
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease attributed to the synergistic effects of genetic risk and environmental stimuli. Although PD is characterized by motor dysfunction resulting from intraneuronal alpha-synuclein accumulations, termed Lewy bodies, and dopaminergic neuronal degeneration in the substantia nigra, multiple systems are involved in the disease process, resulting in heterogenous clinical presentation and progression. Genetic predisposition to PD regarding aberrant immune responses, abnormal protein aggregation, autophagolysosomal impairment, and mitochondrial dysfunction leads to vulnerable neurons that are sensitive to environmental triggers and, together, result in neuronal degeneration. Neuropathology studies have shown that, at least in some patients, Lewy bodies start from the enteric nervous system and then spread to the central dopaminergic neurons through the gut–brain axis, suggesting the contribution of an altered gut microenvironment in the pathogenesis of PD. A plethora of evidence has revealed different gut microbiomes and gut metabolites in patients with PD compared to unaffected controls. Chronic gut inflammation and impaired intestinal barrier integrity have been observed in human PD patients and mouse models of PD. These observations led to the hypothesis that an altered gut microenvironment is a potential trigger of the PD process in a genetically susceptible host. In this review, we will discuss the complex interplay between genetic factors and gut microenvironmental changes contributing to PD pathogenesis.
Similar content being viewed by others
Background
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting 1–3% of the population over 60 years of age worldwide, with increasing prevalence attributed to the global increase in life expectancy [1, 2]. The classical scenario of PD is characterized by progressive motor symptoms, including rest tremor, bradykinesia, rigidity, and postural instability [3]. Nevertheless, recent research has shown that PD is a heterogenous disorder involving multiple system degeneration and leading to various motor and non-motor manifestations that compromise quality of life, such as sleep disorder, neuropsychiatric symptoms, autonomic malfunction, and sensory symptoms [4]. The pathological hallmark of PD is intraneuronal accumulations of misfolded alpha-synuclein, termed Lewy bodies, along with neuroinflammation, leading to dopaminergic neuronal degeneration in the substantia nigra [3]. Intriguingly, in some patients, Lewy body pathology was first detected outside the brain, starting from neurons in Auerbach’s and Meissner’s plexuses of the gut enteric nervous system (ENS), as well as the olfactory bulbs [5]. This distribution of alpha-synuclein pathology could explain early non-motor symptoms of PD, especially constipation, which can precede motor symptoms by decades [6]. Based on these pathological findings, Braak and colleagues suggested active retrograde transport of alpha-synuclein from the ENS to the brainstem via the dorsal motor nucleus of the vagus nerve [7]. This hypothesis is supported by data from rodent and non-human primate experimental models [8] in which different forms of misfolded alpha-synuclein were transported via the vagus nerve, reaching the dorsal motor nucleus of the vagus nerve in the brainstem, after injection into the intestinal wall [9,10,11]. Moreover, misfolded alpha-synuclein has been proposed to be capable of self-propagation in a prion-like manner, transferring the pathology to unaffected cells through cell-to-cell transmission through different routes, including endoplasmic reticulum-associated vesicle secretion, exosomes, tunneling nanotubes, and passive diffusion, by promoting the misfolding and spread of alpha-synuclein [12]. However, the molecular and cellular processes that trigger the misfolding and fibrillization, Lewy body formation, and spread of alpha-synuclein from the ENS into the brain remain poorly understood.
The complex interplay between aging and genetic and environmental risk factors collaboratively promotes the development of PD [3]. Over the past few decades, the advent of genetic technology has identified more than 20 PD-causative genes in familial forms of PD [13]. The identification of these genes shed light on the molecular mechanisms causing alpha-synuclein aggregation and neuronal degeneration, including mitochondrial dysfunction, increased oxidative stress, autophagolysosomal impairment, and aberrant intracellular vesical trafficking and neuroinflammation [14]. Among these PD-causative genes, mutations in the gene encoding leucine-rich repeat kinase 2 (LRRK2) is the most prevalent for both familial and sporadic forms of PD. However, fewer than 50% of the mutation carriers will end up developing the disease by 80 years of age [15], indicating that factors other than the genetic mutation are needed to trigger the PD process. The gastrointestinal tract is one of the routes by which environmental substances enter the body, and the enteric neurons residing in proximity are protected only by a thin epithelial cell layer [7]. Given the neuropathological evidence that Lewy bodies may start in the ENS [5], it is reasonable to raise the hypothesis that an altered gut microenvironment is a potential trigger of the PD process in genetically susceptible hosts.
In this review, we summarize recent evidence of altered gut microenvironments in the PD process. We also discuss the complex interplay between genetic predisposition and potential gut microenvironmental triggers in the pathogenesis of PD. As the gastrointestinal tract is involved in the early disease stage, elucidating the pathophysiological process that takes place in the very beginning gut prodromal stage of PD will not only unveil the disease mechanism, but provide future directions in the development of novel diagnostic biomarkers and disease-modifying therapy that could mitigate disease progression.
Clinical evidence of gastrointestinal system involvement in PD
Although the hallmark clinical symptom of PD is motor dysfunction (i.e., rigidity, bradykinesia, resting tremor) when there is at least 60% neuronal loss in the substantia nigra (SN), clinical features of patients with PD also present with versatile non-motor symptoms, such as REM sleep behavior disorder, depression, idiopathic anosmia, and constipation [4]. These non-motor PD symptoms can occur years before the onset of classical motor symptoms [16]. Thus, they are now considered prodromal clinical markers of early disease stages according to the International Movement Disorders Society [17]. Gastrointestinal dysfunction has been well-described, and constipation is the most prevalent and earliest pre-motor symptom in the prodromal phase of the disease [18]. Prospective studies have consistently revealed that populations with constipation have a long-term increased risk of PD, by up to one to threefold over 10 years [6, 19,20,21,22,23,24]. In a systemic analysis, the odds ratio for developing PD was estimated to be 2 to 2.5 in a group of participants with constipation compared to those without constipation [6]. Notably, the prevalence and severity of constipation worsens as disease progresses, with one study reporting up to 82% of advanced-stage patients having constipation [24]. Although the frequency of constipation varies greatly between studies depending on the diagnostic criteria applied and study design, it estimated to be 27 to 66% in PD patients and 5 to 34% in the general population [6, 24]. These clinical observations are in line with the neuropathological findings that Lewy body pathology is first detected in the neurons of the ENS in the gut [5]. In addition to the clinical symptom of constipation, recent studies have identified that inflammatory markers are elevated in both the colonic tissues and feces of patients with PD compared to controls, indicating evidence of intestinal inflammation in PD [25,26,27,28]. Epidemiology studies have revealed an increased risk of PD in patients with inflammatory bowel disease (IBD) [29, 30] and reduced risk of developing PD if they receive anti-inflammatory treatment [31]. These observations suggest that, in addition to a vagal nerve connection, the gut could also participate in the PD process through the inflammatory response [25]. These pieces of epidemiological evidence have led to the hypothesis that gastrointestinal dysfunction contributes to PD. The close connection of the gastrointestinal tract with the environment offers a gateway between environmental triggers and the brain [32].
Histopathological evidence of gastrointestinal involvement in PD
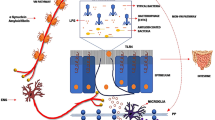
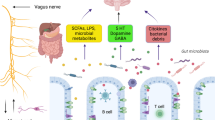
The gastrointestinal tract is one of the largest interfaces between the host and environment in humans. The gut is also referred to as the second brain in the human body. Large numbers of neurons are contained in the gut ENS, a count that is similar to the number of neurons in the spinal cord, with 20 neuronal and 4–7 glial cell types [33]. This gut neural network extends beyond enteric neurons and is comprised of enteric glia, neurons of peripheral ganglia innervating the gut, intrinsic neurons, and specialized innervated epithelial sensors, such as enteroendocrine cells, which constitute the “gut connectome” [34]. The gut neurons are divided into two systems. One is an intrinsic ENS comprising the myenteric and submucosal plexuses, and the other is extrinsic innervation that emanates from the central nervous system (CNS) and communicates with the gut via sympathetic and parasympathetic inputs [34]. The interaction between the gut and brain is a bi-directional communication between the ENS and CNS [35]. The gut–brain axis is composed of neuroendocrine and neuroimmune systems, including the hypothalamic–pituitary–adrenal axis, sympathetic and parasympathetic arms of the autonomic nervous system (including the ENS and vagus nerve), and the residents within the gut lumen, the gut microbiota [35]. Several pathological findings could provide explicit evidence that the gastrointestinal tract and ENS are involved in the disease process of PD, including aggregation of misfolded alpha-synuclein within the ENS, intestinal inflammation, and impaired gut barrier integrity. These manifestations develop in the early stage of the disease and are attributed to the pathogenesis of PD (Fig. 1).

Schematic of gut microenvironmental changes in PD and their contribution to disease pathophysiology. Enteric α-synuclein deposition, gastrointestinal inflammation, and intestinal epithelial barrier dysfunction are observed in PD. Pathological α-synuclein is found in enteroendocrine cells (EECs) and enteric neurons, which propagates through the vagus nerve to the brainstem, resulting in Lewy pathology in the central nervous system. Enteric α-synuclein also induces inflammatory responses involving activation of the caspase-1-dependent inflammasome and production of pro-inflammatory cytokines, leading to intestinal barrier malfunction [87]. Meanwhile, alteration of the gut microbiota composition in PD may accelerate α-synuclein aggregation, partly through secretion of bacterial amyloid [101, 117, 197]. Gut dysbiosis may also trigger enteric inflammation and increased intestinal permeability with systemic infiltration of microbial toxins and metabolites that activate and modulate immune responses and promote PD pathogenesis [85, 103]. Intestinal and systemic inflammation involving increased expression of TLR-4, T cells, and associated pro-inflammatory cytokines (e.g., TNF, IL-1ß, IL-6) results in greater permeability of the blood–brain barrier, neuroinflammation, and dopaminergic neuronal degeneration [64, 74, 166, 198]. BA, bile acid; HA, hippuric acid; IL-1ß, interleukin-1ß; TNF, tumor necrosis factor; TLR, toll-like receptor; TMA, trimethylamine; TMAO, trimethylamine N-oxide; Trp, tryptophan; LPS, lipopolysaccharide; SCFAs, short chain fatty acids
Alpha-synucleinopathy in the gastrointestinal tract
Post-mortem pathology studies have reported widespread Lewy body pathology in enteric neurons at the intestinal mucosa, submucosa, and myenteric plexus interconnecting the vagus nerve and dorsal motor nucleus of the vagus nerve in the brainstem in both prodromal and symptomatic PD patients [7, 36]. These findings led to Braak’s hypothesis that Lewy body pathology in PD may initiate in the gastrointestinal tract and then propagate to the CNS through the vagus nerve in some patients [7, 36]. Subsequent histopathological studies demonstrated the deposition of pathological alpha-synuclein in the intestinal tissues of both the upper and lower gastrointestinal tract of prodromal or pre-clinical PD patients up to 20 years before the onset of motor symptoms [37,38,39]. In addition, the dorsal motor nucleus of the vagus nerve was not involved in widespread Lewy body pathology in only 7 to 8.3% of PD patients. In contrast, more than half of the patients with PD had pathological progress adhering to Braak’s theory [40]. Furthermore, inter-neuronal spread of pathological alpha-synuclein has been observed in PD patients with an accumulation of Lewy body in grafted dopaminergic neurons 10 years after receiving fetal dopaminergic neuronal transplantation [41,42,43]. Evidence from animal and cell models also support the cell-to-cell prion-like transmission of pathological alpha-synuclein that leads to Lewy pathology and dopaminergic neuronal loss [44, 45]. Supporting these findings from human PD patients, data from rodent and non-human primate models have shown that intra-intestinal injection of alpha-synuclein fibrils triggers aggregation of phosphorylated alpha-synuclein at the dorsal motor nucleus of the vagus nerve, followed by the locus coeruleus and substantia nigra via the vagus nerve in a longitudinal follow-up [32, 46, 47]. The pathological alpha-synuclein spread, dopaminergic neuronal degeneration, and motor dysfunction were mitigated by vagotomy [46, 48, 49]. Consistently, epidemiological studies using Swedish and Danish registries have demonstrated that truncal vagotomy decreases the risk of developing PD by 15–22%, further supporting that the vagus nerve is involved in the transmission of pathological alpha-synuclein in the pathogenesis of PD [50, 51].
