
Abstract
Background
Diabetes mellitus (DM) is a major risk factor for tuberculosis (TB). Evidence has linked the DM-related dysbiosis of gut microbiota to modifiable host immunity to Mycobacterium tuberculosis infection. However, the crosslinks between gut microbiota composition and immunological effects on the development of latent TB infection (LTBI) in DM patients remain uncertain.
Methods
We prospectively obtained stool, blood samples, and medical records from 130 patients with poorly-controlled DM (pDM), defined as ever having an HbA1c > 9.0% within previous 1 year. Among them, 43 had LTBI, as determined by QuantiFERON-TB Gold in-Tube assay. The differences in the taxonomic diversity of gut microbiota between LTBI and non-LTBI groups were investigated using 16S ribosomal RNA sequencing, and a predictive algorithm was established using a random forest model. Serum cytokine levels were measured to determine their correlations with gut microbiota.
Results
Compared with non-LTBI group, the microbiota in LTBI group displayed a similar alpha-diversity but different beta-diversity, featuring decrease of Prevotella_9, Streptococcus, and Actinomyces and increase of Bacteroides, Alistipes, and Blautia at the genus level. The accuracy was 0.872 for the LTBI prediction model using the aforementioned 6 microbiome-based biomarkers. Compared with the non-LTBI group, the LTBI group had a significantly lower serum levels of IL-17F (p = 0.025) and TNF-α (p = 0.038), which were correlated with the abundance of the aforementioned 6 taxa.
Conclusions
The study results suggest that gut microbiome composition maybe associated with host immunity relevant to TB status, and gut microbial signature might be helpful for the diagnosis of LTBI.
Similar content being viewed by others

Introduction
With the acceleration of the global burden of type 2 diabetes mellitus (DM) among people older than 40 years, DM has become a major threat for tuberculosis (TB) control [1, 2] because it compromises host immunity and facilitates either primary infection by Mycobacterium tuberculosis (Mtb) or reactivation from latent TB infection (LTBI) [3]. According to estimations, individuals with DM had a 3- and 1.18-fold higher risks of having active TB [4] or LTBI [5], respectively. Furthermore, every 1% increment of glycosylated hemoglobin (HbA1c) level can result in a 1.13-fold higher risk of LTBI [6]. A Taiwanese study revealed that approximately 26.7% of patients with poorly controlled diabetes, defined as ever having an HbA1c level > 9.0% in the past year, had LTBI. The prevalence was even higher than that among TB close contacts (15%) [7].
The effects of DM on host immune responses are complicated and may differ based on different TB statuses. Studies have suggested that although DM may augment systemic inflammation in patients with active TB, it attenuates mycobacteria-induced immune responses in those with LTBI by diminishing CD4-positive lymphocytes [8] and decreasing circulating levels of pro-inflammatory cytokines and anti-inflammatory cytokines [9]. The exact mechanism of disrupted host defense in DM patients with LTBI remains unknown, and host immunity enhancement might be a future strategy for preventing TB reactivation.
Recent advances in microbiome research indicate that gut microbiome–immune interactions in hosts are systemic, dynamic, and context dependent [10]. The transition and imbalance of gut microbiota can be observed under different DM statuses, which subsequently alters host immunity and homeostasis [11]. Additionally, growing evidence highlights the bidirectional modulation of lung immune responses through the enhancement of the niche-specific functions of the gut microbiome and its metabolites [12, 13]. Relevant gut microbiota alternations were associated with the disruption of host responses against TB infection in a healthy population [14], indicating that microbiota features could be modified to provide a critical asset in host responses to Mtb infection and serve as biomarkers to differentiate between different stages of TB.
This prospective cohort study aimed to investigate whether gut microbiota signatures can be used to identify LTBI status and reveal whether gut microbiota composition is correlated with cytokine levels in patients with poorly controlled DM (pDM).
Materials and methods
Study population and sample collection
This prospective study recruited patients with pDM, defined as those aged ≥ 45 years with a maximum HbA1c level of > 9.0% within the previous year before enrollment, between October 2019 and December 2020. Each patient with pDM received LTBI screening by using QuantiFERON-TB Gold In-tube (Cellestis/Qiagen, Carnegie, Australia). This study excluded patients with culture-confirmed active TB, history of LTBI treatment, malignancy, HIV infection, pregnancycy, concomitant acute or chronic inflammatory disease, and who used systemic antibiotics, immunosuppressive agents, or probiotics within 3 months prior to enrolment. Patients’ baseline characteristics and clinical medical records were obtained. The study was approved by the Institutional Ethics Committees of Kaohsiung Medical University Hospital (IRB: KMUIRB-G(II)-20170033, KMUHIRB-G(I)-20190035). Each participant provided informed consent before enrollment.
