
Abstract
The pathogenesis of systemic autoimmune diseases such as systemic lupus erythematosus (SLE) is based on the loss of self-tolerance against ubiquitous autoantigens involving all mechanisms of adaptive immunity. However, data accumulating over the last decade imply an important role also for numerous elements of innate immunity, namely the Toll-like receptors in the pathogenesis of SLE. Here we discuss their role in the most common organ complication of SLE, i.e. lupus nephritis. We summarize experimental and clinical data on the expression and functional contribution of the Toll-like receptors in immune complex glomerulonephritis, and intrarenal inflammation. Based on these discoveries Toll-like receptors are evolving as therapeutic targets for the treatment of SLE and lupus nephritis.
Similar content being viewed by others
Background
Toll-like receptors (TLR) have an essential role in innate immunity during host defense which impacts also on auto-inflammatory and autoimmune diseases [1]. Once Tlr-deficient mice became available the hypothesis of TLRs as boosters of systemic autoimmunity [2] including disease such as systemic lupus erythematosus (SLE) became possible. In the first part of this review, we discuss the role of TLRs in SLE and in the second part their contribution to intrarenal inflammation in lupus nephritis (LN). Further, we have also briefly mentioned about TLR role in immune tolerance break and germinal center formation in the second part.
Toll-like receptors in systemic autoimmunity upstream to nephritis
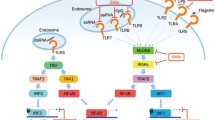
Based on the location of expression, TLRs are classified into extra-cellular and intra-cellular (Fig. 1).

Toll-like receptor (TLR) signalling pathways. TLR1, TLR2, TLR4, TLR5, TLR6, TLR11, TLR12 as homo or heterodimers bind to their respective ligands at the cell surface, whereas TLR3, TLR7/TLR8, TLR9, and TLR13 sense nucleic acids in endosomes. TLR4 localizes at both the plasma membrane and the endosomes. The Toll–IL-1-resistence (TIR) domains of TLRs-dimers bind TIR domain-containing adaptor proteins (either myeloid differentiation primary-response protein 88 (MYD88), or TIR domain-containing adaptor protein inducing IFNβ (TRIF) and TRIF-related adaptor molecule (TRAM)). Downstream signalling pathways of TLRs involve IL-1R-associated kinases (IRAKs), IκB kinase -alpha, beta, gamma and epsilon (IKK αβγε-) and the adaptor molecules TNF receptor-associated factors (TRAFs). These protein complexes activate p50 and p65, as well as various transcription factors such as interferon-regulatory factors (IRFs) and activator protein 1 (AP1). TLR signalling leads to the induction of pro-inflammatory cytokines or the induction of type I interferon (IFN)
Extracellular TLRs
Toll-like receptors 2 and 4
TLR-2 and TLR-4 are membrane-bound receptors that recognize bacterial wall components and endogenous components of dying cells, so-called damage-associated molecular patterns (DAMPs) [3, 4]. In C57BL6lpr/lpr mice, a model of spontaneous autoimmunity, TLR2 or TLR4 deficiency limits the expansion of the marginal zone B cell compartment and attenuates the levels of antinuclear-antibodies and rheumatoid factors [5]. However, it should be noted that C57BL6lpr/lpr mice are a very mild strain in developing the disease compared with MRL-lpr strain. Interestingly, anti-nucleosome antibodies are diminished in the absence of TLR2 but not of TLR4 [5]. Administration of pristine a natural saturated terpenoid alkane to genetically intact mice results in the development of hypergammaglobulinaemia with the production of lupus-like autoantibodies and proliferative glomerulonephritis, with similarities to human lupus nephritis. However, administration of the pristine in Tlr4- deficient mice demonstrated a global decrease in both Th1, IFNγ, and Th17 associated IL-17A and IL-6 cytokine production. Also, autoantibody levels of anti-dsDNA and anti-RNP were both decreased indicating the requirement of TLR4 for the full-blown autoimmunity [6]. Furthermore, exposing anti-ds DNA transgenic mice to TLR4 agonistic LPS aggravates systemic autoimmunity and LN [7].
Using transgenic mouse called TCr-5, whose genome contains multiple copies of the tlr4 gene on a TLR4-deficient C57/BL10ScCr (deletion of tlr4 gene and point mutation in IL-12Rβ 2gene) background, it has been shown that up-regulation TLR4 is pathogenic both at the protein level and gene level and triggered spontaneous SLE-like autoimmunity and lupus nephritis [8]. Mice deficient in Lyn, a serin threonine kinase with an inhibitory function on TLR4/IL-1 signaling in dendritic cells, develop spontaneous lymphoproliferation, nephritis and autoantibody production. Indeed, the effects of Lyn-deficiency could be restored by simultaneous deletion of MyD88 in dendritic cells [9]. Obviously, the immunostimulatory functions of TLR4 can trigger the initiation of autoimmunity in permissive genetic constellations [10] even when aberrant TLR-4/IL-1 signaling is restricted only to dendritic cells [9]. However, Tlr2 or − 4-deficiency in MRL/lpr mice did not significantly affect disease severity [11].
In human overt SLE, TLR2 mRNA is upregulated in PBMCs [12] but in young lupus patients (median age of 13 years at the time of enrolment) monocyte TLR2 and-4 cell surface expression can be even reduced when examined by flow cytometry [13]. Stimulation of PBMCs from SLE patients with TLR2 or − 4 agonists triggers more cytokine production as compared to healthy subjects [14]. TLR2 is also increased on CD4+ and CD8+ positive T cells and B cells from SLE patients, which is associated with the increased in-vitro production of IL-17 upon activation with TLR-2 agonists [15]. South Indians bearing the TLR4 promoter polymorphisms D299G and T399I show an increased risk for specific autoantibody production (Table 1) [16]. Together, accumulating data suggest an involvement of TLR2 and − 4 in the pathogenesis of systemic autoimmunity in mice and humans.