Notably, Lewy body pathology also spreads by a non-vagal route. Transmission of pathological alpha-synuclein through sympathetic nerves and the peripheral circulation has been observed in rodent and non-human primate models [8, 47, 52]. An increase in plasma alpha-synuclein correlates with disease duration and motor symptom severity in PD patients and to the extent of enteric alpha-synuclein deposition in primate models of PD [8, 53, 54]. In addition, enteric alpha-synuclein is a pivotal modulator of immune responses. Accumulation of intestinal alpha-synuclein occurs most abundantly in the appendix, which is full of lymphatic tissues and responsible for preserving the intestinal immune system in both neurologically normal subjects and patients with PD [55, 56]. Appendectomy was associated with a reduced risk of developing PD in a long-term follow-up but, intriguingly, increased the occurrence of PD shortly after surgery, which may be attributed to intestinal inflammation [57,58,59,60,61,62]. Studies in cellular and animal models have revealed that alpha-synuclein possesses chemotactic features [63]. Misfolded alpha-synuclein could activate innate immune responses, up-regulate the expression of pro-inflammatory cytokines (e.g., tumor necrosis factor-α (TNFα), interleukin-1ß (IL-1ß)) and promote maturation of antigen-presenting cells, leading to adaptive immune responses [63]. A recent in vivo PD rodent model study further demonstrated that the enteric alpha-synuclein aggregations cause chronic intestinal inflammation and impair gut barrier junction integrity, which may promote neurodegeneration through systemic inflammatory processes [64]. Thus, pathological alpha-synuclein aggregations are observed in the gastrointestinal tract in the early stage of the PD process and may affect the CNS through the vagus nerve and non-vagal immune responses.
Chronic intestinal inflammation
Intestinal inflammation has been identified as another gastrointestinal feature of PD based on colonic tissue findings and increased inflammatory markers in feces from patients with PD [25,26,27,28]. Mild chronic colitis has been detected in the early disease stage [28, 65]. Studies of colon biopsies have shown that the level of expression of inflammatory markers, including toll-like receptor (TLR)-2, TLR4, CD3+ T cells, T helper (Th)1, Th17, and related pro-inflammatory cytokines and chemokines CCL2, CCL5, CCR5, IL-1ß, IL-6, IL-8, IL-17A, interferon (IFN)-ß, IFN-γ, TNF-α associated with glial cell markers is elevated in PD patients compared to controls [28, 66]. Examination of stool immune profiles also found that inflammatory signatures, including calprotectin, IL-1α, IL-1ß, CXCL8, and C-reactive protein, are higher in PD patients than unaffected controls, independent of disease duration, suggesting the participation of intestinal inflammation throughout the disease course [26, 27, 67]. In addition, epidemiological studies have shown that, in both Western and Asian populations, the risk of developing PD is higher in patients with IBD than those without IBD [29, 31, 68,69,70,71]. The risk of PD is increased by 28% in patients with Crohn’s disease and 30% in patients with ulcerative colitis, whereas the risk is reduced by 78% in IBD patients who receive anti-TNF-α therapy [30, 31]. Furthermore, recent genome-wide association studies have revealed that PD and IBD share several common pleiotropic genetic loci related to microbial sensing, immunity, and autophagolysosomal function, including LRRK2 [72, 73].
The mechanism by which chronic colitis promotes PD pathogenesis remains elusive. In vivo studies have demonstrated that intestinal inflammation induces and exacerbates neuroinflammation and dopaminergic neuronal degeneration with or without Lewy pathology in the CNS [74,75,76,77]. It is though that TLR4, a pattern recognition receptor, plays a crucial role in mediating the disease process because an elevation of TLR4 in colonic tissues and TLR4-downstream inflammatory mediators [e.g., nuclear factor-κB (NF-κB) and TNF-α] has been observed in PD patients and mouse models of PD. Knockout of TLR4 or treatment with TNF-α antagonist or TLR4 blockers mitigates neuroinflammation, dopaminergic neuronal loss, and motor dysfunction in both cellular and rodent models of PD [66, 76, 78, 79]. As previously mentioned, intestinal inflammation could trigger the expression of enteric alpha-synuclein and vice versa [62, 80], with intestinal inflammation per se increasing the permeability of the blood–brain barrier (BBB), promoting microgliosis, and aggravating neuronal loss through upregulation of systemic pro-inflammatory cytokines and neuroinflammation [74]. Not surprisingly, impaired intestinal barrier and gut dysbiosis are also closely related to enteric inflammation [81, 83]. Notably, enteric gliosis correlating with intestinal inflammation has been observed in human PD patients and animal models of PD [28, 66]. Although the role of enteric glia in the PD disease process is still unclear, they are known to participate in the maintenance of intestinal homeostasis through modulation of gut immunity, intestinal barrier function, and the gut microbiota [83]. Thus, multiple lines of evidence indicate that chronic intestinal inflammation with or without enteric Lewy body pathology promotes neuropathology in the disease process of PD.
Increased intestinal permeability (leaky gut phenomenon)
The intestinal epithelium forms a defensive physical barrier with a mucus layer to protect the host against environmental toxins and pathogens [81]. The function of the intestinal barrier is regulated by epithelial and immune cells, associated cytokines, and gut microbiota that, when impaired, leads to increased penetration of luminal materials, including bacteria, toxins, and metabolites, into the body, termed “leaky gut syndrome”, which may trigger detrimental systemic responses and endotoxin invasion of the blood circulation [81, 82, 84]. PD patients have reduced expression of tight junctional proteins, including zonula occludens-1 and occludin, in colonic epithelial cells [66, 85, 86]. In a functional study, the concentrations of urine sucralose excreted after oral ingestion of a sugar cocktail were higher in patients with PD than healthy controls, suggesting increased intestinal permeability, possibly due to disrupted intestinal epithelium integrity [66, 85]. Leaky gut syndrome in PD could be an effect of complicated interactions between enteric inflammation, alpha-synucleinopathy, and an altered gut microbiota as increased intestinal permeability and reduced tight junction proteins have been observed to correlate with colonic alpha-synuclein accumulation, fecal inflammatory markers, and gut dysbiosis in PD patients and animal models of PD [66, 85, 87, 88]. Furthermore, disrupted intestinal permeability has been observed early in the disease course before the occurrence of brain pathology in peripheral endotoxin-induced and transgenic models of PD [88, 89]. This leaky gut phenomenon would allow the entry of intestinal luminal contents into the systemic circulation and then across the BBB to promote PD neuropathology in the CNS. Numerous studies have shown higher systemic concentrations of microbial metabolites and elevated endotoxin-related inflammatory markers in PD patients compared to controls, and the levels have been associated with PD severity, supporting the “leaky gut phenomenon” and associated consequences in the PD process [90,91,92].
Metagenomic and metabolomic evidence of gastrointestinal involvement in PD
Trillions of bacteria, viruses, protozoa, and fungi residing in the gastrointestinal tract comprise a vast, dynamic ecosystem that interacts with the host [93]. They participate in maintaining homeostasis of the intestinal barrier, immune maturation, nutrition absorption, and metabolism [93]. Recent evidence indicates that the gut microbiota communicates with the brain and is associated with several neurodegenerative disorders, including PD [94]. Alterations in the composition of bacteria, fungi, and viruses and their related metabolites have been described in PD patients, with most studies focusing on bacteria [95,96,97]. Small intestinal bacterial overgrowth compared to unaffected controls is a common presentation in PD patients, with an estimated prevalence of 46% in a meta-analysis and this associates with worse motor function and complications [98,99,100]. 16S rRNA gene amplicon surveys to study the bacterial and archaeal community structures and shotgun metagenomic sequencing to carry out non-targeted sequencing of the gut microbiota community are widely used techniques for investigating the associations between changes in the gut microbiota and the development of PD.
Altered gut microbiota in PD
A plethora of studies have investigated changes in the gut microbiota in patients with PD compared to healthy controls [101,102,103,104,105]. The results of various studies regarding the gut microbiota in PD are heterogenous, possibly due to differences in the study design, genetic background of enrolled participants, geographical region, dietary habits, lifestyle, disease status, co-morbidities, and use of medication [103]. Meta-analyses showed that the relative abundance of anti-inflammatory and short chain fatty acid (SCFA)-producing bacteria at the phylum level, including Blautia, Coprococcus, Roseburia, Lachnospira, Fusicatenibacter, and Faecalibacterium, are reduced in PD patients compared to controls, whereas, the amount of Lactobacillus, Bifidobacterium, and Akkermansia is higher in PD patients than unaffected participants from different ethnicities [101,102,103,104]. Notably, opportunistic pathogens and pro-inflammatory bacteria at the phylum level, including Corynebacterium, Porphyromonas, Alistipes, Bacteroides, Escherichia, Megasphaera and Desulfovibrio are also reportedly enriched in PD [103, 106, 107]. In metagenomic research, gene markers from the gut microbiome were found to accurately discriminate PD patients from healthy controls, with most of the identified markers belonging to Bacteroides and Escherichia species [108]. Although PD medications do affect the structure of the gut microbiota, as levodopa and entacapone correlate with the degree of reduction in SCFA-producing bacteria, these changes have also been observed in drug-naïve early-stage PD patients [96, 106, 109]. Furthermore, the gut microbiota can also influence the metabolism of anti-PD medication levodopa by using an alternative metabolic pathway and potentially reducing the bioavailability of levodopa [110, 111]. A study reported that Enterococcus faecalis harbors the gut microbial tyrosine decarboxylase enzyme, which is responsible for the decarboxylation of levodopa into dopamine in the gut [110]. In this regard, the abundance of Enterococcus faecalis compromised levodopa uptake and, consistently, the relative abundance of gut microbial tyrosine decarboxylase gene in fecal samples from PD patients was positively correlated with a higher daily levodopa dosage requirement [110]. Another study reported that Eggerthella lenta would metabolize dopamine into m-tyramine, thus reducing dopamine bioavailability [111]. These observations suggest that that levels of Enterococcus faecalis and Eggerthella lenta in the gut may affect the tratment efficacy of levodopa in PD patients.