Fresh fecal samples were obtained from the recruited participants at enrollment for microbiota analysis, and blood samples were simultaneously collected for cytokine measurements. The fecal samples were stored in collection tubes with a DNA stabilizer, and the plasma isolated from blood samples was frozen at − 80 °C until further processing.
Fecal sample processing and sequencing
Bacterial genomic DNA was extracted from the fecal samples using the QIAmp fast DNA Stool Mini Kit (Qiagen, Germany) according to the manufacturer’s protocol, and DNA was quantified using NanoDrop 2000 (Thermo Fisher Scientific, USA).
DNA libraries were produced by polymerase chain reaction amplicons targeting the V3 and V4 hypervariable regions of the 16S rRNA gene by using 341F and 805R primers, and were sequenced using the Illumina MiSeq (Illumina, USA) 2 × 300-bp paired-end reads platform (details in Additional file 1).
Bioinformatics analysis
Demultiplexed sequencing reads were trimmed using QIIME2.0 and filtered for quality using a DADA2 pipeline; subsequently, the reads were merged into amplicon sequence variants (ASVs) for downstream analysis. Taxonomy identification was mapped based on the SILVA 16S rRNA gene reference database (version 132) [15].
Alpha-diversity metrics, including observed ASVs, Faith’s phylogenetic diversity (PD), Shannon index, and Pielou’s evenness index were compared between groups using Mann–Whitney U tests with correction for multiple comparison using the Benjamini–Hochberg method [16]. For the beta-diversity analysis, UniFrac distances were used to illustrate microbial community structures, and permutational multivariate analysis of variance (PERMANOVA) was used to determine taxonomic differences between LTBI and non-LTBI groups. Principal coordinate analysis (PCoA) enabled the visualization of unweighted and weighted UniFrac distances [17]. Linear discriminant analysis (LDA) and LDA with effect size measurements (LEfSe) were used to evaluate the influence of each differentially abundant taxon between groups [18]. Pathway enrichment analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and SILVA reference database by PICRUSt2 [19].
Plasma cytokine measurement
The serum concentrations (pg/mL) of interleukin (IL)-17A, IL-17F, IL-22, interferon gamma (IFN-γ), IL-2, IL-10, IL-22, and tumor necrosis factor-alpha (TNF-α) were measured using enzyme‐linked immunofluorescence assay (ELISA) kits according to the manufacturer’s protocol (R&D Systems Inc., Minneapolis, Minnesota, USA).
Statistical analysis
Statistical analysis was performed using SPSS version 22 (IBM, Armonk, NY, USA). Continuous data were expressed as mean ± standard error of mean or standard deviation and compared using Student’s t test or Mann–Whitney U test based on the normality of data distribution. Categorical data were expressed as percentages and compared using chi-squared test. A linear regression model was applied to analyze the correlation between microbial relative abundance and cytokine level. Statistical significance was denoted by p < 0.05.
Random forest model creation for predicting LTBI in patients with pDM
A random forest model was constructed using the RandomForest Classifier from the scikit-learn Python library. The study cohort was randomly divided into training and test sets (70:30). Each decision tree in the forest classified samples as being from participants with or without LTBI based on differential taxa abundance, and the model parameters included number of trees (1000), number of top genera (from 2 to 26), and other default parameters to retain consistency among comparisons; the parameters were systemically tested using bootstrapping to avoid overfitting. The feature importance score was computed based on Gini index [20] (details in Additional file 1). A confusion matrix was constructed to calculate the accuracy, sensitivity, specificity and F1 score to predict LTBI status based on the relative abundance of top genera. The areas under the receiver operating characteristic curve (AUROC) was used for evaluating the discriminative ability of the model.
Results
Characteristics of enrolled participants
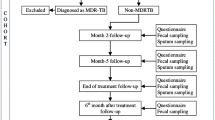
Figure 1 presents the patient selection process. In total, 130 participants with pDM (LTBI = 43; non-LTBI = 87) were included in the final analysis.

