
Abstract
The influenza A virus was isolated for the first time in 1931, and the first attempts to develop a vaccine against the virus began soon afterwards. In addition to causing seasonal epidemics, influenza viruses can cause pandemics at random intervals, which are very hard to predict. Vaccination is the most effective way of preventing the spread of influenza infection. However, seasonal vaccination is ineffective against pandemic influenza viruses because of antigenic differences, and it takes approximately six months from isolation of a new virus to develop an effective vaccine. One of the possible ways to fight the emergence of pandemics may be by using a new type of vaccine, with a long and broad spectrum of action. The extracellular domain of the M2 protein (M2e) of influenza A virus is a conservative region, and an attractive target for a universal influenza vaccine. This review gives a historical overview of the study of M2 protein, and summarizes the latest developments in the preparation of M2e-based universal influenza vaccines.
Similar content being viewed by others

Introduction
The extremely high variability in the antigenic properties of influenza virus is related to the structure of its genome that allows reassortment and the structural flexibility of the viral glycoproteins, which tolerate amino acid residue substitutions in the major antigenic sites without loss of function. Often immunity raised against previously circulating variants is not capable of protecting against newly emerging drift variants. In addition, completely new antigenic variants of the influenza virus are occasionally introduced into the human population; since the population is immunologically naive to them, these viruses spread easily and can cause pandemics. These features of influenza infection explain why there is interest around the world in developing a universal influenza vaccine, which could induce a cross-reactive immune response to the most conservative parts of the viral proteins.
Classic influenza vaccines commonly induce antibodies to the viral surface antigens, hemagglutinin (HA) and neuraminidase (NA), mainly to their immunodominant hypervariable regions. Constant antigenic drift allows the virus easily to escape the action of these antibodies, reducing the effectiveness of vaccination and leading to a need for annual updating of vaccine strains in the seasonal influenza vaccines [95, 129]. The performance of seasonal influenza vaccines can be improved by increasing the speed of vaccine production and using new adjuvants and new vaccination strategies. However, these vaccines will not be able to protect against newly emerging pandemic influenza viruses because of significant antigenic differences. Potentially pandemic viruses have been identified and many vaccines, based on different approaches and platforms, have been developed against them [133, 134]. However, these vaccines also have narrow specificity and may not have cross-reactivity, even within a single subtype. There has, therefore, been significant interest in the development of new vaccines that would have a longer and wider spectrum of action [9], and which would provide long-term protection not only against drifted variants of influenza A viruses of one subtype but also against viruses of other subtypes. One approach to expanding the spectrum of the protective action of influenza vaccines is to enhance the induction of cross-reacting immune response factors that target highly conserved antigens in influenza viruses of various subtypes [61, 62]. The influenza virion contains multiple conservative domains that, because of their functional significance, are rather weak immunogens; classical approaches to immunization are not able to induce a strong immune response to these sites.
The broadly protective vaccines currently being developed can be divided into two groups.
-
(1)
Vaccines that induce antibodies to structurally conserved regions of viral proteins, such as the stalk domain of the hemagglutinin which is important for the penetration of the influenza virus into the cell [63]; broadly immunogenic epitopes located at the contact surface between HA head domains [5, 130]; the enzymatic site of NA, where the surface cell sialic acids are cut [23]; and the ectodomain of the M2 protein (reviewed in this manuscript). Induced antibodies can both have a neutralizing function and participate in the process of antibody-dependent cellular cytotoxicity (ADCC) or phagocytosis (ADCP), thereby accelerating the elimination of the virus from an infected organism [126].
-
(2)
Vaccines that induce a cross-reactive T-cell immune response to the conserved epitopes of the virion’s internal proteins, such as nucleoprotein (NP) and M1 [27]. This is supported by a strong correlation of CD8+ T cells specific to conserved viral epitopes with cross-protection against symptomatic influenza in the absence of cross-reactive neutralizing antibodies [111].
Historical aspects of studying M2 protein
Most attempts to create a universal influenza vaccine have been based on the M2e epitopes. The first evidence of the existence of M2 protein was published in 1981 [65]. Several groups had already shown that the influenza A virus has a segmented genome consisting of 8 elements [84, 90, 98]. It was also known that the 8th mRNA encodes two proteins, NS1 and NS2 [66], and that the nucleotide sequence of the 7th mRNA contains two open reading frames [135], which were thought to encode the already discovered M1 protein and a previously unknown protein consisting of 97 amino acid residues [67].
Further study using viral lysate labelling with [35S] methionine and subsequent separation in a polyacrylamide gel detected a new protein with greater electrophoretic mobility than the already known influenza A protein. Isolation of different mRNA segments followed by peptide synthesis in vitro produced a new peptide of similar molecular weight; it was concluded that the 7th segment of the mRNA of the influenza A genome and the new M2 protein were genetically related. RNA-RNA hybridization established that the 7th segment encodes M1 and M2 proteins [65].
At first, M2 was detected only in infected cells [65], and not in the influenza virions themselves, which made it possible to put forward an assumption about the specific location of this protein. On the basis of the amino acid sequence of M2 [67], a hydropathy graph was constructed indicating the presence of a hydrophobic domain. Further study of the protein led to the conclusion that the C-terminus is located in the cytoplasmic space, while the N-terminal part of M2 is exposed at the viral surface [69].
The first monoclonal antibody to M2 (14C2) was obtained in 1988 as a result of the immunization of BALB/c mice with purified M2 protein along with Freund’s adjuvant. A subsequent study of 14C2 showed that the antibody binds to the extracellular N-terminal region of M2 (M2e) [140]. Currently, there is evidence that the binding site is a fragment of M2e from the 6th to the 15th residue [128]. In 1998, it was shown that the 14C2 antibody is not able to inhibit the adsorption and penetration of influenza virus into cells, but is able to limit the growth of influenza A virus in vitro, though this restriction is highly strain dependent [140]. Another M2e-specific monoclonal antibody was obtained in 1996. To this end, splenocytes from BALB/c mice immunized with the keyhole limpet hemocyanin-linked M2 peptide SLLTEVETPIRNEWGCRCND were fused with F23.1 hybridoma cells producing an immunoglobulin G2a (IgG2a) monoclonal antibody. The authors selected a monoclonal antibody (3F12), which not only had cross-reactivity to different influenza A strains, but could also bind to the T-cell receptor. It was also shown that such a bispecific antibody is able to limit the growth of influenza A virus in vitro (with less efficiency than 14C2) and redirect activated T-cells to eliminate infected cells [30].
In 2003, another study was published in which rabbits were immunized with 4 peptides from the N-terminal section of M2, with overlapping areas: а.a. 2–24, а.a. 2–12, а.a. 8–18 and а.a. 13–24. Further study showed that they all produced antibodies capable of recognizing each of the four peptides. It has also been shown that antibodies recognizing the N-terminus of M2 (a.a 2–12) have virus-neutralizing activity against both influenza A and B in vitro [77]. The authors suggested that this part of M2 may contain one epitope that can induce antibodies with inhibitory activities against both viruses, but there is no additional evidence that could confirm that anti-M2e immunity can suppress influenza B virus replication. Later, the same group of scientists reported that a small area of M2 EVETPIRN (a.a. 6–13) can produce antibodies 8C6 and 1B12, but their protective effect has not yet been sufficiently studied [78].
One of the potential therapeutic antibodies is TCN-032, which has passed phase I and II clinical trials. TCN-032 recognizes the M2e2–6 epitope [37] and may provide immediate immunity against influenza A infection [99]. Another potential therapeutic antibody is anti-M2e antibody Z3G1, which has been shown to protect mice applied therapeutically at different time points after infection [128].
Mutational analysis, crystallography analysis of the structure and nuclear magnetic resonance have established that M2 is a transmembrane protein, consisting of four α-helices. Each helix has two conservative residues (His37 and Trp41), which play an important role in the life cycle of influenza virus [115]. Highly conserved and oxidized cysteine residues at positions 17 and 19 are responsible for the stabilization of the structure [44].
The entire M2 protein can be divided into 3 parts: the N-terminal or extracellular domain (23 a.a., excluding the 1st methionine); the hydrophobic transmembrane domain (19 a.a.); and the C-terminal domain (54 a.a.). The extracellular domain of M2 (M2e) consists of 23 amino acid residues and is a highly conserved region in all influenza A viruses. One of the reasons for the low variability of M2e is its inability to induce an immune response in an infected organism [11], so there is no selection pressure on the site. A more important reason is believed to be the genetic relationship between M2e and M1: a.a. 1–9 of M2e and M1 are encoded by the same nucleotides in the same reading frame. Amino acids 10–23 of M2e and a.a. 239–252 of M1 are also encoded by the same RNA sequence but are translated in different reading frames [51]. M1, in turn, is a highly conserved matrix protein with a countable number of currently known mutations [31].
