetes versus controls, but because of the expense and time it takes for the analysis on the confocal microscope, we only looked at a random number of 15 diabetes patents that we additionally exposed to LBP and/or Ergothioneine. We also added LPS followed by LBP to purified fibrinogen to confirm this principle that LPS presence in T2D blood is one of (the main) causes of an amyloidogenic fibrin(ogen) structure (see methods in next section).
The LPS used was from E. coli O111:B4 (Sigma, L2630). A final LPS exposure concentration to plasma and purified fibrinogen of 0.4 ng L−1, and final LPS-binding protein (LBP) exposure concentrations of 8, 20 and 30 ng L−1 were used. A final exposure concentration of 250 µM ergothioneine (Sigma, E7521) was used. Purified fibrinogen (Sigma, F3879) was made up to 0.166 mg mL−1.
Confocal and super-resolution structured illumination (SR-SIM) microscopy of platelet poor plasma (PPP) clots
Platelet poor plasma (PPP) of all healthy and T2D individuals were prepared (as mentioned above), followed by storage at − 80 °C. On the day of analysis, the − 80 °C-stored PPPs were brought to room temperature, followed by a 30-min incubation with ThT at a final concentration of 5 µM and Amytracker™ 480 and 680 (0.1 µL into 100 µL PPP). LBP (8, 20 and 30 ng L−1 final exposure concentration), and ergothioneine (250 µM, final exposure concentration) were also added before addition of fluorescent markers. LBP and/or ergothioneine incubation time was 1 h. Before viewing clots on the confocal microscope, thrombin was added in the ratio 1:2, (5 µL thrombin: 10 µL) to create extensive fibrin networks. One clot was prepared for each individual and different random areas were imaged multiple times. Thrombin was provided by the South African National Blood Service, and the thrombin solution was at a concentration of 20 U mL−1 and made up in PBS containing 0.2% human serum albumin (see https://www.sigmaaldrich.com/content/dam/sigma-aldrich/docs/Sigma/Product_Information_Sheet/1/t6884pis.pdf for a description of how thrombin units are calculated). A coverslip was placed over the prepared clot, and samples were viewed using a Zeiss LSM 780 with ELYRA PS1 confocal microscope with a Plan-Apochromat 63x/1.4 Oil DIC objective. The following settings were used:
-
For ThT: the 488 nm excitation laser was used, with emission measured at 508–570 nm;
-
For Amytracker™ 480: the 405 nm excitation laser was used, with emission measured at 478–539 nm; and,
-
For Amytracker™ 680: the 561 nm excitation laser was used, with emission measured at 597–695 nm.
A selection of micrographs of the prepared clots were captured. Gain settings were kept the same during all data capture and used for statistical analyses; however, brightness and contrast were slightly adjusted for figure preparation. We captured the fluorescent signal of each of the three fluorescent markers as a composite.czi file in the Zeiss ZEN software and then used ImageJ (FIJI) to split and analyse the RGB channels.
Quantification of fluorescent staining of clots
We assessed the variance between (black) background and the presence of fluorescent pixels (binary comparison) for each of the three fluorescent markers in the clots. See [10, 11] for a detailed explanation of the methods. We used the histogram function in ImageJ (FIJI) and calculated the coefficient of variation (CV) (as SD/mean) of the histogram of different pixel intensities as our metric to quantify and discriminate between clots of healthy (age-controlled) naïve PPP and clots from T2D individuals. Figure 2 gives an example of the 3 marker histograms (Amytracker™ 480, 680 and ThT). CVs were calculated from the data shown at the bottom of each histogram.

Example of the histograms generated from diabetes clots with added fluorescent markers. (The x-axis represents the grey value range (8-bit greyscale [0, 255]) and the y-axis indicates the number of total pixels for each grey value.) The histograms were generated using FIJI and every composite confocal micrograph contains data of all 3 fluorescent markers. In FIJI the channels can be split and analysed separately, using the histogram function. a Amytracker™ 480; b Amytracker™ 680; c ThT
Structured illumination super-resolution (SR-SIM) microscopy
We also prepared structured illumination super-resolution (SR-SIM) microscopy [60] Z-stacks of T2D clots and compared the T2D clots to clots where we previously added known amyloidogenic molecules (iron, LPS, LTA1 and LTA2) to PPP from healthy donors (unpublished data from [11]). Previously, we incubated the four candidate amyloidogenic molecules for 1 h before adding the three fluorescent markers.
