
Abstract
The existence of an independent association between elevated triglyceride (TG) levels, cardiovascular (CV) risk and mortality has been largely controversial. The main difficulty in isolating the effect of hypertriglyceridemia on CV risk is the fact that elevated triglyceride levels are commonly associated with concomitant changes in high density lipoprotein (HDL), low density lipoprotein (LDL) and other lipoproteins. As a result of this problem and in disregard of the real biological role of TG, its significance as a plausible therapeutic target was unfoundedly underestimated for many years. However, taking epidemiological data together, both moderate and severe hypertriglyceridaemia are associated with a substantially increased long term total mortality and CV risk. Plasma TG levels partially reflect the concentration of the triglyceride-carrying lipoproteins (TRL): very low density lipoprotein (VLDL), chylomicrons and their remnants. Furthermore, hypertriglyceridemia commonly leads to reduction in HDL and increase in atherogenic small dense LDL levels. TG may also stimulate atherogenesis by mechanisms, such excessive free fatty acids (FFA) release, production of proinflammatory cytokines, fibrinogen, coagulation factors and impairment of fibrinolysis. Genetic studies strongly support hypertriglyceridemia and high concentrations of TRL as causal risk factors for CV disease. The most common forms of hypertriglyceridemia are related to overweight and sedentary life style, which in turn lead to insulin resistance, metabolic syndrome (MS) and type 2 diabetes mellitus (T2DM). Intensive lifestyle therapy is the main initial treatment of hypertriglyceridemia. Statins are a cornerstone of the modern lipids-modifying therapy. If the primary goal is to lower TG levels, fibrates (bezafibrate and fenofibrate for monotherapy, and in combination with statin; gemfibrozil only for monotherapy) could be the preferable drugs. Also ezetimibe has mild positive effects in lowering TG. Initial experience with en ezetimibe/fibrates combination seems promising. The recently released IMPROVE-IT Trial is the first to prove that adding a non-statin drug (ezetimibe) to a statin lowers the risk of future CV events. In conclusion, the classical clinical paradigm of lipids-modifying treatment should be changed and high TG should be recognized as an important target for therapy in their own right. Hypertriglyceridemia should be treated.
Similar content being viewed by others

Introduction
The independent association between elevated triglycerides (TG), cardiovascular (CV) risk and mortality has been largely controversial [1],[2]. The main difficulty in isolating the effect of hypertriglyceridemia on CV is the fact that elevated TG levels are commonly associated with concomitant changes in high density lipoprotein (HDL), low density lipoprotein (LDL) and other lipoproteins. Although the majority of studies found a substantial direct association between TG and adverse outcomes [3]-[12], this association sometimes became nonsignificant after multivariate adjustment including other lipids and weight-related variables [13]-[16]. For example, in the largest till now meta-analysis [13], TG were associated with an increased risk of coronary artery disease (CAD) after adjustment for age and sex, but this association was abolished following an additional adjustment for HDL and non-HDL cholesterol. Correlations with lower HDL cholesterol led to studies in which the authors mathematically “adjusted” for these relationships, suggesting that the HDL values could be invoked as more likely contributors to risk than the TG themselves. As a result of these mathematical over-adjustment exercises and in disregard of the real biological role of TG, the significance of hypertriglyceridemia as a plausible therapeutic target was unfoundedly underestimated for many years. However, epidemiology can be a poor guide to clinical decisions and gives us only limited insight into the mechanisms of atherogenesis and their relative importance in this process. On the other hand, classification of elevated TG as a major CV risk factor is clinically important since it determines whether high TG should be a target for therapy in their own right.
Hypertriglyceridemia and CV events
Serum TG are routinely measured under fasting conditions to obtain more stable concentrations and to enable the physician to calculate LDL cholesterol levels. In addition, hypertriglyceridemia and postprandial lipidemia may affect the measurement of HDL cholesterol and therefore the calculation of non-HDL cholesterol.
The NCEP ATP III arbitrarily divided fasting serum TG into four different classes [17]. Classification of serum TG levels greater than 150 mg/dl (1.7 mmol/liter) as elevated is mainly based on large prospective observational studies. However, the exact level at which serum TG start to confer risk or become a marker for CV disease is unknown, but it may be even lower than 150 mg/dl (1.7 mmol/liter) [18]. Serum TG are higher in men and increase with age in both sexes [19].
Very high TG (correspondent to severe hypertriglyceridemia) are defined as serum TG > or = 500 mg/dl [3], whereas the Endocrine Society Clinical Practice Guideline [20] labeled as very severe hypertriglyceridemia serum TG > or = 2000 mg/dl. The common view was that severe and very severe hypertriglyceridemia increase the risk for pancreatitis, whereas mild or moderate hypertriglyceridemia may be a risk factor for CV disease [20]. In patients with very high TG levels – i.e. more than 25 mmol/L - and patients with the familial chylomicronemia syndrome the risk for atherosclerosis is attenuated, perhaps because their plasma lipoprotein particles are too large to enter into the arterial intima [21] and [22]. However, there are many indications that there is an increased risk of CV disease in the marked or severe hypertriglyceridemia (fasting triglyceride concentration exceeding 5.6 mmol/L and 11.2 mmol/L) as well [23],[24]. Moreover, even in the prominent old study of Assmann et al. [21] which emphasized a J-shape for TG cardiovascular risk, TG above 800 mg/dl were still associated with significantly greater risk than TG less than 200 mg/dl, albeit it was decreased in comparison to the 400–799 mg/dl levels.
Prospective studies have indicated that, compared with fasting levels, nonfasting serum TG levels may be a better or similar predictor of CV events in the general population [25]-[29]. In a number of studies using standardized meals, greater CV risk was found to be associated with increased hypertriglyceridemia [27],[28].
The serum TG concentration is often more strongly correlated with future CAD incidence in univariate analysis than is serum cholesterol. However, in multiple logistic regression analysis, particularly when HDL cholesterol is included, the strength of the apparent independent relationship between TG and CAD incidence is weakened often to the point of insignificance in individual trials. The erosion of the relationship between TG and CAD incidence when HDL is included in multiple logistic regression analysis is to some extent an artifact of the greater biological variation of TG concentrations compared with HDL cholesterol. When allowance is made, TG can have more predictive power than HDL [30]. Taking epidemiological data together, both moderate and severe hypertriglyceridaemia are associated with a substantially increased long term total mortality and CV risk.
Triglycerides and atherogenesis
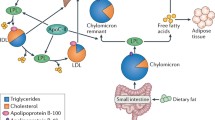
Currently LDL is considered as the major atherogenic lipoprotein; however other lipoproteins size is of key importance in determining whether particles can penetrate the arterial wall. The plasma TG level represents in part the concentration of the TRL: VLDL, chylomicrons and their remnants. Although chylomicrons and probably VLDL are both too large to penetrate the arterial wall, their remnants are small enough to do this, and have been demonstrated in human and animal atherosclerotic plaques [31].
Physiologically TG are the densest form of calories and serve as an important source of energy. Dietary TG are assembled in the gut into chylomicrons. Their interaction with lipoprotein lipase (LpL) located on the luminal surface of capillary endothelial cells leads to liberation of free fatty acids from TG; free fatty acids are able to traverse cell membranes. Only 50% of chylomicron’s TG is estimated to be lost in this process, and the remainder of the lipoprotein, called a chylomicron remnant, contains lipids such as cholesteryl esters, retinylesters, and apoB-48 [20].