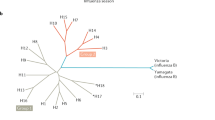
The whole of M2 is highly conserved, but its extracellular part is of the greatest interest as a potential antigen. We analyzed 27,253 human, 15,367 avian and 5379 swine individual sequences of influenza A virus M gene (obtained from the Influenza Research Database (fludb.org)) and generated a phylogenetic tree using MAFFT [55]. The tree was visualized using the Interactive Tree Of Life (iTOL) server [76]. As shown on Fig. 1a, the M2e sequences of influenza A viruses have evolutionary diverged into several lineages mainly related to host species. The generated phylogenetic tree basically repeats the published analysis of Furuse et al. [33], but in contrast to this study published before the 2009 pandemic, our analysis included a large number of sequences of H1N1pdm09 virus M gene, which is known to originate from the Eurasian avian-like swine H1N1 lineage [109]. We selected six different lineages of M2e protein based on their phylogenetic relatedness and obtained consensus sequences for each lineage using UGENE Multiple Alignment software (Fig. 1b). As mentioned above, the first nine amino acids have the lowest variability for each analyzed group. The remaining residues have different percentage mutations for each origin; however, the residues Arg12, Trp15, Cys17, Cys19, Ser22 and Asp24 have the lowest frequency of mutations, which may reflect their functional significance. The only exception is the residue at 19th position in the swine influenza viruses that fall into lineage 2, in which Tyr dominated in this position (Fig. 1b). The slight difference between the consensus sequences is also significant for the choice of the appropriate sequence for a universal vaccine. For example, immunization of pigs with human M2e using different carriers did not protect the animals from the lethal swine subtype of influenza A virus [41]. It has also been shown that monoclonal antibody 8C6, which protects mice against lethal challenge virus [78], is able to recognize the consensus part of human M2e “EVETPIRN” sequence (а.a. 6–13), but not the consensus fragment of avian M2e “EVETPTRN” (a.a. 6–13). Thus, it is possible that an amino acid substitution at the 11th position in avian M2e may allow avian influenza A virus to escape the immune response in humans [79]. Based on our analysis of all available M2e sequences, in order to develop a M2e-based universal influenza vaccine we propose using four different consensus M2e protein sequences that will cover most human, avian and swine influenza A viruses (Fig. 1c). In contrast to previously published studies, we suggest using two different M2e sequences for human influenza viruses due to the significant variations between the human viruses isolated before the 2009 pandemic and the H1N1pdm09 viruses.

Analysis of M2e sequences of human, swine and avian influenza viruses. a Phylogenetic tree was generated from 27,253 human, 15,367 avian and 5379 swine individual sequences of influenza A virus M gene (obtained from the Influenza Research Database (fludb.org)) using MAFFT. The tree was visualized using the Interactive Tree Of Life (iTOL) server. b Prevalence of amino acid residues in M2e protein of selected M2e lineages. The consensus sequences for each lineage were obtained using UGENE Multiple Alignment software. Residues with a frequency below 3% are indicated with (*). c Proposed consensus M2e sequences for the development of a universal influenza vaccine
The structure of free M2e is currently unknown. However, a number of studies have obtained crystallographic data on the structures of “M2e + antibody”. Using the example of MAb65, MAb148, Fab148 and Fab65 antibodies, it has been shown that M2e can take at least two different conformations to increase its affinity. For example, interaction with Fab148 and MAb148 occurs through residues 2–5, which play an important role in the interaction and form a β-turn. For interaction with MAb65, not only do residues 5–8 form a β-turn, but also the whole M2e takes a U-shaped formation with a central Trp15 [15]. For the formation of a stable complex with the Fab65 antibody, the importance of residues 10–11 in M2e has been shown, while residues 6, 8 and 14 are responsible for hydrophilic interactions with MAb65 [16].
Study of the structural features of the transmembrane domain of M2 (a.a. 18–60) and nearby areas at pH 7.5 revealed the presence of a nonstructural N-terminal region (a.a. 18–23), a channel-forming transmembrane helix (a.a. 25–46), a short flexible loop (a.a. 47–50) and a C-terminal aliphatic helix (a.a. 51–59) [104].
The 7th segment of the influenza A virus genome can be subjected to alternative splicing with the formation of M2 mRNA and mRNA3; the formation of M4 mRNA is also possible in some strains [100]. This process can proceed independently of other influenza A virus proteins [68] using only the host cell machinery. However, a number of papers have reported the control of M1 mRNA splicing using other influenza A virus proteins [10, 100, 106, 107]. Currently, it is known that NS1 provides the main regulatory function of the amount of M2 in infected cells [10, 100], rather than polymerase proteins, as was previously thought [106, 107].
Functions of M2 protein
Currently, several biological functions of M2 protein are known. The first and most important is as a proton-selective channel or so-called viroporin [97], which is formed by four fragments of H37xxxW41 [4, 118, 125]. Shortly after the influenza virus enters a cell as part of the endosome [42, 75, 132], M2 goes into an activated state triggered by the acidic environment inside the endosomes. Using the M2 protein, H+ ions are transferred through the viral membrane into the virion (proton transfer occurs through the formation of the protonated state of His37 [35, 125] and rotation of the indole of Trp41 [118]). The influx of protons also leads to an influx of potassium and sodium ions (K+ and Na+), which in turn lead to a change in the M1 conformation, and the viral RNA goes into a relaxed state [75, 112]. The low endosomal pH leads to conformational changes in the HA molecule, which in turn triggers the fusion of the virion membrane with the endosomal membrane. Further, the interaction between M1 and the ribonucleoprotein complex is weakened, which leads to the release of viral RNA into the cytosol [14]. It has also been shown that the ionic imbalance in the cell, resulting from the operation of the M2 protein, is a signal for the formation of inflammatory activity in myeloid cells such as macrophages and dendritic cells [49].
Another function of M2 is participation in the formation and budding of new influenza A virions. The study of this feature began with the discovery that viral replication is inhibited by antibodies that recognize the extracellular domain of M2, despite the small amount of M2 on the surface of influenza A virus [83]. Using reverse genetics and mutational analysis, it has been shown that truncated forms of M2, or forms with mutations in the cytoplasmic tail, further reduce the level of free M1 in the cellular environment, which is required for the assembly and budding of new virions [12]. It has also been shown that M2 protein mediates membrane budding and scission processes via a pathway that is independent of ESCRT (endosomal sorting complexes required for transport), unlike many other enveloped viruses [101]. The major role in M2-mediated budding and scission processes is played by the amphipathic region (residues 45–62), which interacts with various host cell proteins involved in remodeling the cell membrane (reviewed in [81]).
Furthermore, the M2 of influenza A virus is a mediator of macroautophagy inhibition at the stage of fusion of autophagosomes and lysosomes, which causes a subsequent increase in cell death as a result of influenza infection and an increase in viral antigen yield [7, 34].
The functional ortholog of influenza A virus M2 is M2 from influenza B virus (BM2), which also carries electrons through the membrane [86], interacts with the influenza B matrix 1 protein [127], crucial for virus assembly [50], and has an HxxxW motif in its transmembrane part [93]. However, the sequences of M2 and BM2 have significant differences. For example, the extracellular part of BM2 consists of only seven amino acid residues [102] (the consensus for human BM2e according to analysis of 9011 sequences from the Influenza Research Database is “MLEPFQI”). Because of its small size, BM2e is most likely not able to establish a protective immune responses [102].
A functional analogue of M2 is the M42 protein, which is obtained by splicing the 7th segment of the influenza A virus genome in some influenza strains [136].
Despite the importance of the M2 functions, it is not an essential protein for influenza A virus. In laboratory practice, so-called “M2 null” viruses have been obtained by various modifications of stop codons [13, 48, 131]. All of these viruses were viable but had a reduced replication level [13, 48, 116, 131, 136, 137]. Moreover, an M2-deficient single replication (M2SR) virus has been generated and evaluated as a possible candidate for live influenza vaccine. The M2SR vaccine was able to infect cells and express all viral proteins except M2, without generating progeny virus [103]. This vaccine platform proved efficient in protecting animals (mice and ferrets) against heterologous/heterosubtypic influenza virus infections [39, 40], and is now undergoing phase I clinical trials (clinicaltrials.gov NCT03553940; NCT02822105).
Protection mechanisms of M2e-based vaccines
Despite the small size of M2e, many questions about this protein remain unanswered. For example, the defense mechanism of M2e-containing vaccines has not been fully clarified [71]. Since anti-M2e antibodies were discovered [140], it has been believed that they are the main mechanism of the protective action that has been repeatedly shown in various animal models [19, 22, 59]. Importantly, several groups have noted that M2e-based influenza vaccines induced a long-lasting M2e-specific antibody response [56, 105, 110]. The presence of antibody 14C2 reduces the expression level of viral protein M2 [45], thereby indirectly affecting the formation of new viral particles. However, it has been shown that influenza viruses that have mutations in the C-terminal region of M2 and the N-terminal region of M1 (even one amino acid substitution could be sufficient, for example, Pro10His, Val31Ile or Ala41Val) are resistant to the action of antibody 14C2 [139, 142].