Statistical analysis
Sample analysis was performed by the Mann–Whitney U test, using the STATSDIRECT (version 2.8.0) software and with GraphPad Prism (version 5.0) and one-way ANOVA with the Kruskal–Wallis non-parametric test and the Dunns post-test.
Results
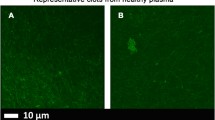
Table 1 shows demographics of our control groups and T2D sample, while Table 2 shows CV results which are calculated by using the mean and SD of the fluorescence for each of the three fluorescent markers in the different clots (Materials and Methods). There were no significant differences in CVs for the three markers when we compared the young and the old control PPP clot structure, and we therefore conclude age does not appear to influence our experimental design. However, there were significant differences in CVs for all three markers when the healthy (age-matched) individuals and T2D clots were compared. Figures 3 and 4 show confocal microscopy of PPP for representative clots from healthy and T2D samples. Figure 5 shows SR-SIM z-stacks of a representative T2D clot and also that of a healthy individual with added LPS, LTA1 and LTA2 (unpublished data from [11]). In addition, we did not find significant differences between our normal and overweight BMI control groups, therefore in this paper, we group both the normal BMI and overweight individuals as the control sample for the T2D sample.

Clots from representative healthy individuals with added amyloid stains. a young individual (median: 22 years) and b age-controlled individual (median: 61 years) and from left to right Amytracker 680 (red), Amytracker 480 (blue) and ThT (green)

Clots from three type 2 diabetes individuals with added amyloid–specific fluorescent markers (rows a to c represent 3 different individuals with type 2 diabetes); columns from left to right Amytracker 680 (red), Amytracker 480 (blue) and ThT (green)

Z-stacks from a type 2 diabetes, b healthy clot with added LPS (E. coli), c healthy clot with added LTA1 (S. aureus), and d healthy clot with added LTA2 (S. pyogenes)
The clots from the T2D patients most resemble that of the controls incubated with LPS. We have previously suggested that the potent inflammagens LPS and LTA can cause aberrant clot formation where the fibrin(ogen) protein becomes amyloid(ogenic) after exposure to these inflammagens [11]. In this paper, our results show that in T2D, amyloidogenesis is also present, as confirmed by the 2 LCO markers, and are similar to clots from healthy individuals but where LPS and LTA had been added prior to coagulation. We thus suggest that this aberrant morphology may (at least in part), be due to the presence of bacterial inflammagens in T2D blood.
We also added various concentrations of LPS-binding protein (LBP) and the antioxidant ergothioneine to T2D plasma to determine if the amyloid signal from the LCO dyes will be reduced. See Table 2 for the results as well as Fig. 6 for confocal data. These results show a dose-response in the reduction of fluorescent signal as seen with CV data. LBP breaks up the amyloid signal, where fewer large fluorescent amyloid clumps are noted, but rather more small fluorescent spots are seen (see Fig. 6). These data also show rather clearly the differences of detail between the binding (sites) of the three fluorophores. Ergothioneine does not significantly reduce the amyloid by itself; however, the effects of the addition of other antioxidants needs to be explored further (Fig. 7). Figure 8 shows a comparison of the confocal results [(CVs) of the various LBP exposures to T2D plasma (8, 20 and 30 ng L−1 LBP], using a one-way ANOVA with the Kruskal–Wallis non-parametric test and the Dunns post-test.

Clots from a representative type 2 diabetes individual with added LPS-binding protein (LBP) and ergothioneine, and fluorescent markers. a Naïve T2D clot; b 8 ng L−1 LBP and 250 µM ergothioneine; c 20 ng L−1 LBP and 250 µM ergothioneine d 30 ng L−1 LBP. Fluorescence shown from left to right Amytracker 680 (red), Amytracker 480 (blue) and ThT (green)

T2D with added 250 µM ergothioneine. Fluorescence shown from left to right Amytracker 680 (red), Amytracker 480 (blue) and ThT (green)

Graph drawn from the confocal results [(CVs) of the various LBP exposures to type 2 diabetes (8, 20 and 30 ng L−1 LBP]. Sample analysis was performed with GraphPad Prism (version 5.0) and one-way ANOVA with the Kruskal–Wallis non-parametric test and the Dunns post-test
Purified fibrinogen with added 0.4 ng L−1 LPS followed by increasing concentrations of LBP with and without ergothioneine showed the same trends, where the amyloid signal was dispersed and appeared as small fluorescent spots (see Fig. 9).