Toll-like receptor-5
Little is known about TLR5 in lupus but some contribution to SLE seems possible. Renal TLR5 expression is upregulated in nephritic MRL/lpr mice, probably because of immune cell infiltrates. In vitro treatment of murine macrophages with the TLR5 ligand flagellin induces IL-6 production [17] indicating that microbial components can efficiently ligate with activated immune cells present in immune complex glomerulonephritis. In humans, TLR5 maps to a q41-q42 locus on chromosome 1 that includes strong SLE susceptibility genes. Transmission disequilibrium testing in a cohort of 199 SLE patients versus 75 healthy siblings implies a protective role for SNP1174T in the TLR5 gene, probably because SNP1174T transmits a stop codon abrogating TLR5 signaling [18]. The rs5744168 is most studied SNP for TLR5 and found no genotype-phenotype association in SLE however, the T allele and the TT genotype were raised significantly in LN patients group indicating the putative role of TLR5 in the progression of LN [19]. Even though the available data on TLR5 with respect to SLE are limited the existing data suggest a contribution of TLR5 to SLE manifestations.
Intracellular TLRs
Toll-like receptor-3
TLR3 receptor localizes to intracellular endosomes and senses double-stranded RNA via the adaptor TRIF instead of MyD88 [20]. This way TLR3 employs the transcription factor IRF3 to induce type I interferon secretion, a central element in the pathogenesis of SLE [10]. Although single vaccination-like doses of the TLR3 ligand polyI:C RNA do not exacerbate autoimmunity in MRL/lpr or FcγR2b−/− mice with SLE-like autoimmunity, repetitive doses of polyI:C RNA do aggravate kidney disease in MRL/lpr mice but independently from B cell-driven immune complex disease [21, 22]. It, therefore, was concluded that viral dsRNA triggers disease activity of lupus nephritis by mechanisms that are different from those of bacterial DNA. The gene product of the ITGAM gene, CD11b, is an endogenous negative regulator of TLR3 signaling and gene variants in the ITGAM gene affect the risk for SLE in humans [23] suggesting activation of CD11b could be a potential mechanism for developing SLE therapeutics. In SLE patients also TLR3 expression is upregulated in total PBMCs, T- and B cells [24, 25]. The TLR3 gene rs3775296-T risk allele was found to be associated with anaemia and photosensitivity in Taiwanese SLE patients [26] indicating that TLR3 involved the development of different SLE phenotypes, and in antiviral responses that trigger expression of pro-inflammatory genes. Taken together these data suggest that viral ds RNA trigger disease activity through TLR3 in a MyD88 independent pathway involving various innate immune cells.
Toll-like receptor-7
TLR7 locates together with TLR3 (and TLR9) inside intracellular endosomes. TLR7 senses viral single-stranded RNAs and triggers cytokine and type I interferon production for antiviral host defense but endogenous nucleic acids can have the same effect [27, 28]. TLR7 activation with imiquimod activates systemic type I interferon production and aggravates lupus nephritis in MRL/lpr mice [29]. Monoclonal antibodies targeting TLR7 get internalized after binding to a surface-bound TLR7 fraction [30]. TLR7 activity mediates the formation of anti-RNA autoantibodies and glomerulonephritis in MRL/lpr mice as well as in pristane-induced SLE [31, 32]. MRL/lpr mice deficient for TLR7- and TLR9-signalling do not develop antinuclear antibodies [33]. TLR9 activity inhibits TLR7-mediated autoimmunity [33]. Therefore, MRL/lpr mice lacking TLR9 develop increased autoantibody levels directed against RNA autoantigens such as anti-Sm or anti-ribosomal P Ag [33].
TLR7 overexpression in B6.yaa mice illustrates that increased TLR7 activity alone is not sufficient to trigger autoimmunity without gene variants in other susceptibility genes [34]. The y-linked autoimmune accelerator (yaa) allele is the most potent disease allele on the Y chromosome detected in BXSB mouse strain involved in the pathogenesis of lupus nephritis; however, yaa could produce disease only when combined with other autoimmune promoting genes [35, 36].TLR7-driven type I interferon secretion can also promote hematopoietic abnormalities [37]. TLR3/TLR7 co-activation has synergistic immunostimulatory effects in MRL/lpr mice [38].
TLR7 is expressed in several B cell subtypes, macrophages, and plasmacytoid dendritic cells [39]. In MRL/lpr mice dendritic cells promote expansion and differentiation of T cells, but not their activation [40]. Interestingly, in MRL/lpr mice TLR/MyD88 signaling in dendritic cells is dispensable for the development of autoantibody production and lupus nephritis [41]. TREML4 augments TLR7 signaling during autoimmune disease. Indeed, Treml4-deficient MRL/lpr mice have reduced T cell numbers, lower systemic IFN-α levels, show less nephritis [42]. Mice, lacking the negative regulator Lyn, spontaneously develop autoimmunity [43] and conditional deletion of MyD88 in dendritic cells in Lyn-deficient mice decreases germinal center formation, DNA autoantibody production, and immune complex glomerulonephritis [44]. A similar phenotype is present in mice deficient for the negative regulator interleukin-1 receptor-associated kinase-M [45] and single Ig IL-1-related receptor [46]. In addition, abolishing the function of plasmacytoid dendritic cells in TLR7.Tg mice, a model of TLR7-driven autoimmunity, attenuated glomerular immune complex deposits and prolonged lifespan of the mice [47].
In contrast, genetic depletion of MyD88 in B cells impairs the secretion of antinuclear antibodies and abrogates lupus nephritis because activated B cells contribute to both antibody-dependent and -independent renal T cell infiltrates in MRL/lpr mice [41]. Indeed, in Tlr7 transgenic mice autophagy in B cells is an initiating event triggering TLR7-dependent autoantibody production [48]. B cell receptor-driven uptake of immune complexes stimulates TLR7 and − 9 inside B cells and drives RNA- and DNA-autoantibody production, respectively [33, 49,50,51,52,53,54]. In this context, germinal center formation in health and disease involves B cell TLR7 signaling [55]. Thus, normalizing CD19+ B cell TLR7 expression in transgenic Tg7Sle1B6 mice overexpressing TLR7 in a lupus-prone background normalized lymphocyte activation but not proliferation. Nevertheless, less production of RNA autoantibodies somewhat reduced the activity of immune complex glomerulonephritis [56].