Changes in the gut microbiota also correlate with disease progression in PD. A decrease in the SCFA-producing microbiota and increase in pro-inflammatory bacteria correlate with motor and cognitive severity in patients with PD [101, 103, 112]. Transplanting the fecal gut microbiota from PD patients leads to worsened motor symptoms in a transgenic rodent model of PD compared to transplantation from healthy donors [113]. A 3-year longitudinal follow-up study of PD patients revealed that a reduced amount of Roseburia species predicted faster progression in both motor and non-motor symptoms of PD [114]. SCFA-producing bacteria are considered beneficial to hosts due to their anti-inflammatory effects in the intestine and their role in maintaining gut and BBB homeostasis [115]. Correspondingly, a reduced amount of SCFA-producing bacteria, including Fusicatenibacter and Faecalibacterium, correlates with elevated fecal inflammatory calprotectin levels in PD patients [116]. Furthermore, enrichment in Bacteroides and Bifidobacterium has been linked to elevated expression of systemic and fecal inflammatory markers IFN-γ, TNF-α, and neutrophil gelatinase-associated lipocalin in patients with PD [87, 105]. The increased levels of Lactobacillaceae and Bifidobacteriaceae in PD patients seem paradoxical and require further investigation, as they are usually recognized as probiotics and are used in some clinical trials to treat constipation [117]. In transgenic mouse models of PD, the gut microbiota promotes pathological alpha-synuclein accumulation, neuroinflammation, and dopaminergic neuronal degeneration, along with aggravation of motor and non-motor symptoms, which were alleviated in germ-free conditions [113]. In addition, recent research has shown that Akkermansia induces intestinal alpha-synuclein aggregation as a result of mitochondrial calcium overload in intestinal enteroendocrine cells, which were hypothesized to be an initiating site for alpha-synucleinopathy in response to environmental factors because they are connected to enteric nerves and found to have adjacent accumulation of alpha-synuclein proteins [118, 119].
Notably, emerging evidence indicates that protein nucleation and aggregation may be influenced by an extracellular amyloid protein called “curli”, which is secreted by Escherichia coli, inducing neuronal deposition of alpha-synuclein in the gut and promoting neurodegeneration [120, 121]. The abundance of E. coli at the colonic mucosa correlates with enteric alpha-synuclein deposition in PD patients [85]. Curli is an amyloid protein that bacteria secrete for surface adherence and biofilm formation [122]. Induction of abnormal alpha-synuclein aggregation by curli has been demonstrated in vitro, and ingestion of curli and curli-producing bacteria accelerates enteric and central alpha-synucleinopathy, as well as neuroinflammation, dopaminergic neuronal degeneration, and motor dysfunction, in transgenic rodent models of PD [121, 122]. Therefore, distinct gut microbiota species result in a pro-inflammatory status in the intestinal tract or promote enteric alpha-synuclein aggregation to facilitate the occurrence and progression of PD.
Altered gut metabolites in PD
Intestinal microorganisms interact with the host through secreted toxins, by-products, and metabolites to modulate immune responses, endocrine secretion, metabolism, and neurotransmission [123]. Analyses of predicted functional pathways of the gut microbiome have shown that metabolism involving lipopolysaccharide (LPS), carbohydrates, amino acids, lipids, energy, cofactors, and vitamins is altered in PD [96, 108, 109, 124,125,126]. Consistently, gut microbial metabolites have been observed to be different between PD patients and controls in different biofluids or samples, including feces, plasma, and cerebral spinal fluid (CSF) (Table 1). Some gut metabolites could discriminate PD patients from unaffected controls, whereas some correlate with disease severity and progression, suggesting their participation in the disease mechanism and the potential to serve as disease biomarkers.
LPS is an endotoxin from the outer membrane of Gram-negative bacteria that activates surface pattern recognition receptors (e.g., TLR4) on epithelial and immune cells with the assistance of LPS-binding protein (LBP) to generate innate immune responses and modulate amyloidogenesis of alpha-synuclein through direct molecular interactions [127,128,129]. Studies in PD patients have shown that bacteria with endotoxin (e.g., Escherichia and Bacteroides) are enriched compared to controls, and the increased colonic E. coli correlated with reduced plasma levels of LBP along with upregulated enteric alpha-synuclein expression, oxidative stress, and elevated intestinal permeability [85]. Intra-peritoneal injection of LPS triggers intestinal pathology, including increased intestinal permeability and enteric alpha-synuclein accumulation, before the onset of brain pathology in rodent models of PD [88, 130]. In addition, systemic endotoxin injection evokes chronic systemic inflammation, microglia-mediated neuroinflammation, and dopaminergic neuronal degeneration at the substantia nigra [88, 130]. Exposure to endotoxin triggers the production of LBP, and elevated circulating levels of LBP are associated with chronic systemic inflammation [131]. In PD patients, although plasma LBP levels are reduced at an early disease stage, higher plasma LBP concentrations were observed in the later stage to correlate with systemic pro-inflammatory cytokines TNF-α and IL-6, motor symptom severity, and progression [90]. Taken together, these findings indicate the participation of gut dysbiosis-related endotoxin-mediated inflammation in PD pathogenesis and disease progression.
The major SCFAs are butyrate, propionate, and acetate, which are microbial metabolites and bacterial fermentation products of dietary fibers produced in the colon [115]. Being a major energy source of epithelial cells, most intestinal SCFAs are absorbed by colonocytes, leaving only a minority to enter the systemic circulation and pass across the BBB to modulate systemic immune responses and microgliosis [115]. As SCFA-producing bacteria are reduced in PD patients, it is not surprising to find that fecal levels of SCFAs are decreased in PD patients. The fecal levels of SCFAs positively correlate with the abundance of SCFA-producing microorganisms, including Butyricicoccus and Roseburia, but inversely correlate with Escherichia, Shigella, Akkermansia, and Bifidobacterium. Furthermore, reduced fecal levels of SCFAs further correlate with motor and cognitive severity in patients with PD [87, 91, 92, 97, 125, 132]. SCFAs are activators of G protein-coupled receptors and serve as inhibitors of histone deacetylases [115]. They are considered beneficial to the intestinal epithelium because they promote intestinal barrier integrity, mucosal healing, and anti-inflammatory effect [115]. Deficiency in enteric SCFAs is associated with intestinal inflammation, which may be one of the routes by which gut dysbiosis triggers PD pathogenesis [133]. Intriguingly, though SCFA levels in feces are reduced in PD patients, their concentrations in plasma, urine, and saliva were elevated, which may be attributed to increased intestinal permeability, as a higher plasma to fecal SCFA ratio was observed in PD patients and correlated with a pro-inflammatory gut microbiota [91, 92, 134,135,136,137]. Associations between increased plasma SCFA concentrations and disease severity were also observed in PD [91, 92, 138]. In addition, oral intake of SCFAs induces microglia activation, neuronal alpha-synuclein accumulation, and dopaminergic neuronal degeneration with motor impairment in transgenic mouse models of PD [113, 139]. Taken together, the evidence shows that excessive SCFAs in the systemic circulation, possibly resulting from the leaky gut phenomenon frequently observed in PD patients, may be detrimental to the disease process through activation of neuroinflammation [91]. Nevertheless, some studies have shown improved dopaminergic neuronal survival in response to SCFA treatment in rodent models [138, 140,141,142,143]. As evidence suggests a crucial role of SCFAs in the interconnection between the gut microbiota and neuropathology of PD, further investigations are warranted to explore the molecular mechanisms of SCFAs in the disease process of PD.
Tryptophan is an essential amino acid that is mostly absorbed and catabolized through the kynurenine pathway in the liver, with the remaining 5–10% passing into the colon for degradation by the microbiota into indole derivatives, which are interspecies signaling molecules that modulate gut hormone secretion, intestinal mucosal homeostasis, and systemic inflammation [144]. Studies have shown that plasma levels of indole-3-acetic acid (IAA) are decreased, whereas indole-3-propionic acid (IPA) is elevated, in PD patients [109, 145, 146]. IAA is a ligand of aryl hydrogen receptor (AHR), which regulates innate and adaptive immune responses, including attenuation of pro-inflammatory cytokine expression upon LPS stimulation and T-cell differentiation [144]. In addition to serving as an AHR ligand, IPA also activates pregnane X receptor and enhances intestinal barrier function [144]. In contrast, indoxyl sulfate is the only indole derivative considered toxic with pro-inflammatory features and is decreased in the systemic circulation and elevated in the urine of PD patients [109, 144, 147]. Intriguingly, the concentrations of indole-3-lactic acid have been found to be reduced in the plasma but increased in the urine of PD patients [148, 149]. Although altered microbial catabolism of tryptophan is observed in PD, its exact role in the disease process needs to be further elucidated.
Bile acids (BAs) synthesized in the liver and secreted into the intestine are mostly taken back up in the ileum, whereas those remaining in the gastrointestinal tract are deconjugated and/or dehydroxylated by the gut microbiota to form secondary BAs [150]. Studies have shown that the level of primary BAs is comparable between PD patients and controls. Secondary BAs deoxycholic acid (DCA) and lithocholic acid are increased and correlate with an increase in BA-metabolizing bacteria and reduced expression of BA re-uptake enzymes in the ileum [151]. Higher plasma levels of DCA and its glycine conjugated form (GDCA) are consistently observed in PD patients compared to controls in both Asian and Western populations [146, 152, 153]. Higher GDCA levels in the CSF were also reported in a small cohort of PD patients [154]. In addition to the emulsification of lipid products, BAs are regulators of the gut microbiota, inflammation, and energy metabolism [133]. As conjugated BAs can cross the BBB with the assistance of transporters, deconjugated BAs could enter the CNS by passive diffusion [155]. BAs could also enhance the permeability of the BBB and regulate neurotransmitters [155]. Dysregulated BA levels are generally considered cytotoxic and pro-inflammatory and have been observed in various neurodegenerative diseases, including Alzheimer’s disease and Huntington’s disease, in addition to PD [151, 155, 156].