Although the anti-M2e antibody are not neutralizing [28, 32, 89], their significant protective effect was demonstrated in multiple experiments, including studies on passive transfer of M2e immune serum or anti-M2e monoclonal antibodies [28, 60, 88, 121]. In addition, Eliasson et al. [26] showed that B cell-deficient mice are very poorly protected by a mucosal M2e-based vaccine even though these mice mount a considerable CD4 T cell response against M2e. In 2011, El Bakkouri et al. [24] demonstrated the crucial role of Fc gamma receptors (FcγR) in the in vivo protection afforded by M2e-specific IgG isotypes. In this study, wild-type and FcR γ−/− BALB/c mice were passively immunized with anti-M2e immune serum, followed by lethal challenge with a mouse-adapted virus. Despite similar distribution of anti-M2e IgG titers and antibody isotype in both mouse strains, the FcR γ−/− mice were significantly less protected than wild-type animals. Further experiments demonstrated that the activating receptor FcγRIII associated with the common γ-chain is required for anti-M2e IgG1 isotype-mediated in vivo immune protection [24]. Similar results were yielded in study by Lee et al. [72]: although the wild-type mice and FcR γ−/− genotype mice had similar levels of antibodies (IgG1 and IgG2a) after immunization with M2e5x virus-like particles (VLP), the vaccine was significantly less protective in mice without an FcR γ-chain than in wild-type mice. Van den Hoecke et al. [124] showed that protection against influenza virus with different IgG antibody subclasses requires different FcR subtypes: protection with IgG1 requires FcγRIII, while IgG2a requires all three activating FcγRs.
It is known that M2 protein is expressed abundantly on the surface of infected cells, whereas only a few molecules are incorporated into the virion [140], therefore anti-M2e IgG antibody provide protection by interacting with virus-infected cells and triggering immune effector cell activation through its Fc region, resulting in killing and/or phagocytosis of the infected cells [24]. Many studies attempted to identify the effector cells which are responsible for elimination of M2-expressing cells in the presence of anti-M2e IgGs. Controversial results were yielded with respect to the formation of natural killer (NK)-mediated ADCC in response to immunization with M2e-containing vaccines. A number of studies have shown that the ADCC mechanism has an essential role of [53, 108, 128], whereas experiments with peptide-specific monoclonal antibodies have refuted this [32]. These studies [32, 53, 128] looked at the effect of NK-mediated ADCC during passive immunization, which introduces additional difficulties in identifying the true mechanism of protection. Using a conditional cell depletion protocol, El Bakkouri et al. [24] demonstrated that alveolar macrophages (AM) play a critical role in the protection mediated by anti-M2e IgG antibody. These results are of particular interest because AM are resident in the lung and are considered one the first immune cells to interfere with respiratory pathogens in the airways. The contribution of these components of the immune system alone is not sufficient for a complete defense, but it can influence innate immunity and the first stages of the adaptive immune response [110]. The activation of the complement system as a result of viral elimination after immunization with M2e-containing vaccines also raises questions [71]. The complement system can bind to influenza virions in the presence of virus-specific antibodies [8, 43]. A study by Wang et al. [128] showed the importance of the C3 complement system in reducing viral titer in the lungs of mice after challenge, although Jegerlehner et al. [53] found that the system does not play a significant role in protection.
Various studies have also demonstrated the importance of the M2e-specific CD4+ and CD8+ responses [25, 26, 57, 72, 119, 138]. CD8 T cells are known to kill target cells via perforin and FasL-mediated cytotoxicity pathways, thereby providing viral clearance [120], while CD4 cells influence the production of IFN-γ, which is involved in reducing viral titer [6]. Experiments on T-cell depletion showed the joint importance of CD4+ and CD8+ T cells in heterosubtypic cross-protection [57, 119], however different studies yielded discrepant results in M2e-specific T-cell responses, most probably due to the differences in immunization protocols, vaccine platforms and adjuvants, strains of animals, and routes of immunization [71]. A study by Eliasson et al. [26] demonstrated a critical role of M2e-specific lung-resident memory CD4, but not CD8, T cells induced by mucosal CTA1-3M2e-DD vaccine in protecting mice against a lethal influenza virus infection. Importantly, these CD4 T-cell responses were long-lasting, protecting animals even 12 months after vaccination [26]. In the study by Lee et al., the M2e5x VLP-immunized FcR γ−/− mice recovered more quickly after viral exposure than naïve FcR γ−/− mice, most probably due to the prevalence of IFN-γ-producing CD4 and CD8 T cells in the lungs of immunized FcR γ−/− mice [72].
In summary, no complete defense mechanism valid for all M2e-containing vaccines has been found so far. All of the contradictory data mentioned can be explained by unaccounted mechanisms in the formation of protection or by the additional influence of the M2e carrier (studies with contradictory results used different types of carriers). Another factor may be the lack of a unified methodology. However, all researchers agree that the response to M2e-containing vaccines involves many key parts of the immune system, which can have a significant protective effect.
Possible modifications of M2e
Most studies aimed at preparing a universal M2e-based influenza vaccine have used the whole M2 ectodomain (a.a. 2–24). However, truncated forms of M2e (Table 1) can be used to induce an immune response to important areas of M2e, reduce the cost of vaccine production and allow the use of new vectors that have a small capacity. For example, there are a number of antibodies (14C2, 3F12, 8C6, 1B12, Z3G1) [30, 78, 140] that recognize only a fraction of the total M2e, and that find wide application in practice.
Induction of all kinds of anti-M2e antibodies is required for maximum protection of M2e-containing vaccines. However, these antibodies have different protective characteristics, and an optimum universal influenza vaccine may use only some of the M2e sites that induce the most effective antibodies (Table 1).
Fu et al. [32] compared different anti-M2e antibodies, and showed that the protective properties of L18 antibody (which recognizes the M2e 4–13 site) were superior to those of O19 (which recognizes the M2e 2–9 site), while S1 (recognizes the M2e 7–13 site) did not protect mice against lethal influenza A virus challenge. Data from other groups indicate that the M2e 4–13 region contains a B-cell epitope and a sequence recognized by the 14C2 antibody (M2e 6–13) [79], as well as an additional potential epitope (M2e 8–12) [94].
It is worth noting that, when shortened forms of the ectodomain M2 are used, and with regard to various defense mechanisms, the carrier of M2e epitopes plays an important role in immunogenicity. For example, immunization with a shortened form of M2e (2–16 amino acid residues) along with Freund’s adjuvant did not lead to the formation of a high level of anti-M2e antibodies [94], unlike when phage f88 was used as a carrier of M2e antigen [20]. It was concluded that a shortened form of M2e will produce a high level of anti-M2e antibodies only with a support system capable of inducing a T-cell response. This study also revealed the presence in M2e (17–24 a.a.) of amino acids important for the formation of a T-cell response.
Development of M2e-based vaccines
The inhibition of replication of some influenza A strains by 14C2 antibody [140] merited further study since the extracellular part of the M2 protein is very conservative compared with HA and NA, and the cross-protective effect can be directed precisely at M2e. Study of the protective properties of 14C2 in BALB/c mice showed that it significantly reduced the replication of influenza A (but not influenza B) virus in lung tissue. This prompted the suggestion that the higher immunity of adults who have previously had influenza A infection compared with children is provided by the presence of M2e antibodies [121]. However, subsequent studies have shown that M2e itself is a weak immunogen and that only about half the people infected with influenza A virus are able to produce anti-M2e antibodies [143]. There is no reliable evidence that such a low concentration of antibodies plays any protective role [11, 143]. It has also been suggested that the introduction of M2e epitopes into the influenza vaccine will have a priming effect on the production of antibodies before subsequent influenza A virus infection [143].
Despite numerous reports of the low immunogenicity of M2e because of its small size, small number of copies in the virion, and the possible shielding effect of larger surface proteins of influenza A virus [29, 47, 140], it has been shown that immunization with free synthetic M2e peptide together with an adjuvant can produce high titers of anti-M2e antibodies that protect mice from lethal influenza challenge [138]. However, most attempts to create a universal M2e-based influenza vaccine have used different carriers. One of the first studies used a recombinant baculovirus containing M2 from influenza virus A/Ann Arbor/6/60 (Bac-AM2), to infect the Spodoptera frugipedra (Sf9) cell line. 14C2 antibody detection revealed the presence of M2 on the surface of infected cells. This allowed the use of Sf9 lysates infected with Bac-AM2 as a source of antigen to study the antibody response to M2 in people previously infected with influenza A virus [11].
