
Abstract
Translational research plays a key role in drug development and biomarker discovery for hepatocellular carcinoma (HCC). However, unique challenges exist in this field because of the limited availability of human tumor samples from surgery, the lack of homogenous oncogenic driver mutations, and the paucity of adequate experimental models. In this review, we provide insights into these challenges and review recent advancements, with a particular focus on the two main agents currently used as mainstream therapies for HCC: anti-angiogenic agents and immunotherapy. First, we examine the pre-clinical and clinical studies to highlight the challenges of determining the optimal therapeutic combinations with biologically effective dosage for HCC. Second, we discuss biomarker studies focusing on anti-PD1/anti-PD-L1-based combination therapy. Finally, we discuss the progress made in our collective understanding of tumor immunology and in multi-omics analysis technology, which enhance our understanding of the mechanisms underlying immunotherapy, characterize different patient subgroups, and facilitate the development of novel combination approaches to improve treatment efficacy. In summary, this review provides a comprehensive overview of efforts in translational research aiming at advancing our understanding of and improving the treatment of HCC.
Similar content being viewed by others


Introduction
The development of new oncology drugs for unresectable HCC has been hindered by limited accessibility to early-phase clinical trials [1] as well as concerns of adverse events associated with chronic liver diseases or loco-regional therapy [2, 3]. Despite these challenges, the introduction of new treatment regimens, including multi-kinase inhibitors (MKIs), immune checkpoint inhibitors (ICI), and their combinations, has not only expanded the treatment options available for patients with advanced-stage HCC but also introduced new prospects of multi-modality treatments for patients with earlier stage diseases [4, 5].
Anti-PD1/ anti-PD-L1 ICI-based combination therapy is regarded as the most noteworthy breakthrough in systemic therapy for unresectable HCC. Although findings from recent, pivotal phase III randomized clinical trials play key roles in shaping the future development of novel systemic therapy [6,7,8,9,10,11,12,13,14,15], translational research aiming at elucidating antitumor mechanisms and characterizing patient subgroups who are most likely to benefit from specific treatments is also essential. In this review, we explore the progress of translational research on drug development for HCC treatment from three distinct perspectives, namely, the reader, the interpreter, and the creator, to illustrate how translational research may aid in advancing the understanding and improving the treatment of HCC. As outlined by Dr. Bijay Kumar Das, an Indian literature critic, “A translator is a reader, an interpreter and a creator all in one”. Researchers must be aware of the available literature on HCC treatment and drug development (as readers), capable of analyzing and understanding the complex clinical and pre-clinical data (as interpreters), and, ideally, able to use this knowledge and understanding to design new experiments, develop new drugs, and ultimately advance the field of HCC treatment (as creators).
In this review we focus on the aspects of drug development and biomarker discovery for the 2 major classes of agents that are currently the mainstream therapies for HCC, namely anti-angiogenic agents and ICIs. As readers, we examine how translational research is involved in the development of new drugs for HCC. We also reviewed the preclinical studies focusing on the immunomodulatory effects of anti-angiogenic agents for HCC, highlighting the potential benefits and challenges of using in vivo and in vitro models. As interpreters, we review the correlative biomarker studies from randomized trials of anti-PD1/ anti-PD-L1-based therapy and single-arm cohort studies. We also discuss the benefits and challenges of developing tissue- and blood-based predictive biomarkers and the confounding effects exerted by the underlying etiologies of liver diseases. As creators, we discussed recent advancements in the multi-omics analyses of the HCC micro-environment. Specifically, we focus on advancements in computational biology, which enhance our collective understanding of the complex interactions of immune cells in the tumor microenvironment (TME), and on the implications of these advancements for both efficacy and adverse events of immunotherapy. We also emphasize the need for developing novel pre-clinical models to support mechanistic exploration and biomarker identification. In summary, translational research is a complex and multifaceted process that requires researchers to be readers, interpreters, and creators.
Readers: lessons learned from translational research on anti-angiogenic therapy for HCC
Traditionally, translational research aimed at developing new drugs for HCC has had 2 primary objectives: establishing reliable predictive biomarkers to develop tailored treatment options for specific patient populations and understanding the underlying mechanisms of the new drugs. Nevertheless, achieving these objectives has been challenging for HCC. Clinical diagnosis of HCC, based on clinical and imaging characteristics rather than histological proof, is standard for patients with established risk factors (cirrhosis, chronic viral hepatitis) [16]. In addition, phase III randomized trials of systemic therapy for unresectable HCC have often not required a histological diagnosis, thereby resulting in a lack of adequate tumor samples for correlative biomarker analysis. Moreover, almost all of the molecular aberrations found in HCC, which have been primarily identified in studies using tumor samples obtained from patients who underwent surgery, are not typical drivers of the carcinogenesis process and are undruggable through either monoclonal antibodies or small molecule inhibitors [17]. Although molecular classifications of HCC based on genetic or epigenetic features of the tumors have been proposed to predict clinical outcome, they may not help patient categorization for the development of specific targeted therapy [18, 19].
Anti-angiogenesis: a plausible yet elusive drug target
Generally, current targeted agents used for HCC treatment primarily exert their anticancer effects through the inhibition of angiogenesis. Although extensive clinical and pre-clinical studies have been conducted, no reliable set of predictive biomarkers has been established for identifying patients who may benefit from antiangiogenic therapy. In addition, no reliable pharmacodynamic markers have been identified to monitor the extent of angiogenesis inhibition and to determine the correlation between anti-angiogenic effects and clinical efficacy. The difficulty of preclinical experimental models to recapitulate the clinical features of HCC emerging from an inflammatory or cirrhotic background further widens the gap between preclinical mechanistic research and clinical application [20, 21].
In terms of the development of the anti- vascular endothelial growth factor (anti-VEGF) antibodies, early studies of bevacizumab in murine models suggested that doses of 2.5 mg/kg twice weekly or higher may achieve adequate plasma concentrations and anti-angiogenic effects [22]. Multiple randomized phase 2 and 3 trials have examined the dose–response effects of bevacizumab, either as single-agent therapy or in combination with chemotherapy, in different types of cancer. In these trials, higher doses of bevacizumab were associated with a trend of better treatment benefit, in terms of superior objective response rate or survival, and higher risks of adverse events, including hypertension, proteinuria and vascular events [23,24,25,26]. Since most of the adverse events were generally well tolerated by the patients, a high dosage of bevacizumab (5.0 mg/kg/ week) was eventually used in almost all subsequent clinical trials to develop new combination regimens. Overall, these findings underscore the limitations of pre-clinical models for dose determination in real-world clinical trials.
Pharmacodynamic biomarkers for anti-angiogenic therapy, including functional imaging, immunohistochemistry, and levels of circulating cytokines or angiogenic progenitor cells, have been widely tested but none of them have achieved the reproducibility and robustness required as a companion diagnostic in clinical practice [27, 28]. For example, in developing the MKI regorafenib, which inhibits VEGF receptor (VEGFR), biomarker experiments, including DCE-MRI functional imaging and circulating VEGFR, indicated that daily regorafenib dosage of 120 mg or higher was necessary to elicit anti-angiogenic effects [29]. This finding laid the foundation for subsequent clinical trials on HCC and other types of cancer, leading to the current recommended dosage of 160 mg per day, 3-week on and 1-week off. However, this dosage was not well tolerated by most patients. A dose-escalation strategy for regorafenib, starting from 80 mg per day (half of the recommended dose of 160 mg per day), with incremental adjustments depending on patient tolerance until a median daily dosage of 100 mg to 120 mg was reached, has been proposed to achieve similar progression-free survival to that of patients who received the standard-dosage of regorafenib [30].
The aforementioned challenges are also present in the development of other anti-angiogenic strategies, such as in the modulation of pericyte function. Pericytes play a key role in the stabilization and maturation of vascular sprouts, a process that involves multiple signalling pathways, including the VEGF, platelet-derived growth factor, and angiopoietin/ Tie-2 pathways [31, 32]. Translational research platforms to characterize the interaction among multiple relevant mechanisms and to minimize the gaps between pre-clinical evidence and clinical efficacy/ safety are urgently need.
Complex interaction between anti-angiogenetic agents and ICIs
Mechanistic exploration became much more complicated when researchers attempted to address the immune modulatory effects of anti-angiogenic therapy [33]. Pre-clinical studies revealed that VEGF-targeting therapy can activate antitumor immunity in many aspects, including increasing antigen presentation, activating effector T cells, and counteracting immune suppressor cells in the TME. In addition to VEGF-targeting, tumor angiogenesis can also be indirectly modulated by targeting various immune cells (e.g., tumor-associated macrophages, TAMs) or stromal cells (e.g., pericytes) in the TME. Specific targeting agents and epigenetic-modifying agents are under development to modulate these cells [34,35,36]. Given that many plausible targets are available, developing predictive biomarkers for patient selection and pharmacodynamic monitoring has become more challenging.