A shift toward increased proteolysis has been observed in PD; higher concentrations of detrimental amino acid metabolites with pro-inflammatory features, p-cresol and p-cresol sulfate, have been noted in the plasma and CSF of patients with PD. These levels correlate with an increased abundance of pro-inflammatory bacteria and symptoms of constipation [124, 157]. Benzoate-related metabolites (benzoate, hippuric acid, and catechol sulfate) from dietary polyphenol compounds and choline-associated metabolites (choline, phosphocholine, trimethylamine, and trimethylamine-N-oxide) in plasma, CSF, urine, and saliva are also different between PD patients and controls. These levels also correlate with disease severity, though some conflicting results have been reported [109, 125, 135, 137, 143, 146, 158,159,160,161,162,163,164,165].
Altered microbial metabolism may reflect diverse interactions between the heterogenous host genetic background and gut environmental factors contributing to disease formation in individual patients. Notably, altered metabolomes are not unique findings in PD, but are also observed in other neurodegenerative diseases, including Alzheimer’s disease and atypical parkinsonism, with some similar and different results indicating common and distinct disease processes of neurodegeneration [152, 166, 167].
Interplay between genetic predisposition and gut microenvironmental changes in PD
Genetic mutations contribute to approximately 5 to 15% of patients with PD [13]. Identifying the protein function of these PD-causative genes would lead to a better understanding of the molecular mechanism of dopaminergic neuronal degeneration coming from multiple pathways, including perturbed alpha-synuclein homeostasis, mitochondrial dysfunction, autophagolysosomal impairment, and aberrant immune responses [168]. Though these genetic mutations often cause familial PD, disease penetrance varies widely between mutation carriers, suggesting that non-genetic environmental factors or other genetic modifiers may affect the occurrence of the disease [168]. Notably, the gut microenvironment could be one of the factors that modulates the manifestation of symptoms in genetic forms of PD (Fig. 2). The complex interplay between genetic risk factors in the host and environmental factors, especially gut microorganisms or gut metabolites, has been examined in multiple rodent models of PD (Table 2) [76, 77, 113, 122, 169,170,171,172,173]. In addition, genome-wide association studies have identified numerous genetic risks that increase susceptibility to sporadic PD, and several genes are related to gut microbial regulation and intestinal inflammation, including pattern recognition receptors TLR1 and TLR2, peptidoglycan recognition protein, and MUC2, a component of the mucosal layer that protects the intestinal epithelial barrier [174]. Some of the identified genetic susceptibility to sporadic PD also increases the risk of IBD, among which, NOD2 is known to be a strong predictor of IBD and to interact with LRRK2 [175].

Genetic predisposition and gut microenvironmental changes collaboratively contribute to PD pathogenesis. Genetic susceptibility to PD leads to vulnerable neurons related to aberrant immune responses, abnormal protein aggregation, autophagolysosomal impairment, and mitochondrial dysfunction that together with gut microenvironmental changes attributed to intestinal stimuli (e.g., bacterial infection, exposure to pesticide or herbicide, gut dysbiosis, and altered bacterial amyloid and metabolites) contribute to PD pathogenesis
LRRK2
Mutation of glycine to serine (p.G2019S) in LRRK2 is the most prevalent PD-causative mutation in both familial and sporadic PD [176, 177]. However, fewer than 50% of LRRK2 p.G2019S carriers will end up developing the disease at 80 years of age [15], indicating that factors other than the genetic mutation are needed to trigger the PD process. Interestingly, genome-wide association studies have also identified LRRK2 as a major susceptibility gene for Crohn’s disease, one of the inflammatory bowel disorders [178]. LRRK2 knock-out mice have prominent colitis compared to wild-type mice upon exposure to chemical stimulants [179]. In addition to high expression in neuronal tissues, LRRK2 is primarily expressed in immune cells, including macrophages, dendritic cells, and B lymphocytes, in the lamina propria of the gastrointestinal tract [180]. LRRK2 expression is induced by IFN-γ, consistent with the idea that LRRK2 plays a role in the pathogenesis of gut inflammatory disorder. LRRK2 protein is known to participate in a wide variety of cellular processes, including vesicle trafficking, autophagolysosomal function, and cell proliferation [181]. It can promote the lysozyme-sorting process in Paneth cells and enhance bacterial clearance in Salmonella and Listeria infection [182, 183]. LRRK2 is also highly expressed in peripheral immune cells and up-regulated during inflammation in response to pro-inflammatory cytokines and activated TLRs [184, 185]. Elevated LRRK2 kinase activity promotes inflammatory responses, including activation of the inflammasome and NF-κB pathway, leading to increased secretion of pro-inflammatory cytokines (e.g., TNF-α and IL-1ß), which assist in pathogen clearance but also worsen colitis, chronic neuroinflammation, and dopaminergic neuronal loss in rodent models [171, 182, 186, 187]. Furthermore, treatment with anti-TNF-α monoclonal antibody greatly reduces gut inflammation and neuroinflammation, mitigates dopaminergic neuronal loss, and alleviates motor symptoms exacerbated by chronic colitis in a transgenic LRRK2 p.G2019S mouse model [187]. Most of the pathogenic LRRK2 mutations relating to PD result in hyperactivated kinase activity of LRRK2 protein, and patients carrying the mutation have higher concentrations of pro-inflammatory cytokines in their peripheral circulation [175, 188, 189]. In addition, histopathological studies have demonstrated that LRRK2 expression is up-regulated in colonic biopsy tissue from patients with PD or IBD compared to controls and the expression correlates with disease severity [190,191,192]. Thus, exaggerated inflammatory responses attributed to pathogenic LRRK2 mutation upon immune triggers from the gastrointestinal tract contribute to the pathogenesis of PD. Further studies exploring the role of LRRK2 in the immune system, especially gut innate immunity, are needed to untangle the mechanism of PD beginning from the very early gut prodromal stage.
SNCA
Missense mutations and multiplication of SNCA, the gene encoding alpha-synuclein protein, were among the first identified causes of monogenic PD [168]. Patients carrying pathogenic SNCA mutations often have familial PD with early onset and that involves multiple systems with rapid progression [168]. Abundant Lewy body pathology and prominent neuronal loss have also been identified in these patients [168]. Although a detailed mechanism linking pathogenic SNCA mutations to PD onset is still being investigated, studies have shown that mutated genes change the structure, aggregation rate, post-translational modification, expression levels, and toxicity of alpha-synuclein, which leads to Lewy body pathology and dopaminergic neuronal loss [193]. In addition to brain pathology, recent studies have found that SNCA polymorphisms in PD patients modulate alpha-synuclein aggregation at the ENS before the occurrence of parkinsonian pathological changes in the brain in transgenic mouse models expressing pathogenic SNCA, consistent with Braak’s hypothesis [194, 195]. Intriguingly, SNCA also regulates the microbiota–gut–brain axis and SNCA polymorphisms are associated with alterations in opportunistic pathogens in PD [196]. In support of this finding, intestinal stimulation with oral paraquat, SCFAs, and microbiota transplant resulted in aggravated clinical and pathological severity, reflecting increased alpha-synuclein aggregations and neuroinflammation in both the ENS and CNS of transgenic SNCA mouse models [113, 169].
PRKN, PINK1, DJ-1
PRKN, PINK1, and DJ-1 are the main disease-causative genes for autosomal recessive parkinsonism [168]. They function in maintaining mitochondrial homeostasis. Parkin, which is encoded by PRKN, is a E3 ubiquitin ligase that can be activated by PTEN-induced kinase 1 (PINK1) in response to damaged mitochondria to ubiquitinate mitochondrial surface proteins, leading to mitophagy [197]. DJ-1 is a sensor of oxidative stress that works in parallel to Parkin and PINK1 to protect against mitochondrial fragmentation [197]. Although mutations in PRKN and PINK1 cause high penetrance of PD in mutation carriers, transgenic animal models with mutant PRKN and PINK1 present minimal motor dysfunction, Lewy body pathology, and neuronal loss [198]. Intriguingly, a recent study demonstrated that intestinal bacterial infection triggers parkinsonian pathological changes and clinical motor symptoms in PINK1 knock out (PINK1−/−) mice through neuroinflammation related to mitochondrial antigen presentation in immune cells, which were usually inhibited by PRKN and PINK1 expression [173, 199]. Furthermore, synaptic dysfunction was observed in response to systemic rotenone intoxication in PINK1± mice, possibly resulting from increased oxidative stress [172]. Thus, genetic susceptibility leading to mitochondrial dysfunction alters cellular responses to stress from environmental pathogens and toxins, which further enhances neuroinflammation and neurodegeneration. In addition, alteration of the gut microbiota and microbial metabolites in PD patients with PRKN or LRRK2 mutation and in DJ-1-deficienct rodent models suggests an interplay between host genetics and microbiota in the pathophysiology of PD [164, 200, 201].
Conclusions
Two decades after the dual-hit hypothesis proposed by Braak and colleagues, rapidly emerging evidence has validated the essential role of the gut microenvironment in the pathophysiology of PD as either one of the triggers or an aggravator of disease pathophysiology in those with “body-first” subgroup of PD patients. With advances in technology, elucidating the complex interaction between genetic risk factors with disease-prone gut microenvironmental changes unveils the heterogeneous features and pathophysiology of PD. Interventions targeting gut microbial changes could potentially modify the course of PD through gut-brain axis. A recent in vitro study provided evidence of two gut bacterial strains, Parabacteroides distasonis (MRX0005) and Megasphaera massiliensis (MRX0029) having anti-inflammatory and antioxidant capacity to mitigate neurodegeneration. The pleiotropic nature of these two potentially live biotherapeutic strains promoted a step into a Phase I clinical trial in PD patients in the coming future (https://parkinsonsnewstoday.com/news/phase-1-trial-will-evaluate-first-live-biotherapeutics-parkinsons-patients/). Because of the accumulating evidence from human and animal studies showing alterations in the gut microbiota in PD and the importance of the gut–brain axis in the PD process, future gut-oriented strategies to harmonize the gut microenvironment may have the potential to stop or halt the disease process at the very beginning of the prodromal gut stage of PD.
Availability of data and materials
Any data not published within the article are available from the corresponding author on reasonable request.
Abbreviations
- PD:
-
Parkinson’s disease
- AA:
-
Acetic acid
- BA:
-
Butyric acid
- CA:
-
Cholic acid
- CDCA:
-
Chenodeoxycholic acid
- CSF:
-
Cerebrospinal fluid
- DCA:
-
Deoxycholic acid
- GCA:
-
Glycocholic acid
- GCDCA:
-
Glycochenodeoxycholic acid
- GDCA:
-
Glycodeoxycholic acid
- GDCS:
-
Glycodeoxycholic acid 3-sulfate
- GLCAS:
-
Glycolithocholic acid 3-sulfate
- GUDCA:
-
Glycoursodeoxycholic acid
- HA:
-
Hippuric acid
- 3-HK:
-
3-Hydroxykynurenine
- H-Y stage:
-
Hoehn and Yahr stage
- IAA:
-
Indole-3-acetic acid
- ILA:
-
Indolelactic acid
- IS:
-
Indoxyl sulfate
- LCA:
-
Lithocholic acid
- PA:
-
Propionic acid
- TCA:
-
Taurocholic acid
- TCAS:
-
Taurocholic acid 3-sulfate
- TCDCA:
-
Taurochenodeoxycholic acid
- TDCA:
-
Taurodeoxycholic acid
- TMA:
-
Trimethylamine
- TMAO:
-
Trimethylamine N-oxide
- Trp:
-
Tryptophan
- TUDCA:
-
Tauroursodeoxycholic acid
- UDCA:
-
Ursodeoxycholic acid
- VA:
-
Valeric acid
References
de Lau LM, Breteler MM. Epidemiology of Parkinson’s disease. Lancet Neurol. 2006;5:525–35.