One of the first prototypes of a universal influenza vaccine used hepatitis B core protein combined with the M2e peptide (M2HBc) as a carrier. In this design, the natural position of the N-terminal region of M2, located in the extracellular space, was simulated. Immunization of BALB/c mice with M2HBc resulted in a high level of protection against the lethal dose of influenza A virus, and led to the formation of anti-M2e antibodies, which were shown to be effective in passive immunization experiments [88]. Later various liposomal carriers [1], tobacco mosaic virus surface protein [96], and rotavirus NSP4 [3] were used to induce anti-M2e antibodies. Another carrier used was the GCN4 protein, which is a eukaryotic transcriptional protein activator. The use of M2e-tGCN4 resulted in significant production of M2e-specific antibodies, which protected vaccinated mice from a lethal dose of mouse-adapted influenza virus [17].
The most effective and cost-effective method is the creation of recombinant virus-like particles (VLPs), on the surface of which M2e would be represented [19]. Because M2 is a homotetramer consisting of two subunits linked by a disulfide bond, held together by covalent interactions [88], it has been suggested that M2e could also be used as a tetramer in the creation of recombinant constructs. In this approach, M2e will form a compactly folded protein, thereby ensuring the correct geometry of the virus particles. In addition, there are confirmations of the creation of VLPs consisting of five M2e tandem repeats (M2e5x). The intramuscular administration of M2e5x protected mice from influenza A viruses of different serotypes [56, 58, 74].
There are data on the highly conservative nature of Cys17 and Cys19 residues, although, in many studies, these residues were replaced by Ser17 and Ser19, respectively, in order to avoid protein aggregation due to the formation of disulfide bonds between M2e sites. Such substitutions are widely used because it has been shown that Cys17 and Cys19 residues do not affect the expression of M2e [44], and the double substitution does not affect the immunogenic properties of M2e epitopes [1].
It is interesting to note that in the vast majority of successful M2e vaccine studies mouse models were used, while only a few studies were conducted on other animals (pigs, ferrets, monkeys, dogs), where protection against challenge, if studied, was modest at best. One possible explanation for such diverse results is that mice of various inbred (including knockout and knock-in) strains are readily available at relatively low cost, whereas larger animals are more expensive and genetically outbred, resulting in significant host response variability. Even ferrets within commercial populations have distinct patterns of T-cell reactivity as a result of the heterogeneity at the MHC locus [21]. Furthermore, primary structure, cellular specificity and binding properties of Fc receptors can vary considerably among different mammalian species [54]. For example, a recent study found that pig Fc receptors do not bind human IgG1, which limits the use of this animal model to study human broadly protective monoclonal antibodies [85].
In summary, there are many ways to increase the immunogenicity of M2e. The most significant findings from preclinical studies of these universal vaccine prototypes are shown in Table 2. However, their further use in clinical practice is limited by the lack of knowledge about the safety of the vectors for humans. Currently, the search for new carriers continues, in order to increase the immunogenicity of M2e, while new immunization strategies are also being sought, such as priming with live attenuated influenza virus, and boosting with M2e VLP [73].
Clinical studies of M2e-based influenza vaccines
M2e-based vaccines are not available commercially, but some are currently undergoing clinical trials (Table 3). For example, the ACAM-FLU-A vaccine, an M2e-HBc fusion protein, based on an idea by Neirynck [88], has already passed the first phase of double-blind placebo-controlled clinical trial (NCT00819013, Sanofi). Intramuscular administration of ACAM-FLU-A promoted the formation of anti-M2e antibodies in blood sera in 90% of cases and was well tolerated when given alone or with aluminum hydroxide or QS-21 Stimulon adjuvant (www.biocentury.com/bc-week-review/clinical-results/2008-01-07/acam-flu-phase-i-data). However, there has been no further progress with this vaccine since then, probably because of the rapid decline in M2e-specific antibody over time [ clinicaltrials.gov NCT00819013]. A similar recombinant protein HBc-based prototype universal influenza vaccine, “Uniflu”, is currently being evaluated in a single-site, randomized, double-blind, placebo-controlled study in Saint Petersburg, Russia [ clinicaltrials.gov NCT03789539]. The vaccine contains four copies of human M2e fused within the immunodominant loop of the HBc antigen [122]. This study involves 54 healthy adult subjects 18–60 years, and the vaccine is administered intramuscularly in two different doses (20 and 40 μg) twice at a 3-week interval. The results of the trial are anticipated in the near future.
Another influenza vaccine based on the use of flagellin to increase the immunogenicity of M2e [46] was shown to be safe in a phase I clinical trial after intramuscular administration in doses of 0.3 and 1.0 μg. However, administration in doses of 3.0 μg or 10.0 μg was accompanied by undesirable symptoms (fever, diarrhea, fatigue, headache and muscle pain), although this vaccine demonstrated high immunogenicity (NCT00921206, VaxInnate) [123]. The phase II trial of this vaccine ended in 2011 (NCT00921947, VaxInnate), but the test results are not yet available.
One more vaccine, containing M2 peptides and various conservative CTL epitopes [114], also passed the first phase of clinical trials (NCT01181336, Imutex Limited). However, this vaccine stimulates cellular immunity and is HLA-specific, which may be a reason for its narrow focus. Also, the cellular immune response is slower than the humoral response, and in the event of a pandemic, it would not effectively prevent infection [59].
Clinical trials to study the effect of the presence of additional M2e antigens in the trivalent vaccine have shown that, in the case of a weak response to the main vaccine, the M2e component can significantly enhance the immune response [117]. Thus, additional M2e epitopes will be able to have a “safety net” function when such vaccines are introduced into wide practice.
The TCN-032 antibody passed phase I of clinical trials. Volunteers were given a single dose of TCN-032 intravenously (1, 3, 10, 20, or 40 mg/kg of body weight), and no severe events related to the study drug were observed. A phase II clinical trial showed that non-neutralizing antibody TCN-032 has a therapeutic effect, as parenteral administration provided immediate immunity and therapeutic benefit in influenza A infection [99].
Overall, many prototype universal influenza vaccines contain an increased number of M2e epitopes. The data suggest that there is potential to use such vaccines, but that they need to be improved, in order to overcome the existing shortcomings.
Conclusions
Vaccination is the safest and most effective way to prevent the spread of influenza. However, seasonal vaccination is ineffective against pandemic influenza viruses, as it is often antigenically different from the pandemic virus, and it takes an average of six months to prepare a new vaccine candidate for deployment. The development of a universal influenza vaccine, which would have a long and broad spectrum of action, is an urgent issue of practical public health.
Currently, new universal influenza vaccines are being developed using various strategies, but none of these vaccines has been licensed so far. The development of a new universal influenza vaccine will both avoid the need for annual updates to the composition of seasonal vaccines, and reduce the risk of public health disaster in the event of a new pandemic. However, it is not yet clear whether such vaccines will induce long-lasting protection. It is also unknown how the effects of influenza viruses and previous vaccinations will affect the performance of new vaccines [91].
Ideally, a universal influenza vaccine should induce both a humoral (antibodies) and a T-cell immune response to conservative epitopes of the influenza virus. M2e and HA stalk domain are the most widely used viral targets for a universal influenza vaccine design due to their conserved nature and the proven ability of their specific antibody to protect against heterologous viral infections. Many vaccination strategies have been explored to elicit potent antibody responses against these naturally weak immunogens. The vaccine-induced anti-HA stalk antibodies target either group 1 or group 2 HAs, with limited cross-reactivity between the two HA groups, suggesting that a cHA-based universal influenza vaccine has to include three components: a group 1 HA, a group 2 HA, and an influenza B stalk-based antigen [64, 82]. In contrast, a properly designed M2e-based vaccine should cover all influenza A virus subtypes circulating in both human and animal reservoirs. Since it is not possible to predict which virus will cause the next pandemic, we recommend that four different M2e consensus sequences be included in the new M2e-based vaccines to maximize cross-reactivity of the M2e-targeted antibody against all circulating influenza A viruses (Fig. 1). Licensing of a new M2e-based vaccine is challenging due to the lack of clearly defined correlates of protections of such vaccines for humans. In addition, the M2e-specific protection is of lower potency compared to HA-targeted neutralizing immunity, therefore the problem of proving non-inferiority over existing influenza vaccines remains to be solved. Nevertheless, the conserved M2 ectodomains can be used as a supplement to overcome strain specificity and improve long-term cross-protection of currently licensed seasonal influenza vaccines [57, 87].