VLDL particles are the main TG carrier in the circulation, being produced by the liver, whereas the VLDL TG content is derived from a variety of substrates including lipoprotein TG and FFA. VLDL TG lose FFA by the action of LpL as well, leading to production of VLDL remnants, also referred to as intermediate-density lipoproteins (IDL), and eventually to conversion to LDL. The concentration of VLDL cholesterol and apolipoprotein B (apoB) is at least 10 times higher than the corresponding chylomicron concentration, even after consumption of a large amount of fat [32]-[35]. These lipoproteins contain at least as much cholesterol per particle as does LDL. TG itself is not a component of arterial plaque, but cholesterol within TG -rich particles contributes to plaque development [36],[37].
VLDL can be divided into large, TG -rich VLDL1 and small, dense VLDL2. VLDL1 has a higher TG content and exhibit abundant apolipoprotein CIII (apoCIII) and apolipoprotein E [38],[39].
An increase in TG-rich lipoproteins is commonly associated with a reduction in HDL and an increase in small dense LDL levels. Hypertriglyceridemia stimulates the enzymatic activity of cholesteryl ester transfer protein (CETP), which facilitates the transfer of TG from TG -rich lipoproteins to HDL and LDL in exchange for cholesteryl esters [40]. This leads to an increase in TG content of HDL and LDL. TG -enriched HDL particles are subject to increased catabolism; consequently, they have a short plasma half-life. TG-enriched LDL particles undergo subsequent hydrolysis via LpL or hepatic lipase, thereby reducing LDL particle size. In addition, the difference in metabolic fate between VLDL1 and VLDL2 may also account for the increased formation of small dense LDL. Kinetic data show that large TG -rich VLDL1 particles yield small dense LDL whereas smaller and denser VLDL2 particles are metabolized to normal sized LDL [41].
TG may also stimulate atherogenesis by other mechanisms, which include the production of proinflammatory cytokines, fibrinogen and coagulation factors and impairment of fibrinolysis. Therefore, their roles in atherogenesis have a basic biological plausibility.
Hypertriglyceridemia as a major component of atherogenic dyslipidemia related to insulin resistance, MS and T2DM
Hypertriglyceridemia results from increased TG production, or reduced TG catabolism, or both. Drugs such as bile acid resins, estrogens, isotretinoin and steroids; marked alcohol and fat ingestion in a susceptible patient; or conditions such as poorly controlled diabetes or pregnancy can result in high triglyceride levels [42],[43].
One of the reason for hypertriglyceridemia is alcohol consumption. Alcohol intake increases hepatic fatty acid synthesis and decreases fatty acid oxidation, with a net effect to stimulate hepatic VLDL TG secretion. The effects of alcohol are dose-dependent [44],[45]. However the most common forms of hypertriglyceridemia are related to overweight and sedentary life style which leads to insulin resistance. This setting of hypertriglyceridemia is typical for MS and T2DM. The increase in TG production may be due to excess FFA returning to the liver, particularly in the setting of visceral obesity and insulin resistance, and increased de novo TG production due to hyperinsulinemia [46]-[48]. In hypertriglyceridemia, more VLDL particles, as measured by apoB, and larger and more TG- and apoC-III-enriched lipoproteins are found [49]-[51]. Hepatic insulin resistance may contribute to a high production rate of VLDL because insulin reduces apoB synthesis and VLDL secretion in the liver [52],[53]. Although insulin resistance is associated with high triglycerides, VLDL and TG concentrations can be similar in patients with widely divergent insulin sensitivity [54],[55].
Hypertriglyceridemia, as one of the components of the MS, is closely related to a constellation of metabolic risk factors including a central distribution of adiposity or visceral obesity, insulin resistance, impaired glucose tolerance, hypertension, and high TG and/or low HDL-C, associated with an atherogenic, procoagulant, and proinflammatory state [56]-[64].
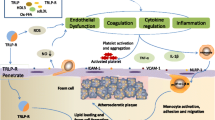
One could thought that TG are not directly involved in the development of atherosclerotic lesions because FFA released from TG by lipoprotein lipase (LpL) act either as an active energy source or stored energy reserve. However, there are several plausible mechanisms by which FFA might cause CV disease and death [65]-[71]. Furthermore, an increase in plasma FFA leads to endothelial activation, inflammation and thrombosis which may initiate early vascular abnormalities that promote atherosclerosis [72]-[77]. Elevation of plasma FFA, in addition to producing peripheral and hepatic insulin resistance, also activates the proinflammatory NFκB pathway [73]-[84] resulting in increased hepatic expression of several proinflammatory cytokines including TNF-α, IL1-β, IL6, matrix metalloproteinases and an increase in circulating MCP-1 [85]-[89], supporting the notion that FFA is an important link between hypertriglyceridemia and the development of inflammatory changes [90]-[99]. Moreover, elevated plasma FFA levels, via producing insulin resistance and hyperinsulinemia, promote a state of increased tendency for thrombosis and decreased ability to fibrinolysis. Together, this substantially increases the risk for acute atherothrombotic events [100]-[105].
Therefore, elevated FFA are not only an independent risk factor for the development of T2DM, but also give rise to metabolic derangements in organs such as the liver and pancreas. Hypertriglyceridemia, FFAs overload and lipid accumulation in non-adipose tissues influence both insulin action and insulin secretion and are frequently associated with IR and the development of T2DM [106]-[109]. However the role of FFAs extends beyond their ability to induce or exacerbate insulin resistance: they may contribute directly to the deterioration of beta cell function that accompanies the development of diabetes [109]-[113]. Both acute stimulatory and long-term detrimental effects of FFAs overloading on pancreatic beta-cell have long been recognized. Chronic exposure of the pancreatic beta-cell to FFA results in desensitization and suppression of secretion (lipotoxicity) as a consequence of TG accumulation in the islets of Langerhans [109].
Since evidence indicates that multiple aberrations in lipids metabolism play a pivotal role in the pathophysiology of diabetes, it was suggested to drop the adjective “mellitus” from diabetes and then to consider the introduction of a new adjective “lipidus” or “lipomellitus” [114] (Figure 1).

Hypertriglyceridemia, FFAs overload and lipid accumulation in nonadipose tissues (so called lipotoxicity) are the key events in the pathogenesis of T2DM, mainly via insulin resistance and pancreatic beta-cell failure.
TG and HDL
Raised TG concentrations are strongly associated with low concentrations of HDL cholesterol, and the past 25 years have been dominated by HDL research, with less focus on TG. The hypothesis that HDL is protective against atherosclerosis was supported by a series of animal studies in the 1980s and 1990s. Badimon and colleagues [115] infused HDL into rabbits and reported inhibition of atherosclerosis. Rubin and colleagues [116] showed that mice overexpressing the major HDL protein apolipoprotein A-I (apoA-I) are protected from atherosclerosis. Viral overexpression of apoA-I in mice with pre-existing atherosclerosis resulted in regression of pre-existing atherosclerotic disease [117]. These preclinical data matched the epidemiological data and strongly reinforced the HDL hypothesis, making HDL a major target for novel therapeutic approaches to decrease atherosclerosis.
Consequently, HDL has long been regarded as the “good” lipoprotein because epidemiological and clinical studies have identified an inverse association between HDL concentration and CV disease [118],[119]. The most important antiatherogenic function of HDL is reverse cholesterol transport [120]. HDL also exhibits other potential cardioprotective functions such as anti-oxidative, anti-inflammatory and endothelium-dependent vasodilatory effects [121]-[123].