Hypoxia in the TME plays a key role in the immunomodulatory effects of anti-angiogenic agents. Although HCC is typically a hypervascular tumor, the high interstitial pressure resulting from its aberrant vasculature may paradoxically induce hypoxia and immune suppression in the TME [37, 38]. This hypoxia-induced immune suppression involves complex interactions among different immune cells, the stroma, and the cytokine network in the TME [39,40,41,42]. Therefore, to improve anti-tumor immunity, multiple agents targeting tumor-associated hypoxia have been studied [43, 44]. According to the theory of vascular normalization in anti-angiogenic therapy, using excessively high doses of anti-angiogenic agents may induce hypoxia, acidosis and immune suppression in the TME, whereas using low-doses of anti-angiogenic therapy may enhance antigen presentation and improve T cell trafficking and function [38]. Pre-clinical studies have also indicated that using lower doses of anti-angiogenic MKI may induce vascular normalization, reduce hypoxia, and improve antitumor immunity, whereas using higher doses of anti-angiogenic MKIs may paradoxically increase hypoxia and promote immune suppression [45].
Understanding the biologically effective dosage of targeted agents and their relevant antitumor mechanisms is essential for developing optimal anti-angiogenic regimens. In our pre-clinical studies on regorafenib, we used regorafenib at a dosage of 5 mg/kg/day in animal models to mimic the half daily recommended dose of regorafenib (i.e., 80 mg per day) in human, in accordance with the aforementioned pharmacokinetic study. We found that this low-dose of regorafenib was associated with enhanced interferon-gamma response, M1 macrophage polarization, and antitumor immunity, independent of its anti-angiogenic effects. Regorafenib inhibits the p38 kinase/ Creb1/Klf4 signaling pathway in macrophages, which may explain its macrophage-polarizing effects [46]. According to Shigeta et al. (2020), regorafenib at a dosage of 10 mg/kg/day in mouse liver cancer models may result in optimal vascular normalization and increased T-cell infiltration in the TME. Regorafenib may also increase the expression of CXCL10 by HCC cells and the intratumoral infiltration of CD8 + CXCR3 + T cells through the inhibition of STAT3 activity. These two mechanisms may account for the antitumor synergy observed between regorafenib and anti-PD1 therapy [47]. Overall, these studies have demonstrated how pre-clinical research can elucidate the optimal biologically effective dosage of targeted agents and their mechanisms of action.
In conclusion, the challenges and complexity in drug development and biomarker discovery are significant and must be addressed through reliable pre-clinical studies and solid mechanistic understanding. Overcoming these challenges ca aid in achieve actual progress in the clinical management of HCC, an unmet need that demands urgent attention.
Interpreters: biomarker studies for the prediction of treatment efficacy and mechanistic exploration
Biomarkers are used clinically in risk stratification, early detection, diagnosis, prognosis, and treatment response prediction. Clinical parameters such as tumor size, tumor number, and liver functional reserves are incorporated in major HCC practice guidelines to recommend the choice of liver-directed therapy, such as chemo-embolization [4, 48,49,50]. For patients who require systemic therapy, no reliable set of biomarkers is yet validated for currently available treatment options. Treatment recommendations are typically based on the clinical and laboratory parameters defined in the pivotal clinical trials and on the safety concerns of specific agents and patient preferences [51].
Traditionally, biomarkers are developed per the principle of Occam’s razor, which posits that natural phenomena should be explained in the simplest form possible, with minimal assumptions [52]. This is done to ensure test robustness, reduce intra- and inter-observer variations, and facilitate external validation in diverse patient populations [53]. The same principle is also used in the development of biomarkers for HCC. Currently the only predictive biomarker with level 1 evidence (proven by randomized trial (s) designed to test biomarker performance and clinical impact, according to the International Liver Cancer Association (ILCA) white paper [54] is alpha-fetoprotein (AFP) in selecting HCC patients for ramucirumab therapy (an anti-VEGFR antibody) in the second-line setting [55], although its usefulness is limited given the relatively low absolute survival gain by ramucirumab treatment.
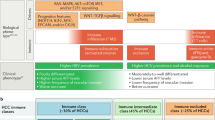
In ICI therapy, tumor PD-L1 expression and tumor mutation burden (TMB) are the most validated predictive biomarker for advanced cancers. More recently, multi-omics approaches are increasingly use to explore the mechanistic interaction among hosts, immune cells, and tumors for biomarker development (Fig. 1 and Table 1) [56]. Expression patterns or ‘signature’ of immune related genes in tumor tissue, particularly those related to inflammation and T cell function, may serve both for prediction of treatment efficacy and for mechanistic exploration [57, 58]. Biomarker studies using archival tumor tissues from HCC patients who received anti-PD1/ anti-PDL1 based therapy identified genes associated with inflammation, antigen presentation, interferon responses and cytokine signaling (ILCA level 2–3 evidence) [59,60,61,62,63]. However, findings from these translational studies cannot be easily validated externally because of difficulties in ensuring methodological standardization.

Approaches of biomarker development for immunotherapy in HCC. Summary of multi-omics profiling approaches of biomarker development. The biomarkers of DNA, RNA, and proteins are intergraded from different modalities. Each modality exhibits advantages and disadvantages for constructing the entire picture of tumors and microenvironments. WES, whole-exome sequencing, WGS, whole-genome sequencing
The role of epigenetic aberrations, including non‐coding RNA expression, DNA promoter hypo‐ or hyper‐methylation, and histone modifications (e.g., acetylation), in hepatocarcinogenesis and their potential as prognostic or predictive biomarkers have been extensively studied [64]. Epigenetic aberrations not only contribute to carcinogenesis but also are involved in TME remodeling, immune evasion, and effector T cell exhaustion [65,66,67]. Reversing epigenetic aberrations using de-methylating agents, histone deacetylase (HDAC) inhibitors, or enhancer of zeste homologue 2 (EZH2) inhibitors, may increase the efficacy of ICI therapy [68,69,70]. Therefore, developing epigenetic biomarkers and targets for immune modulation is another promising approach for enhancing ICI-based combination therapy.
Tissue-based biomarker exploration
Tumor PD-L1 expression is associated with a favorable objective response to anti-PD1/ anti-PD-L1 therapy in both the preclinical models of liver cancer [71] and in clinical trials on patients with HCC [12, 59, 63]. As shown in Table 1, higher density of infiltrating T cells, particularly CD8 + T cells, CD3 + T cells, GZMB + CD3 + T cells, as well as MHC class I protein expression were observed in patients responding to combination immunotherapy with atezolizumab and bevacizumab [63]. Because HCC is associated with a lower TMB compared with other types of cancer, TMB is not useful for predicting immunotherapeutic response in HCC [60, 72]. Another likely explanation for this phenomenon is the high intra-tumoral heterogeneity of HCC, which makes obtaining an accurate measurement of the TMB from a single biopsy sample difficult [73, 74]. These findings clearly underscore the limitations of the minimalist approach for accurately predicting the therapeutic response in highly heterogeneous types of cancer such as HCC. Activation of the WNT/ β-catenin pathway was associated with inferior treatment efficacy in some [75, 76] but not all [63] studies of patients who received anti-PD1 therapy.
Overall, the composition of genes used to represent specific immune related pathways have varied from one study to another, rendering cross comparisons difficult. For instance, in the CheckMate-040 study of nivolumab, 4 genes, namely CD274 (PD-L1), CD8A, LAG3, and STAT1, were selected to constitute an inflammation-related gene signature [59]. By contrast, in the CheckMate459 trial of nivolumab versus sorafenib, the Gajewski inflammation signature [77] was used to identify patients with better objective response and survival after nivolumab therapy [60].
The biomarker study for the atezolizumab plus bevacizumab combination therapy integrated data from the IMbrave150 randomized trial (atezolizumab plus bevacizumab versus sorafenib) and an earlier phase I trial to explore predictors of efficacy of the combination treatment and the synergistic immune modulatory mechanisms of anti-VEGF agent [63]. The team found that a higher objective response rate and longer survival were associated with higher PD-L1 expression, stronger effector T cell signatures (CXCL9, PRF1, and GZMB), and lower expression of certain metabolism-related pathways (e.g., bile acid, fatty acid). The additional therapeutic benefit of bevacizumab was associated with increased expression of genes related to regulatory T (Treg) cells (CCR8, BATF, CTSC, TNFRSF4, FOXP3, TNFRSF18, IKZF2, and IL2RA) and myeloid inflammation (CXCL1, CXCL2, CXCL3, CXCL8, IL6, PTGS1). Consistently, elevated expressions of effector T cells and myeloid inflammation signatures have been correlated with improved efficacy of atezolizumab plus bevacizumab for patients with advanced renal cell carcinoma [78].