PBMCs from different ethnic SLE populations consistently overexpress TLR7 along with a type I interferon transcript signature [57, 58]. TLR7 gene variants can be risk factors for human SLE [26, 59]. A recent meta-analysis comprising 11,984 patients documented a link between the TLR7 variants rs179008 and the risk of SLE in African populations and between rs3853839 and SLE in Asians [60]. Japanese male SLE patients carrying a single nucleotide polymorphism in the 3’UTR of the TLR7 gene corresponds to an increased TLR7 gene dose and type-I interferon gene signature [61]. Furthermore, a Mexican case-control study of 328 childhood-onset SLE patients and 403 controls displayed an increase in TLR7 copy numbers as an SLE risk factor [62]. It is of note that, another study could not confirm these in adult-onset SLE [63]. Interestingly, the gene product of the ITGAM gene, CD11b, is an endogenous negative regulator of TLR7 signaling and gene variants in the ITGAM gene affect the risk for SLE in humans [23]. Intrinsic IgE seems to be another endogenous inhibitor of TLR7-mediated IFN-α induction in dendritic cells isolated from SLE patients [64]. In summary, TLR7 is involved in the pathogenesis of murine and human SLE and LN by sensing endogenous ribonucleoprotein antigens for the activation of antigen presenting cells such as B cells and dendritic cells [10, 65, 66]. In this process, the nucleic acid component acts as an endogenous adjuvant to boost immunity against ribonucleoproteins [67]. The phenomenon of TLR7 sensing danger signals of RNA autoantigens is similar to sensing signals delivered during viral infection and could therefore be referred to as molecular mimicry in innate immunity.
Toll-like receptor-8
In humans, TLR8 senses ssRNA [68,69,70] but its role in autoimmunity is less clear. 564Igi mice, a knock-in strain expressing an autoreactive anti-RNA-antibody, require TLR8 and MyD88 to break tolerance and to induce a type-I interferon signature [71]. Autoantibody secretion is abrogated upon simultaneous deletion of TLR7 and TLR8 but not of TLR7 and TLR9 or other combinations [71]. However, TLR8 deletion of C57BL/6 genetic background triggers anti-dsDNA antibody secretion and glomerulonephritis, an effect abrogated by co-deletion of TLR7 suggesting rather an immunoregulatory role of TLR8 in TLR7 [72]. Indeed, dendritic cells isolated from Tlr8−/− mice present increased TLR7 expression and are hyper-responsive to TLR7 agonists [72]. In contrast to TLR9, which mainly executes its regulatory effect on TLR7 in B cells, TLR8 inhibits TLR7 mostly in dendritic cells [10, 73]. Evidently, TLR9 and TLR8 inhibit TLR7-mediated autoimmunity and renal inflammation in a synergistic manner [71, 73].
TLR8 is also present in humans [26, 74, 75] and TLR8 expression levels are upregulated in blood cells of paediatric and adult lupus patients [76]. Females express higher levels of TLR8 mRNA in blood cells in the presence of 17β-estradiol as a potential factor in gender disparity of SLE [74]. In addition, the TLR8 single nucleotide polymorphism rs3764880 is associated with an increased risk for SLE in females but not in males [26]. As another aspect, certain antiphospholipid antibodies upregulate miR-146a-3p in trophoblast cells, which enhances TLR8-mediated IL-8-secretion [75]. Together, also TLR8 seems to have an immunoregulatory role in systemic autoimmunity, especially in females.
Toll-like receptor-9
AlsoTLR9 localizes to the endosomal compartment where it senses unmethylated CpG motifs in the DNA sequence of viruses or bacteria [10]. However, endogenous TLR9 ligands exist, e.g. hypomethylated self-DNA within immune complexes, neutrophil extracellular traps, oxidized mitochondrial nucleoids or other chromatin formats [10, 28, 77,78,79]. Defects in lysosomal maturation endorse the activation of TLR9 [80]. TLR9 ligation is followed by type I interferon production in immune cells especially plasmacytoid dendritic cells [81]. Indeed, TLR9 and TLR7 in SLE partially mimic a state of anti-viral host defense referred to as “pseudoantiviral immunity” [82, 83]. Type I interferons promote the responsiveness to B-cell receptor crosslinking in B cells and render dendritic cells responsive to endogenous nucleic acids after upregulation of TLR7 and TLR9 [84, 85]. TLR9-dependent type I interferon production depends on a lipid kinase - PIKfyve, a mechanism that can neutralized by apilimod [86]. However, not all features of human SLE are attributable to type I Interferons as longitudinal studies do not always show I interferon signatures to indicate disease activity in patients [87, 88]. Blocking endogenous TLR9 ligands with a TLR9 antagonist in MRL/lpr mice ameliorated systemic autoimmunity and immune complex glomerulonephritis [89]. Consistently, injections with CpG and LPS to anti-dsDNA transgenic mice exacerbated disease [7]. In line, Tlr9-deficient C57BL/6 mice display less autoimmunity and nephritis due to less glomerular Ig-deposits and complement activation in pristane-induced autoimmunity [6]. However, BALB/c mice injected with pristane show different effects, highlighting the contribution of additional genetic factors [90]. Nevertheless, dual inhibition of both TLR7 and TLR9 did not elicit additive effects beyond single blockade of TLR7 in MRL/lpr mice. TLR9 inhibition even neutralized the beneficial effects of the TLR7 blockade on systemic autoantibody levels without affecting the effects of the TLR7 blockade on nephritis [91]. Hence, the regulatory role of TLR9 is context-dependent.