However, a failing initial experience with CETP inhibitors has been most problematic for the HDL hypothesis [124]-[126]. Moreover, reports from several randomized clinical trials of HDL-raising drugs have failed to show a reduction in CV events. Particularly, two recent trials of niacin (using extended-release niacin; AIM-HIGH [127] and HPS2-THRIVE [128]) were done on the background of statin therapy and were primarily designed to show the benefit of the HDL-raising effects of niacin. Neither trial met its primary endpoint and niacin failed to reduce cardiovascular events in both trials. Based on this, extended-release niacin added to a statin in patients with reasonably controlled LDL-C concentrations does not confer a cardiovascular benefit despite an increase in HDL-C concentrations. As a result, niacin at present should not be considered a therapeutic option for raising HDL-C concentrations.
In accordance with the HDL function hypothesis, it is not HDL cholesterol itself that has a causal relation to atheroprotection, but rather HDL function, which cannot be reliably estimated through the simple measurement of HDL-C [129],[130].
Interesting lessons could be derived from the Bezafibrate Infarction Prevention (BIP) study with bezafibrate and basically low HDL in all patients: despite of a significant HDL raising, the overall benefits were non-significant. However, the benefit of bezafibrate in the subgroup of patients with high TG levels was extremely impressive.
Additional challenges to the HDL hypothesis are driven by data derived from human genetic studies and randomized controlled trials. Taken together, genetic studies strongly support the theory that high concentrations of TG-rich lipoproteins or remnant cholesterol are causal risk factors for cardiovascular disease and all-cause mortality [2],[131]-[138], and that low HDL cholesterol is probably an innocent bystander. Low HDL cholesterol might merely be a long-term marker of raised TG and remnant cholesterol. Alternatively, HDL cholesterol might be a marker of cardiovascular health but is non-causal in the atherogenesis [130].
Management of hypertriglyceridemia
Intensive lifestyle therapy, including dietary counseling to achieve appropriate diet composition, physical activity, and a program to achieve weight reduction in overweight and obese individuals are the main initial treatment of hypertriglyceridemia and described elsewhere [20],[139]-[141].
Elevated levels of TG (and TG -rich lipoproteins) are increasingly being recognized as treatment targets to lower CV risk in certain patient subgroups, including individuals receiving statins – a cornerstone of the modern lipids-modifying therapy. The choice of statin should depend on the needs of the individual patient. In this context, the potential benefits of pitavastatin versus other statins in the treatment of patients with dyslipidemia and insulin resistance, metabolic syndrome or type 2 diabetes should be emphasized [142]-[144]. Moreover, although some statins are associated with increased haemoglobin A1C levels in patients receiving intensive but not moderate therapy, pitavastatin has demonstrated neutral or even favourable effects on glucose control in patients with and without T2DM or MS [145],[146]. However, intensive statin therapy with the most potent statins (atorvastatin, rosuvastatin and even pitavastatin) does not completely eliminate the residual cardiovascular risk associated with high TG.
At present the number of drug classes (fibrates, niacin, n-3 fatty acids, CETP –inhibitors, ezetimibe, glitazars, etc.) alone or in combination with statins have been considered as treatment options in patients with moderate to severe TG levels. However, many of these agents are currently under serious concerns: niacin after the negative AIM HIGH study and HPS-2 THRIVE trial results [127],[128]. CETP inhibitors and glitazars are still in controversial developments and not available for clinical use. Supplemental n-3 polyunsaturated fatty acids (PUFAs), mainly eicosapentaenoic acid and docosahexaenoic acid, are well known to reduce hypertriglyceridemia [147]. In addition to hypotriglyceridemic effects, omega-3 fatty acids may attenuate inflammation, improve endothelial function and reduce thrombus formation [148],[149]. However, recent clinical outcome trials with have failed to show significant CV benefits in high risk subjects [150]-[152].
Ezetimibe inhibits intestinal cholesterol absorption and primarily lowers LDL cholesterol via the Niemann-Pick C1- Like 1 protein. Ezetimibe has slight positive effects in lowering plasma fasting TG (8%) [153]. In addition, ezetimibe reduces the cholesterol content of both fasting and postprandial TG -rich lipoproteins, thereby lowering the concentrations of atherogenic remnant particles [154]. Initial experience of ezetimibe/fibrates combination seems promising [155],[156]. The recently released IMPROVE-IT (IMProved Reduction of Outcomes: Vytorin Efficacy International Trial) is the first to prove that adding a non-statin drug (ezetimibe) to a statin (simvastatin) lowers the risk of future CV events. Compared to patients with CAD on simvastatin plus a placebo, those on both simvastatin and ezetimibe, had a 6.4% lower risk of all CV events, a 14% lower risk of all heart attacks, a 14% lower risk of stroke, and a 21% lower risk of ischemic stroke. Deaths from CV disease were statistically the same in both groups. Patients were followed an average of approximately six years, and some as long as 8.5 years. Approximately 2 patients out of every 100 patients treated for 7 years avoided a heart attack or stroke. The Number Needed to Treat was = 50 [157]. This result represents a strong evidence-based support for the concept of the benefits of an appropriate statin/non-statin combination therapy.
Fibrates enhance the oxidation of fatty acids in liver and muscle and reduce the rate of hepatic lipogenesis, thereby reducing secretion of VLDL TG. The increased uptake of t TG -derived fatty acids in muscle cells results from an increase in LpL activity in adjacent capillaries and a decrease in the apolipoprotein CIII (apo CIII) concentration mediated transcriptionally by peroxisome proliferator activated receptor (PPAR) alpha. The decrease in apolipoprotein CIII reduces the inhibition of LpL activity. The enhanced catabolism of VLDL generates surface remnants, which are transferred to HDL. HDL concentrations are further augmented by an increase in PPAR alpha - mediated transcription of apoAI) and apo AII. Ultimately, the rate of HDL-mediated reverse cholesterol transport may increase. Fibrates activate PPAR alpha, which binds to a PPAR alpha response element in conjunction with the retinoid X receptor. Other effects of fibrates include an increase in the size of LDL particles, increased removal of LDL, and a reduction in the levels of plasminogen activator inhibitor type I [158]-[162].
From a clinical point of view, in all available 5 randomized control trials the beneficial effects of major fibrates (gemfibrozil, fenofibrate, bezafibrate) were clearly demonstrated and were highly significant in patients with hypertriglyceridemia [163]-[168].
In a meta-analysis of five dyslipidemic subgroups totaling 4726 patients, a 35% relative risk reduction in CV events was observed compared with a non- significant 6% reduction in those without dyslipidemia [169]. Meta-analysis performed in a so called "general population" [170] reflecting a blend of effects in patients with and without atherogenic dyslipidemia - a mean diluted effect of fibrate therapy was reduced, producing only 13% RR reduction for coronary events (p < 0.0001). Therefore, in patients with high triglycerides, fibrates - either as monotherapy or combined with statins - are consistently associated with reduced risk of cardiovascular events [171],[172]. Therefore, if the primary goal is to lower TG levels, fibrates (bezafibrate and fenofibrate for monotherapy and combination with statin; gemfibrozil only for monotherapy) now are the preferable drugs [173],[174].
Conclusions
Taking epidemiological data together, both moderate and severe hypertriglyceridaemia are associated with a substantially increased long term total mortality and CV disease risk. The plasma TG level represents in part the concentration of the TRL: VLDL, chylomicrons and their remnants. TG may also stimulate atherogenesis by other mechanisms, which include the production of proinflammatory cytokines, fibrinogen and coagulation factors and impairment of fibrinolysis. The most common forms of hypertriglyceridemia are related to overweight and sedentary life style which leads to insulin resistance and typical for MS and T2DM. Therefore, the role of hypertriglyceridemia in atherogenesis has a multifactorial biological plausibility. Also genetic studies strongly support the theory that hypertriglyceridemia and high concentrations of TRL are causal risk factors for CV disease and mortality.