The aforementioned findings support the potential use of transcriptomic markers for investigating the mechanisms of ICI-based combination therapy across different types of cancer. Recent advancements in epigenetic signatures have propelled multi-omics analysis into a new frontier [79, 80]. Some studies on HCC have established links between epigenetic-related gene signature (extracted from bulk RNA sequencing data) and immunotherapeutic responses [81, 82]. Although these studies have provided insights into the complex interactions between different immune cells in regulating antitumor immunity in the TME, a dauntingly high level of analytic expertise is required. In addition, classifying patients into subgroups of high- versus low-expression of specific signatures, based usually on median expression values of the particular patient cohorts, may hinder external validation in different patient cohorts.
Blood-based biomarker exploration
Blood-based biomarker analysis enables non-invasive, real-time monitoring of treatment effects. In patients who received ICI-based therapy, real-time monitoring may aid in the development of pharmacodynamic markers to characterize immune activation after treatment [83] and differentiate between true and pseudo-progression after treatment [84]. Biomarkers detected in patients’ blood may reflect the tumor burden [85, 86], status of systemic inflammation (e.g., neutrophil–lymphocyte ratio (NLR) and platelet–lymphocyte ratio (PLR)), and the genetic features associated with specific biological behaviors [59, 87,88,89]. NLR and AFP levels were reported to predict response to ICI-based therapy [59, 85]. The CRAFITY score, consisting of serum C-reactive protein and AFP levels [90], may serve as both a prognostic factor and a predictor of efficacy for ICI-based systemic therapy for patients with advanced HCC [91, 92]. These data offer level 2 evidence, in accordance with ILCA guidelines, regarding the use of blood-based biomarkers in patients with advanced HCC. Circulating immune cells, particularly CD8 effector memory T cells and antigen presenting cells (APCs) are linked to objective immunotherapeutic response [93].
In addition to circulating immune cells, circulating tumor DNA (ctDNA) and cell-free DNA (cfDNA) are regarded as potential peripheral biomarkers for predicting immunotherapeutic response (Table 1). In patients with advanced HCC who received ICI-based therapy, high ctDNA levels and TERT mutations detected in ctDNA are associated with poor survival outcome [88, 89]. However, these biomarkers exhibit complex interactions with each other, and correlation with the same markers at tissue level needs further clarification. For example, genetic studies of the IMbrave150 trial indicated that patients with TERT promoter mutation in tumors are more likely to benefit from the combination therapy [63], but in another case series HCC patients with TERT mutation detected in circulatory DNA had inferior survival compared with patients without detectable TERT mutation [89].
The pharmacodynamic monitoring of ICI-based therapy may include analysis of T-cell activation or exhaustion by flow cytometry and T-cell clonality by T-cell receptor sequencing [83]. A dose optimization study of durvalumab (anti-PDL1) plus tremelimumab (anti-CTLA4) for patients with non-small-cell lung cancer revealed an increasing trend of T cell proliferation and activation in peripheral blood with increasing tremelimumab dosage. The dosage of 1 mg/kg was finally selected based on safety data [94]. In another similar study on patients with advanced HCC, tremelimumab (300 mg, single-dose) plus durvalumab induced a significant increase in the number of CD8 + Ki67 + T cells, informing the optimal dosage for the HIMALAYA trial [95]. These pharmacodynamic markers may oversimplify the immune regulatory effects of anti_CTLA4 agents. Preclinical studies suggest that the antitumor efficacy of anti-CTLA4 ICIs involves the inhibition of Treg cells [96, 97]. Therefore, more sophisticated technology are required to capture the complex interaction among immune cells in the TME. High dimensional immune-monitoring technologies such as mass cytometry by time-of-flight (CyTOF) combined with single-cell RNA sequencing (scRNA seq) can be used to identify specific immune subsets related to response and immune-related adverse events (irAEs), indicating the possibility of targeting novel immune regulatory pathways to uncouple treatment efficacy and irAEs [93].
Confounders in the interpretation of biomarker studies
The etiologies of the underlying liver diseases have been extensively studied as confounding factors for the interpretation of clinical trial results. Meta-analyses of clinical trials on sorafenib indicated that patients with HCC and hepatitis C infection may benefit more from sorafenib treatment [98, 99]. Although molecular pathogenesis studies have suggested that HCC with different etiologies is associated with different patterns of molecular aberrations, these aberrations are not directly related to the antitumor mechanisms of sorafenib and may not explain the difference in survival benefit among different sub-groups [100]. For HCC patients with different etiologies, no evident difference in survival benefit was noted for other targeted therapeutic regimens, including lenvatinib, regorafenib, cabozantinib, and ramucirumab [55, 101,102,103].
In ICI therapy, etiology-related debates have focused on non-viral etiologies, particularly non-alcoholic steatohepatitis (NASH) [104]. Pre-clinical models indicated that diet-induced NASH may compromise T cell function in the liver microenvironment and confer resistance to anti-PD1 therapy [104,105,106]. In response to metabolic stimuli, a subgroup of CXCR6 + CD8 T cells was identified to induce liver damage (‘auto-aggressive’) and may induce resistance to antiPD1/ antiPD-L1 therapy [104, 106]. A meta-analysis of ICI-based systemic therapy for advanced HCC suggested that patients with hepatitis B (HBV)-related HCC demonstrated more prominent survival benefit, whereas patients with non-viral HCC appeared to benefit the least (Table 2) [104].
However, the difference in survival benefit among HCC patients with viral versus non-viral etiologies was not consistently seen [9, 14, 107, 108]. The non-viral subgroups included in HCC clinical trials encompass a heterogeneous population of patients with different etiologies or underlying liver diseases which are usually less stringently diagnosed on the basis of current clinical practice guidelines [109]. In addition, the co-existence of metabolic dysfunction-associated fatty/steatotic liver disease (MAFLD or MASLD) is often overlooked in these trials [110,111,112]. MAFLD may coexist in about 10–20% patients with chronic viral hepatitis. It may also exacerbate liver inflammation and fibrosis, leading to poorer clinical outcomes than those of patients without MAFLD [111,112,113,114]. Therefore, it is reasonable to hypothesize that the presence of MAFLD with chronic viral hepatitis may modulate the immune microenvironment of HCC and complicate the interpretation of biomarkers for immunotherapy.
In addition to the underlying liver diseases, tumor-related features such as hypoxia and epithelial-mesenchymal transitions (EMT) also play important role in determining treatment responses. In a previous study we described the effect of hypoxia on the enrichment of and interaction between immunosuppressive dendritic cells (DCs) and Treg [39]. Hypoxia may serve as a confounding factor that further attenuates immunotherapeutic responses because of its immunosuppressive effect. Consistent with our findings, those of Kopecka et al. [115] suggested hypoxia is a potential driver of resistance to immunotherapy. EMT is associated with tumor immune escape [116], which may regulate the expression of immune checkpoint molecules [117]. Further research is required to determine the impact of this phenomenon on HCC immunotherapy.
In summary, development of an improved technology or system is required to address the limitations of current biomarkers and the potential confounding effects from underlying etiologies and tumor characteristics.
Creators: advancement in multi-omics approach for translational research in HCC
In published clinical trials of anti-PD1/ anti-PD-L1-based combination therapy for unresectable HCC, efficacy appears to a plateau, with an overall survival of 20 months, a progression-free survival of 7–8 months, and an objective tumor response approximately 25% based on RECIST 1.1 (response evaluation criteria in solid tumors, Table 3) [48]. Several approaches can be considered to enhance the efficacy of systemic therapy. The first approach is combination with agents targeting other immune checkpoints, such as TIGIT (T Cell Immunoreceptor with Ig and ITIM Domains), to enhance T-cell function [118]. The second approach is targeting mechanisms of resistance to anti-PD1/ anti-PD-L1 therapy identified in pre-clinical research, such as tumor-infiltrating Treg cells [39, 119], epigenetic control of immune function [120, 121], and other immune-related signaling pathways [122]. The third approach is exploring novel targets for immune modulation. Further research is required to comprehensively understand the phenotypes and functions of various immune cell subsets within the TME. In recent years, various multi-omics approaches, particularly the single-cell omics (SC-omics) technologies, have been developed, providing a more in-depth understanding of the heterogenous and complex dynamics between different sub-populations within the TME.
SC-omics technologies enable high-throughput profiling of individual cells and play a key role in elucidating the complex interplay between different immune subsets within the TME. These technologies, which encompass proteomics transcriptomics, genomics and even epigenomics, offer valuable insights into the immune evasion mechanisms used by cancer cells and potential targets for immunotherapy by identifying distinct immune cell populations and their associated functional states. Recent developments in single-cell epigenomics analysis, such as in single-cell transposase-accessible chromatin with sequencing [123] and spatial transcriptomics [124], have revolutionized our understanding of the TME and its response to immunotherapy. SC-omics approaches are particularly useful in identifying rare cell types, capturing transcriptional heterogeneity within cell populations, and unveiling the dynamic changes in cell states over time. Integrating SC-omics data with other omics approaches can provide a more comprehensive understanding of the molecular mechanisms driving cancer development and progression, thereby guiding the development of personalized cancer therapies.