a Purified fibrinogen with 0.4 ng L−1 LPS and added thrombin b Purified fibrinogen with 0.4 ng L−1 LPS followed by 30 ng L−1 LBP and thrombin c Purified fibrinogen with 0.4 ng L−1 LPS and 250 µM ergothioneine followed by thrombin. Fluorescence shown from left to right Amytracker 680 (red), Amytracker 480 (blue) and ThT (green)
Discussion
T2D is one of many chronic, inflammatory diseases, and as such, shares a variety of hallmarks with these other conditions. These include the presence of inflammatory cytokines, iron dysregulation, and various coagulopathies [61,62,63,64,65]. What has not been clear is the actual cause of this inflammation; that is, what are the stimulating molecules to which inflammation is a response? Such a cause must exist, as there is otherwise little reason why the inflammation might happen ‘spontaneously’ in a way that it does not in healthy individuals. A strong candidate cause of such systemic inflammation may lie in a dormant microbiome that can shed inflammagens. In nature, most microbes exist in a dormant, non-replicating state, and as such are typically difficult to culture using standard microbiological culture techniques (e.g. [66, 67]). A lack of culturability may mean that a cell is non-viable under the circumstances tested, but they might be culturable in that they may be induced to return to a state of culturability (by a process or processes typically referred to as ‘resuscitation’) [66, 67].
We have suggested [68,69,70,71] that the source of these inflammagens is in fact populations of dormant microbes that are resident in blood and tissues, and that can occasionally ‘wake up’, often in response to bioavailable iron, whereupon they shed known, potent inflammagens such as LPS and LTA [11]. LPS has also been specifically implicated in T2D pathology [18,19,20,21,22,23,24,25, 72]. It is suggested that LPS may contribute to low-grade systemic inflammation in insulin-resistant states, and it is also now accepted that specifically gut bacteria is the sources of LPS [24]. In T2D it is well-known that there is an increased intestinal permeability in the genesis of T2D [73,74,75], and that this can be the origin for LPS.
We have also shown, using scanning electron microscopy (e.g. [1, 8, 69, 76,77,78], that in many cases a particular manifestation of the coagulopathies accompanying these diseases is the clotting of blood into a highly anomalous form. The above two strands of work led to the idea that the anomalous clotting might in fact be caused by the presence of low concentrations of bacterial–derived LPS, and this turned out to be the case when we tested the addition of extremely low concentrations of LPS to PPP from healthy individuals [9]. Further [9, 77], the fact that these clots could be stained with the amyloid stain thioflavin T, and blocked by the addition of LBP, strongly suggested (i) that the anomalous clotting was amyloid in nature, and (ii) that LPS (and the potent inflammagen LTA from Gram-positive bacteria) could indeed be a typical culprit. We recently showed that both LPS and LTA and also iron, cause healthy fibrin(ogen) to become amyloidogenic, and we used Amytracker™ 480, 680 and ThT in these experiments [11]. We found that the nature of the staining efficiencies also varied depending on the inflammagen added, with LTA particularly leading to preferential staining by Amytracker™ 680.
A logical corollary of the above, then, was that the anomalous clotting seen in PPP from patients with chronic inflammatory diseases might also be due (at least in part) to the presence of LPS, and that this too might be reversed by the addition of LBP. We had illustrated this previously using the common stain thioflavin T [59, 77] and here show it further using a variety of microscopic techniques, together with two novel stains, viz. Amytracker™ 480 and Amytracker™ 680. In all cases, there was a very substantial staining of the PPP from T2D patients, and this was removed, in a dose-dependent fashion, by the pre-incubation of the PPP with relatively low concentrations of lipopolysaccharide binding protein. In addition, the detailed nature of the staining varied for the three stains, suggested that they had both common and separate binding sites, with the two Amytracker™ stains being the most potent stains.