Collaborators GBDPsD. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018;17:939–53.
Kalia LV, Lang AE. Parkinson’s disease. Lancet. 2015;386:896–912.
Duncan GW, Khoo TK, Yarnall AJ, O’Brien JT, Coleman SY, Brooks DJ, Barker RA, Burn DJ. Health-related quality of life in early Parkinson’s disease: the impact of nonmotor symptoms. Mov Disord. 2014;29:195–202.
Goedert M, Spillantini MG, Del Tredici K, Braak H. 100 years of Lewy pathology. Nat Rev Neurol. 2013;9:13–24.
Adams-Carr KL, Bestwick JP, Shribman S, Lees A, Schrag A, Noyce AJ. Constipation preceding Parkinson’s disease: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2016;87:710–6.
Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614.
Arotcarena ML, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, Thiolat ML, Tasselli M, Aubert P, Kruse N, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. 2020;143:1462–75.
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, et al. Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627-641.e627.
Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, Mazmanian SK, Volpicelli-Daley LA, Gradinaru V. Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. 2020;23:327–36.
Manfredsson FP, Luk KC, Benskey MJ, Gezer A, Garcia J, Kuhn NC, Sandoval IM, Patterson JR, O’Mara A, Yonkers R, Kordower JH. Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis. 2018;112:106–18.
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Chu YT, Tai CH, Lin CH, Wu RM. Updates on the genetics of Parkinson’s disease: clinical implications and future treatment. Acta Neurol Taiwan. 2021;30(3):83–93.
Tran J, Anastacio H, Bardy C. Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. NPJ Parkinson’s Dis. 2020;6:8.
Lee AJ, Wang Y, Alcalay RN, Mejia-Santana H, Saunders-Pullman R, Bressman S, Corvol JC, Brice A, Lesage S, Mangone G, et al. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord. 2017;32:1432–8.
Chaudhuri KR, Schapira AH. Non-motor symptoms of Parkinson’s disease: dopaminergic pathophysiology and treatment. Lancet Neurol. 2009;8:464–74.
Berg D, Postuma RB, Adler CH, Bloem BR, Chan P, Dubois B, Gasser T, Goetz CG, Halliday G, Joseph L, et al. MDS research criteria for prodromal Parkinson’s disease. Mov Disord. 2015;30:1600–11.
Warnecke T, Schäfer KH, Claus I, Del Tredici K, Jost WH. Gastrointestinal involvement in Parkinson’s disease: pathophysiology, diagnosis, and management. NPJ Parkinsons Dis. 2022;8:31.
Abbott RD, Petrovitch H, White LR, Masaki KH, Tanner CM, Curb JD, Grandinetti A, Blanchette PL, Popper JS, Ross GW. Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology. 2001;57:456–62.
Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. Prediagnostic presentations of Parkinson’s disease in primary care: a case-control study. Lancet Neurol. 2015;14:57–64.
Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, Schrag A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol. 2012;72:893–901.
Lin CH, Lin JW, Liu YC, Chang CH, Wu RM. Risk of Parkinson’s disease following severe constipation: a nationwide population-based cohort study. Parkinsonism Relat Disord. 2014;20:1371–5.
Savica R, Carlin JM, Grossardt BR, Bower JH, Ahlskog JE, Maraganore DM, Bharucha AE, Rocca WA. Medical records documentation of constipation preceding Parkinson disease: a case-control study. Neurology. 2009;73:1752–8.
Knudsen K, Krogh K, Østergaard K, Borghammer P. Constipation in Parkinson’s disease: subjective symptoms, objective markers, and new perspectives. Mov Disord. 2017;32:94–105.
Rolli-Derkinderen M, Leclair-Visonneau L, Bourreille A, Coron E, Neunlist M, Derkinderen P. Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J Neurol. 2019;267:2207–13.
Houser MC, Chang J, Factor SA, Molho ES, Zabetian CP, Hill-Burns EM, Payami H, Hertzberg VS, Tansey MG. Stool immune profiles evince gastrointestinal inflammation in Parkinson’s disease. Mov Disord. 2018;33:793–804.
Schwiertz A, Spiegel J, Dillmann U, Grundmann D, Bürmann J, Faßbender K, Schäfer K-H, Unger MM. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord. 2018;50:104–7.
Devos D, Lebouvier T, Lardeux B, Biraud M, Rouaud T, Pouclet H, Coron E, Bruley des Varannes S, Naveilhan P, Nguyen J-M, et al. Colonic inflammation in Parkinson’s disease. Neurobiol Dis. 2013;50:42–8.
Villumsen M, Aznar S, Pakkenberg B, Jess T, Brudek T. Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut. 2019;68:18–24.
Zhu F, Li C, Gong J, Zhu W, Gu L, Li N. The risk of Parkinson’s disease in inflammatory bowel disease: a systematic review and meta-analysis. Dig Liver Dis. 2019;51:38–42.
Peter I, Dubinsky M, Bressman S, Park A, Lu C, Chen N, Wang A. Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol. 2018;75:939–46.
Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Bjorklund T, Wang ZY, Roybon L, Melki R, Li JY. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–20.
Zeisel A, Hochgerner H, Lonnerberg P, Johnsson A, Memic F, van der Zwan J, Haring M, Braun E, Borm LE, La Manno G, et al. Molecular architecture of the mouse nervous system. Cell. 2018;174:999-1014.e1022.
Bohorquez DV, Liddle RA. The gut connectome: making sense of what you eat. J Clin Invest. 2015;125:888–90.
Carabotti M, Scirocco A, Maselli MA, Severi C. The gut–brain axis: interactions between enteric microbiota, central and enteric nervous systems. Ann Gastroenterol. 2015;28:203–9.
Braak H, de Vos RA, Bohl J, Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett. 2006;396:67–72.
Hilton D, Stephens M, Kirk L, Edwards P, Potter R, Zajicek J, Broughton E, Hagan H, Carroll C. Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol. 2014;127:235–41.
Shannon KM, Keshavarzian A, Dodiya HB, Jakate S, Kordower JH. Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord. 2012;27:716–9.
Stokholm MG, Danielsen EH, Hamilton-Dutoit SJ, Borghammer P. Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol. 2016;79:940–9.
Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol. 2008;34:284–95.
Li JY, Englund E, Holton JL, Soulet D, Hagell P, Lees AJ, Lashley T, Quinn NP, Rehncrona S, Björklund A, et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–3.
Kordower JH, Chu Y, Hauser RA, Freeman TB, Olanow CW. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med. 2008;14:504–6.
Li JY, Englund E, Widner H, Rehncrona S, Björklund A, Lindvall O, Brundin P. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord. 2010;25:1091–6.
Choi YR, Cha S-H, Kang S-J, Kim J-B, Jou I, Park SM. Prion-like propagation of a-synuclein is regulated by the FcγRIIB-SHP-1/2 signaling pathway in neurons. Cell Rep. 2018;22:136–48.
Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Kim S, Kwon SH, Kam TI, Panicker N, Karuppagounder SS, Lee S, Lee JH, Kim WR, Kook M, Foss CA, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron. 2019;103:627-641.e627.
Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, Tsung-Pin P, Riess O, Nyengaard JR, Tamgüney G, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019;138:535–50.
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O’Sullivan GA, Pal A, Said J, Marsico G, Verbavatz J-M, Rodrigo-Angulo M, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Anselmi L, Bove C, Coleman FH, Le K, Subramanian MP, Venkiteswaran K, Subramanian T, Travagli RA. Ingestion of subthreshold doses of environmental toxins induces ascending Parkinsonism in the rat. NPJ Parkinson’s Dis. 2018;4:30–30.
Svensson E, Horváth-Puhó E, Thomsen RW, Djurhuus JC, Pedersen L, Borghammer P, Sørensen HT. Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol. 2015;78:522–9.
Liu B, Fang F, Pedersen NL, Tillander A, Ludvigsson JF, Ekbom A, Svenningsson P, Chen H, Wirdefeldt K. Vagotomy and Parkinson disease: a Swedish register-based matched-cohort study. Neurology. 2017;88:1996–2002.
Sui Y-T, Bullock KM, Erickson MA, Zhang J, Banks WA. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides. 2014;62:197–202.
Foulds PG, Diggle P, Mitchell JD, Parker A, Hasegawa M, Masuda-Suzukake M, Mann DM, Allsop D. A longitudinal study on α-synuclein in blood plasma as a biomarker for Parkinson’s disease. Sci Rep. 2013;3:2540.
Chang C-W, Yang S-Y, Yang C-C, Chang C-W, Wu Y-R. Plasma and serum Alpha-synuclein as a biomarker of diagnosis in patients with Parkinson’s disease. Front Neurol. 2020;10:1388. https://doi.org/10.3389/fneur.2019.01388.
Gray MT, Munoz DG, Gray DA, Schlossmacher MG, Woulfe JM. Alpha-synuclein in the appendiceal mucosa of neurologically intact subjects. Mov Disord. 2014;29:991–8.
Chung SJ, Kim J, Lee HJ, Ryu H-S, Kim K, Lee JH, Jung KW, Kim MJ, Kim M-J, Kim YJ, et al. Alpha-synuclein in gastric and colonic mucosa in Parkinson’s disease: limited role as a biomarker. Mov Disord. 2016;31:241–9.
Marras C, Lang AE, Austin PC, Lau C, Urbach DR. Appendectomy in mid and later life and risk of Parkinson’s disease: a population-based study. Mov Disord. 2016;31:1243–7.
Svensson E, Horváth-Puhó E, Stokholm MG, Sørensen HT, Henderson VW, Borghammer P. Appendectomy and risk of Parkinson’s disease: a nationwide cohort study with more than 10 years of follow-up. Mov Disord. 2016;31:1918–22.
Palacios N, Hughes KC, Cereda E, Schwarzschild MA, Ascherio A. Appendectomy and risk of Parkinson’s disease in two large prospective cohorts of men and women. Mov Disord. 2018;33:1492–6.
Mendes A, Gonçalves A, Vila-Chã N, Moreira I, Fernandes J, Damásio J, Teixeira-Pinto A, Taipa R, Lima AB, Cavaco S. Appendectomy may delay Parkinson’s disease Onset. Mov Disord. 2015;30:1404–7.
Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, Lindqvist D, Chen H, Thomas PM, Brundin P, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. 2018;10:eaar5280.
Stolzenberg E, Berry D, Yang D, Lee EY, Kroemer A, Kaufman S, Wong GCL, Oppenheim JJ, Sen S, Fishbein T, et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun. 2017;9:456–63.
Alam MM, Yang D, Li XQ, Liu J, Back TC, Trivett A, Karim B, Barbut D, Zasloff M, Oppenheim JJ. Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep. 2022;38:110090.
Pellegrini C, D’Antongiovanni V, Miraglia F, Rota L, Benvenuti L, Di Salvo C, Testa G, Capsoni S, Carta G, Antonioli L, et al. Enteric alpha-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. NPJ Parkinsons Dis. 2022;8:9.
Kang X, Ploner A, Roelstraete B, Khalili H, Williams DM, Pedersen NL, Ludvigsson JF, Wirdefeldt K. Association between microscopic colitis and Parkinson’s disease in a Swedish population. Mov Disord. 2021;36:1919–26.
Perez-Pardo P, Dodiya HB, Engen PA, Forsyth CB, Huschens AM, Shaikh M, Voigt RM, Naqib A, Green SJ, Kordower JH, et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: a translational study from men to mice. Gut. 2019;68:829–43.
Dumitrescu L, Marta D, Dănău A, Lefter A, Tulbă D, Cozma L, Manole E, Gherghiceanu M, Ceafalan LC, Popescu BO. Serum and fecal markers of intestinal inflammation and intestinal barrier permeability are elevated in Parkinson’s disease. Front Neurosci. 2021;15:689723.
Lin JC, Lin CS, Hsu CW, Lin CL, Kao CH. Association between Parkinson’s disease and inflammatory bowel disease: a Nationwide Taiwanese Retrospective Cohort Study. Inflamm Bowel Dis. 2016;22:1049–55.
Weimers P, Halfvarson J, Sachs MC, Saunders-Pullman R, Ludvigsson JF, Peter I, Burisch J, Olén O. Inflammatory bowel disease and Parkinson’s disease: a nationwide Swedish cohort study. Inflamm Bowel Dis. 2019;25:111–23.
Park S, Kim J, Chun J, Han K, Soh H, Kang EA, Lee HJ, Im JP, Kim JS. Patients with inflammatory bowel disease are at an increased risk of Parkinson’s disease: a South Korean nationwide population-based study. J Clin Med. 2019;8:1191.
Zhu Y, Yuan M, Liu Y, Yang F, Chen W-Z, Xu Z-Z, Xiang Z-B, Xu R-S. Association between inflammatory bowel diseases and Parkinson’s disease: systematic review and meta-analysis. Neural Regen Res. 2022;17:344–53.
Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, Thompson WK, Hernandez DG, Djurovic S, Schork AJ, et al. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 2017;74:780–92.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18:1091–102.
Villarán RF, Espinosa-Oliva AM, Sarmiento M, De Pablos RM, Argüelles S, Delgado-Cortés MJ, Sobrino V, Van Rooijen N, Venero JL, Herrera AJ, et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: potential risk factor in Parkinson`s disease. J Neurochem. 2010;114:1687–700.
Garrido-Gil P, Rodriguez-Perez AI, Dominguez-Meijide A, Guerra MJ, Labandeira-Garcia JL. Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson’s disease models. Mol Neurobiol. 2018;55:7297–316.
Lin C-H, Lin H-Y, Ho E-P, Ke Y-C, Cheng M-F, Shiue C-Y, Wu C-H, Liao P-H, Hsu AY-H, Chu L-A, et al. Mild chronic colitis triggers parkinsonism in LRRK2 mutant mice through activating TNF-α pathway. Mov Disord. 2022;37:745–57.
Kishimoto Y, Zhu W, Hosoda W, Sen JM, Mattson MP. Chronic mild gut inflammation accelerates brain neuropathology and motor dysfunction in α-synuclein mutant mice. Neuromol Med. 2019;21:239–49.
Houser MC, Caudle WM, Chang J, Kannarkat GT, Yang Y, Kelly SD, Oliver D, Joers V, Shannon KM, Keshavarzian A, Tansey MG. Experimental colitis promotes sustained, sex-dependent, T-cell-associated neuroinflammation and parkinsonian neuropathology. Acta Neuropathol Commun. 2021;9:139.
Chung LY, Lin YT, Liu C, Tai YC, Lin HY, Lin CH, Chen CC. Neuroinflammation upregulated neuronal toll-like receptors 2 and 4 to drive synucleinopathy in neurodegeneration. Front Pharmacol. 2022;13:845930.
Prigent A, Lionnet A, Durieu E, Chapelet G, Bourreille A, Neunlist M, Rolli-Derkinderen M, Derkinderen P. Enteric alpha-synuclein expression is increased in Crohn’s disease. Acta Neuropathol. 2019;137:359–61.
Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50:1–9.
Zeng MY, Inohara N, Nuñez G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017;10:18–26.
Rosenberg HJ, Rao M. Enteric glia in homeostasis and disease: from fundamental biology to human pathology. iScience. 2021;24:102863.
Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut. 2019;68:1516.
Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, Estes JD, Dodiya HB, Keshavarzian A. Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS ONE. 2011;6:e28032.
Clairembault T, Leclair-Visonneau L, Coron E, Bourreille A, Le Dily S, Vavasseur F, Heymann MF, Neunlist M, Derkinderen P. Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun. 2015;3:12.
Aho VTE, Houser MC, Pereira PAB, Chang J, Rudi K, Paulin L, Hertzberg V, Auvinen P, Tansey MG, Scheperjans F. Relationships of gut microbiota, short-chain fatty acids, inflammation, and the gut barrier in Parkinson’s disease. Mol Neurodegener. 2021;16:6.
Kelly LP, Carvey PM, Keshavarzian A, Shannon KM, Shaikh M, Bakay RAE, Kordower JH. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord. 2014;29:999–1009.
Pellegrini C, D’Antongiovanni V, Miraglia F, Rota L, Benvenuti L, Di Salvo C, Testa G, Capsoni S, Carta G, Antonioli L, et al. Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. NPJ Parkinson’s Dis. 2022;8:9.
Chen SJ, Chi YC, Ho CH, Yang WS, Lin CH. Plasma lipopolysaccharide-binding protein reflects risk and progression of Parkinson’s disease. J Parkinsons Dis. 2021;11:1129–39.
Chen SJ, Chen CC, Liao HY, Lin YT, Wu YW, Liou JM, Wu MS, Kuo CH, Lin CH. Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in patients with Parkinson disease. Neurology. 2022;98:e848–58.
Yang X, Ai P, He X, Mo C, Zhang Y, Xu S, Lai Y, Qian Y, Xiao Q. Parkinson’s disease is associated with impaired gut-blood barrier for short-chain fatty acids. Mov Disord. 2022. https://doi.org/10.1002/mds.29063.
Claus SP, Guillou H, Ellero-Simatos S. The gut microbiota: a major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes. 2016;2:16003.
Morais LH, Schreiber HL, Mazmanian SK. The gut microbiota–brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021;19:241–55.
Cirstea MS, Sundvick K, Golz E, Yu AC, Boutin RCT, Kliger D, Finlay BB, Appel-Cresswell S. The gut mycobiome in Parkinson’s disease. J Parkinsons Dis. 2021;11:153–8.
Bedarf JR, Hildebrand F, Coelho LP, Sunagawa S, Bahram M, Goeser F, Bork P, Wüllner U. Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 2017;9:39.
De Pablo-Fernandez E, Gebeyehu GG, Flain L, Slater R, Frau A, Ijaz UZ, Warner T, Probert C. The faecal metabolome and mycobiome in Parkinson’s disease. Parkinsonism Relat Disord. 2022;95:65–9.
Li X, Feng X, Jiang Z, Jiang Z. Association of small intestinal bacterial overgrowth with Parkinson’s disease: a systematic review and meta-analysis. Gut Pathog. 2021;13:25.
Tan AH, Mahadeva S, Thalha AM, Gibson PR, Kiew CK, Yeat CM, Ng SW, Ang SP, Chow SK, Tan CT, et al. Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord. 2014;20:535–40.
Fasano A, Bove F, Gabrielli M, Petracca M, Zocco MA, Ragazzoni E, Barbaro F, Piano C, Fortuna S, Tortora A, et al. The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord. 2013;28:1241–9.
Nishiwaki H, Ito M, Ishida T, Hamaguchi T, Maeda T, Kashihara K, Tsuboi Y, Ueyama J, Shimamura T, Mori H, et al. Meta-analysis of gut dysbiosis in Parkinson’s disease. Mov Disord. 2020;35:1626–35.
Romano S, Savva GM, Bedarf JR, Charles IG, Hildebrand F, Narbad A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinson’s Dis. 2021;7:27.
Toh TS, Chong CW, Lim SY, Bowman J, Cirstea M, Lin CH, Chen CC, Appel-Cresswell S, Finlay BB, Tan AH. Gut microbiome in Parkinson’s disease: new insights from meta-analysis. Parkinsonism Relat Disord. 2022;94:1–9.
Shen T, Yue Y, He T, Huang C, Qu B, Lv W, Lai HY. The association between the gut microbiota and Parkinson’s disease, a meta-analysis. Front Aging Neurosci. 2021;13:636545.
Lin CH, Chen CC, Chiang HL, Liou JM, Chang CM, Lu TP, Chuang EY, Tai YC, Cheng C, Lin HY, Wu MS. Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J Neuroinflamm. 2019;16:129.
Wallen ZD, Appah M, Dean MN, Sesler CL, Factor SA, Molho E, Zabetian CP, Standaert DG, Payami H. Characterizing dysbiosis of gut microbiome in PD: evidence for overabundance of opportunistic pathogens. NPJ Parkinsons Dis. 2020;6:11.
Murros KE, Huynh VA, Takala TM, Saris PEJ. Desulfovibrio bacteria are associated with Parkinson’s disease. Front Cell Infect Microbiol. 2021;11:652617.
Qian Y, Yang X, Xu S, Huang P, Li B, Du J, He Y, Su B, Xu LM, Wang L, et al. Gut metagenomics-derived genes as potential biomarkers of Parkinson’s disease. Brain. 2020;143:2474–89.
Rosario D, Bidkhori G, Lee S, Bedarf J, Hildebrand F, Le Chatelier E, Uhlen M, Ehrlich SD, Proctor G, Wüllner U, et al. Systematic analysis of gut microbiome reveals the role of bacterial folate and homocysteine metabolism in Parkinson’s disease. Cell Rep. 2021;34:108807.
van Kessel SP, Frye AK, El-Gendy AO, Castejon M, Keshavarzian A, van Dijk G, El Aidy S. Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun. 2019;10:310.
Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, Balskus EP. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science. 2019;364:eaau6323.