Multi-omics approaches for understanding the immune mechanisms in HCC
SC-omics analysis has not only facilitated the characterization of the diverse immune cell subsets within the TME but also substantially contributed to the understanding of immune profiles in different disease states and biomarker discovery for immunotherapy in HCC. In an early SC-transcriptomic study, Zheng et al. [125] analyzed the landscape of T cells in individually sorted CD4+ and CD8+ T cells from TME, non-TME and peripheral blood of patients with HCC. They comprehensively examined various T-cell populations and identified LAYN as the key gene associated with the suppressive function of Treg cells and exhausted CD8 T cells within the TME. In addition to Treg and exhausted CD8 + T cells, they discovered a unique TME-specific CD8+FOXP3+ regulatory-like cell population, confirmed by multi-color immunohistochemistry (Fig. 2). They indicated that this Foxp3+CD8+ Treg cell subset was characterized by the expression of typical Treg genes, including FOXP3, CTLA4, TNFRSF9, and TNFRSF18, and cytolytic-related genes, including PRF1, GZMA, and NKG7 [125]. Earlier and subsequent studies on Foxp3+CD8+ Treg cells have suggested an immunosuppressive phenotype [126, 127]. Zhang et al. [128] used a combination of two single-cell RNA sequencing technologies (10 × Genomics and SMART-seq2) to comprehensively analyze the CD45+ immune landscapes of five compartments (tumor, adjacent liver, hepatic lymph node, blood, and ascites) from 16 treatment-naive patients with HCC. Focusing primarily on the role of DCs and TAMs in regulating the functions of lymphocytes in the TME of HCC, the authors examined the key roles of the LAMP3+ DCs and GPNMB- or SLC40A1-expressing TAMs (Fig. 2) [128]. They reported that the LAMP3+ DCs were more likely associated with T-cell dysfunction. In addition, GPNMB+ TAMs promoted TNF-α production, whereas SLC40A1+ TAMs promoted pro-inflammatory cytokines such as IL-23 and IL-6 but suppressed IL1b production (Fig. 2). The latest addition to this series of single-cell RNA sequencing studies from the same group focused on tumor-infiltrating neutrophils (TANs). They found that the CCL4+ and PD-L1+ TANs were both immunosuppressive and associated with poor prognosis in patients with HCC (Fig. 2) [129]. This series of single-cell analyses of the TME of HCC has unveiled the complex composition and dynamic interaction of tumor-infiltrating immune cells, thereby providing a valuable resource for understanding and developing strategies aimed at modifying the TME to enhance antitumor immunity.

Multi-omics analyses of complex dynamics within the TME of HCC. Several immune subsets, which are either immunosuppressive, pro-inflammatory, or cytotoxic, as observed in multi-omics analyses of HCC. Non-immune cells such as the endothelial and tumor cells also play an key role in the TME of HCC. Treg, regulatory T cells; DC, dendritic cells; TAN, tumor-associated neutrophils; TAM, tumor-associated macrophages; APC, antigen-presenting cells; TEM, T-effector memory cells
In addition to the immunophenotyping of the TME of HCC, SC-omics analysis has played a key role in elucidating various immune profiles across different disease states. Nguyen et al. [158] reported distinct immune landscapes with HCC progression, with the peak of immune evasion observed at the intermediate stage, characterized by accumulation of exhausted CD8+ T cells and Treg cells. Sun et al. [130] reported an increase in DCs with decreasing antigen-presentation capability and reduced Treg cells in early-relapse HCC cases. They also discovered a unique CD8+ T-cell population enriched in early-relapsed HCC, which expressed KLRB1 (CD161) and displayed an innate-like, low cytotoxic and clonal expansion phenotype (Fig. 2). These two studies have indicated that immune evasion is a dynamic process that occurs at different time points along tumor progression and relapse, indicating that anti-CD161 may serve as a potential novel checkpoint target, particularly for relapsed HCC [131].
Multi-omics analysis has achieved great progress in biomarker discovery for therapeutic response [132,133,134]. Ma et al. [135, 136] examined the clonal evolution of tumor cells and their interaction with immune cells in patients with HCC and cholangiocarcinoma who received immunotherapy (Fig. 2). Sharma et al. [137] reported a similarity between the immune modulation of fetal liver and the TME of HCC and discovered that VEGF and NOTCH signalling play a functional role in maintaining immune-suppressive onco-fetal reprogramming (Fig. 2). This study provides valuable insights into the potential mechanisms and targets of anti-VEGF therapy in HCC. Other studies have indicated that the interaction between effector T cells and DCs [93, 138], macrophages, and cancer-associated fibroblasts [139] regulate immunotherapeutic response. In a study based primarily on the peripheral blood SC transcriptomic analysis, CXCR3+ effector memory CD8+ T cells and HLA-DR+ APCs were identified as two key potentially interacting immune cells linked to distinct clinical fates of either response or immune-related adverse effects (irAEs) in patients with HCC who underwent anti-PD-1 therapy (Fig. 2) [64]. Subsequently, a therapeutic strategy was designed to uncouple response and irAEs for optimal therapeutic outcome [64]. In a more recent study, scRNA-seq and spatial transcriptomic analyses were used to identify a triadic interaction among granzyme K + PD-1 + effector-like CD8 + T cells, CXCL13 + CH25H + IL-21 + PD-1 + CD4 + T helper cells, and LAMP3 + mature DCs enriched in immunoregulatory molecules (mregDC), which are linked to therapeutic response in HCC patients treated with neoadjuvant anti-PD-1 ICIs [138].
In summary, multi-omics studies have offered a comprehensive and multi-dimensional understanding of the TME, thereby laying the foundation for the discovery of novel therapeutic targets for next-generation immunotherapy [132, 140].
Development in multi-omics driven therapeutic design for HCC
At present, the most popular approach in clinical trials is to combine anti-PD1/ anti-PDL1 ICIs with another ICI agent, such as anti-TIM3 [141], anti-LAG3 [142], or anti-TIGIT [143], to enhance the re-invigorating effects of antiPD1/ anti-PDL1 ICIs on exhausted CD8 T cells [144]. Chiu et al. [145] compared human HCC to adjacent non-tumor liver tissues and observed an increase in PVRL1, which stabilizes cell surface poliovirus receptor (PVR) that interacts with TIGIT. They reported that TIGIT inhibition or genetic ablation of PVRL1 increased ratio of cytotoxic CD8 + T cells to Treg cells in murine liver cancer models and sensitized the mice to anti-PD1 therapy (Table 4). Wei et al. [146] identified a signaling pathway linking protein kinase C alpha (PKCα), the transcription factor ZPF64, and colony-stimulating factor-1 (CSF-1), which plays a key role in polarization of TAMs towards an immunosuppressive M2 phenotype in the TME of HCC and resistance to anti-PD-1 therapy. They also discovered potent antitumoral activity in preclinical models when inhibitors targeting PKCα (Gö6976) or CSF1 (BLZ945) were combined with anti-PD-1 therapy, suggesting new options for reversing resistance to anti-PD-1 therapy (Table 4).
Because the efficacy and adverse events of ICI therapy are both immune-related, uncoupling these events to enhance efficacy without aggravating adverse events will greatly improve the therapeutic index of new combination regimens. Chuah et al. [93] identified the interaction between CXCR3+ effector memory CD8+ T cells and HLA-DR+ APCs as a key mechanism determining response versus irAEs in patients with HCC treated with anti-PD-1 ICI. They identified TNFR2 as a key biomarker specifically linked to clinical response but not irAEs, and demonstrated enhanced therapeutic response without increased irAEs in preclinical models by combination of anti-TNFR2 and anti-PD-1. They also discovered that TNFR2 was specifically enriched in Treg cells within the TME of HCC, indicating a potential tumor Treg-specific target (Fig. 2). Overall, these findings may facilitate the development of therapeutic strategies aimed at uncoupling therapeutic response and irAEs to optimize therapeutic outcome (Table 4).
Multiple studies have examined the mechanisms underlying the immune-suppressive TME associated with NASH- or MASLD-related HCC [147]. According to preclinical models, the NASH microenvironment may induce CD8 T-cell subpopulations that caused liver damage [106] or even promote HCC development [104]. Wabitsch et al. [105] reported that the metabolic reprogramming of hepatic CD8+ T cells resulted in impaired motility and resistance to anti-PD-1 therapy in murine NASH-HCC models. They indicated that this dysfunctional CD8+ T-cell phenotype was reversed by metformin treatment (Table 4). Many studies have extensively examined the cancer-preventing effects of metformin, and numerous mechanisms have been proposed [148,149,150,151]. Leslie et al. [147] identified TANs, which over-expressed the neutrophil receptor CXCR2, as key factors underlying the inferior efficacy of anti-PD-1 therapy in NASH-related HCC. They reported that combining anti-PD-1 with AZD5069, a CXCR2 inhibitor, led to the reprogramming of TANs to a more proliferative and inflammatory phenotype, increased intra-tumoral XCR1+ DCs and CD8+ T-cell infiltration, and enhanced anti-tumor response in NASH-HCC models (Table 4). In summary, multi-omics approaches can be used to clarify the immune modulatory mechanisms of the underlying liver diseases and to identify novel therapeutic targets in the TME of HCC.