Ergothioneine is a potent antioxidant that is resistant to autoxidation [53, 54]. However, it was without effect on the amyloid staining, suggesting that there is no redox-dependent basis for these effects. This said, the effects of the addition of other antioxidants and/or polyphenolic compound need to be explored further.
Overall, we have shown very clearly that there is a substantial potential for amyloidogenesis when the plasma of individuals with T2D is clotted, and that this can be prevented by preincubation of the PPP with lipopolysaccharide-binding protein. This suggests very strongly that there is indeed a microbial component involved in the development of T2D and its sequelae. Of course there are potentially other circulating inflammatory molecules in T2D blood that might contribute to the amyloidogenic fibrin(ogen) structure, and some may include iron, LTA, SAA and upregulated cytokines. However, in this paper, we show that LPS/LTA is one of the important inflammagens in T2D, and the LBP removes most of the molecules that causes the amyloid formation in fibrin(ogen). Considering that amyloids can be cytotoxic, and many of the sequelae of chronic T2D involve damage to cells of other tissues such as the kidney (nephropathies) and the eye (retinopathies), it is at least reasonable that treatments designed either to remove the dormant microbes or the use of LBP to remove their cell wall products might be of therapeutic benefit. This is an important question for the future.
References
Pretorius E, Kell DB. Diagnostic morphology: biophysical indicators for iron-driven inflammatory diseases. Integr Biol. 2014;6:486–510.
Bester J, Pretorius E. Effects of IL-1β, IL-6 and IL-8 on erythrocytes, platelets and clot viscoelasticity. Sci Rep. 2016;6:32188.
Pretorius E, du Plooy JN, Bester J. A comprehensive review on eryptosis. Cell Physiol Biochem. 2016;39(5):1977–2000.
Pretorius E, Olumuyiwa-Akeredolu OO, Mbotwe S, Bester J. Erythrocytes and their role as health indicator: using structure in a patient-orientated precision medicine approach. Blood Rev. 2016;30(4):263–74.
Gasparyan AY, Ayvazyan L, Pretorius E, Kitas GD. Platelets in rheumatic diseases: friend or foe? Curr Pharm Des. 2014;20(4):552–66.
Soma P, Swanepoel AC, du Plooy JN, Mqoco T, Pretorius E. Flow cytometric analysis of platelets type 2 diabetes mellitus reveals ‘angry’ platelets. Cardiovasc Diabetol. 2016;15(1):52.
Van Der Spuy WJ, Pretorius E. A place for ultrastructural analysis of platelets in cerebral ischemic research. Microsc Res Tech. 2013;76(8):795–802.
Kell DB, Pretorius E. The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr Biol. 2015;7:24–52.
Pretorius E, Mbotwe S, Bester J, Robinson CJ, Kell DB. Acute induction of anomalous and amyloidogenic blood clotting by molecular amplification of highly substoichiometric levels of bacterial lipopolysaccharide. J R Soc Interface. 2016;13(122):20160539.
Pretorius E, Mbotwe S, Kell DB. Lipopolysaccharide-binding protein (LBP) reverses the amyloid state of fibrin seen in plasma of type 2 diabetics with cardiovascular co-morbidities. Sci Rep 2017;7:9680
Pretorius E, Page MJ, Hendricks L, Nkosi NB, Benson SR, Kell DB. Both lipopolysaccharide and lipoteichoic acids potently induce anomalous fibrin amyloid formation: assessment with novel Amytracker™ stains. R Soc Inter. 2017 (submitted).
Banerjee M, Saxena M. Interleukin-1 (IL-1) family of cytokines: role in type 2 diabetes. Clin Chim Acta. 2012;413(15–16):1163–70.
Qiao YC, Shen J, He L, Hong XZ, Tian F, Pan YH, Liang L, Zhang XX, Zhao HL. Changes of Regulatory T cells and of proinflammatory and immunosuppressive cytokines in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. J Diabetes Res. 2016;2016:3694957.
Biancalana M, Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochem Biophys Acta. 2010;1804(7):1405–12.
Biancalana M, Makabe K, Koide A, Koide S. Molecular mechanism of thioflavin-T binding to the surface of beta-rich peptide self-assemblies. J Mol Biol. 2009;385:1052–63.