It is of note that TLR9 deletion in MRL/lpr mice aggravates systemic autoimmunity and immune complex glomerulonephritis, a phenomenon that can be neutralized co-deletion of TLR7 [33]. Obviously, TLR9 and TLR7 normally compete for endosomal trafficking implying that increased TLR7 signaling driving autoimmunity can outweigh the pro-inflammatory effects of TLR9 [92, 93]. In addition, Tlr9- and Tlr8-double-deficient C57BL/6 mice develop spontaneous signs of autoimmunity such as splenomegaly, autoantibody production, and glomerulonephritis. Here, TLR9 seems to restrict B cell TLR7 signaling, while TLR8 suppresses TLR7-driven activation of dendritic cells [73]. However, B cell TLR9 is required for the generation of DNA antibodies in mice and promotes the differentiation of autoantibody-secreting B and plasma cells in humans [94, 95]. TLR9 mRNA expression is also upregulated in PBMCs from SLE patients, correlates with lupus nephritis [57, 96], and correlates with circulating anti-DNA autoantibody levels [96]. B cell TLR9 expression is associated with serum creatinine levels in SLE patients [25]. Intrinsic IgE inhibits also TLR9-mediated IFN-α induction in dendritic cells isolated from SLE patients [64]. Together, TLR has a complex role in SLE as it promotes DNA autoantibody production as an important component of lupus nephritis. However, TLR9 also inhibits TLR7 signaling as another central pathway in the pathogenesis of SLE. Therefore, therapeutic TLR9 inhibition may cause unexpected effects in human SLE depending on the predominant role of these different elements of disease pathogenesis.
Toll-like receptors inside the nephritic kidney
How do TLRs maintain tissue inflammation inside the kidney once lupus nephritis has established? Extrinsic and intrinsic ligands can ligate TLRs on infiltrating monocytes, dendritic cells and B cells to enhance cytokine secretion [17, 97, 98]. Furthermore, mesangial cells and other parenchymal cells express TLR1–4, and TLR6 [17]. In culture, renal cells respond to TLR activation and secrete interleukins and chemokines [17, 99]. Also, immune complex deposits containing TLR agonists activate glomerular mesangial cells and contribute to immune complex glomerulonephritis [100, 101].
Inside the kidney, TLR2 and TLR4 are expressed not only by parenchymal cells but also by infiltrating neutrophils and mononuclear phagocytes including macrophages and dendritic cells [10, 102]. Murine macrophages respond to TLR2 and − 4 agonists by secretion of pro-inflammatory cytokines [17]. Also, HMGB1, a DNA-binding protein, and lupus autoantigen, released under inflammatory conditions can induce NF-κB activation in a TLR2 and TLR4-RAGE-dependent manner in mononuclear phagocytes and neutrophils [103, 104] as well as in mesangial cells [103, 105]. When HMGB1 combines with the nephritogenic anti-DNA antibody 1A3F they form complexes that activate the TLR2 axis in mesangial cells via the FoxO1 signaling pathway [83, 105, 106]. Mesangial cells and podocytes express also TLR4 in the nephritic environment [17, 107]. Mesangial cells isolated from NZB/W mice with spontaneous autoimmunity express significantly higher levels of TLR4 and produce significantly more pro-inflammatory chemokines under baseline and LPS-stimulated conditions [108]. Also, necrotic cell debris is enriched with endogenous TLR ligands that stimulate cytokine release via TLR2/MyD88 depend on the way in mesangial cells, a finding that probably implies also to other intrarenal cell types expressing TLR2 [109]. Intrarenal TLR2 and TLR4 expression are also enhanced in kidneys from FcgammaRIIb-deficient mice suffering from cryoglobulinemic glomerulonephritis [107]. In MRL/lpr mice bacterial lipopeptide aggravates albuminuria in nephritic animals via TLR2-mediated activation of podocytes and endothelial cells at the glomerular filtration barrier [110]. These data help to understand how TLR2 and − 4 on infiltrating immune cells and parenchymal cells amplify local inflammation and barrier dysfunction by sensing extrinsic and intrinsic danger signals serving as TLR agonists.
An inflammatory microenvironment stimulates TLR3 expression in murine and human mesangial cells [17, 111]. Repetitive injections with the TLR3 agonist poly I:C RNA aggravates glomerulonephritis in MRL/lpr mice without affecting systemic autoantibody levels, hence, circulating viral double-stranded promote lupus nephritis mainly via local cytokine production by infiltrating macrophages and resident renal mesangial cells [22]. It is of note that mesangial cells from female animals express more TLR3 copies and secrete more IL-6 production upon TLR3 activation, providing another explanation for gender disparity in lupus. Indeed, deficiency of estrogen receptor-α in mesangial cells abrogates TLR3 signaling in mesangial cells [112]. Also, chloroquine attenuates TLR3-mediated type I interferon induction in human mesangial cells [113, 114]. TLR3 ligation in human mesangial cells promotes the secretion of the neutrophil chemoattractant CXCL1 and CXCL1 is also expressed in diagnostic biopsies from patients with lupus nephritis but not in biopsies of patients with IgA nephropathy [99]. These data support the concept of pseudo-antiviral immunity also at the tissue level because type I interferons from resident renal cells drive renal damage in experimental antibody-triggered glomerulonephritis [98, 114, 115]. Tubuloreticular inclusions represent a morphological imprint of local type I interferon activity and can be found only in lupus nephritis, viral nephropathies, and upon IFN-α treatment [116]. It is of note that type I interferon production in mesangial cells does not require TRIF or MyD88 when it involves non-TLR cytosolic RNA recognition receptors [117, 118].
In murine and human lupus nephritis plasmacytoid dendritic cells type recognize ssRNA, CpG DNA from bacteria and viruses as well as altered eucaryotic nucleic acids via TLR7 and − 9 [119], which induces the release of type I interferons and foster local and systemic immune responses via enhanced expression of costimulatory molecules [116, 119]. Specific deletion of plasmacytoid dendritic cells in a murine TLR7-driven lupus model (Tlr7.Tg) or a polygenic model (B6.Sle1.Sle3) attenuates not only systemic autoimmunity but also lupus nephritis [47]. Transient depletion of plasmacytoid dendritic cells can somewhat suppress glomerulonephritis [120]. Also, conventional dendritic cells express TLR7 in Sle1Tg7 mice. TLR7 overexpression is associated with an accumulation of CD11b + conventional dendritic cells inside the kidneys. Correcting the abnormal expression levels of TLR7 selectively in CD11c + conventional dendritic cells protected kidneys from autoimmune inflammation [39]. Thus, TLR7-mediated RNA sensing in conventional dendritic cells is an essential component in lupus nephritis in mice [39]. Similar studies were conducted to evaluate the alterations in circulating or possibly tissue infiltrating DCs in human lupus nephritis patients and found that conventional and plasmacytoid dendritic cells decreased in circulation enriched in both, the glomerular and tubulointerstitial, compartments in diagnostic kidney biopsies of SLE patients with lupus nephritis suggesting locally recruited DCs might play a pathogenic role in LN [121].