Research into the immunomodulatory effects of MKIs should not be limited to their anti-angiogenic properties. Lenvatinib may inhibit the PKCα/ZFP64/CSF1 [146] and transforming growth factor-β signaling pathways in the TME of HCC (Table 4) [152]. Cabozantinib may also increase neutrophil chemotaxis, induce infiltration of TANs [153] with a more cytotoxic N1 phenotype [154], and reduce intra-tumoral myeloid-derived suppressor cells (MDSCs) (Table 4) [155]. Although these immunomodulatory mechanisms of MKIs may enhance effector T-cell infiltration and response to anti-PD1 therapy in preclinical HCC models, the lack of additional survival benefits provided by combination therapy in randomized clinical trials indicates that additional comprehensive mechanistic studies are required to determine whether and how these mechanisms enhance antitumor immunity in clinical settings.
Epigenetic regulation plays a key role in modulating antitumor immune response through both innate and adaptive immunity. Epigenetic modifiers such as de-methylating agents, HDAC inhibitors, and EZH2 inhibitors, can activate NK cells and macrophages, reverse CD8 T-cell exhaustion, and suppress Treg-mediated immune suppression [67, 70]. Among all types of epigenetic modifiers, HDAC inhibitors are the most widely evaluated in pre-clinical models of HCC [120, 156, 157]. Nevertheless, identifying the most relevant cellular and molecular targets of HDAC inhibitors is a challenging task. Therefore, conducting multi-omics analyses at the single-cell level can aid in elucidating the evolution of immune cells, dissecting the intra-tumor heterogeneity, and identifying rare but functionally essential cell populations [21].
Future perspectives
In this review, we highlighted the potential of advanced technologies in addressing the limitations in the current process of drug development and biomarkers discovery for HCC. These technologies can provide a more comprehensive understanding of the heterogeneity and complexity of the TME, which can consequently clarify the mechanisms underlying various treatment options for HCC. As creators, translational researchers should be aware of the most recent advance of the novel technologies to rapidly and accurately identify new biomarkers and treatment options.
Conclusion
Integrating advanced multi-omics technologies into clinical trials, from early proof-of-concept trials involving novel combination strategies to pivotal trials versus the current standard of care, requires close collaboration between translational researchers and clinical trial specialists to push the frontiers of HCC treatment toward a definitive cure.
Availability of data and materials
Not applicable.
References
Kurzrock R, Lin C-C, Wu T-C, Hobbs BP, Roberto Carmagnani Pestana M, Hong DS. Moving Beyond 3+3: the future of clinical trial design. Am Soc Clin Oncol Educ Book. 2021;41:e133–44.
Kudo M, Han KH, Ye SL, Zhou J, Huang YH, Lin SM, et al. A changing paradigm for the treatment of intermediate-stage hepatocellular carcinoma: Asia-pacific primary liver cancer expert consensus statements. Liver Cancer. 2020;9(3):245–60.
Hsu C. Success is not final, failure Is not fatal: the changing landscape of systemic therapy for advanced hepatocellular carcinoma. J Cancer Res Pract. 2021;8:7.
Reig M, Forner A, Rimola J, Ferrer-Fàbrega J, Burrel M, Garcia-Criado Á, et al. BCLC strategy for prognosis prediction and treatment recommendation: the 2022 update. J Hepatol. 2022;76(3):681–93.
Chen LT, Martinelli E, Cheng AL, Pentheroudakis G, Qin S, Bhattacharyya GS, et al. Pan-Asian adapted ESMO Clinical Practice Guidelines for the management of patients with intermediate and advanced/relapsed hepatocellular carcinoma: a TOS-ESMO initiative endorsed by CSCO, ISMPO, JSMO, KSMO, MOS and SSO. Ann Oncol. 2020;31(3):334–51.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N Engl J Med. 2020;382(20):1894–905.
Cheng A-L, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, et al. Updated efficacy and safety data from IMbrave150: atezolizumab plus bevacizumab vs. sorafenib for unresectable hepatocellular carcinoma. J Hepatol. 2021.
Ren Z, Xu J, Bai Y, Xu A, Cang S, Du C, et al. Sintilimab plus a bevacizumab biosimilar (IBI305) versus sorafenib in unresectable hepatocellular carcinoma (ORIENT-32): a randomised, open-label, phase 2–3 study. Lancet Oncol. 2021;22(7):977–90.
Abou-Alfa GK, Lau GK, Kudo M, Chan SL, Kelley RK, Furuse J, et al. Tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. N Engl J Med Evid. 2022;1(8)
Finn RS, Ryoo BY, Merle P, Kudo M, Bouattour M, Lim HY, et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind. Phase III Trial J Clin Oncol. 2020;38(3):193–202.
Kelley RK, Rimassa L, Cheng AL, Kaseb A, Qin S, Zhu AX, et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23(8):995–1008.
Yau T, Park JW, Finn RS, Cheng AL, Mathurin P, Edeline J, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, phase 3 trial. Lancet Oncol. 2022;23(1):77–90.
Qin S, Kudo M, Meyer T, et al. Final analysis of RATIONALE-301: Randomized, phase III study of tislelizumab versus sorafenib as first-line treatment for unresectable hepatocellular carcinoma. ESMO 2022; Paris, France: Ann Oncol; 2022. p. S808–69.
Qin S, Chan SL, Gu S, Bai Y, Ren Z, Lin X, et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet. 2023;402(10408):1133–46.
Llovet JM, Kudo M, Merle P, Meyer T, Qin S, Ikeda M, et al. Lenvatinib plus pembrolizumab versus lenvatinib plus placebo for advanced hepatocellular carcinoma (LEAP-002): a randomised, double-blind, phase 3 trial. Lancet Oncol. 2023;24(12):1399–410.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391(10127):1301–14.
(TCGA) CGARN. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell. 2017;169(7):1327–41.
Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. 2015;149(5):1226-39.e4.
Llovet JM, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer. 2022;3(4):386–401.
Brown ZJ, Heinrich B, Greten TF. Mouse models of hepatocellular carcinoma: an overview and highlights for immunotherapy research. Nat Rev Gastroenterol Hepatol. 2018;15(9):536–54.
Heinrich S, Craig AJ, Ma L, Heinrich B, Greten TF, Wang XW. Understanding tumour cell heterogeneity and its implication for immunotherapy in liver cancer using single-cell analysis. J Hepatol. 2021;74(3):700–15.
Warren RS, Yuan H, Matli MR, Gillett NA, Ferrara N. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J Clin Invest. 1995;95(4):1789–97.
Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, et al. A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer. N Engl J Med. 2003;349(5):427–34.
Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2004;22(11):2184–91.
Miles DW, Chan A, Dirix LY, Cortés J, Pivot X, Tomczak P, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28(20):3239–47.
Iwamoto S, Takahashi T, Tamagawa H, Nakamura M, Munemoto Y, Kato T, et al. FOLFIRI plus bevacizumab as second-line therapy in patients with metastatic colorectal cancer after first-line bevacizumab plus oxaliplatin-based therapy: the randomized phase III EAGLE study. Ann Oncol. 2015;26(7):1427–33.
Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10(2):145–7.
Jain RK, Duda DG, Willett CG, Sahani DV, Zhu AX, Loeffler JS, et al. Biomarkers of response and resistance to antiangiogenic therapy. Nat Rev Clin Oncol. 2009;6(6):327–38.
Mross K, Frost A, Steinbild S, Hedbom S, Buchert M, Fasol U, et al. A phase I dose-escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res. 2012;18(9):2658–67.
Bekaii-Saab TS, Ou FS, Ahn DH, Boland PM, Ciombor KK, Heying EN, et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): a randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019;20(8):1070–82.
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8(8):592–603.
Harrell CR, Simovic Markovic B, Fellabaum C, Arsenijevic A, Djonov V, Volarevic V. Molecular mechanisms underlying therapeutic potential of pericytes. J Biomed Sci. 2018;25(1):21.
Hegde PS, Wallin JJ, Mancao C. Predictive markers of anti-VEGF and emerging role of angiogenesis inhibitors as immunotherapeutics. Semin Cancer Biol. 2018;52(Pt 2):117–24.
Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1):75.
Berndsen RH, Abdul UK, Weiss A, Zoetemelk M, Te Winkel MT, Dyson PJ, et al. Epigenetic approach for angiostatic therapy: promising combinations for cancer treatment. Angiogenesis. 2017;20(2):245–67.