Kell DB, Pretorius E. On the translocation of bacteria and their lipopolysaccharides between blood and peripheral locations in chronic, inflammatory diseases: the central roles of LPS and LPS-induced cell death. Integr Biol. 2015;7:1339–77.
Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by toll-like receptor 2. J Biol Chem. 1999;274(25):17406–9.
Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M, Evans M, Harte AL, Kumar S. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740–7.
Hawkesworth S, Moore SE, Fulford AJ, Barclay GR, Darboe AA, Mark H, Nyan OA, Prentice AM. Evidence for metabolic endotoxemia in obese and diabetic Gambian women. Nutr Diabetes. 2013;3:e83.
Nymark M, Pussinen PJ, Tuomainen AM, Forsblom C, Groop PH, Lehto M. Serum lipopolysaccharide activity is associated with the progression of kidney disease in finnish patients with type 1 diabetes. Diabetes Care. 2009;32(9):1689–93.
Vergès B, Duvillard L, Lagrost L, Vachoux C, Garret C, Bouyer K, Courtney M, Pomié C, Burcelin R. Changes in lipoprotein kinetics associated with type 2 diabetes affect the distribution of lipopolysaccharides among lipoproteins. J Clin Endocrinol Metab. 2014;99(7):E1245–53.
Zaman GS, Zaman F. Relationship between postprandial endotoxemia in nonobese postmenopausal women and diabetic nonobese postmenopausal women. J Nat Sci Biol Med. 2015;6(1):89–93.
Al-Attas OS, Al-Daghri NM, Al-Rubeaan K, da Silva NF, Sabico SL, Kumar S, McTernan PG, Harte AL. Changes in endotoxin levels in T2DM subjects on anti-diabetic therapies. Cardiovasc Diabetol. 2009;8:20.
Jayashree B, Bibin YS, Prabhu D, Shanthirani CS, Gokulakrishnan K, Lakshmi BS, Mohan V, Balasubramanyam M. Increased circulatory levels of lipopolysaccharide (LPS) and zonulin signify novel biomarkers of proinflammation in patients with type 2 diabetes. Mol Cell Biochem. 2014;388(1–2):203–10.
Pussinen PJ, Havulinna AS, Lehto M, Sundvall J, Salomaa V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care. 2011;34(2):392–7.
Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, Kung HF, Lam KS, Oddo S, Laferla FM, et al. Congo red and thioflavin-T analogs detect Abeta oligomers. J Neurochem. 2008;104(2):457–68.
Chang WM, Dakanali M, Capule CC, Sigurdson CJ, Yang J, Theodorakis EA. ANCA: a family of fluorescent probes that bind and stain amyloid plaques in human tissue. ACS Chem Neurosci. 2011;2(5):249–55.
Giorgadze TA, Shiina N, Baloch ZW, Tomaszewski JE, Gupta PK. Improved detection of amyloid in fat pad aspiration: an evaluation of Congo red stain by fluorescent microscopy. Diagn Cytopathol. 2004;31(5):300–6.
Nilsson KPR, Hammarström P, Ahlgren F, Herland A, Schnell EA, Lindgren M, Westermark GT, Inganäs O. Conjugated polyelectrolytes-conformation-sensitive optical probes for staining and characterization of amyloid deposits. ChemBioChem. 2006;7(7):1096–104.
Rajamohamedsait HB, Sigurdsson EM. Histological staining of amyloid and pre-amyloid peptides and proteins in mouse tissue. Methods Mol Biol. 2012;849:411–24.
Volkova KD, Kovalska VB, Balanda AO, Vermeij RJ, Subramaniam V, Slominskii YL, Yarmoluk SM. Cyanine dye-protein interactions: looking for fluorescent probes for amyloid structures. J Biochem Biophys Methods. 2007;70(5):727–33.
Mishra R, Sjolander D, Hammarström P. Spectroscopic characterization of diverse amyloid fibrils in vitro by the fluorescent dye Nile red. Mol BioSyst. 2011;7(4):1232–40.
Shen D, Coleman J, Chan E, Nicholson TP, Dai L, Sheppard PW, Patton WF. Novel cell- and tissue-based assays for detecting misfolded and aggregated protein accumulation within aggresomes and inclusion bodies. Cell Biochem Biophys. 2011;60(3):173–85.