Ren T, Gao Y, Qiu Y, Jiang S, Zhang Q, Zhang J, Wang L, Zhang Y, Wang L, Nie K. Gut microbiota altered in mild cognitive impairment compared with normal cognition in sporadic Parkinson’s disease. Front Neurol. 2020;11:137.
Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell. 2016;167:1469-1480.e1412.
Cilia R, Piatti M, Cereda E, Bolliri C, Caronni S, Ferri V, Cassani E, Bonvegna S, Ferrarese C, Zecchinelli AL, et al. Does gut microbiota influence the course of Parkinson’s Disease? a 3-year prospective exploratory study in de novo patients. J Parkinsons Dis. 2021;11:159–70.
Parada Venegas D, De la Fuente MK, Landskron G, González MJ, Quera R, Dijkstra G, Harmsen HJM, Faber KN, Hermoso MA. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019;10:277.
Weis S, Schwiertz A, Unger MM, Becker A, Faßbender K, Ratering S, Kohl M, Schnell S, Schäfer KH, Egert M. Effect of Parkinson’s disease and related medications on the composition of the fecal bacterial microbiota. NPJ Parkinsons Dis. 2019;5:28.
Cassani E, Privitera G, Pezzoli G, Pusani C, Madio C, Iorio L, Barichella M. Use of probiotics for the treatment of constipation in Parkinson’s disease patients. Minerva Gastroenterol Dietol. 2011;57:117–21.
Amorim Neto DP, Bosque BP, Pereira de Godoy JV, Rodrigues PV, Meneses DD, Tostes K, Costa Tonoli CC, Faustino de Carvalho H, González-Billault C, de Castro Fonseca M. Akkermansia muciniphila induces mitochondrial calcium overload and α -synuclein aggregation in an enteroendocrine cell line. iScience. 2022;25:103908.
Chandra R, Hiniker A, Kuo YM, Nussbaum RL, Liddle RA. alpha-Synuclein in gut endocrine cells and its implications for Parkinson’s disease. JCI Insight. 2017;2:e92295.
Wang C, Lau CY, Ma F, Zheng C. Genome-wide screen identifies curli amyloid fibril as a bacterial component promoting host neurodegeneration. Proc Natl Acad Sci USA. 2021;118:e2106504118.
Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, Jagadapillai R, Liu R, Choe K, Shivakumar B, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged fischer 344 rats and Caenorhabditis elegans. Sci Rep. 2016;6:34477.
Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG, Thron T, Needham BD, Horvath I, Debelius JW, et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife. 2020;9:e53111.
Rosario D, Boren J, Uhlen M, Proctor G, Aarsland D, Mardinoglu A, Shoaie S. Systems biology approaches to understand the host-microbiome interactions in neurodegenerative diseases. Front Neurosci. 2020;14:716.
Cirstea MS, Yu AC, Golz E, Sundvick K, Kliger D, Radisavljevic N, Foulger LH, Mackenzie M, Huan T, Finlay BB, Appel-Cresswell S. Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov Disord. 2020;35:1208–17.
Tan AH, Chong CW, Lim SY, Yap IKS, Teh CSJ, Loke MF, Song SL, Tan JY, Ang BH, Tan YQ, et al. Gut microbial ecosystem in Parkinson disease: new clinicobiological insights from multi-omics. Ann Neurol. 2021;89:546–59.
Keshavarzian A, Green SJ, Engen PA, Voigt RM, Naqib A, Forsyth CB, Mutlu E, Shannon KM. Colonic bacterial composition in Parkinson’s disease. Mov Disord. 2015;30:1351–60.
Kieser KJ, Kagan JC. Multi-receptor detection of individual bacterial products by the innate immune system. Nat Rev Immunol. 2017;17:376–90.
Guo S, Nighot M, Al-Sadi R, Alhmoud T, Nighot P, Ma TY. Lipopolysaccharide regulation of intestinal tight junction permeability is mediated by TLR4 signal transduction pathway activation of FAK and MyD88. J Immunol. 2015;195:4999.
Bhattacharyya D, Mohite GM, Krishnamoorthy J, Gayen N, Mehra S, Navalkar A, Kotler SA, Ratha BN, Ghosh A, Kumar R, et al. Lipopolysaccharide from gut microbiota modulates α-synuclein aggregation and alters its biological function. ACS Chem Neurosci. 2019;10:2229–36.
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S, Knapp DJ, Crews FT. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–62.
Lakatos PL, Kiss LS, Palatka K, Altorjay I, Antal-Szalmas P, Palyu E, Udvardy M, Molnar T, Farkas K, Veres G, et al. Serum lipopolysaccharide-binding protein and soluble CD14 are markers of disease activity in patients with Crohn’s disease. Inflamm Bowel Dis. 2011;17:767–77.
Unger MM, Spiegel J, Dillmann KU, Grundmann D, Philippeit H, Burmann J, Fassbender K, Schwiertz A, Schafer KH. Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord. 2016;32:66–72.
Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17:223–37.
Shin C, Lim Y, Lim H, Ahn T-B. Plasma short-chain fatty acids in patients with Parkinson’s disease. Mov Disord. 2020;35:1021–7.
Kumari S, Goyal V, Kumaran SS, Dwivedi SN, Srivastava A, Jagannathan NR. Quantitative metabolomics of saliva using proton NMR spectroscopy in patients with Parkinson’s disease and healthy controls. Neurol Sci. 2020;41:1201–10.
Kumari S, Kumaran SS, Goyal V, Sharma RK, Sinha N, Dwivedi SN, Srivastava AK, Jagannathan NR. Identification of potential urine biomarkers in idiopathic Parkinson’s disease using NMR. Clin Chim Acta. 2020;510:442–9.
Kim C-H, Jung J, Lee Y-U, Kim K-H, Kang S, Kang G-H, Chu H, Kim S-Y, Lee S. Comparison of metabolites and gut microbes between patients with Parkinson’s disease and healthy individuals-a pilot clinical observational study (STROBE Compliant). Healthcare (Basel, Switzerland). 2022;10:302.
Wu G, Jiang Z, Pu Y, Chen S, Wang T, Wang Y, Xu X, Wang S, Jin M, Yao Y, et al. Serum short-chain fatty acids and its correlation with motor and non-motor symptoms in Parkinson’s disease patients. BMC Neurol. 2022;22:13.
Qiao C-M, Sun M-F, Jia X-B, Li Y, Zhang B-P, Zhao L-P, Shi Y, Zhou Z-L, Zhu Y-L, Cui C, Shen Y-Q. Sodium butyrate exacerbates Parkinson’s disease by aggravating neuroinflammation and colonic inflammation in MPTP-induced mice model. Neurochem Res. 2020;45:2128–42.
Paiva I, Pinho R, Pavlou MA, Hennion M, Wales P, Schutz AL, Rajput A, Szego EM, Kerimoglu C, Gerhardt E, et al. Sodium butyrate rescues dopaminergic cells from alpha-synuclein-induced transcriptional deregulation and DNA damage. Hum Mol Genet. 2017;26:2231–46.
Liu J, Wang F, Liu S, Du J, Hu X, Xiong J, Fang R, Chen W, Sun J. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J Neurol Sci. 2017;381:176–81.
Ostendorf F, Metzdorf J, Gold R, Haghikia A, Tönges L. Propionic acid and fasudil as treatment against rotenone toxicity in an in vitro model of Parkinson’s disease. Molecules. 2020;25:2502.
Ahmed SS, Santosh W, Kumar S, Christlet HT. Metabolic profiling of Parkinson’s disease: evidence of biomarker from gene expression analysis and rapid neural network detection. J Biomed Sci. 2009;16:63.
Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9:3294.
Hatano T, Saiki S, Okuzumi A, Mohney RP, Hattori N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J Neurol Neurosurg Psychiatry. 2016;87:295–301.
Chen SJ, Chen CC, Liao HY, Wu YW, Liou JM, Wu MS, Kuo CH, Lin CH. Alteration of gut microbial metabolites in the systemic circulation of patients with Parkinson’s disease. J Parkinsons Dis. 2022;12:1219–30.
Cassani E, Barichella M, Cancello R, Cavanna F, Iorio L, Cereda E, Bolliri C, Zampella Maria P, Bianchi F, Cestaro B, Pezzoli G. Increased urinary indoxyl sulfate (indican): new insights into gut dysbiosis in Parkinson’s disease. Parkinsonism Relat Disord. 2015;21:389–93.
Shao Y, Le W. Recent advances and perspectives of metabolomics-based investigations in Parkinson’s disease. Mol Neurodegener. 2019;14:3.
Luan H, Liu LF, Meng N, Tang Z, Chua KK, Chen LL, Song JX, Mok VC, Xie LX, Li M, Cai Z. LC-MS-based urinary metabolite signatures in idiopathic Parkinson’s disease. J Proteome Res. 2015;14:467–78.
Kiriyama Y, Nochi H. The biosynthesis, signaling, and neurological functions of bile acids. Biomolecules. 2019;9:232.
Li P, Killinger BA, Ensink E, Beddows I, Yilmaz A, Lubben N, Lamp J, Schilthuis M, Vega IE, Woltjer R, et al. Gut microbiota dysbiosis is associated with elevated bile acids in Parkinson’s disease. Metabolites. 2021;11:29.
Shao Y, Li T, Liu Z, Wang X, Xu X, Li S, Xu G, Le W. Comprehensive metabolic profiling of Parkinson’s disease by liquid chromatography-mass spectrometry. Mol Neurodegener. 2021;16:4.
Yakhine-Diop SMS, Morales-García JA, Niso-Santano M, González-Polo RA, Uribe-Carretero E, Martinez-Chacon G, Durand S, Maiuri MC, Aiastui A, Zulaica M, et al. Metabolic alterations in plasma from patients with familial and idiopathic Parkinson’s disease. Aging. 2020;12:16690–708.
Yilmaz A, Ugur Z, Ustun I, Akyol S, Bahado-Singh RO, Maddens M, Aasly JO, Graham SF. Metabolic profiling of CSF from people suffering from sporadic and LRRK2 Parkinson’s disease: a pilot study. Cells. 2020;9:2394.
MahmoudianDehkordi S, Arnold M, Nho K, Ahmad S, Jia W, Xie G, Louie G, Kueider-Paisley A, Moseley MA, Thompson JW, et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—an emerging role for gut microbiome. Alzheimers Dement. 2019;15:76–92.
Chen ML, Takeda K, Sundrud MS. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019;12:851–61.
Willkommen D, Lucio M, Moritz F, Forcisi S, Kanawati B, Smirnov KS, Schroeter M, Sigaroudi A, Schmitt-Kopplin P, Michalke B. Metabolomic investigations in cerebrospinal fluid of Parkinson’s disease. PLoS ONE. 2018;13:e0208752.