Intensive lifestyle therapy is the main initial treatment of hypertriglyceridemia. If the primary goal is to lower TG levels, fibrates (bezafibrate and fenofibrate for monotherapy and combination with statin; gemfibrozil only for monotherapy) now are the preferable drugs. Finally, the clinical paradigm of lipids-modifying treatment should be changed and high TG should be recognized as an important target for therapy in their own right. Hypertriglyceridemia should be treated.
Abbreviations
- apoA-I:
-
Apolipoprotein A-I
- apoB:
-
Apolipoprotein B
- apo CIII:
-
Apolipoprotein CIII
- CAD:
-
Coronary artery disease
- CETP:
-
Cholesteryl ester transfer protein
- CV:
-
Cardiovascular
- FFA:
-
Free fatty acids
- HDL:
-
High density lipoprotein
- LDL:
-
Low density lipoprotein
- LpL:
-
Lipoprotein lipase
- MS:
-
Metabolic syndrome
- PPAR:
-
Peroxisome proliferator activated receptor
- TG:
-
Triglyceride, triglycerides
- TRL:
-
Triglyceride-carrying lipoproteins
- T2DM:
-
Type 2 diabetes mellitus
References
Miller M, Stone NJ, Ballantyne C, Bittner V, Criqui MH, Ginsberg HN, Goldberg AC, Howard WJ, Jacobson MS, Kris-Etherton PM, Lennie TA, Levi M, Mazzone T, Pennathur S: Triglycerides and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. 2011, 123: 2292-2333.
Nordestgaard BG, Varbo A: Triglycerides and cardiovascular disease. Lancet. 2014, 384: 626-635.
Criqui MH, Heiss G, Cohn R, Cowan LD, Suchindran CM, Bangdiwala S, Kritchevsky S, Jacobs DR, O’Grady HK, Davis CE: Plasma triglyceride level and mortality from coronary heart disease. N Engl J Med. 1993, 328: 1220-1225.
Laakso M, Lehto S, Penttila I, Pyorala K: Lipids and lipoproteins predicting coronary heart disease mortality and morbidity in patients with non-insulin-dependent diabetes. Circulation. 1993, 88: 1421-1430.
West KM, Ahuja MM, Bennett PH, Czyzyk A, De Acosta OM, Fuller JH, Grab B, Grabauskas V, Jarrett RJ, Kosaka K: The role of circulating glucose and triglyceride concentrations and their interactions with other “risk factors” as determinants of arterial disease in nine diabetic population samples from the WHO multinational study. Diabetes Care. 1983, 6: 361-369.
Fontbonne A, Eschwege E, Cambien F, Richard JL, Ducimetiere P, Thibult N, Warnet JM, Claude JR, Rosselin GE: Hypertriglyceridaemia as a risk factor of coronary heart disease mortality in subjects with impaired glucose tolerance or diabetes: results from the 11-year follow-up of the Paris Prospective Study. Diabetologia. 1989, 32: 300-304.
Bass KM, Newschaffer CJ, Klag MJ, Bush TL: Plasma lipoprotein levels as predictors of cardiovascular death in women. Arch Intern Med. 1993, 153: 2209-2216.
Austin MA, McKnight B, Edwards KL, Bradley CM, McNeely MJ, Psaty BM, Brunzell JD, Motulsky AG: Cardiovascular disease mortality in familial forms of hypertriglyceridemia: a 20-year prospective study. Circulation. 2000, 101: 2777-2782.
He Y, Lam TH, Li LS, He SF, Liang BQ: Triglyceride and coronary heart disease mortality in a 24-year follow-up study in Xi’an, China. Ann Epidemiol. 2004, 14: 1-7.
Almeda-Valdes P, Cuevas-Ramos D, Mehta R, Muñoz-Hernandez L, Cruz-Bautista I, Perez-Mendez O, Tusie-Luna MT, Gomez-Perez FJ, Pajukanta P, Matikainen N, Taskinen MR, Aguilar-Salinas CA: Factors associated with postprandial lipemia and apolipoprotein A-V levels in individuals with familial combined hyperlipidemia.BMC Endocr Disord 2014, 14:90.,
Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A: Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007, 298: 299-308.
Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM: Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007, 298: 309-316.
Di Angelantonio E, Sarwar N, Perry P, Kaptoge S, Ray KK, Thompson A, Wood AM, Lewington S, Sattar N, Packard CJ, Collins R, Thompson SG, Danesh J: Major lipids, apolipoproteins, and risk of vascular disease. JAMA. 2009, 302: 1993-2000.
Schwartz GG, Olsson AG, Szarek M, Sasiela WJ: Relation of characteristics of metabolic syndrome to short-term prognosis and effects of intensive statin therapy after acute coronary syndrome: an analysis of the Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial. Diabetes Care. 2005, 28: 2508-2513.
Kasai T, Miyauchi K, Kurata T, Ohta H, Okazaki S, Miyazaki T, Kajimoto K, Kubota N, Daida H: Prognostic value of the metabolic syndrome for long-term outcomes in patients undergoing percutaneous coronary intervention. Circ J. 2006, 70: 1531-1537.
Anderson JL, Horne BD, Jones HU, Reyna SP, Carlquist JF, Bair TL, Pearson RR, Lappe DL, Muhlestein JB: Which features of the metabolic syndrome predict the prevalence and clinical outcomes of angiographic coronary artery disease?. Cardiology. 2004, 101: 185-193.
Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002, 106: 3143-3421.
Heiss G, Tamir I, Davis CE, Tyroler HA, Rifkand BM, Schonfeld G, Jacobs D, Frantz ID: Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation. 1980, 61: 302-315.
Ford ES, Li C, Zhao G, Pearson WS, Mokdad AH: Hypertriglyceridemia and its pharmacologic treatment among US adults. Arch Intern Med. 2009, 169: 572-578.
Berglund L, Brunzell JD, Goldberg AC, Goldberg IJ, Sacks F, Murad MH, Stalenhoef AF: Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012, 97: 2969-2989.
Assmann G, Schulte H, von Eckardstein A: Hypertriglyceridemia and elevated lipoprotein(a) are risk factors for major coronary events in middle-aged men. Am J Cardiol. 1996, 77: 1179-1184.
Kasai T, Miyauchi K, Yanagisawa N, Kajimoto K, Kubota N, Ogita M, Tsuboi S, Amano A, Daida H: Mortality risk of triglyceride levels in patients with coronary artery disease. Heart. 2013, 99: 22-29.
Neil HA, Cooper J, Betteridge DJ, Capps N, McDowell IF, Durrington PN, Seed M, Mann JI, Humphries SE: All-cause and cardiovascular mortality in treated patients with severe hypertriglyceridemia: A long-term prospective registry study. Atherosclerosis. 2010, 211: 618-623.
Miselli MA, Nora ED, Passaro A, Tomasi F, Zuliani G: Plasma triglycerides predict ten-years all-cause mortality in outpatients with type 2 diabetes mellitus: a longitudinal observational study.Cardiovasc Diabetol 2014, 13:135.,
Mora S, Rifai N, Buring JE, Ridker PM: Fasting compared with nonfasting lipids and apolipoproteins for predicting incident cardiovascular events. Circulation. 2008, 118: 993-1001.
Stalenhoef AF, de Graaf J: Association of fasting and nonfasting serum triglycerides with cardiovascular disease and the role of remnant-like lipoproteins and small, dense LDL. Curr Opin Lipidol. 2008, 19: 355-361.
Patsch JR, Miesenbock G, Hopferwieser T, Mu hlberger V, Knapp E, Dunn JK, Gotto AM, Patsch W: Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler Thromb. 1992, 12: 1336-1345.