Easterhoff D, DiMaio JTM, Liyanage W, Lo CW, Bae W, Doran TM, Smrcka A, Nilsson BL, Dewhurst S. Fluorescence detection of cationic amyloid fibrils in human semen. Bioorg Med Chem Lett. 2013;23(18):5199–202.
Kovalska VB, Losytskyy MY, Tolmachev OI, Slominskii YL, Segers-Nolten GM, Subramaniam V, Yarmoluk SM. Tri- and pentamethine cyanine dyes for fluorescent detection of alpha-synuclein oligomeric aggregates. J Fluoresc. 2012;22(6):1441–8.
Ono M, Watanabe H, Kimura H, Saji H. BODIPY-based molecular probe for imaging of cerebral beta-amyloid plaques. ACS Chem Neurosci. 2012;3(4):319–24.
Rajasekhar K, Narayanaswamy N, Murugan NA, Kuang G, Agren H, Govindaraju T. A high affinity red fluorescence and colorimetric probe for amyloid beta aggregates. Sci Rep. 2016;6:23668.
Watanabe H, Ono M, Matsumura K, Yoshimura M, Kimura H, Saji H. Molecular imaging of beta-amyloid plaques with near-infrared boron dipyrromethane (BODIPY)-based fluorescent probes. Mol Imaging. 2013;12(5):338–47.
Yuan L, Lin W, Zheng K, He L, Huang W. Far-red to near infrared analyte-responsive fluorescent probes based on organic fluorophore platforms for fluorescence imaging. Chem Soc Rev. 2013;42(2):622–61.
Guo Z, Park S, Yoon J, Shin I. Recent progress in the development of near-infrared fluorescent probes for bioimaging applications. Chem Soc Rev. 2014;43(1):16–29.
Staderini M, Martin MA, Bolognesi ML, Menéndez JC. Imaging of beta-amyloid plaques by near infrared fluorescent tracers: a new frontier for chemical neuroscience. Chem Soc Rev. 2015;44(7):1807–19.
Zhang X, Tian Y, Zhang C, Tian X, Ross AW, Moir RD, Sun H, Tanzi RE, Moore A, Ran C. Near-infrared fluorescence molecular imaging of amyloid beta species and monitoring therapy in animal models of Alzheimer’s disease. Proc Natl Acad Sci USA. 2015;112(31):9734–9.
Klingstedt T, Nilsson KP. Luminescent conjugated poly- and oligo-thiophenes: optical ligands for spectral assignment of a plethora of protein aggregates. Biochem Soc Trans. 2012;40(4):704–10.
Magnusson K, Simon R, Sjölander D, Sigurdson CJ, Hammarström P, Nilsson KPR. Multimodal fluorescence microscopy of prion strain specific PrP deposits stained by thiophene-based amyloid ligands. Prion. 2014;8(4):319–29.
Shirani H, Linares M, Sigurdson CJ, Lindgren M, Norman P, Nilsson KPR. a palette of fluorescent thiophene-based ligands for the identification of protein aggregates. Chemistry (Weinheim an Der Bergstrasse, Germany). 2015;21(43):15133–7.
Åslund A, Sigurdson CJ, Klingstedt T, Grathwohl S, Bolmont T, Dickstein DL, Glimsdal E, Prokop S, Lindgren M, Konradsson P, et al. Novel pentameric thiophene derivatives for in vitro and in vivo optical imaging of a plethora of protein aggregates in cerebral amyloidoses. ACS Chem Biol. 2009;4(8):673–84.
Berg I, Nilsson KPR, Thor S, Hammarström P. Efficient imaging of amyloid deposits in Drosophila models of human amyloidoses. Nat Protoc. 2010;5(5):935–44.
Klingstedt T, Åslund A, Simon RA, Johansson LBG, Mason JJ, Nyström S, Hammarström P, Nilsson KPR. Synthesis of a library of oligothiophenes and their utilization as fluorescent ligands for spectral assignment of protein aggregates. Org Biomol Chem. 2011;9(24):8356–70.
Klingstedt T, Blechschmidt C, Nogalska A, Prokop S, Haggqvist B, Danielsson O, Engel WK, Askanas V, Heppner FL, Nilsson KPR. Luminescent conjugated oligothiophenes for sensitive fluorescent assignment of protein inclusion bodies. ChemBioChem. 2013;14(5):607–16.