Chen S-J, Kuo C-H, Kuo H-C, Chen C-C, Wu W-K, Liou J-M, Wu M-S, Lin C-H. The gut metabolite trimethylamine n-oxide is associated with Parkinson’s disease severity and progression. Mov Disord. 2020;35:2115–6.
Chung SJ, Rim JH, Ji D, Lee S, Yoo HS, Jung JH, Baik K, Choi Y, Ye BS, Sohn YH, et al. Gut microbiota-derived metabolite trimethylamine N-oxide as a biomarker in early Parkinson’s disease. Nutrition. 2021;83:111090.
Sankowski B, Księżarczyk K, Raćkowska E, Szlufik S, Koziorowski D, Giebułtowicz J. Higher cerebrospinal fluid to plasma ratio of p-cresol sulfate and indoxyl sulfate in patients with Parkinson’s disease. Clin Chim Acta. 2020;501:165–73.
Wuolikainen A, Jonsson P, Ahnlund M, Antti H, Marklund SL, Moritz T, Forsgren L, Andersen PM, Trupp M. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol BioSyst. 2016;12:1287–98.
Trivedi DK, Sinclair E, Xu Y, Sarkar D, Walton-Doyle C, Liscio C, Banks P, Milne J, Silverdale M, Kunath T, et al. Discovery of volatile biomarkers of Parkinson’s disease from sebum. ACS Cent Sci. 2019;5:599–606.
LeWitt PA, Li J, Lu M, Guo L, Auinger P. Metabolomic biomarkers as strong correlates of Parkinson disease progression. Neurology. 2017;88:862–9.
Okuzumi A, Hatano T, Ueno S-I, Ogawa T, Saiki S, Mori A, Koinuma T, Oji Y, Ishikawa K-I, Fujimaki M, et al. Metabolomics-based identification of metabolic alterations in PARK2. Ann Clin Transl Neurol. 2019;6:525–36.
Burté F, Houghton D, Lowes H, Pyle A, Nesbitt S, Yarnall A, Yu-Wai-Man P, Burn DJ, Santibanez-Koref M, Hudson G. Metabolic profiling of Parkinson’s disease and mild cognitive impairment. Mov Disord. 2017;32:927–32.
Marizzoni M, Cattaneo A, Mirabelli P, Festari C, Lopizzo N, Nicolosi V, Mombelli E, Mazzelli M, Luongo D, Naviglio D, et al. Short-chain fatty acids and lipopolysaccharide as mediators between gut dysbiosis and amyloid pathology in Alzheimer’s disease. J Alzheimers Dis. 2020;78:683–97.
Del Rio D, Zimetti F, Caffarra P, Tassotti M, Bernini F, Brighenti F, Zini A, Zanotti I. The gut microbial metabolite trimethylamine-N-oxide is present in human cerebrospinal fluid. Nutrients. 2017;9:1053.
Blauwendraat C, Nalls MA, Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020;19:170–8.
Naudet N, Antier E, Gaillard D, Morignat E, Lakhdar L, Baron T, Bencsik A. Oral exposure to paraquat triggers earlier expression of phosphorylated α-synuclein in the enteric nervous system of A53T mutant human α-synuclein transgenic mice. J Neuropathol Exp Neurol. 2017;76:1046–57.
La Vitola P, Balducci C, Baroni M, Artioli L, Santamaria G, Castiglioni M, Cerovic M, Colombo L, Caldinelli L, Pollegioni L, Forloni G. Peripheral inflammation exacerbates α-synuclein toxicity and neuropathology in Parkinson’s models. Neuropathol Appl Neurobiol. 2021;47:43–60.
Kozina E, Sadasivan S, Jiao Y, Dou Y, Ma Z, Tan H, Kodali K, Shaw T, Peng J, Smeyne RJ. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. 2018;141:1753–69.
Martella G, Madeo G, Maltese M, Vanni V, Puglisi F, Ferraro E, Schirinzi T, Valente EM, Bonanni L, Shen J, et al. Exposure to low-dose rotenone precipitates synaptic plasticity alterations in PINK1 heterozygous knockout mice. Neurobiol Dis. 2016;91:21–36.
Matheoud D, Cannon T, Voisin A, Penttinen A-M, Ramet L, Fahmy AM, Ducrot C, Laplante A, Bourque M-J, Zhu L, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature. 2019;571:565–9.
Gorecki AM, Bakeberg MC, Theunissen F, Kenna JE, Hoes ME, Pfaff AL, Akkari PA, Dunlop SA, Kõks S, Mastaglia FL, Anderton RS. Single nucleotide polymorphisms associated with gut homeostasis influence risk and age-at-onset of Parkinson’s disease. Front Aging Neurosci. 2020;12:603849.
Lee HS, Lobbestael E, Vermeire S, Sabino J, Cleynen I. Inflammatory bowel disease and Parkinson’s disease: common pathophysiological links. Gut. 2020. https://doi.org/10.1136/gutjnl-2020-322429.
Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7.
Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600.
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet. 2008;40:955–62.
Liu Z, Lenardo MJ. The role of LRRK2 in inflammatory bowel disease. Cell Res. 2012;22:1092–4.
Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol. 2010;185:5577–85.
Alessi DR, Sammler E. LRRK2 kinase in Parkinson’s disease. Science. 2018;360:36–7.
Liu W, Liu X, Li Y, Zhao J, Liu Z, Hu Z, Wang Y, Yao Y, Miller AW, Su B, et al. LRRK2 promotes the activation of NLRC4 inflammasome during Salmonella Typhimurium infection. J Exp Med. 2017;214:3051–66.
Zhang Q, Pan Y, Yan R, Zeng B, Wang H, Zhang X, Li W, Wei H, Liu Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat Immunol. 2015;16:918–26.
Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. 2014;23:4201–14.
Thévenet J, PesciniGobert R, Hooft van Huijsduijnen R, Wiessner C, Sagot YJ. Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS ONE. 2011;6:e21519.
Takagawa T, Kitani A, Fuss I, Levine B, Brant SR, Peter I, Tajima M, Nakamura S, Strober W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci Transl Med. 2018;10:eaan8162.
Lin CH, Lin HY, Ho EP, Ke YC, Cheng MF, Shiue CY, Wu CH, Liao PH, Hsu AY, Chu LA, et al. Mild chronic colitis triggers Parkinsonism in LRRK2 mutant mice through activating TNF-alpha pathway. Mov Disord. 2022;37:745–57.
Dzamko N, Rowe DB, Halliday GM. Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord. 2016;31:889–97.
Qin XY, Zhang SP, Cao C, Loh YP, Cheng Y. Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: a systematic review and meta-analysis. JAMA Neurol. 2016;73:1316–24.
Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, Podolsky DK. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol (Baltimore, Md: 1950). 2010;185:5577–85.
Liao P-H, Chiang H-L, Shun C-T, Hang J-F, Chiu H-M, Wu M-S, Lin C-H. Colonic Leucine-Rich Repeat Kinase 2 expression is increased and associated with disease severity in patients with Parkinson’s disease. Front Aging Neurosci. 2022;13:819373.
Liao PH, Chiang HL, Shun CT, Hang JF, Chiu HM, Wu MS, Lin CH. Colonic Leucine-Rich Repeat Kinase 2 expression is increased and associated with disease severity in patients with Parkinson’s disease. Front Aging Neurosci. 2021;13:819373.
Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med. 2017;23:1–13.
Rota L, Pellegrini C, Benvenuti L, Antonioli L, Fornai M, Blandizzi C, Cattaneo A, Colla E. Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl Neurodegener. 2019;8:5.
Chung SJ, König IR, Lohmann K, Hinrichs F, Kim J, Ryu HS, Lee HJ, Kim K, Lee JH, Jung KW, et al. Association of SNCA variants with α-synuclein of gastric and colonic mucosa in Parkinson’s disease. Parkinsonism Relat Disord. 2019;61:151–5.
Wallen ZD, Stone WJ, Factor SA, Molho E, Zabetian CP, Standaert DG, Payami H. Exploring human-genome gut-microbiome interaction in Parkinson’s disease. NPJ Parkinson’s Dis. 2021;7:74.
McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7:531–2.
Oliveras-Salvá M, Van Rompuy AS, Heeman B, Van den Haute C, Baekelandt V. Loss-of-function rodent models for parkin and PINK1. J Parkinsons Dis. 2011;1:229–51.
Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, Fazel A, Bergeron JJ, Trudeau L-E, Burelle Y, et al. Parkinson’s disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–27.
Singh Y, Trautwein C, Dhariwal A, Salker MS, Alauddin M, Zizmare L, Pelzl L, Feger M, Admard J, Casadei N, et al. DJ-1 (Park7) affects the gut microbiome, metabolites and the development of innate lymphoid cells (ILCs). Sci Rep. 2020;10:16131.
Johansen KK, Wang L, Aasly JO, White LR, Matson WR, Henchcliffe C, Beal MF, Bogdanov M. Metabolomic profiling in LRRK2-related Parkinson’s disease. PLoS ONE. 2009;4:e7551.
Amorim Neto DP, Bosque BP, Pereira de Godoy JV, Rodrigues PV, Meneses DD, Tostes K, Costa Tonoli CC, Faustino de Carvalho H, González-Billault C, de Castro Fonseca M. Akkermansia muciniphila induces mitochondrial calcium overload and a-synuclein aggregation in an enteroendocrine cell line. iScience. 2022;25:103908.
Toczylowska B, Zieminska E, Michałowska M, Chalimoniuk M, Fiszer U. Changes in the metabolic profiles of the serum and putamen in Parkinson’s disease patients—in vitro and in vivo NMR spectroscopy studies. Brain Res. 2020;1748:147118.
Zhao H, Wang C, Zhao N, Li W, Yang Z, Liu X, Le W, Zhang X. Potential biomarkers of Parkinson’s disease revealed by plasma metabolic profiling. J Chromatogr B Analyt Technol Biomed Life Sci. 2018;1081–1082:101–8.
Acknowledgements
The authors are grateful to the Second Core Lab, Department of Medical Research, National Taiwan University Hospital, for technical support during the study.
Funding
This work was supported by Grants from National Health Research Institute (NHRI-EX111-11136NI), Ministry of Science and Technology (MOST 109-2320-B-002-073 –), and National Taiwan University (NTU-110-A-CC-5400-64841).
Author information
Authors and Affiliations
Contributions
SJC and CHL conceived and designed the study. SJC acquired the data. SJC and CHL analyzed and interpreted the data. SJC drafted the manuscript. CHL performed critical revision of the manuscript for important intellectual content. SJC and CHL obtained funding. CHL supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was reviewed by the Institutional Research Board Committee at National Taiwan University Hospital and carried out in accordance with relevant regulations.
Consent for publication
Not applicable.
Competing interests
All authors report no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, SJ., Lin, CH. Gut microenvironmental changes as a potential trigger in Parkinson’s disease through the gut–brain axis. J Biomed Sci 29, 54 (2022). https://doi.org/10.1186/s12929-022-00839-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-022-00839-6