Circulating nucleic acids inside immune complexes can ligate TLR9 inside antigen-presenting cells (mononuclear phagocytes and B cells) to trigger pro-inflammatory cytokines as well as antibody secretion [10, 17]. TLR9 is absent in mesangial cells of MRL/lpr mice but other groups found TLR9 in tubular epithelial cells of NZBxNZW mice and in diagnostic biopsies of patients with lupus nephritis [17, 122,123,124]. In childhood-onset lupus nephritis injured podocytes stained positive for TLR9, while TLR9 was absent in podocytes of healthy control kidneys or during remission of lupus nephritis [125, 126]. BXSB-Yaa mice with SLE express TLR8 mRNA and protein in podocytes and TLR8 expression was found to correlate with podocyte injury and negatively correlate with albuminuria [127]. Overall, TLR7, − 8, and 9 drive lupus nephritis because endogenous (and during infections possibly exogenous) nucleic acid-based TLR agonists activate infiltrating antigen-presenting cells inside the nephritic kidney.
TLRs in immune tolerance break and germinal center formation
Other aspects of TLR involvement in autoimmune diseases are immune tolerance and germinal centers formation. Here we would like to provide readers a brief summary of these two aspects. Failure or loss of central and peripheral immune system results in the uncontrolled activation of self-reactive B and T cells and hence results in autoimmunity. Different TLRs have been shown to involve in this breakdown of tolerance [128,129,130]. For example, repeated stimulation of TLRs induces unresponsiveness to the same TLR ligand in cell lines [131, 132], B cells [133] and plasmacytoid DCs [134, 135].In some observations, it was found that repeated stimulation of a specific TLR can even result in cross-tolerance but also in enhanced responsiveness to other specific TLR ligands [131].This discrepancy might be due to differences in the heterogeneity and activation status of target cells [132].
Various TLRs expressed on B cells are not only capable of promoting antibody responses in the absence of T cells help [136, 137], but also promote germinal center (GC) responses very strongly [137,138,139,140].TLRs promote GC formation in an antigen oligomerization degree dependent manner [141](82). For example, viral particles with high oligomerized antigen proteins stimulate GC formation using TLR7 or TLR9, this response could be speculated as an adaptive mechanism to promote the generation of protective antibodies against viral infections [141].The TLR7 intrinsic expression is required for effective GC response to generate neutralizing antibodies against Friend virus as retrovirus [142] or LCMV clone 13 [143, 144].Therefore TLRs ability to promote GC formation evolved as a protective mechanism against viral infections however this could also promote lupus-like autoantibodies in a genetically susceptible background [140].
Conclusion
SLE is a consequence of genetic variants that promote a loss-of-tolerance and systemic autoimmunity against nuclear autoantigens. Half of the SLE patients suffer from some sort of renal involvement. Endogenous nucleic acids do not only serve as autoantigens but also as auto-adjuvants in the pathogenesis of lupus and lupus nephritis because they drive the activation of antigen-presenting cells, and type I interferon release, and consequently adaptive immunity and immune complex disease, e.g. lupus nephritis. Targeting these mechanisms is possible in several ways: 1) avoiding cell death, e.g. sunburns by applying sunscreens, 2) avoiding drugs that endorse hypomethylation of endogenous nucleic acids, e.g. dihydralazine, 3) scavenging nucleic acid debris [145], 4) blocking TLR activation with chloroquine or more specific endosomal TLR antagonists, 5) blocking co-stimulation or 6) blocking type I interferon signaling [146, 147]. Other endogenous triggers of innate immunity activate TLRs on also renal parenchymal cells, e.g. TLR3 in mesangial cells or TLR2 and TLR4 on endothelial cells and podocytes at the glomerular filtration barrier [148]. Altogether, TLRs elicit major contributions to the pathogenesis of SLE and lupus nephritis and their associated pathways remain attractive targets for therapeutic interventions.
Abbreviations
- AP1:
-
Activator protein 1
- APRIL:
-
A proliferation-inducing ligand
- BLyS:
-
B lymphocyte stimulator
- DC:
-
Dendritic cells
- dsDNA:
-
Double stranded deoxyribonucleic acid
- IC:
-
Immune complex
- IFN:
-
Type I interferon
- IKK:
-
IκB kinase (αβγε-apha,beta,gamma, epsilon)
- IRAKs:
-
IL-1R-associated kinases
- IRFs:
-
Interferon-regulatory factors
- LN:
-
Lupus nephritis
- MYD88:
-
Myeloid differentiation primary-response protein 88
- RIP:
-
Receptor-interacting protein kinases
- SLE:
-
Systemic lupus erythematosus
- ssRNA:
-
Single stranded ribonucleic acid
- TAK1:
-
Transforming growth factor beta-activated kinase 1
- TIR domain-containing adaptor protein:
-
Tirap toll-interleukin 1 receptor (TIR) domain-containing adaptor protein
- TLRs:
-
Toll like receptors
- TNF-α:
-
Tumor necrosis factor-α
- TRAFs:
-
TNF receptor-associated factors
- TRIF:
-
Related adaptor molecule TRAM
References
Weidenbusch M, Kulkarni OP, Anders HJ. The innate immune system in human systemic lupus erythematosus. Clin Sci (Lond). 2017;131:625–34.