Karpe F: Postprandial lipoprotein metabolism and atherosclerosis. J Intern Med. 1999, 246: 341-355.
Eberly LE, Stamler J, Neaton JD: Relation of triglyceride levels, fasting and nonfasting, to fatal and nonfatal coronary heart disease. Arch Intern Med. 2003, 163: 1077-1083.
Durrington PN: Triglycerides are more important in atherosclerosis than epidemiology has suggested. Atherosclerosis. 1998, 141 (Supp l 1): S57-S62.
Deedwania P, Barter P, Carmena R, Fruchart JC, Grundy SM, Haffner S, Kastelein JJ, LaRosa JC, Schachner H, Shepherd J, Waters DD: Reduction of low-density lipoprotein cholesterol in patients with coronary heart disease and metabolic syndrome: analysis of the Treating to New Targets study. Lancet. 2006, 368: 919-928.
Cohn JS, McNamara JR, Cohn SD, Ordovas JM, Schaefer EJ: Plasma apolipoprotein changes in the triglyceride-rich lipoprotein fraction of human subjects fed a fat-rich meal. J Lipid Res. 1988, 29: 925-936.
Karpe F, Bell M, Bjorkegren J, Hamsten A: Quantification of postprandial triglyceride-rich lipoproteins in healthy men by retinyl ester labeling and simultaneous measurement of apolipoproteins B-48 and B-100. Arterioscler Thromb Vasc Biol. 1995, 15: 199-207.
Campos H, Khoo C, Sacks FM: Diurnal and acute patterns of postprandial apolipoprotein B-48 in VLDL, IDL, and LDL from normolipidemic humans. Atherosclerosis. 2005, 181: 345-351.
Nakano T, Tanaka A, Okazaki M, Tokita Y, Nagamine T, Nakajima K: Particle size of apoB-48 carrying lipoproteins in remnant lipoproteins isolated from postprandial plasma. Ann Clin Biochem. 2011, 48: 57-64.
Tabas I, Williams KJ, Boren J: Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007, 116: 1832-1844.
Kannel WB, Vasan RS: Triglycerides as vascular risk factors: new epidemiologic insights. Curr Opin Cardiol. 2009, 24: 345-350.
Wu L, Parhofer KG: Diabetic dyslipidemia. Metabolism. 2014
Adiels M, Boren J, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson SO, Packard C, Taskinen MR: Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2005, 25: 1697-1703.
Guerin M, Le Goff W, Lassel TS, Van Tol A, Steiner G, Chapman MJ: Atherogenic role of elevated CE transfer from HDL to VLDL(1) and dense LDL in type 2 diabetes : impact of the degree of triglyceridemia. Arterioscler Thromb Vasc Biol. 2001, 21: 282-288.
Berneis KK, Krauss RM: Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002, 43: 1363-1379.
Donahoo WT, Kosmiski LA, Eckel RH: Drugs causing dyslipoproteinemia. Endocrinol Metab Clin North Am. 1998, 27: 677-697.
Brunzell JD: Clinical practice. Hypertriglyceridemia. N Engl J Med. 2007, 357: 1009-1017.
Brinton EA: Effects of ethanol intake on lipoproteins and atherosclerosis. Curr Opin Lipidol. 2010, 21: 346-351.
Taskinen MR, Nikkilä EA, Välimäki M, Sane T, Kuusi T, Kesäniemi A, Ylikahri R: Alcohol-induced changes in serum lipoproteins and in their metabolism. Am Heart J. 1987, 113 (2 Pt 2): 458-464.
Ginsberg HN: New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002, 106: 2137-2142.
Batal R, Tremblay M, Barrett PH, Jacques H, Fredenrich A, Mamer O, Davignon J, Cohn JS: Plasma kinetics of apoC-III and apoE in normolipidemic and hypertriglyceridemic subjects. J Lipid Res. 2000, 41: 706-718.
Cohn JS, Patterson BW, Uffelman KD, Davignon J, Steiner G: Rate of production of plasma and very-low-density lipoprotein (VLDL) apolipoprotein C-III is strongly related to the concentration and level of production of VLDL triglyceride in male subjects with different body weights and levels of insulin sensitivity. J Clin Endocrinol Metab. 2004, 89: 3949-3955.
Campos H, Moye LA, Glasser SP, Stampfer MJ, Sacks FM: Low density lipoprotein size, pravastatin treatment, and coronary events. JAMA. 2001, 286: 1468-1474.
Zheng C, Khoo C, Ikewaki K, Sacks FM: Rapid turnover of apolipoprotein C-III-containing triglyceride-rich lipoproteins contributing to the formation of LDL subfractions. J Lipid Res. 2007, 48: 1190-1203.
Ooi EM, Barrett PH, Chan DC, Watts GF: Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci. 2008, 114: 611-624.
Malmstrom R, Packard CJ, Caslake M, Bedford D, Stewart P, Yki-Jarvinen H, Shepherd J, Taskinen MR: Defective regulation of triglyceride metabolism by insulin in the liver in NIDDM. Diabetologia. 1997, 40: 454-462.
Kamagate A, Dong HH: FoxO1 integrates insulin signaling to VLDL production. Cell Cycle. 2008, 7: 3162-3170.
Lee SJ, Moye LA, Campos H, Williams GH, Sacks FM: Hypertriglyceridemia but not diabetes status is associated with VLDL containing apolipoprotein CIII in patients with coronary heart disease. Atherosclerosis. 2003, 167: 293-302.
Cheal KL, Abbasi F, Lamendola C, McLaughlin T, Reaven GM, Ford ES: Relationship to insulin resistance of the adult treatment panel III diagnostic criteria for identification of the metabolic syndrome. Diabetes. 2004, 53: 1195-1200.
Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Spertus JA, Costa F: Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung and Blood Institute scientific statement. Circulation. 2005, 112: 2735-2752.
Won KB, Chang HJ, Kim HC, Jeon K, Lee H, Shin S, Cho IJ, Park SH, Lee SH, Jang Y: Differential impact of metabolic syndrome on subclinical atherosclerosis according to the presence of diabetes.Cardiovasc Diabetol 2013, 12:41.,
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC: Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009, 120: 1640-1645.
An X, Yu D, Zhang R, Zhu J, Du R, Shi Y, Xiong X: Insulin resistance predicts progression of de novo atherosclerotic plaques in patients with coronary heart disease: a one-year follow-up study.Cardiovasc Diabetol 2012, 11:71.,
Nieves DJ, Cnop M, Retzlaff B, Walden CE, Brunzell JD, Knopp RH, Kahn SE: The atherogenic lipoprotein profile associated with obesity and insulin resistance is largely attributable to intraabdominal fat. Diabetes. 2003, 52: 172-179.
Novo S, Peritore A, Trovato RL, Guarneri FP, Di Lisi D, Muratori I, Novo G: Preclinical atherosclerosis and metabolic syndrome increase cardio- and cerebrovascular events rate: a 20-year follow up.Cardiovasc Diabetol 2013, 12:155.,
Feinberg MS, Schwartz R, Tanne D, Fisman EZ, Hod H, Zahger D, Schwammenthal E, Eldar M, Behar S, Tenenbaum A: Impact of the metabolic syndrome on the clinical outcomes of non-clinically diagnosed diabetic patients with acute coronary syndrome. Am J Cardiol. 2007, 99: 667-672.
Brunzell JD, Ayyobi AF: Dyslipidemia in the metabolic syndrome and type 2 diabetes mellitus. Am J Med. 2003, 115 (Suppl 8A): 24S-28S.