Török O, Schreiner B, Schaffenrath J, Tsai H-C, Maheshwari U, Stifter SA, et al. Pericytes regulate vascular immune homeostasis in the CNS. Proc Natl Acad Sci. 2021;118(10): e2016587118.
Cramer T, Vaupel P. Severe hypoxia is a typical characteristic of human hepatocellular carcinoma: scientific fact or fallacy? J Hepatol. 2022;76(4):975–80.
Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc Natl Acad Sci U S A. 2012;109(43):17561–6.
Suthen S, Lim CJ, Nguyen PHD, Dutertre CA, Lai HLH, Wasser M, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology. 2022;76(5):1329–44.
Mpekris F, Voutouri C, Baish JW, Duda DG, Munn LL, Stylianopoulos T, et al. Combining microenvironment normalization strategies to improve cancer immunotherapy. Proc Natl Acad Sci U S A. 2020;117(7):3728–37.
Schwörer S, Cimino FV, Ros M, Tsanov KM, Ng C, Lowe SW, et al. Hypoxia potentiates the inflammatory fibroblast phenotype promoted by pancreatic cancer cell-derived cytokines. Can Res. 2023;83(10):1596–610.
de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF treatment enhances CD8(+) T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res. 2020;8(6):806–18.
Cowman SJ, Koh MY. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends in Cancer. 2022;8(1):28–42.
Finisguerra V, Dvorakova T, Formenti M, Van Meerbeeck P, Mignion L, Gallez B, et al. Metformin improves cancer immunotherapy by directly rescuing tumor-infiltrating CD8 T lymphocytes from hypoxia-induced immunosuppression. J Immunother Cancer. 2023;11(5):e005719.
Lin YY, Tan CT, Chen CW, Ou DL, Cheng AL, Hsu C. Immunomodulatory effects of current targeted therapies on hepatocellular carcinoma: implication for the future of immunotherapy. Semin Liver Dis. 2018;38(4):379–88.
Ou DL, Chen CW, Hsu CL, Chung CH, Feng ZR, Lee BS, et al. Regorafenib enhances antitumor immunity via inhibition of p38 kinase/Creb1/Klf4 axis in tumor-associated macrophages. J Immunother Cancer. 2021;9(3):e001657.
Shigeta K, Matsui A, Kikuchi H, Klein S, Mamessier E, Chen IX, et al. Regorafenib combined with PD1 blockade increases CD8 T-cell infiltration by inducing CXCL10 expression in hepatocellular carcinoma. J Immunother Cancer. 2020;8(2):e001435.
Singal AG, Llovet JM, Yarchoan M, Mehta N, Heimbach JK, Dawson LA, et al. AASLD Practice Guidance on prevention, diagnosis, and treatment of hepatocellular carcinoma. Hepatology. 2023;78(6):1922–65.
Lee IC, Hung YW, Liu CA, Lee RC, Su CW, Huo TI, et al. A new ALBI-based model to predict survival after transarterial chemoembolization for BCLC stage B hepatocellular carcinoma. Liver Int. 2019;39(9):1704–12.
Hung YW, Lee IC, Chi CT, Lee RC, Liu CA, Chiu NC, et al. Redefining tumor burden in patients with intermediate-stage hepatocellular carcinoma: the seven-eleven criteria. Liver Cancer. 2021;10(6):629–40.
Bruix J, Chan SL, Galle PR, Rimassa L, Sangro B. Systemic treatment of hepatocellular carcinoma: an EASL position paper. J Hepatol. 2021;75(4):960–74.
Kroemer G, Zitvogel L. Immune checkpoint inhibitors. J Exp Med. 2021;218(3).
In vitro companion diagnostic devices: guidance for industry and Food and Drug Administration staff [Internet]. U.S. Food and Drug Administration. 2014 [cited January 19, 2023]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-companion-diagnostic-devices.
Singal AG, Hoshida Y, Pinato DJ, Marrero J, Nault JC, Paradis V, et al. International Liver Cancer Association (ILCA) white paper on biomarker development for hepatocellular carcinoma. Gastroenterology. 2021;160(7):2572–84.
Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, et al. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased alpha-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96.
Kroemer G, McQuade JL, Merad M, André F, Zitvogel L. Bodywide ecological interventions on cancer. Nat Med. 2023;29(1):59–74.
Danaher P, Warren S, Lu R, Samayoa J, Sullivan A, Pekker I, et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J Immunother Cancer. 2018;6(1):63.
Litchfield K, Reading JL, Puttick C, Thakkar K, Abbosh C, Bentham R, et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184(3):596-614.e14.
Sangro B, Melero I, Wadhawan S, Finn RS, Abou-Alfa GK, Cheng AL, et al. Association of inflammatory biomarkers with clinical outcomes in nivolumab-treated patients with advanced hepatocellular carcinoma. J Hepatol. 2020;73(6):1460–9.
Neely J, Yao J, Kudo M, Finn RS, Sangro B, Melero I, et al. Abstract 2145: genomic and transcriptomic analyses related to the clinical efficacy of first-line nivolumab in advanced hepatocellular carcinoma from the phase 3 CheckMate 459 trial. Cancer Res. 2022;82(12_supplement):2145.
Haber PK, Castet F, Torres-Martin M, Andreu-Oller C, Puigvehí M, Miho M, et al. Molecular markers of response to anti-PD1 therapy in advanced hepatocellular carcinoma. Gastroenterology. 2023;164(1):72-88.e18.
Hsu CL, Ou DL, Bai LY, Chen CW, Lin L, Huang SF, et al. Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer. 2021;10(4):346–59.
Zhu AX, Abbas AR, de Galarreta MR, Guan Y, Lu S, Koeppen H, et al. Molecular correlates of clinical response and resistance to atezolizumab in combination with bevacizumab in advanced hepatocellular carcinoma. Nat Med. 2022;28(8):1599–611.
Braghini MR, Lo Re O, Romito I, Fernandez-Barrena MG, Barbaro B, Pomella S, et al. Epigenetic remodelling in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41(1):107.
Villanueva L, Álvarez-Errico D, Esteller M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol. 2020;41(8):676–91.
Cheng B, Yu Q, Wang W. Intimate communications within the tumor microenvironment: stromal factors function as an orchestra. J Biomed Sci. 2023;30(1):1.
Tien F-M, Lu H-H, Lin S-Y, Tsai H-C. Epigenetic remodeling of the immune landscape in cancer: therapeutic hurdles and opportunities. J Biomed Sci. 2023;30(1):3.
Ny L, Jespersen H, Karlsson J, Alsén S, Filges S, All-Eriksson C, et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat Commun. 2021;12(1):5155.
Mei M, Chen L, Godfrey J, Song J, Egelston C, Puverel S, et al. Pembrolizumab plus vorinostat induces responses in patients with Hodgkin lymphoma refractory to prior PD-1 blockade. Blood. 2023;142(16):1359–70.
Akce M, El-Rayes BF, Wajapeyee N. Combinatorial targeting of immune checkpoints and epigenetic regulators for hepatocellular carcinoma therapy. Oncogene. 2023;42(14):1051–7.
Ou DL, Lin YY, Hsu CL, Lin YY, Chen CW, Yu JS, et al. Development of a PD-L1-expressing orthotopic liver cancer model: implications for immunotherapy for hepatocellular carcinoma. Liver Cancer. 2019;8(3):155–71.
Greten TF, Abou-Alfa GK, Cheng AL, Duffy AG, El-Khoueiry AB, Finn RS, et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immunotherapy for the treatment of hepatocellular carcinoma. J Immunother Cancer. 2021;9(9).
Losic B, Craig AJ, Villacorta-Martin C, Martins-Filho SN, Akers N, Chen X, et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat Commun. 2020;11(1):291.
Zhai W, Lai H, Kaya NA, Chen J, Yang H, Lu B, et al. Dynamic phenotypic heterogeneity and the evolution of multiple RNA subtypes in hepatocellular carcinoma: the PLANET study. Natl Sci Rev. 2021;9(3).
Harding JJ, Nandakumar S, Armenia J, Khalil DN, Albano M, Ly M, et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin Cancer Res. 2019;25(7):2116–26.
Kudo M, Ueshima K, Nakahira S, Nishida N, Ida H, Minami Y, et al. Adjuvant nivolumab for hepatocellular carcinoma (HCC) after surgical resection (SR) or radiofrequency ablation (RFA) (NIVOLVE): a phase 2 prospective multicenter single-arm trial and exploratory biomarker analysis. J Clin Oncol. 2021;39(15_suppl):4070.
Trujillo JA, Sweis RF, Bao R, Luke JJ. T cell-inflamed versus Non-T cell-inflamed tumors: a conceptual framework for cancer immunotherapy drug development and combination therapy selection. Cancer Immunol Res. 2018;6(9):990–1000.
McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24(6):749–57.