Overview of case selection, sample collection, and analysis strategies. QFT: QuantiFERON-TB Gold In-tube. * Poorly controlled diabetes mellitus was defined as ever having a glycated hemoglobin (HbA1c) level of ≥ 9.0% within 1 year before enrollment. # Two patients had bronchitis and one had urinary tract infection. $ One patient had influenza A and one had urinary tract infection
Table 1 and Additional file 1: Table S1 present the clinical, laboratory, and immunological profiles of the 130 patients with pDM. Among them, 51% were male. The mean age was 67.1 years, mean body mass index (BMI) was 26.6 kg/m2, 80.0% were nonsmokers, and the mean maximum HbA1c level within 1 year was 10.7%. The baseline characteristics (age, sex, BMI, diet, and use of antihyperglycemic and lipid-lowering drugs) were similar between LTBI and non-LTBI groups. The mean HbA1c level at enrollment was 8.8% and 8.9% in the LTBI and non-LTBI groups, respectively (p = 0.723).
Sequencing data and microbiome characteristics
In total, 16,147,438 16S rRNA reads were generated from fecal samples provided by the 130 participants, and 6,813,529 reads and 4371 ASVs were obtained after denoising and filtering were conducted. We obtained a median of 49,423 sequences (range: 32,136–121,226) per sample and used 45,000 sequences per sample for rarefaction, revealing that saturation was reached (see Additional file 1: Fig. S1). Following taxonomic assignment, all ASVs were aligned to 366 genera and 871 species. Figure 2A/2B and Additional file 1: Tables S2–S5 reveal the relatively abundant composition of gut microbiota at phylum and genus level in LTBI and non-LTBI groups. Bacteroides was more abundant in the LTBI group (37.79% vs. 29.72%, p = 0.001), whereas Prevotella_9 was more abundant in the non-LTBI group (2.53% vs. 8.95%, p < 0.001).

Altered biodiversity and the major components of gut microbial communities in diabetes mellitus (DM) patients with or without latent tuberculosis infection (LTBI). A The top 10 phyla in LTBI and non-LTBI groups are shown with their abundance. B The top 10 genera present in both LTBI and non-LTBI groups (254 genera; center panel), only in the non-LTBI group (84 genera; left panel), and only in the LTBI group (28 genera; right panel) are presented with their abundance. C The alpha-diversity, namely the richness of gut microbes, was determined by the observed amplicon sequence variants (ASVs), Shannon index, Pielou’s evenness index, and Faith’s phylogenetic diversity (PD). D Beta-diversity was determined by a principal coordinates analysis (PCoA) plot based on the weighted UniFrac distance. Each dot represents one sample from each group. The relevant p values were calculated using a Mann–Whitney U test
Biodiversity of gut microbiota is associated with LTBI status in patients with pDM
Figure 2C indicates that no differences exist between LTBI and non-LTBI groups in terms of alpha-diversity based on observed ASVs (p = 0.988), Shannon index (p = 0.853), evenness (p = 0.669), and Faith’s PD (p = 0.377). By contrast, beta-diversity was significantly different between the 2 groups, as determined using PCoA plots based on the unweighted and weighted UniFrac distance (p = 0.007 and p = 0.002 by PERMANOVA, respectively) (Fig. 2D and Additional file 1: Fig. S2A). The LTBI group had a significantly lower Prevotella/Bacteroides ratio than non-LTBI group did (0.251 vs. 0.724, p < 0.001, Additional file 1: Fig. S2B). Additionally, there was no differences in beta-diversity among subgroups stratified based on different clinical characteristics, including sex, obesity (body-mass-index ≥ 27 kg/m2), use of various antidiabetic and antihyperlipidemic drugs (Additional file 1: Fig. S3).
LEfSe revealed 41 discriminating taxon features between LTBI and non-LTBI groups across different phylogenetic levels, including 3 classes, 5 orders, 7 families, and 26 genera (LDA score (log10) > 2.0, p < 0.05; Fig. 3A and B and Additional file 1: Table S6). Compositional differences in gut microbiota at the genus level (LDA score (log10) > 3.5) were primarily driven by the enrichment of Bacteroides, Alistipes and Blautia in the LTBI group and the enrichment of Prevotella_9, Streptococcus, and Actinomyces in the non-LTBI group.