Lleo A, Invernizzi P, Gao B, Podda M, Gershwin ME. Definition of human autoimmunity--autoantibodies versus autoimmune disease. Autoimmun Rev. 2010;9:A259–66.
Anders HJ. Toll-like receptors and danger signaling in kidney injury. J Am Soc Nephrol. 2010;21:1270–4.
Anders HJ, Schaefer L. Beyond tissue injury-damage-associated molecular patterns, toll-like receptors, and inflammasomes also drive regeneration and fibrosis. J Am Soc Nephrol. 2014;25:1387–400.
Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, Tron F, Gilbert D, Musette P. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol. 2009;183:6207–16.
Summers SA, Hoi A, Steinmetz OM, O'Sullivan KM, Ooi JD, Odobasic D, Akira S, Kitching AR, Holdsworth SR. TLR9 and TLR4 are required for the development of autoimmunity and lupus nephritis in pristane nephropathy. J Autoimmun. 2010;35:291–8.
Lee TP, Huang JC, Liu CJ, Chen HJ, Chen YH, Tsai YT, Yang W, Sun KH. Interactions of surface-expressed TLR-4 and endosomal TLR-9 accelerate lupus progression in anti-dsDNA antibody transgenic mice. Exp Biol Med (Maywood). 2014;239:715–23.
Liu B, Yang Y, Dai J, Medzhitov R, Freudenberg MA, Zhang PL, Li Z. TLR4 up-regulation at protein or gene level is pathogenic for lupus-like autoimmune disease. J Immunol. 2006;177:6880–8.
Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci U S A. 2013;110:E3311–20.
Lorenz G, Anders HJ. Neutrophils, dendritic cells, toll-like receptors, and interferon-alpha in lupus nephritis. Semin Nephrol. 2015;35:410–26.
Freeley SJ, Giorgini A, Tulone C, Popat RJ, Horsfield C, Robson MG. Toll-like receptor 2 or toll-like receptor 4 deficiency does not modify lupus in MRLlpr mice. PLoS One. 2013;8:e74112.
Komatsuda A, Wakui H, Iwamoto K, Ozawa M, Togashi M, Masai R, Maki N, Hatakeyama T, Sawada K. Up-regulated expression of toll-like receptors mRNAs in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Clin Exp Immunol. 2008;152:482–7.
Kirchner M, Sonnenschein A, Schoofs S, Schmidtke P, Umlauf VN, Mannhardt-Laakmann W. Surface expression and genotypes of toll-like receptors 2 and 4 in patients with juvenile idiopathic arthritis and systemic lupus erythematosus. Pediatr Rheumatol Online J. 2013;11:9.
Tsao JT, Hsieh SC, Chiang BL, Yu CL, Lin SC. Altered IL-10 and TNF-alpha production in peripheral blood mononuclear cells of systemic lupus erythematosus patients after toll-like receptor 2, 4, or 9 activation. Clin Exp Med. 2012;12:153–8.
Liu Y, Liao J, Zhao M, Wu H, Yung S, Chan TM, Yoshimura A, Lu Q. Increased expression of TLR2 in CD4(+) T cells from SLE patients enhances immune reactivity and promotes IL-17 expression through histone modifications. Eur J Immunol. 2015;45:2683–93.
Rupasree Y, Naushad SM, Rajasekhar L, Uma A, Kutala VK. Association of TLR4 (D299G, T399I), TLR9 -1486T>C, TIRAP S180L and TNF-alpha promoter (−1031, −863, −857) polymorphisms with risk for systemic lupus erythematosus among south Indians. Lupus. 2015;24:50–7.
Patole PS, Pawar RD, Lech M, Zecher D, Schmidt H, Segerer S, Ellwart A, Henger A, Kretzler M, Anders HJ. Expression and regulation of toll-like receptors in lupus-like immune complex glomerulonephritis of MRL-Fas(lpr) mice. Nephrol Dial Transplant. 2006;21:3062–73.
Hawn TR, Wu H, Grossman JM, Hahn BH, Tsao BP, Aderem A. A stop codon polymorphism of toll-like receptor 5 is associated with resistance to systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2005;102:10593–7.
Elloumi N, Fakhfakh R, Abida O, Ayadi L, Marzouk S, Hachicha H, Fourati M, Bahloul Z, Mhiri MN, Kammoun K, Masmoudi H. Relevant genetic polymorphisms and kidney expression of toll-like receptor (TLR)-5 and TLR-9 in lupus nephritis. Clin Exp Immunol. 2017;190:328–39.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3.
Sadanaga A, Nakashima H, Akahoshi M, Masutani K, Miyake K, Igawa T, Sugiyama N, Niiro H, Harada M. Protection against autoimmune nephritis in MyD88-deficient MRL/lpr mice. Arthritis Rheum. 2007;56:1618–28.
Patole PS, Grone HJ, Segerer S, Ciubar R, Belemezova E, Henger A, Kretzler M, Schlondorff D, Anders HJ. Viral double-stranded RNA aggravates lupus nephritis through toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–38.
Faridi MH, Khan SQ, Zhao W, Lee HW, Altintas MM, Zhang K, Kumar V, Armstrong AR, Carmona-Rivera C, Dorschner JM, Schnaith AM, Li X, Ghodke-Puranik Y, Moore E, Purmalek M, Irizarry-Caro J, Zhang T, Day R, Stoub D, Hoffmann V, Khaliqdina SJ, Bhargava P, Santander AM, Torroella-Kouri M, Issac B, Cimbaluk DJ, Zloza A, Prabhakar R, Deep S, Jolly M, Koh KH, Reichner JS, Bradshaw EM, Chen J, Moita LF, Yuen PS, Li Tsai W, Singh B, Reiser J, Nath SK, Niewold TB, Vazquez-Padron RI, Kaplan MJ, Gupta V. CD11b activation suppresses TLR-dependent inflammation and autoimmunity in systemic lupus erythematosus. J Clin Invest. 2017;127:1271–83.
Klonowska-Szymczyk A, Wolska A, Robak T, Cebula-Obrzut B, Smolewski P, Robak E. Expression of toll-like receptors 3, 7, and 9 in peripheral blood mononuclear cells from patients with systemic lupus erythematosus. Mediat Inflamm. 2014;2014:381418.