Klingstedt T, Shirani H, Åslund KOA, Cairns NJ, Sigurdson CJ, Goedert M, Nilsson KPR. The structural basis for optimal performance of oligothiophene-based fluorescent amyloid ligands: conformational flexibility is essential for spectral assignment of a diversity of protein aggregates. Chemistry. 2013;19(31):10179–92.
Nilsson KP, Lindgren M, Hammarström P. A pentameric luminescent-conjugated oligothiophene for optical imaging of in vitro-formed amyloid fibrils and protein aggregates in tissue sections. Methods Mol Biol. 2012;849:425–34.
Nyström S, Psonka-Antonczyk KM, Ellingsen PG, Johansson LB, Reitan N, Handrick S, Prokop S, Heppner FL, Wegenast-Braun BM, Jucker M, et al. Evidence for age-dependent in vivo conformational rearrangement within Abeta amyloid deposits. ACS Chem Biol. 2013;8(6):1128–33.
Cheah IK, Halliwell B. Ergothioneine; antioxidant potential, physiological function and role in disease. Biochem Biophys Acta. 2012;1822(5):784–93.
Halliwell B, Cheah IK, Drum CL. Ergothioneine, an adaptive antioxidant for the protection of injured tissues? A hypothesis. Biochem Biophys Res Commun. 2016;470(2):245–50.
El Assar M, Angulo J, Rodriguez-Manas L. Oxidative stress and vascular inflammation in aging. Free Radical Biol Med. 2013;65:380–401.
Lichtman MA, Williams WJ. Hematology in the aged. In: Ernest Beutler MAL, Coller BS, Kipps TJ, Seligsohn U, editors. Williams Hematology, Chap 8. 6th ed. USA: McGraw-Hill; 2000
Cox AJ, West NP, Cripps AW. Obesity, inflammation, and the gut microbiota. Lancet Diabetes Endocrinol. 2015;3(3):207–15.
SEMSDA. The 2017 SEMDSA guideline for management of Type 2 diabetes guideline committee. JEMDSA. 2017;21(1):S1–196.
Pretorius E, Mbotwe S, Kell DB. Lipopolysaccharide-binding protein (LBP) can reverse the amyloid state of fibrin seen or induced in Parkinson's Disease: implications for its aetiology. bioRxiv preprint archive 2017. doi:10.1101/124180.
Sivaguru M, Urban MA, Fried G, Wesseln CJ, Mander L, Punyasena SW. Comparative performance of airyscan and structured illumination superresolution microscopy in the study of the surface texture and 3D shape of pollen. Microsc Res Tech. 2016. doi:10.1002/jemt.22732.
Pretorius E, Bester J. Viscoelasticity as a measurement of clot structure in poorly controlled type 2 diabetes patients: towards a precision and personalized medicine approach. Oncotarget. 2016;7(32):50895–907.
Samad F, Ruf W. Inflammation, obesity, and thrombosis. Blood. 2013;122(20):3415–22.
Soma P, Pretorius E. Interplay between ultrastructural findings and atherothrombotic complications in type 2 diabetes mellitus. Cardiovasc Diabetol. 2015;14:96.
Tousoulis D, Papageorgiou N, Androulakis E, Siasos G, Latsios G, Tentolouris K, Stefanadis C. Diabetes mellitus-associated vascular impairment: novel circulating biomarkers and therapeutic approaches. J Am Coll Cardiol. 2013;62(8):667–76.
Tripodi A, Branchi A, Chantarangkul V, Clerici M, Merati G, Artoni A, Mannucci PM. Hypercoagulability in patients with type 2 diabetes mellitus detected by a thrombin generation assay. J Thromb Thrombolysis. 2011;31(2):165–72.
Kaprelyants AS, Kell DB. Dormancy in stationary-phase cultures of micrococcus luteus: flow cytometric analysis of starvation and resuscitation. Appl Environ Microbiol. 1993;59(10):3187–96.
Kell DB, Kaprelyants AS, Weichart DH, Harwood CL, Barer MR. Viability and activity in readily culturable bacteria: a review and discussion of the practical issues. Antonie Van Leeuwenhoek. 1998;73:169–87.
Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, et al. Microbes and Alzheimer’s disease. JAD. 2016;51:979–84.