Differential abundance analysis and identification of representative genera as predictive signatures through linear discriminant analysis (LDA) with effect size measurements (LEfSe) analysis and a random forest model to discriminate between patients with poorly controlled diabetes with and without latent tuberculosis infection (LTBI). A Significantly different taxa in the cladogram according to a LDA score of ≥ 2 (each circle represents phylogenetic levels from phylum to genus [inside to outside], and each diameter is proportional to the taxon’s abundance). B Significantly different genera in terms of relative abundance (LDA score of ≥ 2) between LTBI and non-LTBI groups. C Receiver operating characteristics (ROC) curve analysis of the predictive model using the 6 most differentially abundant genera (Bacteroides, Alistipes, Blautia, Prevotella_9, Streptococcus, and Actinomyces) for discriminating between LTBI and non-LTBI groups (AUROC: area under the ROC curve). D Feature importance of each of the 6 genera in the predictive model
Gut microbiota-based prediction for discriminating between patients with and without LTBI
In accordance with the 6 most differential genera between the LTBI and non-LTBI groups for predicting LTBI status in the 130 patients with pDM, the best random forest model constructed had an AUROC of 0.834, an accuracy of 0.872, sensitivity of 0.769, and specificity of 0.923 (Fig. 3C and Additional file 1: Tables S7–S8). Regarding feature importance in this predictive model, Bacteroides was the genus with the highest weighting, followed by Streptococcus, Alistipes, Blautia, Prevotella_9, and Actinomyces (Fig. 3D).
Plasma cytokine level is correlated with gut microbiota composition
Compared with non-LTBI group, the plasma concentrations of IL-17F and TNF-α, which indicate cytokine responses from T helper-17 (Th17) cells and T helper-1 (Th1) cells, respectively, were significantly lower in the LTBI groups (p = 0.025 and p = 0.038, respectively; Fig. 4A and Additional file 1: Table S1). The 11 potential confounders, including sex, obesity (BMI ≥ 27 kg/m2), use of various antidiabetic medications, and statin, were poorly correlated with plasma cytokine levels of IFN-γ, IL-17A, IL-17F, IL-2, IL-10, IL-22, TNF-α and TGF-β, and the model performance improved after adding the 6 selected genera into the linear regression model (Additional file 1: Tables S9 and S10). Additionally, the 6 gut microbiota signatures accounted for the majority of top 5 important features in linear regression model (Additional file 1: Fig. S4).

Correlation of the plasma levels of individual cytokines with latent tuberculosis infection (LTBI) status and the 6 most differentially abundant genera (Bacteroides, Alistipes, Blautia, Prevotella_9, Streptococcus, and Actinomyces) (* represents p < 0.05). A Plasma cytokine levels (mean ± standard error mean, pg/mL) between LTBI and non-LTBI groups (compared using a Mann–Whitney U test) and the overall performance of the linear regression (LR) models in predicting the individual levels of plasma cytokines. B Beta estimates related to the 6 selected genera in the 6 LR models for predicting individual cytokine levels among overall population. C Gene functions related to immune, D metabolism, and replication pathways in the gut microbiota according to PICRUSt2 with an extended error bar plot to demonstrate the difference between LTBI and non-LTBI groups (the middle value represents the mean inter-group differences with the non-LTBI group used as a reference, and the error bar represents the relevant 95% confidence interval)
Regarding the effect of individual genera, Blautia was positively correlated with all measured cytokines except for TGF-β; Bacteroides had a negative correlation with IFN-γ, IL-17A, IL-10, IL-22, and TNF-α; and Prevotella 9 was negatively associated with IL-22 (Fig. 4B and Additional file 1: Table S9).
Functional analysis
In total, 20 and 43 differentially abundant pathways were significantly modified at KEGG levels 2 and 3, respectively (p < 0.05), suggesting diverse changes in the functions of microbiota between LTBI and non-LTBI groups (Additional file 1: Fig. S5). Most of the predicted microbial functions in patients with LTBI are negatively correlated with immune (Fig. 4C), metabolism, and replication (Fig. 4D) pathways.
Discussion
This is the first study to unravel links between LTBI status, host immunity, and gut microbiome dysbiosis in patients with pDM. We demonstrated the following: (1) pDM patients with LTBI have a markedly different gut microbiome to individuals without LTBI; they had enriched Bacteroides, Alistipes, and Blautia, but depleted Prevotella_9, Streptococcus, and Actinomyces. (2) The proposed microbiota-based model involving the 6 most differential genera in gut microbiota had favorable performance with an accuracy of 0.872 in predicting LTBI status among patients with pDM. (3) The 6 discriminatory genera were correlated with a decrease in Th1/Th17 cell-mediated inflammatory cytokines and indicated to involve in depletion of immune, metabolism, and replication pathways.