It is worth mentioning that it is possible that a new antigenic drift, different from that observed in wild strains, will start to act on conservative epitopes, which will make it necessary to periodically update the composition of any universal vaccine. However, such an update will be required less frequently than is currently the case for seasonal vaccines, as there is evidence that changes affecting the conserved M2e region also require changes in other influenza A viral proteins because of the various functions of M2 in the virus life cycle.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- A.a.:
-
Amino acid
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- Bac-AM2:
-
Baculovirus containing M2 from influenza virus A/Ann Arbor/6/60
- BLS:
-
Brucella abortus lumazine synthase protein
- CTL:
-
Cytotoxic T lymphocyte
- ESCRT:
-
Endosomal sorting complexes required for transport
- FcR:
-
Fc receptor
- HA:
-
Hemagglutinin
- HBc:
-
Hepatitis B core protein
- IM:
-
Intramuscular immunization
- IN:
-
Intranasal immunization
- IP:
-
Intraperitoneal immunization
- KLH:
-
Keyhole limpet hemocyanin
- LLC:
-
Limited Liability Company
- M2e:
-
Extracellular domain of the M2 protein
- M2e5x:
-
Five M2e tandem repeats
- M2HBc:
-
Hepatitis B core protein combined with the M2e peptide
- M2SR:
-
M2-deficient single replication virus
- NA:
-
Neuraminidase
- NK:
-
Natural killer
- NP:
-
Nucleoprotein
- OMPC:
-
Outer membrane protein complex
- SC:
-
Subcutaneous immunization
- Sf9:
-
Spodoptera frugipedra
- VLP:
-
Virus-like particles
References
Adler-Moore JP, Ernst W, Kim H, Ward N, Chiang SM, Do T, Fujii G. Monomeric M2e antigen in VesiVax((R)) liposomes stimulates protection against type a strains of influenza comparable to liposomes with multimeric forms of M2e. J Liposome Res. 2017;27(3):210–20.
Alvarez P, Zylberman V, Ghersi G, Boado L, Palacios C, Goldbaum F, Mattion N. Tandem repeats of the extracellular domain of matrix 2 influenza protein exposed in Brucella lumazine synthase decameric carrier molecule induce protection in mice. Vaccine. 2013;31(5):806–12.
Andersson AM, Hakansson KO, Jensen BA, Christensen D, Andersen P, Thomsen AR, Christensen JP. Increased immunogenicity and protective efficacy of influenza M2e fused to a tetramerizing protein. PLoS One. 2012;7(10):1–11.
Balannik V, Carnevale V, Fiorin G, Levine BG, Lamb RA, Klein ML, Degrado WF, Pinto LH. Functional studies and modeling of pore-lining residue mutants of the influenza a virus M2 ion channel. Biochemistry. 2010;49(4):696–708.
Bangaru S, Lang S, Schotsaert M, Vanderven HA, Zhu X, Kose N, Bombardi R, Finn JA, Kent SJ, Gilchuk P, Gilchuk I, Turner HL, Garcia-Sastre A, Li S, Ward AB, Wilson IA, Crowe JE Jr. A Site of Vulnerability on the Influenza Virus Hemagglutinin Head Domain Trimer Interface. Cell. 2019;177(5):1136–1152 e1118.
Baumgarth N, Kelso A. In vivo blockade of gamma interferon affects the Influenza virus-induced Humoral and the local cellular immune response in lung tissue. J Virol. 1996;70(7):4411–8.
Beale R, Wise H, Stuart A, Ravenhill BJ, Digard P, Randow F. A LC3-interacting motif in the influenza a virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host Microbe. 2014;15(2):239–47.
Beebe DP, Schreiber RD, Cooper NR. Neutralization of Influenza virus by Normal human sera: mechanisms involving antibody and complement. J Immunol. 1983;130(3):1317–22.
Berlanda SF, Tsvetnitsky V, Donnelly JJ. Universal influenza vaccines: shifting to better vaccines. Vaccine. 2016;34(26):2926–33.
Bier K, York A, Fodor E. Cellular cap-binding proteins associate with influenza virus mRNAs. J Gen Virol. 2011;92(Pt 7):1627–34.
Black RA, Rota PA, Gorodkova N, Klenk HD, Kendal AP. Antibody response to the M2 protein of influenza a virus expressed in insect cells. J Gen Virol. 1993;74:143–6.
Chen BJ, Leser GP, Jackson D, Lamb RA. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J Virol. 2008;82(20):10059–70.
Cheung TK, Guan Y, Ng SS, Chen H, Wong CH, Peiris JS, Poon LL. Generation of recombinant influenza a virus without M2 ion-channel protein by introduction of a point mutation at the 5′ end of the viral intron. J Gen Virol. 2005;86(Pt 5):1447–54.
Chizhmakov IV, Geraghty FM, Ogden DC, Hayhurst A, Antoniou M, Hay AJ. Selective proton permeability and pH regulation of the influenza virus M2 channel expressed in mouse erythroleukaemia cells. J Physiol. 1996;494(2):329–36.
Cho KJ, Schepens B, Moonens K, Deng L, Fiers W, Remaut H, Saelens X. Crystal structure of the conserved amino terminus of the extracellular domain of matrix protein 2 of Influenza a virus gripped by an antibody. J Virol. 2016;90(1):611–5.
Cho KJ, Schepens B, Seok JH, Kim S, Roose K, Lee JH, Gallardo R, Van Hamme E, Schymkowitz J, Rousseau F, Fiers W, Saelens X, Kim KH. Structure of the extracellular domain of matrix protein 2 of influenza a virus in complex with a protective monoclonal antibody. J Virol. 2015;89(7):3700–11.
De Filette M, Martens W, Roose K, Deroo T, Vervalle F, Bentahir M, Vandekerckhove J, Fiers W, Saelens X. An Influenza a vaccine based on Tetrameric Ectodomain of matrix protein 2. J Biol Chem. 2008;283(17):11382–7.
De Filette M, Ysenbaert T, Roose K, Schotsaert M, Roels S, Goossens E, Schepens B, Fiers W, Saelens X. Antiserum against the conserved nine amino acid N-terminal peptide of influenza a virus matrix protein 2 is not immunoprotective. J Gen Virol. 2011;92(Pt 2):301–6.
Deng L., Cho K.J., Fiers W. and Saelens X. M2e-Based Universal Influenza A Vaccines. Vaccines 3(1):105–136, 2015.
Deng L, Ibanez LI, Van den Bossche V, Roose K, Youssef SA, de Bruin A, Fiers W, Saelens X. Protection against Influenza a virus challenge with M2e-displaying filamentous Escherichia coli phages. PLoS One. 2015;10(5):e0126650.
DiPiazza A, Richards K, Batarse F, Lockard L, Zeng H, Garcia-Sastre A, Albrecht RA, Sant AJ. Flow Cytometric and cytokine ELISpot approaches to characterize the cell-mediated immune response in ferrets following Influenza virus infection. J Virol. 2016;90(17):7991–8004.
Ebrahimi SM, Tebianian M. Influenza a viruses: why focusing on M2e-based universal vaccines. Virus Genes. 2011;42(1):1–8.
Eichelberger MC, Wan H. Influenza neuraminidase as a vaccine antigen. Curr Top Microbiol Immunol. 2015;386:275–99.
El Bakkouri K, Descamps F, De Filette M, Smet A, Festjens E, Birkett A, Van Rooijen N, Verbeek S, Fiers W, Saelens X. Universal vaccine based on ectodomain of matrix protein 2 of influenza a: fc receptors and alveolar macrophages mediate protection. J Immunol. 2011;186(2):1022–31.
Eliasson DG, El Bakkouri K, Schon K, Ramne A, Festjens E, Lowenadler B, Fiers W, Saelens X, Lycke N. CTA1-M2e-DD: a novel mucosal adjuvant targeted influenza vaccine. Vaccine. 2008;26(9):1243–52.
Eliasson DG, Omokanye A, Schon K, Wenzel UA, Bernasconi V, Bemark M, Kolpe A, El Bakkouri K, Ysenbaert T, Deng L, Fiers W, Saelens X, Lycke N. M2e-tetramer-specific memory CD4 T cells are broadly protective against influenza infection. Mucosal Immunol. 2018;11(1):273–89.
Epstein SL. Universal Influenza vaccines: Progress in achieving broad cross-protection in vivo. Am J Epidemiol. 2018;187(12):2603–14.
Fan J, Liang X, Horton MS, Perry HC, Citron MP, Heidecker GJ, Fu TM, Joyce J, Przysiecki CT, Keller PM, Garsky VM, Ionescu R, Rippeon Y, Shi L, Chastain MA, Condra JH, Davies ME, Liao J, Emini EA, Shiver JW. Preclinical study of influenza virus a M2 peptide conjugate vaccines in mice, ferrets, and rhesus monkeys. Vaccine. 2004;22(23–24):2993–3003.
Feng J, Zhang M, Mozdzanowska K, Zharikova D, Hoff H, Wunner W, Couch RB, Gerhard W. Influenza a virus infection engenders a poor antibody response against the ectodomain of matrix protein 2. Virol J. 2006;3:102.
Fernandez-Sesma A, Schulman JL, Moran TM. A Bispecific antibody recognizing Influenza a virus M2 protein redirects effector cells to inhibit virus replication in vitro. J Virol. 1996;70(7):4800–4.
Fiers W, De Filette M, El Bakkouri K, Schepens B, Roose K, Schotsaert M, Birkett A, Saelens X. M2e-based universal influenza a vaccine. Vaccine. 2009;27(45):6280–3.