Nasr AS, Fawzy SM, Gheita TA, El-Khateeb E. Expression of Tolllike receptors 3 and 9 in Egyptian systemic lupus erythematosus patients. Z Rheumatol. 2016;75(5):502–7.
Wang CM, Chang SW, Wu YJ, Lin JC, Ho HH, Chou TC, Yang B, Wu J, Chen JY. Genetic variations in toll-like receptors (TLRs 3/7/8) are associated with systemic lupus erythematosus in a Taiwanese population. Sci Rep. 2014;4:3792.
Takagi H, Arimura K, Uto T, Fukaya T, Nakamura T, Choijookhuu N, Hishikawa Y, Sato K. Plasmacytoid dendritic cells orchestrate TLR7-mediated innate and adaptive immunity for the initiation of autoimmune inflammation. Sci Rep. 2016;6:24477.
Lech M, Anders HJ. The pathogenesis of lupus nephritis. J Am Soc Nephrol. 2013;24:1357–66.
Pawar RD, Patole PS, Zecher D, Segerer S, Kretzler M, Schlondorff D, Anders HJ. Toll-like receptor-7 modulates immune complex glomerulonephritis. J Am Soc Nephrol. 2006;17:141–9.
Kanno A, Tanimura N, Ishizaki M, Ohko K, Motoi Y, Onji M, Fukui R, Shimozato T, Yamamoto K, Shibata T, Sano S, Sugahara-Tobinai A, Takai T, Ohto U, Shimizu T, Saitoh S, Miyake K. Targeting cell surface TLR7 for therapeutic intervention in autoimmune diseases. Nat Commun. 2015;6:6119.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28.
Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, Krug A. Requirement of toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. 2008;58:1107–15.
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol. 2010;184:1840–8.
Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–5.
Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97:6670–5.
Croker BP, Gilkeson G, Morel L. Genetic interactions between susceptibility loci reveal epistatic pathogenic networks in murine lupus. Genes Immun. 2003;4:575–85.
Zhuang H, Han S, Xu Y, Li Y, Wang H, Yang LJ, Reeves WH. Toll-like receptor 7-stimulated tumor necrosis factor alpha causes bone marrow damage in systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:140–51.
Patole PS, Pawar RD, Lichtnekert J, Lech M, Kulkarni OP, Ramanjaneyulu A, Segerer S, Anders HJ. Coactivation of toll-like receptor-3 and -7 in immune complex glomerulonephritis. J Autoimmun. 2007;29:52–9.
Celhar T, Hopkins R, Thornhill SI, De Magalhaes R, Hwang SH, Lee HY, Yasuga H, Jones LA, Casco J, Lee B, Thamboo TP, Zhou XJ, Poidinger M, Connolly JE, Wakeland EK, Fairhurst AM. RNA sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc Natl Acad Sci U S A. 2015;112:E6195–204.
Teichmann LL, Ols ML, Kashgarian M, Reizis B, Kaplan DH, Shlomchik MJ. Dendritic cells in lupus are not required for activation of T and B cells but promote their expansion, resulting in tissue damage. Immunity. 2010;33:967–78.
Teichmann LL, Schenten D, Medzhitov R, Kashgarian M, Shlomchik MJ. Signals via the adaptor MyD88 in B cells and DCs make distinct and synergistic contributions to immune activation and tissue damage in lupus. Immunity. 2013;38:528–40.
Ramirez-Ortiz ZG, Prasad A, Griffith JW, Pendergraft WF 3rd, Cowley GS, Root DE, Tai M, Luster AD, El Khoury J, Hacohen N, Means TK. The receptor TREML4 amplifies TLR7-mediated signaling during antiviral responses and autoimmunity. Nat Immunol. 2015;16:495–504.
Nishizumi H, Taniuchi I, Yamanashi Y, Kitamura D, Ilic D, Mori S, Watanabe T, Yamamoto T. Impaired proliferation of peripheral B cells and indication of autoimmune disease in lyn-deficient mice. Immunity. 1995;3:549–60.
Hua Z, Gross AJ, Lamagna C, Ramos-Hernandez N, Scapini P, Ji M, Shao H, Lowell CA, Hou B, DeFranco AL. Requirement for MyD88 signaling in B cells and dendritic cells for germinal center anti-nuclear antibody production in Lyn-deficient mice. J Immunol. 2014;192:875–85.
Lech M, Kantner C, Kulkarni OP, Ryu M, Vlasova E, Heesemann J, Anz D, Endres S, Kobayashi KS, Flavell RA, Martin J, Anders HJ. Interleukin-1 receptor-associated kinase-M suppresses systemic lupus erythematosus. Ann Rheum Dis. 2011;70:2207–17.
Lech M, Skuginna V, Kulkarni OP, Gong J, Wei T, Stark RW, Garlanda C, Mantovani A, Anders HJ. Lack of SIGIRR/TIR8 aggravates hydrocarbon oil-induced lupus nephritis. J Pathol. 2010;220:596–607.
Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D'Agati V, Elkon KB, Reizis B. Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med. 2014;211:1969–76.
Weindel CG, Richey LJ, Bolland S, Mehta AJ, Kearney JF, Huber BT. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy. 2015;11:1010–24.
Dominguez-Villar M, Gautron AS, de Marcken M, Keller MJ, Hafler DA. TLR7 induces anergy in human CD4(+) T cells. Nat Immunol. 2015;16:118–28.
Celhar T, Fairhurst AM. Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol. 2014;5:265.
Applequist SE, Wallin RP, Ljunggren HG. Variable expression of toll-like receptor in murine innate and adaptive immune cell lines. Int Immunol. 2002;14:1065–74.
Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–7.
Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature. 2002;416:603–7.
Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199:1631–40.
Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, Rahman ZS. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol. 2014;193:4400–14.