Interferon gamma release assays (IGRAs), based on the detection of IFN-γ responses of peripheral lymphocytes against Mtb-specific antigens, are the current standard diagnostic test for LTBI. However, they have a low sensitivity in patients with DM, most likely because of the attenuation of IFN-γ release [21, 22]. Additionally, the results of sequential IGRAs cannot reflect dynamic changes in Mtb infection and identify subclinical TB [23]. A new diagnostic platform incorporating various diagnostic modalities for LTBI is necessary to guide point-of-care management and the timing of treatment initiation.
Studies have demonstrated that compared with healthy individuals, the gut microbiota of patients with DM have a lower relative abundance of genera Bacteroides [24], Alistepes [25] and Blautia [26] and a higher relative abundance of Actinomyces [24] and Prevotella [27]. We however demonstrated that the DM patients with LTBI had higher abundance of Bacteroides, Alistipes, and Blautia than those without LTBI, implying that LTBI status might be associated with the dysbiotic microbiota of patients with pDM.
Furthermore, growing evidence indicates that the disruption of gut microbiome equilibrium can contribute to changes in TB stage [14, 28, 29], which could provide clinical utility for LTBI diagnosis beyond conventional IGRAs.
The most differential bacterial taxa between pDM patients with and without LTBI identified by the current prediction model have previously been associated with TB susceptibility based on the regulation of Th1/Th17 immune responses and inflammation [14, 28,29,30,31]. Although the role of Streptococcus and Actinomyces had not previously been identified in patients with TB, patients with active TB have a lower phylogenetic diversity and a significantly lower abundance of short chain fatty acid (SCFA)–producing bacteria such as Bacteroidetes, Alistepes, and Prevotella compared with healthy individuals [32,33,34]. Conversely, the unique gut microbiome features with high abundance of SCFA producers (Bacteroides and Alistepes) in current pDM patients with LTBI may enhance TB susceptibility by suppressing B cells and CD4+ and CD8+ lymphocytes, reducing the production of TB-induced IFN-γ and IL-17, increasing Foxp1 expression [14, 28, 30, 31] and elevating the number of T regulatory cells in peripheral blood [29]. Blautia was reported to be more abundant in patients with TB than in symptomatic patients without TB and transcriptome analysis indicated that this abundance may be related to inflammation-modulating pathways [35]. Paradoxically, Prevotella had diverse immunomodulatory properties in different TB stages; it was positively correlated with CD4+ cell counts in patients with newly diagnosed TB but negatively correlation with such cell counts in patients with TB recurrence [33]. Further mechanistic studies are required to confirm the immunomodulatory effect of each relevant taxon.
In accordance with the findings in previous studies [9, 36], this study revealed significantly lower plasma levels of TNF-α and IL-17 in DM patients with LTBI than non-LTBI counterparts, suggesting increased TB susceptibility through the decreasing phagocytic ability of macrophages, interference with granuloma formation [37] and inhibiting Mtb-specific memory responses [38]. We though herein demonstrated the significant correlation between Th1- and Th17-related cytokines and the 6 most differentially abundant taxa of gut microbiota, further studies should be conducted to explore the immunomodulatory effect of gut microbiome on Mtb infection.
Furthermore, the significant downregulation of gene expression in immune, metabolism, and replication pathways in the LTBI group was in accordance with the evasion of immune surveillance through the suppression of host immunity, a reduction in energy expenditure, and the attainment of an intracellularly nonreplicant dormancy status by Mtb [39]. Taken together, the findings of the current study provide preliminary evidence that gut microbiota composition may reflect the immune response of pDM patients with different status of Mtb infection. Furthermore, the findings could provide a first step toward host-directed immunomodulatory therapy through the precise tuning of the enteric microbiome to enhance host immunity against Mtb infection.
This study had several limitations. First, this cross-sectional study could not determine causality or the mechanisms behind the effect of gut microbiota alterations on host immunity during various stages of Mtb infection. Second, using 16s rRNA sequencing rather than shotgun metagenomics may interfere the taxonomic and functional resolution of microbiomes owing to the inadequacy of gene-related information obtained from strains. Third, current study did not reveal the influence of microbiome-derived metabolite alterations on TB pathogenesis. Further metabolomics studies should be conducted to verify this influence. Forth, ethnicity and regional variations among individuals can alter the precision of microbiome-based diagnostics. An external validation of the proposed prediction model is necessary.
Conclusion
Our study indicates that gut microbiome composition could modulate Th1/Th17 cell–mediated immune responses, which are potentially relevant to TB susceptibility among patients with pDM. We provide a gut microbiome-based prediction model for discriminating between pDM patients with and without LTBI status. This study provides a foundation for explorations relevant to gut microbiome-based diagnostic biomarkers and host-directed treatment strategies; such explorations could alleviate the TB–DM co-epidemic.