Fu T-M, Freed DC, Horton MS, Fan J, Citron MP, Joyce JG, Garsky VM, Casimiro DR, Zhao Q, Shiver JW, Liang X. Characterizations of four monoclonal antibodies against M2 protein ectodomain of influenza a virus. Virology. 2009;385(1):218–26.
Furuse Y, Suzuki A, Kamigaki T, Oshitani H. Evolution of the M gene of the influenza a virus in different host species: large-scale sequence analysis. Virol J. 2009;6:67.
Gannage M, Dormann D, Albrecht R, Dengjel J, Torossi T, Ramer PC, Lee M, Strowig T, Arrey F, Conenello G, Pypaert M, Andersen J, Garcia-Sastre A, Munz C. Matrix protein 2 of influenza a virus blocks autophagosome fusion with lysosomes. Cell Host Microbe. 2009;6(4):367–80.
Georgieva ER, Borbat PP, Norman HD, Freed JH. Mechanism of influenza a M2 transmembrane domain assembly in lipid membranes. Sci Rep. 2015;5:11757.
Gianfrani С, Oseroff С, Sidney J, Chesnut RW, Sette A. Human memory CTL response specific for Influenza a virus is broad and multispecific. Hum Immunol. 2000;61:438–52.
Grandea AG, Olsen OA, Cox TC, Renshaw M, Hammond PW, Chan-Hui PY, Mitcham JL, Cieplak W, Stewart SM, Grantham ML, Pekosz A, Kiso M, Shinya K, Hatta M, Kawaoka Y, Moyle M. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc Natl Acad Sci U S A. 2010;107(28):126658–12663.
Hashemi H, Pouyanfard S, Bandehpour M, Noroozbabaei Z, Kazemi B, Saelens X, Mokhtari-Azad T. Immunization with M2e-displaying T7 bacteriophage nanoparticles protects against influenza a virus challenge. PLoS One. 2012;7(9):e45765.
Hatta Y, Boltz D, Sarawar S, Kawaoka Y, Neumann G, Bilsel P. M2SR, a novel live influenza vaccine, protects mice and ferrets against highly pathogenic avian influenza. Vaccine. 2017;35(33):4177–83.
Hatta Y, Boltz D, Sarawar S, Kawaoka Y, Neumann G, Bilsel P. Novel influenza vaccine M2SR protects against drifted H1N1 and H3N2 influenza virus challenge in ferrets with pre-existing immunity. Vaccine. 2018;36(33):5097–103.
Heinen PP, Rijsewijk FA, de Boer-Luijtzev EA, Bianchi AT. Vaccination of pigs with a DNA construct expressing an influenza virus M2–nucleoprotein fusion protein exacerbates disease after challenge with influenza a virus. J Gen Virol. 2002;83:1851–9.
Helenius A. Unpacking the incoming Influenza virus. Cell. 1992;69:577–8.
Hirsch RL, Winkelstein JA, Griffin DE. The role of complement in viral infections. III Activation of the classical and alternative complement pathways by Sindbis virus. J Immunol. 1980;124(5):2507–10.
Holsinger LJ, Lamb RA. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology. 1991;183(1):32–43.
Hughey PG, Roberts PC, Holsinger LJ, Zebedee SL, Lamb RA, Compans RW. Effects of antibody to the influenza a virus M2 protein on M2 surface expression and virus assembly. Virology. 1995;212(2):411–21.
Huleatt JW, Nakaar V, Desai P, Huang Y, Hewitt D, Jacobs A, Tang J, McDonald W, Song L, Evans RK, Umlauf S, Tussey L, Powell TJ. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine. 2008;26(2):201–14.
Hutchinson EC, Charles PD, Hester SS, Thomas B, Trudgian D, Martinez-Alonso M, Fodor E. Conserved and host-specific features of influenza virion architecture. Nat Commun. 2014;5:4816.
Hutchinson EC, Curran MD, Read EK, Gog JR, Digard P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza a virus. J Virol. 2008;82(23):11869–79.
Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11(5):404–10.
Imai M, Watanabe S, Ninomiya A, Obuchi M, Odagiri T. Influenza B virus BM2 protein is a crucial component for incorporation of viral ribonucleoprotein complex into virions during virus assembly. J Virol. 2004;78(20):11007–15.
Ito T, Gorman OT, Kawaoka Y, Bean WJ, Webster RJ. Evolutionary analysis of the influenza a virus M gene with comparison of the M1 and M2 proteins. J Virol. 1991;65:5491–8.
Jameson J, Cruz J, Terajima M, Ennis FA. Human CD8+ and CD4+ T lymphocyte memory to Influenza a viruses of swine and avian. J Immunol. 1999;162:7578–83.
Jegerlehner A, Schmitz N, Storni T, Bachmann MF. Influenza a vaccine based on the extracellular domain of M2: weak protection mediated via antibody-dependent NK cell activity. J Immunol. 2004;172(9):5598–605.
Kacskovics I. Fc receptors in livestock species. Vet Immunol Immunopathol. 2004;102(4):351–62.
Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017.
Kim MC, Lee JS, Kwon YM, O E, Lee YJ, Choi JG, Wang BZ, Compans RW, Kang SM. Multiple heterologous M2 extracellular domains presented on virus-like particles confer broader and stronger M2 immunity than live influenza A virus infection. Antiviral Res. 2013;99(3):328–35.
Kim MC, Lee YN, Ko EJ, Lee JS, Kwon YM, Hwang HS, Song JM, Song BM, Lee YJ, Choi JG, Kang HM, Quan FS, Compans RW, Kang SM. Supplementation of influenza split vaccines with conserved M2 ectodomains overcomes strain specificity and provides long-term cross protection. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(7):1364–74.
Kim MC, Song JM, O E, Kwon YM, Lee YJ, Compans RW, Kang SM. Virus-like particles containing multiple M2 extracellular domains confer improved cross-protection against various subtypes of influenza virus. Mol Ther. 2013;21(2):485–92.
Kolpe A, Schepens B, Fiers W, Saelens X. M2-based influenza vaccines: recent advances and clinical potential. Expert Rev Vaccines. 2017;16(2):123–36.
Kolpe A, Schepens B, Ye L, Staeheli P, Saelens X. Passively transferred M2e-specific monoclonal antibody reduces influenza a virus transmission in mice. Antivir Res. 2018;158:244–54.
Krammer F. Emerging influenza viruses and the prospect of a universal influenza virus vaccine. Biotechnol J. 2015;10(5):690–701.
Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov. 2015;14(3):167–82.
Krammer F, Palese P. Influenza virus hemagglutinin stalk-based antibodies and vaccines. Curr Opin Virol. 2013;3(5):521–30.
Krammer F, Pica N, Hai R, Margine I, Palese P. Chimeric hemagglutinin influenza virus vaccine constructs elicit broadly protective stalk-specific antibodies. J Virol. 2013;87(12):6542–50.
Lamb RA, Choppin PW. Identification of a second protein (M2) encoded by RNA segment 7 of Influenza virus. Virology. 1981;112:729–37.
Lamb RA, Choppin PW. Segment 8 of the influenza virus genome is unique in coding for two polypeptides. Biochemistry. 1979;76(10):4908–12.
Lamb RA, Lai CJ. Conservation of the Influenza virus membrane protein (M1) amino acid sequence and an open Reading frame of RNA segment 7 encoding a second protein (M2) in HlN1 and H3N2 strains. Virology. 1981;112:746–51.
Lamb RA, Lai CJ. Spliced and Unspliced messenger RNAs synthesized from cloned Influenza virus M DNA in an SV40 vector: expression of the Influenza virus membrane protein (M1). Virology. 1982;123:237–56.
Lamb RA, Zebedee SL, Richardson CD. Influenza virus M2 protein is an integral membrane protein expressed on the infected-cell surface. Cell. 1985;40:627–33.
Leclerc D, Rivest M, Babin C, Lopez-Macias C, Savard P. A novel M2e based flu vaccine formulation for dogs. PLoS One. 2013;8(10):e77084.
Lee YN, Kim MC, Lee YT, Kim YJ, Kang SM. Mechanisms of cross-protection by Influenza virus M2-based vaccines. Immune Network. 2015;15(5):213–21.
Lee YN, Lee YT, Kim MC, Hwang HS, Lee JS, Kim KH, Kang SM. Fc receptor is not required for inducing antibodies but plays a critical role in conferring protection after influenza M2 vaccination. Immunology. 2014;143(2):300–9.
Lee YT, Kim KH, Ko EJ, Kim MC, Lee YN, Hwang HS, Lee Y, Jung YJ, Kim YJ, Santos J, Perez DR, Kang SM. Enhancing the cross protective efficacy of live attenuated influenza virus vaccine by supplemented vaccination with M2 ectodomain virus-like particles. Virology. 2019;529:111–21.
Lee YT, Ko EJ, Lee Y, Kim KH, Kim MC, Lee YN, Kang SM. Intranasal vaccination with M2e5x virus-like particles induces humoral and cellular immune responses conferring cross-protection against heterosubtypic influenza viruses. PLoS One. 2018;13(1):e0190868.