Herder M, Arntzen KA, Johnsen SH, Mathiesen EB: The metabolic syndrome and progression of carotid atherosclerosis over 13 years. The Tromsø study.Cardiovasc Diabetol 2012, 11:77.,
Pilz S, Scharnagl H, Tiran B, Seelhorst U, Wellnitz B, Boehm BO, Schaefer JR, Marz W: Free fatty acids are independently associated with all-cause and cardiovascular mortality in subjects with coronary artery disease. J Clin Endocrinol Metab. 2006, 91: 2542-2547.
Oliver MF: Prevention of ventricular fibrillation during acute myocardial ischemia: control of free fatty acids. J Cardiovasc Pharmacol Ther. 2001, 6: 213-217.
Tripathy D, Mohanty P, Dhindsa S, Syed T, Ghanim H, Aljada A, Dandona P: Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003, 52: 2882-2887.
Lee JY, Sohn KH, Rhee SH, Hwang D: Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. 2001, 276: 16683-16689.
Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M: Free fatty acids regulate insulin secretion from pancreatic cells through GPR40. Nature. 2003, 422: 173-176.
Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI: The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003, 278: 11303-11311.
Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G: Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005, 11: 90-94.
Mathew M, Tay E, Cusi K: Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects.Cardiovasc Diabetol 2010, 9:9.,
Boden G, Lebed B, Schatz M, Homko C, Lemieux S: Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001, 50: 1612-1617.
Itani SI, Ruderman NB, Schmieder F, Boden G: Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IκB-α. Diabetes. 2002, 51: 2005-2011.
Barazzoni R, Zanetti M, Gortan Cappellari G, Semolic A, Boschelle M, Codarin E, Pirulli A, Cattin L, Guarnieri G: Fatty acids acutely enhance insulin-induced oxidative stress and cause insulin resistance by increasing mitochondrial reactive oxygen species (ROS) generation and nuclear factor-κB inhibitor (IκB)-nuclear factor-κB (NFκB) activation in rat muscle, in the absence of mitochondrial dysfunction. Diabetologia. 2012, 55: 773-782.
Boden G, She P, Mozzoli M, Cheung P, Gumireddy K, Reddy P, Xiang X, Luo Z, Ruderman N: Free fatty acids produce insulin resistance and activate the proinflammatory nuclear factor-κB pathway in rat liver. Diabetes. 2005, 54: 3458-3465.
Hotamisligil GS: Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005, 54: S73-S78.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H: High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000, 49: 1939-1945.
Shi A, Kokoeva V, Inouye K, Tzameli I, Yin H, Flier JS: TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006, 116: 3015-3025.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI: Mechanism by which fatty acids inhibit activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002, 277: 50230-50236.
Saltiel AR, Kahn CR: Insulin signaling and the regulation of glucose and lipid metabolism. Nature. 2001, 414: 799-806.
Newby AC: Dual role of matrix metalloproteinases (Matrixins) in intimal thickening of atherosclerotic plaque rupture. Physiol Rev. 2005, 85: 1-31.
Galis ZS, Sukhova GK, Lark MW, Libby P: Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994, 94: 2493-2503.
Pasterkamp G, Schoneveld AH, Hijnen DJ, e Kleijn DP, Teepen H, van der Wal AC, Borst C: Atherosclerotic arterial remodeling and the localization of macrophages and matrix metalloproteinases 1, 2 and 9 in the human coronary artery. Atherosclerosis. 2000, 150: 245-253.
Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT: Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002, 110: 625-632.
Long SD, Pekala PH: Regulation of Glut4 gene expression by arachidonic acid. Evidence for multiple pathways, one of which requires oxidation to prostaglandin. J Biol Chem. 1996, 271: 1138-1144.
Armoni M, Harel C, Bar-Yoseph F, Milo S, Karnieli E: Free fatty acids repress the Glut4 gene expression in cardiac muscle via novel response elements. J Biol Chem. 2005, 280: 34786-34795l.
Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H: Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003, 112: 1821-1830.
van Oostrom AJ, van Dijk H, Verseyden C, Sniderman AD, Cianflone K, Rabelink TJ, Castro Cabezas M: Addition of glucose to an oral fat load reduces postprandial free fatty acids and prevents the postprandial increase in complement component 3. Am J Clin Nutr. 2004, 79: 5-10.
Rollins BJ, Walz A, Baggiolini M: Recombinant human MCP-1/JE induces chemotaxis, calcium flux, and the respiratory burst in human monocytes. Blood. 1991, 78: 1112-1116.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW: Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003, 112: 1796-1808.
Gao Z, Zhang X, Zuberi A, Hwang D, Quon MJ, Lefevre M, Ye J: Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004, 18: 2024-2034.
Medzhitov R: Toll-like receptors and innate immunity. Nat Rev Immunol. 2001, 1: 135-145.
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Görgün C, Glimcher LH, Hotamisligil GS: Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004, 306: 457-461.
Tomita T, Hosoda K, Fujikura J, Inagaki N, Nakao K: The G-protein-coupled long-chain fatty acid receptor GPR40 and glucose metabolism.Front Endocrinol (Lausanne) 2014, 5:152.,
Boden G: Free fatty acids and insulin secretion in humans. Curr Diab Rep. 2005, 5: 167-270.
Benjamin SM, Valdez R, Geiss LS, Rolka DB, Narayan KM: Estimated number of adults with prediabetes in the US in 2000: opportunities for prevention. Diabetes Care. 2003, 26: 645-649.
Bamba V, Rader DJ: Obesity and atherogenic dyslipidemia. Gastroenterology. 2007, 132: 2181-2190.
Boden G, Rao AK: Effects of hyperglycemia and hyperinsulinemia on the tissue factor pathway of blood coagulation. Curr Diab Rep. 2007, 7: 223-227.
Boden G, Vaidyula VR, Homko C, Cheung P, Rao AK: Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: Effects of insulin and glucose. J Clin Endocrinol Metab. 2007, 92: 4352-4358.
Vague P, Juhan-Vague I, Aillaud MF, Badier C, Viard R, Alessi MC, Collen D: Correlation between blood fibrinolytic activity, plasminogen activator inhibitor level, plasma insulin level and relative body weight in normal and obese subjects. Metabolism. 1986, 35: 250-253.
Pannacciulli N, De Mitrio R, Giorgino R, De Pergola G: Effect of glucose tolerance status on PAI-1 plasma levels in overweight and obese subjects. Obes Res. 2002, 10: 717-725.
Festa A, D’Agostino R, Tracy RP, Haffner SM: Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the Insulin Resistance Atherosclerosis Study. Diabetes. 2002, 51: 1131-1137.
Sobel BE, Schneider DJ: Platelet function, coagulopathy, and impaired fibrinolysis in diabetes. Cardiol Clin. 2004, 22: 511-526.
Hennekens CH: Increasing burden of cardiovascular disease: current knowledge and future directions for research on risk factors. Circulation. 1998, 97: 1095-1102.
Shimabukuro M, Zhou YT, Levi M, Unger RH: Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci U S A. 1998, 95: 2498-2502.
Unger RH: Lipotoxicity in the pathogenesis of obesity-dependent NIDDM Genetic and clinical implications. Diabetes. 1995, 44: 863-870.
Robertson RP, Harmon J, Tran PO, Poitout V: Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes. 2004, 53 (Suppl 1): S119-S124.
Haber EP, Procopio J, Carvalho CR, Carpinelli AR, Newsholme P, Curi R: New insights into fatty acid modulation of pancreatic beta-cell function. Int Rev Cytol. 2006, 248: 1-41.
Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS: Role of apoptosis in failure of beta cell mass compensation for insulin resistance and beta cell defects in the male Zucker diabetic fatty rat. Diabetes. 1998, 47: 358-364.