Availability of data and materials
Sequencing data for all samples used in this study have been deposited in https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE199810. Deidentified individual participant data that underlie the results reported in this article will be available immediately following publication upon request to the corresponding author.
Abbreviations
- ASVs:
-
Amplicon sequence variants
- AUROC:
-
Areas under the receiver operating characteristic curve
- DM:
-
Diabetes mellitus
- HbA1c:
-
Glycosylated hemoglobin
- IL:
-
Interleukin
- IFN-γ:
-
Interferon gamma
- LTBI:
-
Latent TB infection
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LDA:
-
Linear discriminant analysis
- LEfSe:
-
LDA with effect size measurements
- PCoA:
-
Principal coordinate analysis
- PD:
-
Phylogenetic diversity
- pDM:
-
Poorly controlled diabetes
- PERMANOVA:
-
Permutational multivariate analysis of variance
- RF model:
-
Random forest model
- TB:
-
Tuberculosis
- TNF-α:
-
Tumor necrosis factor-alpha
References
Thomas RL, Halim S, Gurudas S, Sivaprasad S, Owens DR. IDF Diabetes Atlas: A review of studies utilising retinal photography on the global prevalence of diabetes related retinopathy between 2015 and 2018. Diabetes Res Clin Pract. 2019;157:107840.
Ottmani SE, Murray MB, Jeon CY, et al. Consultation meeting on tuberculosis and diabetes mellitus: meeting summary and recommendations. Int J Tuberc Lung Dis. 2010;14(12):1513–7.
Restrepo BI. Diabetes and tuberculosis. Microbiol Spectr. 2016. https://doi.org/10.1128/microbiolspec.TNMI7-0023-2016.
Allin KH, Nielsen T, Pedersen O. Mechanisms in endocrinology: gut microbiota in patients with type 2 diabetes mellitus. Eur J Endocrinol. 2015;172(4):R167–77.
Lee MR, Huang YP, Kuo YT, et al. Diabetes mellitus and latent tuberculosis infection: a systematic review and metaanalysis. Clin Infect Dis. 2017;64(6):719–27.
Martinez L, Zhu L, Castellanos ME, et al. Glycemic control and the prevalence of tuberculosis infection: a population-based observational study. Clin Infect Dis. 2017;65(12):2060–8.
Huang HL, Huang WC, Lin KD, et al. Completion rate and safety of programmatic screening and treatment for latent tuberculosis infection in elderly patients with poorly controlled diabetic mellitus: a prospective multicenter study. Clin Infect Dis. 2021;73(6):e1252–60.
Kumar NP, Moideen K, George PJ, Dolla C, Kumaran P, Babu S. Coincident diabetes mellitus modulates Th1-, Th2-, and Th17-cell responses in latent tuberculosis in an IL-10-and TGF-β-dependent manner. Eur J immunol. 2016;46(2):390–9.
Kumar NP, George PJ, Kumaran P, Dolla CK, Nutman TB, Babu S. Diminished systemic and antigen-specific type 1, type 17, and other proinflammatory cytokines in diabetic and prediabetic individuals with latent Mycobacterium tuberculosis infection. J Infect Dis. 2014;210(10):1670–8.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol. 2021;19(1):55–71.
Dabke K, Hendrick G, Devkota S. The gut microbiome and metabolic syndrome. J Clin Invest. 2019;129(10):4050–7.
Budden KF, Gellatly SL, Wood DL, et al. Emerging pathogenic links between microbiota and the gut–lung axis. Nat Rev Microbiol. 2017;15(1):55–63.
Zheng D, Liwinski T, Elinav E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020;30(6):492–506.
Liu Y, Wang J, Wu C. Microbiota and tuberculosis: a potential role of probiotics, and postbiotics. Front Nutr. 2021;8: 626254.
Pfeiffer S, Pastar M, Mitter B, et al. Improved group-specific primers based on the full SILVA 16S rRNA gene reference database. Environ Microbiol. 2014;16(8):2389–407.
Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75(23):7537–41.
Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–35.
Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60.
Douglas GM, Beiko RG, Langille MGI. Predicting the functional potential of the microbiome from marker genes using PICRUSt. Methods Mol Biol. 2018;1849:169–77.
Menze BH, Kelm BM, Masuch R, et al. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinform. 2009;10:213.