Goltz D, Gevensleben H, Vogt TJ, Dietrich J, Golletz C, Bootz F, et al. CTLA4 methylation predicts response to anti–PD-1 and anti–CTLA-4 immunotherapy in melanoma patients. JCI Insight. 2018;3(13).
Jung H, Kim HS, Kim JY, Sun J-M, Ahn JS, Ahn M-J, et al. DNA methylation loss promotes immune evasion of tumours with high mutation and copy number load. Nat Commun. 2019;10(1):4278.
Jiang H, Ning G, Wang Y, Lv W. Identification of an m6A-related signature as biomarker for hepatocellular carcinoma prognosis and correlates with sorafenib and Anti-PD-1 immunotherapy treatment response. Dis Markers. 2021;2021:5576683.
Cai J, Wu S, Zhang F, Dai Z. Construction and validation of an epigenetic regulator signature as a novel biomarker for prognosis, immunotherapy, and chemotherapy in hepatocellular carcinoma. Front Immunol. 2022;13: 952413.
Agdashian D, ElGindi M, Xie C, Sandhu M, Pratt D, Kleiner DE, et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol Immunother: CII. 2019;68(4):599–608.
Lee JH, Long GV, Menzies AM, Lo S, Guminski A, Whitbourne K, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti-programmed cell death 1 antibodies. JAMA Oncol. 2018;4(5):717–21.
Zhu AX, Dayyani F, Yen C-J, Ren Z, Bai Y, Meng Z, et al. Alpha-fetoprotein as a potential surrogate biomarker for atezolizumab + bevacizumab treatment of hepatocellular carcinoma. Clin Cancer Res. 2022;28(16):3537–45.
Shao Y-Y, Liu T-H, Hsu C, Lu L-C, Shen Y-C, Lin Z-Z, et al. Early alpha-foetoprotein response associated with treatment efficacy of immune checkpoint inhibitors for advanced hepatocellular carcinoma. Liver Int. 2019;39(11):2184–9.
Winograd P, Hou S, Court CM, Lee YT, Chen PJ, Zhu Y, et al. Hepatocellular carcinoma-circulating tumor cells expressing PD-L1 Are prognostic and potentially associated with response to checkpoint inhibitors. Hepatol Commun. 2020;4(10):1527–40.
von Felden J, Craig AJ, Garcia-Lezana T, Labgaa I, Haber PK, D’Avola D, et al. Mutations in circulating tumor DNA predict primary resistance to systemic therapies in advanced hepatocellular carcinoma. Oncogene. 2021;40(1):140–51.
Matsumae T, Kodama T, Myojin Y, Maesaka K, Sakamori R, Takuwa A, et al. Circulating cell-free DNA profiling predicts the therapeutic outcome in advanced hepatocellular carcinoma patients treated with combination immunotherapy. Cancers. 2022;14(14):3367.
Scheiner B, Pomej K, Kirstein MM, Hucke F, Finkelmeier F, Waidmann O, et al. Prognosis of patients with hepatocellular carcinoma treated with immunotherapy—development and validation of the CRAFITY score. J Hepatol. 2022;76(2):353–63.
Teng W, Lin CC, Su CW, Lin PT, Hsieh YC, Chen WT, et al. Combination of CRAFITY score with Alpha-fetoprotein response predicts a favorable outcome of atezolizumab plus bevacizumab for unresectable hepatocellular carcinoma. Am J Cancer Res. 2022;12(4):1899–911.
Hsu WF, Lai HC, Chen CK, Wang HW, Chuang PH, Tsai MH, et al. Combined CRAFITY score and α-fetoprotein response predicts treatment outcomes in patients with unresectable hepatocellular carcinoma receiving anti-programmed death-1 blockade-based immunotherapy. Am J Cancer Res. 2023;13(2):654–68.
Chuah S, Lee J, Song Y, Kim HD, Wasser M, Kaya NA, et al. Uncoupling immune trajectories of response and adverse events from anti-PD-1 immunotherapy in hepatocellular carcinoma. J Hepatol. 2022;77(3):683–94.
Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A, et al. Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol. 2016;17(3):299–308.
Kelley RK, Sangro B, Harris W, Ikeda M, Okusaka T, Kang Y-K, et al. Safety, efficacy, and pharmacodynamics of tremelimumab plus durvalumab for patients with unresectable hepatocellular carcinoma: randomized expansion of a phase I/II study. J Clin Oncol. 2021;39(27):2991–3001.
Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exp Med. 2009;206(8):1717–25.
Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131(1):58–67.
Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of viral status on survival in patients receiving sorafenib for advanced hepatocellular cancer: a meta-analysis of randomized phase III trials. J Clin Oncol. 2017;35(6):622–8.
Shao YY, Shau WY, Chan SY, Lu LC, Hsu CH, Cheng AL. Treatment efficacy differences of sorafenib for advanced hepatocellular carcinoma: a meta-analysis of randomized clinical trials. Oncology. 2015;88(6):345–52.
Llovet JM, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nature Cancer. 2022;3(4):386–401.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–73.
Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, et al. Cabozantinib in patients with advanced and progressing hepatocellular carcinoma. N Engl J Med. 2018;379(1):54–63.
Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M, et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021;592(7854):450–6.
Wabitsch S, McCallen JD, Kamenyeva O, Ruf B, McVey JC, Kabat J, et al. Metformin treatment rescues CD8(+) T-cell response to immune checkpoint inhibitor therapy in mice with NAFLD. J Hepatol. 2022;77(3):748–60.
Dudek M, Pfister D, Donakonda S, Filpe P, Schneider A, Laschinger M, et al. Auto-aggressive CXCR6+ CD8 T cells cause liver immune pathology in NASH. Nature. 2021;592(7854):444–9.
Qin S, Chen M, Cheng AL, Kaseb AO, Kudo M, Lee HC, et al. Atezolizumab plus bevacizumab versus active surveillance in patients with resected or ablated high-risk hepatocellular carcinoma (IMbrave050): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2023;402(10415):1835–47.
Espinoza M, Muquith M, Lim M, Zhu H, Singal AG, Hsiehchen D. Disease etiology and outcomes after atezolizumab plus bevacizumab in hepatocellular carcinoma: post-hoc analysis of IMbrave150. Gastroenterology. 2023;165(1):286-8.e4.
Rinella ME, Neuschwander-Tetri BA, Siddiqui MS, Abdelmalek MF, Caldwell S, Barb D, et al. AASLD Practice Guidance on the clinical assessment and management of nonalcoholic fatty liver disease. Hepatology. 2023;77(5):1797–835.
Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: an international expert consensus statement. J Hepatol. 2020;73(1):202–9.
Kawaguchi T, Tsutsumi T, Nakano D, Eslam M, George J, Torimura T. MAFLD enhances clinical practice for liver disease in the Asia-Pacific region. Clin Mol Hepatol. 2022;28(2):150–63.
Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology. 2023;78(6):1966–86.
Al-Omary A, Byth K, Weltman M, George J, Eslam M. The importance and impact of recognizing metabolic dysfunction-associated fatty liver disease in patients with chronic hepatitis C. J Dig Dis. 2022;23(1):33–43.
van Kleef LA, Choi HSJ, Brouwer WP, Hansen BE, Patel K, de Man RA, et al. Metabolic dysfunction-associated fatty liver disease increases risk of adverse outcomes in patients with chronic hepatitis B. JHEP Rep. 2021;3(5): 100350.
Kopecka J, Salaroglio IC, Perez-Ruiz E, Sarmento-Ribeiro AB, Saponara S, De Las RJ, et al. Hypoxia as a driver of resistance to immunotherapy. Drug Resist Updates. 2021;59: 100787.
Terry S, Savagner P, Ortiz-Cuaran S, Mahjoubi L, Saintigny P, Thiery J-P, et al. New insights into the role of EMT in tumor immune escape. Mol Oncol. 2017;11(7):824–46.
Shrestha R, Bridle KR, Crawford DHG, Jayachandran A. Immune checkpoint molecules are regulated by transforming growth factor (TGF)-β1-induced epithelial-to-mesenchymal transition in hepatocellular carcinoma. Int J Med Sci. 2021;18(12):2466–79.
Finn RS, Ryoo B-Y, Hsu C-H, Li D, Burgoyne A, Cotter C, et al. Results from the MORPHEUS-liver study: Phase Ib/II randomized evaluation of tiragolumab (tira) in combination with atezolizumab (atezo) and bevacizumab (bev) in patients with unresectable, locally advanced or metastatic hepatocellular carcinoma (uHCC). J Clin Oncol. 2023;41(16_suppl):4010.
Tay C, Tanaka A, Sakaguchi S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell. 2023;41(3):450–65.
Yang W, Feng Y, Zhou J, Cheung OK, Cao J, Wang J, et al. A selective HDAC8 inhibitor potentiates antitumor immunity and efficacy of immune checkpoint blockade in hepatocellular carcinoma. Sci Transl Med. 2021;13(588).