Leiding T, Wang J, Martinsson J, DeGrado WF, Arskold SP. Proton and cation transport activity of the M2 proton channel from influenza a virus. Proc Natl Acad Sci U S A. 2010;107(35):15409–14.
Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47(W1):W256–9.
Liu W, Li H, Chen Y-H. N-terminus of M2 protein could induce antibodies with inhibitory activity against influenza virus replication. FEMS Immunol Med Microbiol. 2003;35(2):141–6.
Liu W, Zou P, Chen YH. Monoclonal antibodies recognizing EVETPIRN epitope of influenza a virus M2 protein could protect mice from lethal influenza a virus challenge. Immunol Lett. 2004;93(2–3):131–6.
Liu W, Zou P, Ding J, Lu Y, Chen YH. Sequence comparison between the extracellular domain of M2 protein human and avian influenza a virus provides new information for bivalent influenza vaccine design. Microbes Infect. 2005;7(2):171–7.
Lotfi Z, Golchin M, Khalili-Yazdi A, Khalili M. Immunological properties of the SLLTEVET epitope of Influenza a virus in multiple display on filamentous M13 phage. Comp Immunol Microbiol Infect Dis. 2019;65:76–80.
Manzoor R, Igarashi M, Influenza TA. A Virus M2 Protein: Roles from Ingress to Egress. Int J Mol Sci. 2017;18(12). https://doi.org/10.3390/ijms18122649.
Margine I, Krammer F, Hai R, Heaton NS, Tan GS, Andrews SA, Runstadler JA, Wilson PC, Albrecht RA, Garcia-Sastre A, Palese P. Hemagglutinin stalk-based universal vaccine constructs protect against group 2 influenza a viruses. J Virol. 2013;87(19):10435–46.
McCown MF, Pekosz A. The influenza a virus M2 cytoplasmic tail is required for infectious virus production and efficient genome packaging. J Virol. 2005;79(6):3595–605.
Mcgeoch D., Fellnert P. and Newton C. Influenza virus genome consists of eight distinct RNA species Biochemistry 73(9):3045–3049, 1976.
Morgan SB, Holzer B, Hemmink JD, Salguero FJ, Schwartz JC, Agatic G, Cameroni E, Guarino B, Porter E, Rijal P, Townsend A, Charleston B, Corti D, Tchilian E. Therapeutic Administration of Broadly Neutralizing FI6 antibody reveals lack of interaction between human IgG1 and pig fc receptors. Front Immunol. 2018;9:865.
Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA, Pinto LH. Influenza B virus BM2 protein has Ion Channel activity that conducts protons across membranes. Dev Cell. 2003;5(1):175–84.
Music N, Reber AJ, Kim MC, York IA, Kang SM. Supplementation of H1N1pdm09 split vaccine with heterologous tandem repeat M2e5x virus-like particles confers improved cross-protection in ferrets. Vaccine. 2016;34(4):466–73.
Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM, Fiers W. A universal influenza a vaccine based on the extracellular domain of the M2 protein. Nat Med. 1999;5(10):1157–63.
Okuda K, Ihata A, Watabe S, Okada E, Yamakawa T, Hamajima K, Yang J, Ishii N, Nakazawa M, Okuda K, Ohnari K, Nakajima K, Xin KQ. Protective immunity against influenza a virus induced by immunization with DNA plasmid containing influenza M gene. Vaccine. 2001;19:3681–91.
Palese P, Schulman JL. Differences in RNA patterns of Influenza a viruses. J Virol. 1976;17(3):876–84.
Park JK, Taubenberger JK. Universal Influenza vaccines: to dream the possible dream? ACS Infect Dis. 2016;2(1):5–7.
Park KS, Seo YB, Lee JY, Im SJ, Seo SH, Song MS, Choi YK, Sung YC. Complete protection against a H5N2 avian influenza virus by a DNA vaccine expressing a fusion protein of H1N1 HA and M2e. Vaccine. 2011;29(33):5481–7.
Paterson RG, Takeda M, Ohigashi Y, Pinto LH, Lamb RA. Influenza B virus BM2 protein is an oligomeric integral membrane protein expressed at the cell surface. Virology. 2003;306(1):7–17.
Pejoski D, Zeng W, Rockman S, Brown LE, Jackson DC. A lipopeptide based on the M2 and HA proteins of influenza a viruses induces protective antibody. Immunol Cell Biol. 2010;88(5):605–11.
Petrie JG, Malosh RE, Cheng CK, Ohmit SE, Martin ET, Johnson E, Truscon R, Eichelberger MC, Gubareva LV, Fry AM, Monto AS. The household Influenza vaccine effectiveness study: lack of antibody response and protection following receipt of 2014-2015 Influenza vaccine. Clin Infect Dis. 2017;65(10):1644–51.
Petukhova N, Gasanova T, Stepanova L, Rusova O, Potapchuk M, Korotkov A, Skurat E, Tsybalova L, Kiselev O, Ivanov P, Atabekov J. Immunogenicity and protective efficacy of candidate universal Influenza a Nanovaccines produced in plants by tobacco mosaic virus-based vectors. Curr Pharm Des. 2013;19(31):5587–600.
Pinto LH, Holsinger LJ, Lamb RA. Influenza virus M2 protein has ion channel activity. Cell. 1992;69:517–28.
Pons MW. A reexamination of influenza single- and double-stranded RNAs by gel electrophoresis. Virology. 1976;69:789–92.
Ramos EL, Mitcham JL, Koller TD, Bonavia A, Usner DW, Balaratnam G, Fredlund P, Swiderek KM. Efficacy and safety of treatment with an anti-m2e monoclonal antibody in experimental human influenza. J Infect Dis. 2015;211(7):1038–44.
Robb NC, Fodor E. The accumulation of influenza a virus segment 7 spliced mRNAs is regulated by the NS1 protein. J Gen Virol. 2012;93(Pt 1):113–8.
Rossman JS, Jing X, Leser GP, Lamb RA. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell. 2010;142(6):902–13.
Saelens X. The Role of Matrix Protein 2 Ectodomain in the Development of Universal Influenza Vaccines. J Infect Dis 219(Supplement_1):S68-S74, 2019.
Sarawar S, Hatta Y, Watanabe S, Dias P, Neumann G, Kawaoka Y, Bilsel P. M2SR, a novel live single replication influenza virus vaccine, provides effective heterosubtypic protection in mice. Vaccine. 2016;34(42):5090–8.
Schnell JR, Chou JJ. Structure and mechanism of the M2 proton channel of influenza a virus. Nature. 2008;451(7178):591–5.
Schotsaert M, Ysenbaert T, Smet A, Schepens B, Vanderschaeghe D, Stegalkina S, Vogel TU, Callewaert N, Fiers W, Saelens X. Long-lasting cross-protection against Influenza a by neuraminidase and M2e-based immunization strategies. Sci Rep. 2016;6:24402.
Shih SR, Krug RM. Surprising function of the three Influenza viral polymerase proteins: selective protection of viral mRNAs against the cap-snatching reaction catalyzed by the same polymerase proteins. Virology. 1996;226:430–5.
Shih SR, Nemeroff ME, Krug RM. The choice of alternative 5′ splice sites in influenza virus M1 mRNA is regulated by the viral polymerase complex. Proc Natl Acad Sci U S A. 1995;92:6324–8.
Simhadri VR, Dimitrova M, Mariano JL, Zenarruzabeitia O, Zhong W, Ozawa T, Muraguchi A, Kishi H, Eichelberger MC, Borrego F. A human anti-M2 antibody mediates antibody-dependent cell-mediated cytotoxicity (ADCC) and cytokine secretion by resting and cytokine-Preactivated natural killer (NK) cells. PLoS One. 2015;10(4):e0124677.
Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza a epidemic. Nature. 2009;459(7250):1122–5.
Song JM, Wang BZ, Park KM, Rooijen NV, Quan FS, Kim MC, Jin HT, Pekosz A, Compans RW, Kang SM. Influenza virus-like particles containing M2 induce broadly cross protective immunity. PLoS One. 2011;6(1):14538–145348.
Sridhar S, Begom S, Bermingham A, Hoschler K, Adamson W, Carman W, Bean T, Barclay W, Deeks JJ, Lalvani A. Cellular immune correlates of protection against symptomatic pandemic influenza. Nat Med. 2013;19(10):1305–12.
Stauffer S, Feng Y, Nebioglu F, Heilig R, Picotti P, Helenius A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza a virus cores after penetration. J Virol. 2014;88(22):13029–46.
Stepanova LA, Mardanova ES, Shuklina MA, Blokhina EA, Kotlyarov RY, Potapchuk MV, Kovaleva AA, Vidyaeva IG, Korotkov AV, Eletskaya EI, Ravin NV, Tsybalova LM. Flagellin-fused protein targeting M2e and HA2 induces potent humoral and T-cell responses and protects mice against various influenza viruses a subtypes. J Biomed Sci. 2018;25(1):33.