Chukwudi UK, Chuka EP, MaryAnn ON, Okwudiri IM. Comparative diagnosis of latent tuberculosis infection amongst HIV and diabetic patients attending tertiary hospitals in Anambra State. Int J Trop Dis Health. 2020;41(4):70–9.
Faurholt-Jepsen D, Aabye MG, Jensen AV, et al. Diabetes is associated with lower tuberculosis antigen-specific interferon gamma release in Tanzanian tuberculosis patients and non-tuberculosis controls. Scand J Infect Dis. 2014;46(5):384–91.
Sester M, Sotgiu G, Lange C, et al. Interferon-γ release assays for the diagnosis of active tuberculosis: a systematic review and meta-analysis. Eur Respir J. 2011;37(1):100–11.
Que Y, Cao M, He J, et al. Gut bacterial characteristics of patients with type 2 diabetes mellitus and the application potential. Front Immunol. 2021;12: 722206.
Zhu L, Sha L, Li K, et al. Dietary flaxseed oil rich in omega-3 suppresses severity of type 2 diabetes mellitus via anti-inflammation and modulating gut microbiota in rats. Lipids Health Dis. 2020;19(1):20.
Liu X, Mao B, Gu J, et al. Blautia-a new functional genus with potential probiotic properties? Gut Microbes. 2021;13(1):1–21.
Leite AZ, Rodrigues NC, Gonzaga MI, et al. Detection of increased plasma interleukin-6 levels and prevalence of Prevotella copri and Bacteroides vulgatus in the feces of type 2 diabetes patients. Front Immunol. 2017;8:1107.
Osei Sekyere J, Maningi NE, Fourie PB. Mycobacterium tuberculosis, antimicrobials, immunity, and lung-gut microbiota crosstalk: current updates and emerging advances. Ann N Y Acad Sci. 2020;1467(1):21–47.
Khan N, Vidyarthi A, Nadeem S, Negi S, Nair G, Agrewala JN. Alteration in the gut microbiota provokes susceptibility to tuberculosis. Front Immunol. 2016;7:529.
Maji A, Misra R, Dhakan DB, et al. Gut microbiome contributes to impairment of immunity in pulmonary tuberculosis patients by alteration of butyrate and propionate producers. Environ Microbiol. 2018;20(1):402–19.
Wood MR, Yu EA, Mehta S. The human microbiome in the fight against tuberculosis. Am J Trop Med Hyg. 2017;96(6):1274–84.
Hu Y, Yang Q, Liu B, et al. Gut microbiota associated with pulmonary tuberculosis and dysbiosis caused by anti-tuberculosis drugs. J Infect. 2019;78(4):317–22.
Luo M, Liu Y, Wu P, et al. Alternation of gut microbiota in patients with pulmonary tuberculosis. Front Physiol. 2017;8:822.
Li W, Zhu Y, Liao Q, Wang Z, Wan C. Characterization of gut microbiota in children with pulmonary tuberculosis. BMC Pediatr. 2019;19(1):445.
Naidoo CC, Nyawo GR, Sulaiman I, et al. Anaerobe-enriched gut microbiota predicts pro-inflammatory responses in pulmonary tuberculosis. EBioMedicine. 2021;67: 103374.
Ssekamatte P, Nakibuule M, Nabatanzi R, et al. Type 2 diabetes mellitus and latent tuberculosis infection moderately influence innate lymphoid cell immune responses in Uganda. Front Immunol. 2021;12: 716819.
Bekker LG, Freeman S, Murray PJ, Ryffel B, Kaplan G. TNF-alpha controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol. 2001;166(11):6728–34.
Khader SA, Bell GK, Pearl JE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007;8(4):369–77.
Sundararajan S, Muniyan R. Latent tuberculosis: interaction of virulence factors in Mycobacterium tuberculosis. Mol Biol Rep. 2021;48(8):6181–96.
Acknowledgements
The authors thank the staff of Aging and Disease Prevention Research Center, Department of Medical Laboratory Science and Biotechnology, Fooyin University and the staff of the Eighth Core Lab, Department of Medical Research of National Taiwan University Hospital for the technical support.