Moran B, Davern M, Reynolds JV, Donlon NE, Lysaght J. The impact of histone deacetylase inhibitors on immune cells and implications for cancer therapy. Cancer Lett. 2023;559: 216121.
Xiong Z, Chan SL, Zhou J, Vong JSL, Kwong TT, Zeng X, et al. Targeting PPAR-gamma counteracts tumour adaptation to immune-checkpoint blockade in hepatocellular carcinoma. Gut. 2023;72(9):1758–73.
Cusanovich DA, Hill AJ, Aghamirzaie D, Daza RM, Pliner HA, Berletch JB, et al. A single-cell atlas of in vivo mammalian chromatin accessibility. Cell. 2018;174(5):1309-24.e18.
Marx V. Method of the year: spatially resolved transcriptomics. Nat Methods. 2021;18(1):9–14.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169(7):1342-56.e16.
Lozano T, Conde E, Martin-Otal C, Navarro F, Lasarte-Cia A, Nasrallah R, et al. TCR-induced FOXP3 expression by CD8(+) T cells impairs their anti-tumor activity. Cancer Lett. 2022;528:45–58.
Mishra S, Srinivasan S, Ma C, Zhang N. CD8(+) regulatory T cell—a mystery to be revealed. Front Immunol. 2021;12: 708874.
Zhang Q, He Y, Luo N, Patel SJ, Han Y, Gao R, et al. Landscape and dynamics of single immune cells in hepatocellular carcinoma. Cell. 2019;179(4):829–45.
Xue R, Zhang Q, Cao Q, Kong R, Xiang X, Liu H, et al. Liver tumour immune microenvironment subtypes and neutrophil heterogeneity. Nature. 2022;612(7938):141–7.
Sun Y, Wu L, Zhong Y, Zhou K, Hou Y, Wang Z, et al. Single-cell landscape of the ecosystem in early-relapse hepatocellular carcinoma. Cell. 2021;184(2):404–21.
Zhou X, Du J, Liu C, Zeng H, Chen Y, Liu L, et al. A pan-cancer analysis of CD161, a potential new immune checkpoint. Front Immunol. 2021;12: 688215.
Chuah S, Chew V. High-dimensional immune-profiling in cancer: implications for immunotherapy. J Immunother Cancer. 2020;8(1).
Gohil SH, Iorgulescu JB, Braun DA, Keskin DB, Livak KJ. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat Rev Clin Oncol. 2021;18(4):244–56.
Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24(2):144–53.
Ma L, Wang L, Khatib SA, Chang CW, Heinrich S, Dominguez DA, et al. Single-cell atlas of tumor cell evolution in response to therapy in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J Hepatol. 2021;75(6):1397–408.
Ma L, Heinrich S, Wang L, Keggenhoff FL, Khatib S, Forgues M, et al. Multiregional single-cell dissection of tumor and immune cells reveals stable lock-and-key features in liver cancer. Nat Commun. 2022;13(1):7533.
Sharma A, Seow JJW, Dutertre CA, Pai R, Bleriot C, Mishra A, et al. Onco-fetal reprogramming of endothelial cells drives immunosuppressive macrophages in hepatocellular carcinoma. Cell. 2020;183(2):377–94.
Magen A, Hamon P, Fiaschi N, Soong BY, Park MD, Mattiuz R, et al. Intratumoral dendritic cell–CD4+ T helper cell niches enable CD8+ T cell differentiation following PD-1 blockade in hepatocellular carcinoma. Nat Med. 2023;29(6):1389–99.
Liu Y, Xun Z, Ma K, Liang S, Li X, Zhou S, et al. Identification of a tumour immune barrier in the HCC microenvironment that determines the efficacy of immunotherapy. J Hepatol. 2023;78(4):770–82.
Yofe I, Dahan R, Amit I. Single-cell genomic approaches for developing the next generation of immunotherapies. Nat Med. 2020;26(2):171–7.
Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J Immunother Cancer. 2020;8(1): e000911.
Tawbi HA, Schadendorf D, Lipson EJ, Ascierto PA, Matamala L, Castillo Gutiérrez E, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl J Med. 2022;386(1):24–34.
Rousseau A, Parisi C, Barlesi F. Anti-TIGIT therapies for solid tumors: a systematic review. ESMO Open. 2023;8(2): 101184.
Ge Z, Zhou G, Campos Carrascosa L, Gausvik E, Boor PPC, Noordam L, et al. TIGIT and PD1 Co-blockade restores ex vivo functions of human tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Cell Mol Gastroenterol Hepatol. 2021;12(2):443–64.
Chiu DK, Yuen VW, Cheu JW, Wei LL, Ting V, Fehlings M, et al. Hepatocellular carcinoma cells up-regulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 2020;159(2):609–23.
Wei CY, Zhu MX, Zhang PF, Huang XY, Wan JK, Yao XZ, et al. PKCalpha/ZFP64/CSF1 axis resets the tumor microenvironment and fuels anti-PD1 resistance in hepatocellular carcinoma. J Hepatol. 2022;77(1):163–76.
Leslie J, Mackey JBG, Jamieson T, Ramon-Gil E, Drake TM, Fercoq F, et al. CXCR2 inhibition enables NASH-HCC immunotherapy. Gut. 2022.
Chen HP, Shieh JJ, Chang CC, Chen TT, Lin JT, Wu MS, et al. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: population-based and in vitro studies. Gut. 2013;62(4):606–15.
Kramer JR, Natarajan Y, Dai J, Yu X, Li L, El-Serag HB, et al. Effect of diabetes medications and glycemic control on risk of hepatocellular cancer in patients with nonalcoholic fatty liver disease. Hepatology. 2022;75(6):1420–8.
Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun. 2016;7:13007.
Kasznicki J, Sliwinska A, Drzewoski J. Metformin in cancer prevention and therapy. Ann Transl Med. 2014;2(6):57.
Torrens L, Montironi C, Puigvehí M, Mesropian A, Leslie J, Haber PK, et al. Immunomodulatory effects of lenvatinib plus anti-programmed cell death protein 1 in mice and rationale for patient enrichment in hepatocellular carcinoma. Hepatology. 2021;74(5):2652–69.
Esteban-Fabró R, Willoughby CE, Piqué-Gili M, Montironi C, Abril-Fornaguera J, Peix J, et al. Cabozantinib enhances anti-PD1 activity and elicits a neutrophil-based immune response in hepatocellular carcinoma. Clin Cancer Res. 2022;28(11):2449–60.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94.
Ou DL, Lin L, Hsu CL, Jian CZ, Lee BS, Cheng AL, et al. Cabozantinib may enhance efficacy of anti-PD-1 therapy in hepatocellular carcinoma through suppression of MDSCs. Ann Oncol. 2022;33(suppl 6):S483.
Llopiz D, Ruiz M, Villanueva L, Iglesias T, Silva L, Egea J, et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol Immunother. 2019;68(3):379–93.
Qiu W, Wang B, Gao Y, Tian Y, Tian M, Chen Y, et al. Targeting histone deacetylase 6 reprograms interleukin-17-producing helper T cell pathogenicity and facilitates immunotherapies for hepatocellular carcinoma. Hepatology. 2020;71(6):1967–87.
Nguyen PHD, Wasser M, Tan CT, et al. Trajectory of immune evasion and cancer progression in hepatocellular carcinoma. Nat Commun. 2022;13(1):1441.
Acknowledgements
The authors would like to thank the Liver Disease Prevention & Treatment Foundation, Taiwan, for logistic support.
Funding
This paper was supported by Grants MOHW112-TDU-B-221-124007 (from Ministry of Health and Welfare, Taiwan), 111-2314-B-002 -039 (from National Science & Technology Council, Taiwan), NTUH-112L892101, VN112-12 (from National Taiwan University Hospital) (to Dr. Hsu) and National Medical Research Council (NMRC), Singapore (ref numbers: NMRC/CSA-SI/0013/2017, NMRC/CSA-SI/0018/2017, NMRC/OFLCG/003/2018) as well as Duke-NUS Khoo Bridge Funding Award (Duke-NUS-KBrFA/2022/0058) and International Gilead Sciences Research Scholars Program in Liver Disease—Asia (to Dr. Chew).
Author information
Authors and Affiliations
Contributions
CH and VC conceptualized the overall scope of this review article. VC, CHC, and CH reviewed and analyzed the data reported in the literature. VC and CH drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Dr. Chiun Hsu received research grants from Bristol-Myers Squibb (BMS)/ ONO, Roche, and Ipsen and received honorarium from the following pharmaceutical companies: AstraZeneca, Bayer, BMS/ONO, Eisai, MSD, Novartis, Roche, TTY Biopharm. Dr. Chew and Dr. Chuang declared that they had no competing interests to disclose.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chew, V., Chuang, CH. & Hsu, C. Translational research on drug development and biomarker discovery for hepatocellular carcinoma. J Biomed Sci 31, 22 (2024). https://doi.org/10.1186/s12929-024-01011-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-024-01011-y