Zhou YT, Shimabukuro M, Wang MY, Lee Y, Higa M, Milburn JL, Newgard CB, Unger RH: Role of peroxisome proliferator-activated receptor alpha in disease of pancreatic beta cells. Proc Natl Acad Sci U S A. 1998, 95: 8898-8903.
Arner P: Free fatty acids - do they play a central role in type 2 diabetes?. Diab Obes Metab. 2001, 3 (Suppl 1): 11-19.
Tenenbaum H, Behar S, Boyko V, Adler Y, Fisman EZ, Tanne D, Lapidot M, Schwammenthal E, Feinberg M, Matas Z, Motro M, Tenenbaum A: Long-term effect of bezafibrate on pancreatic beta-cell function and insulin resistance in patients with diabetes. Atherosclerosis. 2007, 194: 265-271.
Shafrir E, Raz I: Diabetes: mellitus or lipidus?. Diabetologia. 2003, 46: 433-440.
Badimon JJ, Badimon L, Fuster V: Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990, 85: 1234-1243.
Rubin E, Krauss R, Spangler E, Verstuyft J, Clift S: Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991, 353: 265-267.
Tangirala RK, Tsukamoto K, Chun SH, Usher D, Pure E, Rader DJ: Regression of atherosclerosis induced by liver-directed gene transfer of apolipoprotein A-I in mice. Circulation. 1999, 100: 1816-1822.
Castelli WP, Garrison RJ, Wilson PW, Abbott RD, Kalousdian S, Kannel WB: Incidence of coronary heart disease and lipoprotein cholesterol levels. The Framingham Study. JAMA. 1986, 256: 2835-2838.
Assmann G, Schulte H, von Eckardstein A, Huang Y: High density lipoprotein cholesterol as a predictor of coronary heart disease risk. The PROCAM experience and pathophysiological implications for reverse cholesterol transport. Atherosclerosis. 1996, 124 (Suppl): S11-S20.
von Eckardstein A, Nofer JR, Assmann G: High density lipoproteins and arteriosclerosis. Role of cholesterol efflux and reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 2001, 21: 13-27.
Leança CC, Nunes VS, Panzoldo NB, Zago VS, Parra ES, Cazita PM, Jauhiainen M, Passarelli M, Nakandakare ER, de Faria EC, Quintão EC: Metabolism of plasma cholesterol and lipoprotein parameters are related to a higher degree of insulin sensitivity in high HDL-C healthy normal weight subjects.Cardiovasc Diabetol 2013, 12:173.,
Annema W, von Eckardstein A: High-density lipoproteins. Multifunctional but vulnerable protections from atherosclerosis. Circ J. 2013, 77: 2432-2448.
Tong X, Peng H, Liu D, Ji L, Niu C, Ren J, Pan B, Hu J, Zheng L, Huang Y: High-density lipoprotein of patients with type 2 diabetes mellitus upregulates cyclooxgenase-2 expression and prostacyclin I-2 release in endothelial cells: relationship with HDL-associated sphingosine-1-phosphate.Cardiovasc Diabetol 2013, 12:27.,
Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez-Sendon J, Mosca L, Tardif JC, Waters DD, Shear CL, Revkin JH, Buhr KA, Fisher MR, Tall AR, Brewer B: Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007, 357: 2109-2122.
Funder JW: Aldosterone, sodium, and hypertension: lessons from torcetrapib?. Hypertension. 2010, 55: 221-223.
Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, Leitersdorf E, McMurray JJ, Mundl H, Nicholls SJ, Shah PK, Tardif JC, Wright RS: Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012, 367: 2089-2099.
Investigators AIM-HIGH, Boden WE, Probstfield JL, Anderson T, Chaitman BR, Desvignes-Nickens P, Koprowicz K, McBride R, Teo K, Weintraub W: Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011, 365: 2255-2267.
Landray MJ, Haynes R, Hopewell JC, Parish S, Aung T, Tomson J, Wallendszus K, Craig M, Jiang L, Collins R, Armitage J: Effects of extended release niacin with laropiprant in high-risk patients. N Engl J Med. 2014, 371: 203-212.
Rader DJ, Tall AR: The not-so-simple HDL story: is it time to revise the HDL cholesterol hypothesis?. Nat Med. 2012, 18: 1344-1346.
Rader DJ, Hovingh GK: HDL and cardiovascular disease. Lancet. 2014, 384: 618-625.
Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, Johansen CT, Fouchier SW, Isaacs A, Peloso GM, Barbalic M, Ricketts SL, Bis JC, Aulchenko YS, Thorleifsson G, Feitosa MF, Chambers J, Orho-Melander M, Melander O, Johnson T, Li X, Guo X, Li M, Shin Cho Y, Jin Go M, Jin Kim Y: Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010, 466: 707-713.
Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A: Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013, 45: 1345-1352.
Varbo A, Benn M, Tybjaerg-Hansen A, Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG: Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013, 61: 427-436.
Thomsen M, Varbo A, Tybjærg-Hansen A, Nordestgaard BG: Low nonfasting triglycerides and reduced all-cause mortality: a mendelian randomization study. Clin Chem. 2014, 60: 737-746.
Calabresi L, Simonelli S, Gomaraschi M, Franceschini G: Genetic lecithin:cholesterol acyltransferase defi ciency and cardiovascular disease. Atherosclerosis. 2012, 222: 299-306.
de Grooth GJ, Klerkx AH, Stroes ES, Stalenhoef AF, Kastelein JJ, Kuivenhoven JA: A review of CETP and its relation to atherosclerosis. J Lipid Res. 2004, 45: 1967-1974.
Adams JN, Cox AJ, Freedman BI, Langefeld CD, Carr JJ, Bowden DW: Genetic analysis of haptoglobin polymorphisms with cardiovascular disease and type 2 diabetes in the Diabetes Heart Study.Cardiovasc Diabetol 2013, 12:31.,
Sarwar N, Sandhu MS, Ricketts SL, Butterworth AS, Di Angelantonio E, Boekholdt SM, Ouwehand W, Watkins H, Samani NJ, Saleheen D, Lawlor D, Reilly MP, Hingorani AD, Talmud PJ, Danesh J: Triglyceride-mediated pathways and coronary disease: collaborative analysis of 101 studies. Lancet. 2010, 375: 1634-1639.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Nordestgaard BG, Ray KK, Reiner Z, Taskinen MR, Tokgözoglu L, Tybjærg-Hansen A, Watts GF: Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011, 32: 1345-1361.
Hegele RA, Ginsberg HN, Chapman MJ, Nordestgaard BG, Kuivenhoven JA, Averna M, Borén J, Bruckert E, Catapano AL, Descamps OS, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Raal FJ, Ray KK, Santos RD, Stalenhoef AF, Stroes E, Taskinen MR, Tybjærg-Hansen A, Watts GF, Wiklund O: The polygenic nature of hypertriglyceridaemia: implications for definition, diagnosis, and management. Lancet Diab Endocrinol. 2014, 2: 655-666.
Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D: ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011, 32: 1769-1818.
Ray K: Statin diabetogenicity: guidance for clinicians.Cardiovasc Diabetol 2013, 12(Suppl 1):S3.,
Mita T, Nakayama S, Abe H, Gosho M, Iida H, Hirose T, Kawamori R, Watada H: Comparison of effects of pitavastatin and atorvastatin on glucose metabolism in type 2 diabetic patients with hypercholesterolemia. J Diabetes Investig. 2013, 4: 297-303.