Pretorius E, Akeredolu OO, Soma P, Kell DB. Major involvement of bacterial components in rheumatoid arthritis and its accompanying oxidative stress, systemic inflammation and hypercoagulability. Exp Biol Med (Maywood, NJ). 2017;242(4):355–73.
Pretorius E, Bester J, Kell DB. A bacterial component to Alzheimer’s-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. JAD. 2016;53(4):1237–56.
Kell DB, Pretorius E. To what extent are the terminal stages of sepsis, septic shock, systemic inflammatory response syndrome, and multiple organ dysfunction syndrome actually driven by a prion/amyloid form of fibrin? Semin Thromb Hemost. 2017. doi:10.1055/s-0037-1604108.
Zhou H, Hu J, Zhu Q, Yang S, Zhang Y, Gao R, Liu L, Wang Y, Zhen Q, Lv Q et al. Lipopolysaccharide-binding protein cannot independently predict type 2 diabetes mellitus: a nested casecontrol study. J Diabetes 2016;8:214–219. doi:10.1111/1753-0407.12281
de Kort S, Keszthelyi D, Masclee AA. Leaky gut and diabetes mellitus: what is the link? Obes Rev. 2011;12(6):449–58.
Slyepchenko A, Maes M, Machado-Vieira R, Anderson G, Solmi M, Sanz Y, Berk M, Kohler CA, Carvalho AF. Intestinal dysbiosis, gut hyperpermeability and bacterial translocation: missing links between depression, obesity and Type 2 diabetes. Curr Pharm Des. 2016;22(40):6087–106.
Sato J, Kanazawa A, Watada H. Type 2 diabetes and bacteremia. Ann Nutr Metab. 2017;71(Suppl 1):17–22.
Kell DB, Pretorius E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics. 2014;6(4):748–73.
Kell DB, Pretorius E. Proteins behaving badly. Substoichiometric molecular control and amplification of the initiation and nature of amyloid fibril formation: lessons from and for blood clotting. Prog Biophys Mol Biol. 2017;123:16–41.
Pretorius E, Bester J, Vermeulen N, Alummoottil S, Soma P, Buys AV, Kell DB. Poorly controlled type 2 diabetes is accompanied by significant morphological and ultrastructural changes in both erythrocytes and in thrombin-generated fibrin: implications for diagnostics. Cardiovasc Diabetol. 2015;14:30.
Authors’ contributions
EP Studyleader, confocal analysis draft and critical revision of paper. MJP Acquisition and analysis of all data, editing of paper. LE Technical assistance on confocal microscope. GCE Endocrinologist; clinical advisor. DBK Co-study leader and critical revision of paper. All authors read and approved the final manuscript.
Acknowledgements
We thank the diabetes clinic and sister Laura Botha for diabetes patient blood collection and Dr Theo Nell for control blood collection, as well as Retha Engelbrecht for data capturing assistance.
Competing interests
The authors declare the following patent application: Method for treating Diabetes (ZA2017/05546).
Availability of data and materials
Raw data, including original micrographs can be accessed at: https://1drv.ms/f/s!AgoCOmY3bkKHvEigbzhPJ-gPv1Vr or https://www.researchgate.net/profile/Etheresia_Pretorius.
Consent for publication
All authors give consent for publication.
Ethics approval and consent to participate
This study was approved by the Ethical Committee of the University of Pretoria (South Africa) and Stellenbosch University: Ethics Clearance Number: 298/2016. We adhered strictly to the Declaration of Helsinki. A written form of informed consent was obtained from all donors (available on request). Blood was collected and methods were carried out in accordance with the relevant guidelines of the ethics committee.
Funding
Biotechnology and Biological Sciences Research Council (Grant BB/L025752/1). National Research Foundation (NRF) of South Africa (91548: Competitive Program) and Medical Research Council (MRC) of South Africa (Self-Initiated Research Program 2017 to 2019). Grant holder: E Pretorius.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pretorius, E., Page, M.J., Engelbrecht, L. et al. Substantial fibrin amyloidogenesis in type 2 diabetes assessed using amyloid-selective fluorescent stains. Cardiovasc Diabetol 16, 141 (2017). https://doi.org/10.1186/s12933-017-0624-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-017-0624-5