Funding
The study was supported by grants from the Ministry of Health and Welfare (MOHW110-CDC-C-114-000108, MOHW111-CDC-C-114-000102), Ministry of Science and Technology (MOST108-2314-B-002-190-MY3, MOST109-2314-B-037-085-MY3, MOST110-2314-B-A49-002), Kaohsiung Municipal Ta-Tung Hospital (KMTTH-110-R002, KMTTH-111-039), The National Chiao Tung University–Kaohsiung Medical University Joint Research Project (NCTUKMU109-AI-02, NYCUKMU-111-I003), the MOST Joint Research Center for AI Technology and All Vista Healthcare (MOST110-2634-F-009-015 and MOST107-2634-F-002-019), "Smart Platform of Dynamic Systems Biology for Therapeutic Development" project, Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
HLH provided the conceptualization, did the formal analysis of this study and wrote the main original draft. YCL did the formal bioinformatics analysis and statistics. PLL, KDL, and MHC collected the clinical data and investigate the progress of this project. MRL collected clinical data and validated the statistical results. CHH and YTY helped to perform the bioinformatics analysis and validated the statistical results. CYK extracted DNA and perform the 16s rRNA sequencing. JMY supervised and validated the bioinformatics analysis and reviewed the methodology in the manuscript. JYW and IWC supervised the progression of this project, reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Declaration of Helsinki and was approved by the Institutional Ethics Committees of Kaohsiung Medical University Hospital (IRB: KMUIRB-G(II)-20170033, KMUHIRB-G(I)-20190035). Each participant provided informed consent before enrollment.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Laboratory and cytokine analysis results for the 130 patients with poorly controlled diabetes mellitus. Table S2. Relative abundance (%) of the top 10 phyla in both the latent tuberculosis infection (LTBI) group and non-LTBI groups. Table S3. Relative abundance (%) of the top 10 genera in both latent tuberculosis infection (LTBI) group and non-LTBI group. Table S4. Relative abundance (%) of the top 10 genera only in the non-latent tuberculosis infection (non-LTBI) group. Table S5. Relative abundance (%) of the top 10 genera only in the latent tuberculosis infection (LTBI) group. Table S6. Relative abundance (%) of the 26 most differential genera between the latent tuberculosis infection (LTBI) and non-LTBI groups. Table S7. Performance of predictive models that included different numbers of the 26 most differential genera between the latent tuberculosis infection (LTBI) and non-LTBI groups. Table S8. Confusion matrix of the classifier involving 6 genera and a test set (39 samples [30%]) for differentiating between latent tuberculosis infection (LTBI) and non-LTBI groups, as determined by a random forest model. Table S9. P values for each of the 6 selected genera in linear regression models for predicting the plasma levels of individual cytokines and the proposed model performance. Table S10. Change of model performance for predicting the plasma levels of individual cytokines before and after including the 6 selected genera into the linear regression models containing 11 potential confounders (sex, body-mass index ≥ 27 kg/m2, and use of metformin, DDP4 inhibitor, SGLT2 inhibitor, sulfonylurea, thiazolidinedione, meglitinides, acarbose, and use of ≥ 3 oral antidiabetic drugs, as well as statin). Figure S1. Rarefaction curve of sequencing data from 130 fecal samples. Samples with fewer than 45,000 sequences were excluded, and the remaining samples were rarefied to 49,423 sequences (range: 32,136 to 121,226) per sample for subsequent ordinations and permutational multivariate ANOVA analysis.Abbreviations: ASVs, amplicon sequence variants. Figure S2. Biodiversity and Prevotella/Bacteroides (P/B) ratio of gut microbial communities in diabetes mellitus (DM) patients with or without latent tuberculosis infection (LTBI). (A) Beta diversity was determined by a principal coordinates analysis plot based on the unweighted UniFrac distance. Each dot represents one sample. (B) Prevotella/Bacteroides (P/B) ratio between the latent tuberculosis infection (LTBI) and non-LTBI groups. *** represents a P value of < 0.001 according to a Mann–Whitney U test. Figure S3. Biodiversity of gut microbial communities in diabetes mellitus (DM) patients with or without potential confounders: (A) sex, (B) BMI ≥ 27kg/m2, (C) Metformin, (D) DDP4 inhibitor, (E) SGLT2 inhibitor, (F) Sulfonylurea, (G) Thiazolidinedione, (H) Meglitinide, (I) Acarbose, (J) Statin. Figure S4. The feature importance of the 6 selected genera and potential confounders in the predicting models for serum cytokine levels. Figure S5. Prediction of gene function of the 6 most differential genera between latent tuberculosis infection (LTBI) and non-LTBI groups using PICRUSt2.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, HL., Luo, YC., Lu, PL. et al. Gut microbiota composition can reflect immune responses of latent tuberculosis infection in patients with poorly controlled diabetes. Respir Res 24, 11 (2023). https://doi.org/10.1186/s12931-023-02312-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-023-02312-w