Ginsberg H: Statins in cardiometabolic disease: what makes pitavastatin different?Cardiovasc Diabetol 2013, 12(Suppl 1):S1.,
Chapman MJ, Orsoni A, Robillard P, Hounslow N, Sponseller CA, Giral P: Effect of high-dose pitavastatin on glucose homeostasis in patients at elevated risk of new-onset diabetes: insights from the CAPITAIN and PREVAIL-US studies. Curr Med Res Opin. 2014, 30: 775-784.
Masana L: Pitavastatin in cardiometabolic disease: therapeutic profile.Cardiovasc Diabetol 2013, 12(Suppl 1):S2.,
Saravanan P, Davidson NC, Schmidt EB, Calder PC: Cardiovascular effects of marine omega-3 fatty acids. Lancet. 2010, 376: 540-550.
Swanson D, Block R, Mousa SA: Omega-3 fatty acids EPA and DHA: health benefits throughout life. Adv Nutr. 2012, 3: 1-7.
Brinton EA, Ballantyne CM, Bays HE, Kastelein JJ, Braeckman RA, Soni PN: Effects of icosapent ethyl on lipid and inflammatory parameters in patients with diabetes mellitus-2, residual elevated triglycerides (200–500 mg/dL), and on statin therapy at LDL-C goal: the ANCHOR study.Cardiovasc Diabetol 2013, 12:100.,
Bosch J, Gerstein HC, Dagenais GR, Díaz R, Dyal L, Jung H, Maggiono AP, Probstfield J, Ramachandran A, Riddle MC, Rydén LE, Yusuf S: n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012, 367: 309-318.
Roncaglioni MC, Tombesi M, Avanzini F, Barlera S, Caimi V, Longoni P, Marzona I, Milani V, Silletta MG, Tognoni G, Marchioli R: n-3 fatty acids in patients with multiple cardiovascular risk factors. N Engl J Med. 2013, 368: 1800-1808.
Rizos EC, Ntzani EE, Bika E, Kostapanos MS, Elisaf MS: Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: a systematic review and meta-analysis. JAMA. 2012, 308: 1024-1033.
Pandor A, Ara RM, Tumur I, Wilkinson AJ, Paisley S, Duenas A, Durrington PN, Chilcott J: Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med. 2009, 265: 568-580.
Bozzetto L, Annuzzi G, Corte GD, Patti L, Cipriano P, Mangione A, Riccardi G, Rivellese AA: Ezetimibe beneficially influences fasting and postprandial triglyceride-rich lipoproteins in type 2 diabetes. Atherosclerosis. 2011, 217: 142-148.
Teramoto T, Abe K, Taneyama T: Safety and efficacy of long-term combination therapy with bezafibrate and ezetimibe in patients with dyslipidemia in the prospective, observational J-COMPATIBLE study.Cardiovasc Diabetol 2013, 12:163.,
Farnier M, Retterstøl K, Steinmetz A, Császár A: Comparative efficacy and safety of fenofibrate/pravastatin plus ezetimibe triple therapy and simvastatin/ezetimibe dual therapy in type 2 diabetic patients with mixed hyperlipidaemia and cardiovascular disease. Diab Vasc Dis Res. 2012, 9: 205-215.
Cannon CP (presenter): Cholesterol-lowering drug with different action adds to statin’s reduction of cardiovascular risk. American Heart Association Meeting Report Abstract LBCT.02. (accessed 21/11/2014)., [http://newsroom.heart.org/news/cholesterol-lowering-drug-with-different-action-adds-to-statins-reduction-of-cardiovascular-risk]
Knopp RH: Drug treatment of lipid disorders. N Engl J Med. 1999, 341: 498-511.
Tenenbaum A, Fisman EZ, Motro M, Adler Y: Optimal management of combined dyslipidemia: what have we behind statins monotherapy?. Adv Cardiol. 2008, 45: 127-153.
Tenenbaum A, Fisman EZ, Boyko V, Benderly M, Tanne D, Haim M, Matas Z, Motro M, Behar S: Attenuation of progression of insulin resistance in patients with coronary artery disease by bezafibrate. Arch Intern Med. 2006, 166: 737-741.
Teramoto T, Shirai K, Daida H, Yamada N: Effects of bezafibrate on lipid and glucose metabolism in dyslipidemic patients with diabetes: the J-BENEFIT study.Cardiovasc Diabetol 2012, 11:29.,
Tenenbaum A, Motro M, Fisman EZ, Tanne D, Boyko V, Behar S: Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med. 2005, 165: 1154-1160.
Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010, 362: 1563-1574.
Tenenbaum A, Fisman EZ: “If it ain”t broke, don’t fix it”: a commentary on the positive–negative results of the ACCORD Lipid study.Cardiovasc Diabetol 2010, 9:24.,
Scott R, O’Brien R, Fulcher G, Pardy C, D’Emden M, Tse D, Taskinen MR, Ehnholm C, Keech A: Effects of fenofibrate treatment on cardiovascular disease risk in 9,795 individuals with type 2 diabetes and various components of the metabolic syndrome: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care. 2009, 32: 493-498.
Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease: the Bezafibrate Infarction Prevention (BIP) study. Circulation. 2000, 102: 21-27.
Manninen V, Tenkanen L, Koskinen P, Huttunen JK, Mänttäri M, Heinonen OP, Frick MH: Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study: implications for treatment. Circulation. 1992, 85: 37-45.
Robins SJ, Collins D, Wittes JT, Papademetriou V, Deedwania PC, Schaefer EJ, McNamara JR, Kashyap ML, Hershman JM, Wexler LF, Rubins HB: Relation of gemfibrozil treatment and lipid levels with major coronary events: VA-HIT: a randomized controlled trial. JAMA. 2001, 285: 1585-1591.
Sacks FM, Carey VJ, Fruchart JC: Combination lipid therapy in type 2 diabetes. N Engl J Med. 2010, 363: 692-694.
Jun M, Foote C, Lv J, Neal B, Patel A, Nicholls SJ, Grobbee DE, Cass A, Chalmers J, Perkovic V: Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet. 2010, 375: 1875-1878.
Tenenbaum A, Medvedofsky D, Fisman EZ, Bubyr L, Matetzky S, Tanne D, Klempfner R, Shemesh J, Goldenberg I: Cardiovascular events in patients received combined fibrate/statin treatment versus statin monotherapy: Acute Coronary Syndrome Israeli Surveys data.PLoS One 2012, 7(4):e35298.,
Klempfner R, Goldenberg I, Fisman EZ, Matetzky S, Amit U, Shemesh J, Tenenbaum A: Comparison of statin alone versus bezafibrate and statin combination in patients with diabetes mellitus and acute coronary syndrome. Am J Cardiol. 2014, 113: 12-16.
Tenenbaum A, Fisman EZ: Balanced pan-PPAR activator bezafibrate in combination with statin: comprehensive lipids control and diabetes prevention?Cardiovasc Diabetol 2012, 11:140.,
Tenenbaum A, Fisman EZ: Fibrates are an essential part of modern anti-dyslipidemic arsenal: spotlight on atherogenic dyslipidemia and residual risk reduction.Cardiovasc Diabetol 2012, 11:125.,
Acknowledgments
This work was supported by the Cardiovascular Diabetology Research Foundation (RA 58-040-684-1), Holon, Israel.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
AT received speaker fee and travel expenses support from Abbott, Tribute, Novartis and Merck. EZF and RK declare that they have no competing interests.
Authors’ contributions
All authors have equally contributed in the conception and drafting of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Tenenbaum, A., Klempfner, R. & Fisman, E.Z. Hypertriglyceridemia: a too long unfairly neglected major cardiovascular risk factor. Cardiovasc Diabetol 13, 159 (2014). https://doi.org/10.1186/s12933-014-0159-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-014-0159-y