Stoloff GA, Caparros-Wanderley W. Synthetic multi-epitope peptides identified in silico induce protective immunity against multiple influenza serotypes. Eur J Immunol. 2007;37(9):2441–9.
Stouffer AL, Acharya R, Salom D, Levine AS, Di Costanzo L, Soto CS, Tereshko V, Nanda V, Stayrook S, DeGrado WF. Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 2008;451(7178):596–9.
Takeda M, Pekosz A, Shuck K, Pinto LH, Lamb RA. Influenza a virus M2 Ion Channel activity is essential for efficient replication in tissue culture. J Virol. 2002;76(3):1391–9.
Talbot HK, Rock MT, Johnson C, Tussey L, Kavita U, Shanker A, Shaw AR, Taylor DN. Immunopotentiation of trivalent influenza vaccine when given with VAX102, a recombinant influenza M2e vaccine fused to the TLR5 ligand flagellin. PLoS One. 2010;5(12):e14442.
Tang Y, Zaitseva F, Lamb RA, Pinto LH. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J Biol Chem. 2002;277(42):39880–6.
Tompkins SM, Zhao ZS, Lo CY, Misplon JA, Liu T, Ye Z, Hogan RJ, Wu Z, Benton KA, Tumpey TM, Epstein SL. Matrix protein 2 vaccination and protection against influenza viruses, including subtype H5N1. Emerg Infect Dis. 2007;13(3):426–35.
Topham DI, Tripp RA, Doherty PC. CD8+ T cells clear Influenza virus by Perforin or Fas-dependent processes. J Immunol. 1997;159(11):5197–200.
Treanor JJ, Tierney EL, Zebedee SL, Lamb RA, Murphy BR. Passively transferred monoclonal antibody to the M2 protein inhibits Influenza a virus replication in mice. J Virol. 1990;64(3):1375–7.
Tsybalova LM, Stepanova LA, Kuprianov VV, Blokhina EA, Potapchuk MV, Korotkov AV, Gorshkov AN, Kasyanenko MA, Ravin NV, Kiselev OI. Development of a candidate influenza vaccine based on virus-like particles displaying influenza M2e peptide into the immunodominant region of hepatitis B core antigen: broad protective efficacy of particles carrying four copies of M2e. Vaccine. 2015;33(29):3398–406.
Turley CB, Rupp RE, Johnson C, Taylor DN, Wolfson J, Tussey L, Kavita U, Stanberry L, Shaw A. Safety and immunogenicity of a recombinant M2e-flagellin influenza vaccine (STF2.4xM2e) in healthy adults. Vaccine. 2011;29(32):5145–52.
Van den Hoecke S, Ehrhardt K, Kolpe A, El Bakkouri K, Deng L, Grootaert H, Schoonooghe S, Smet A, Bentahir M, Roose K, Schotsaert M, Schepens B, Callewaert N, Nimmerjahn F, Staeheli P, Hengel H, Saelens X. Hierarchical and Redundant Roles of Activating FcgammaRs in Protection against Influenza Disease by M2e-Specific IgG1 and IgG2a Antibodies. J Virol. 2017;91(7). https://doi.org/10.1128/JVI.02500-16.
Venkataraman P, Lamb RA, Pinto LH. Chemical rescue of histidine selectivity filter mutants of the M2 ion channel of influenza a virus. J Biol Chem. 2005;280(22):21463–72.
Von Holle TA, Moody MA. Influenza and antibody-dependent cellular cytotoxicity. Front Immunol. 2019;10:1457.
Wang J, Pielak RM, McClintock MA, Chou JJ. Solution structure and functional analysis of the influenza B proton channel. Nat Struct Mol Biol. 2009;16(12):1267–71.
Wang R, Song A, Levin J, Dennis D, Zhang NJ, Yoshida H, Koriazova L, Madura L, Shapiro L, Matsumoto A, Yoshida H, Mikayama T, Kubo RT, Sarawar S, Cheroutre H, Kato S. Therapeutic potential of a fully human monoclonal antibody against influenza a virus M2 protein. Antivir Res. 2008;80(2):168–77.
Wang Y, Chen L, Cheng Y, Zhou S, Pang Y, Zhang J, Greene CM, Song Y, Zhang T, Zhao G. Potential impact of B lineage mismatch on trivalent influenza vaccine effectiveness during the 2015-2016 influenza season among nursery school children in Suzhou. China Hum Vaccin Immunother. 2018;14(3):630–6.
Watanabe A, McCarthy KR, Kuraoka M, Schmidt AG, Adachi Y, Onodera T, Tonouchi K, Caradonna TM, Bajic G, Song S, McGee CE, Sempowski GD, Feng F, Urick P, Kepler TB, Takahashi Y, Harrison SC, Kelsoe G. Antibodies to a Conserved Influenza Head Interface Epitope Protect by an IgG Subtype-Dependent Mechanism. Cell. 2019;177(5):1124–1135 e1116.
Watanabe T, Watanabe S, Ito H, Kida H, Kawaoka Y. Influenza a virus can undergo multiple cycles of replication without M2 ion channel activity. J Virol. 2001;75(12):5656–62.
Wharton SA, Belshe RB, Skehel JJ, Hay AJ. Role of virion M2 protein in influenza virus uncoating: specific reduction in the rate of membrane fusion between virus and liposomes by amantadine. J Gen Virol. 1994;75(Pt 4):945–8.
WHO. Antigenic and genetic characteristics of zoonotic influenza viruses and candidate vaccine viruses developed for potential use in human vaccines. 22 February 2018. Available at: http://www.who.int/entity/influenza/vaccines/virus/201802_zoonotic_vaccinevirusupdate.pdf?ua=1. 2018.
WHO. Tables on clinical evaluation of influenza vaccines. Pandemic and potentially pandemic influenza vaccines. Available at: http://www.who.int/entity/immunization/diseases/influenza/Table_clinical_evaluation_influenza_pandemic.xlsx?ua=1. 2018.
Winter G, Fields S. Cloning of influenza cDNA into M13: the sequence of the RNA segment encoding the a/PR/8/34 matrix protein. Nucleic Acids Res. 1980;8(9):1965–74.
Wise HM, Hutchinson EC, Jagger BW, Stuart AD, Kang ZH, Robb N, Schwartzman LM, Kash JC, Fodor E, Firth AE, Gog JR, Taubenberger JK, Digard P. Identification of a novel splice variant form of the influenza a virus M2 ion channel with an antigenically distinct ectodomain. PLoS Pathog. 2012;8(11):e1002998.
Wohlgemuth N, Lane AP, Pekosz A. Influenza A Virus M2 Protein Apical Targeting Is Required for Efficient Virus Replication. J Virol. 2018;92(22). https://doi.org/10.1128/JVI.01425-18.
Wu F, Huang JH, Yuan XY, Huang WS, Chen YH. Characterization of immunity induced by M2e of influenza virus. Vaccine. 2007;25(52):8868–73.
Zebedee SL, Lamb RA. Growth restriction of influenza a virus by M2 protein antibody is genetically linked to the M1 protein. Proc Natl Acad Sci U S A. 1989;86(3):1061–5.
Zebedee SL, Lamb RA. Influenza a virus M2 protein: monoclonal antibody restriction of virus growth and detection of M2 in virions. J Virol. 1988;62:2762–72.
Zhao G, Sun S, Du L, Xiao W, Ru Z, Kou Z, Guo Y, Yu H, Jiang S, Lone Y, Zheng BZ, Zhou Y. An H5N1 M2e-based multiple antigenic peptide vaccine confers heterosubtypic protection from lethal infection with pandemic 2009 H1N1 virus. Virol J. 2010;7:151–6.
Zharikova D, Mozdzanowska K, Feng J, Zhang M, Gerhard W. Influenza type a virus escape mutants emerge in vivo in the presence of antibodies to the ectodomain of matrix protein 2. J Virol. 2005;79(11):6644–54.
Zhong W, Reed C, Blair PJ, Katz JM, Hancock K, Influenza Serology Working G. Serum antibody response to matrix protein 2 following natural infection with 2009 pandemic influenza A(H1N1) virus in humans. J Infect Dis. 2014;209(7):986–94.
Acknowledgements
We thank Dr. Patricia Butler for editorial assistance.
Funding
This review was prepared with financial support of Russian Science Foundation grant 19–15-00015.
Author information
Authors and Affiliations
Contributions
DM: contribution to conception and design, manuscript writing; IIS: contribution to data collection and manuscript writing; LR: contribution to manuscript writing and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mezhenskaya, D., Isakova-Sivak, I. & Rudenko, L. M2e-based universal influenza vaccines: a historical overview and new approaches to development. J Biomed Sci 26, 76 (2019). https://doi.org/10.1186/s12929-019-0572-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-019-0572-3