Hwang SH, Lee H, Yamamoto M, Jones LA, Dayalan J, Hopkins R, Zhou XJ, Yarovinsky F, Connolly JE, Curotto de Lafaille MA, Wakeland EK, Fairhurst AM. B cell TLR7 expression drives anti-RNA autoantibody production and exacerbates disease in systemic lupus erythematosus-prone mice. J Immunol. 2012;189:5786–96.
Lyn-Cook BD, Xie C, Oates J, Treadwell E, Word B, Hammons G, Wiley K. Increased expression of toll-like receptors (TLRs) 7 and 9 and other cytokines in systemic lupus erythematosus (SLE) patients: ethnic differences and potential new targets for therapeutic drugs. Mol Immunol. 2014;61:38–43.
Pacheco GV, Novelo Noh IB, Velasco Cardenas RM, Angulo Ramirez AV, Lopez Villanueva RF, Quintal Ortiz IG, Alonso Salomon LG, Ruz NP, Rivero Cardenas NA. Expression of TLR-7, MyD88, NF-kB, and INF-alpha in B lymphocytes of Mayan women with systemic lupus erythematosus in Mexico. Front Immunol. 2016;7:22.
Kawasaki A, Furukawa H, Kondo Y, Ito S, Hayashi T, Kusaoi M, Matsumoto I, Tohma S, Takasaki Y, Hashimoto H, Sumida T, Tsuchiya N. TLR7 single-nucleotide polymorphisms in the 3′ untranslated region and intron 2 independently contribute to systemic lupus erythematosus in Japanese women: a case-control association study. Arthritis Res Ther. 2011;13:R41.
Lee YH, Choi SJ, Ji JD, Song GG. Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus. 2016;25:593–601.
Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen JY, Yang W, Wong M, Kawasaki A, Tsuchiya N, Sumida T, Kawaguchi Y, Howe HS, Mok MY, Bang SY, Liu FL, Chang DM, Takasaki Y, Hashimoto H, Harley JB, Guthridge JM, Grossman JM, Cantor RM, Song YW, Bae SC, Chen S, Hahn BH, Lau YL, Tsao BP. Sex-specific association of X-linked toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2010;107:15838–43.
Garcia-Ortiz H, Velazquez-Cruz R, Espinosa-Rosales F, Jimenez-Morales S, Baca V, Orozco L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann Rheum Dis. 2010;69:1861–5.
Kelley J, Johnson MR, Alarcon GS, Kimberly RP, Edberg JC. Variation in the relative copy number of the TLR7 gene in patients with systemic lupus erythematosus and healthy control subjects. Arthritis Rheum. 2007;56:3375–8.
Khoryati L, Augusto JF, Shipley E, Contin-Bordes C, Douchet I, Mitrovic S, Truchetet ME, Lazaro E, Duffau P, Couzi L, Jacquemin C, Barnetche T, Vacher P, Schaeverbeke T, Blanco P, Richez C. IgE inhibits toll-like receptor 7- and toll-like receptor 9-mediated expression of interferon-alpha by Plasmacytoid dendritic cells in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2016;68:2221–31.
Green NM, Moody K-S, Debatis M, Marshak-Rothstein A. Activation of autoreactive B cells by endogenous TLR7 and TLR3 RNA ligands. J Biol Chem. 2012;287:39789–99.
Saito K, Kukita K, Kutomi G, Okuya K, Asanuma H, Tabeya T, Naishiro Y, Yamamoto M, Takahashi H, Torigoe T, Nakai A, Shinomura Y, Hirata K, Sato N, Tamura Y. Heat shock protein 90 associates with toll-like receptors 7/9 and mediates self-nucleic acid recognition in SLE. Eur J Immunol. 2015;45:2028–41.
Anders HJ, Krug A, Pawar RD. Molecular mimicry in innate immunity? The viral RNA recognition receptor TLR7 accelerates murine lupus. Eur J Immunol. 2008;38:1795–9.
Cervantes JL, La Vake CJ, Weinerman B, Luu S, O'Connell C, Verardi PH, Salazar JC. Human TLR8 is activated upon recognition of Borrelia burgdorferi RNA in the phagosome of human monocytes. J Leukoc Biol. 2013;94:1231–41.
Diebold SS. Recognition of viral single-stranded RNA by toll-like receptors. Adv Drug Deliv Rev. 2008;60:813–23.
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–9.
Umiker BR, Andersson S, Fernandez L, Korgaokar P, Larbi A, Pilichowska M, Weinkauf CC, Wortis HH, Kearney JF, Imanishi-Kari T. Dosage of X-linked toll-like receptor 8 determines gender differences in the development of systemic lupus erythematosus. Eur J Immunol. 2014;44:1503–16.
Demaria O, Pagni PP, Traub S, de Gassart A, Branzk N, Murphy AJ, Valenzuela DM, Yancopoulos GD, Flavell RA, Alexopoulou L. TLR8 deficiency leads to autoimmunity in mice. J Clin Invest. 2010;120:3651–62.
Desnues B, Macedo AB, Roussel-Queval A, Bonnardel J, Henri S, Demaria O, Alexopoulou L. TLR8 on dendritic cells and TLR9 on B cells restrain TLR7-mediated spontaneous autoimmunity in C57BL/6 mice. Proc Natl Acad Sci U S A. 2014;111:1497–502.
Young NA, Wu LC, Burd CJ, Friedman AK, Kaffenberger BH, Rajaram MV, Schlesinger LS, James H, Shupnik MA, Jarjour WN. Estrogen modulation of endosome-associated toll-like receptor 8: an IFNalpha-independent mechanism of sex-bias in systemic lupus erythematosus. Clin Immunol. 2014;151:66–77.
Gysler SM, Mulla MJ, Guerra M, Brosens JJ, Salmon JE, Chamley LW, Abrahams VM. Antiphospholipid antibody-induced miR-146a-3p drives trophoblast interleukin-8 secretion through activation of toll-like receptor 8. Mol Hum Reprod. 2016;22(7):465–74.
Guo Y, Chai Q, Zhao Y, Li P, Qiao J, Huang J. Increased activation of toll-like receptors-7 and -8 of peripheral blood mononuclear cells and upregulated serum cytokines in patients with pediatric systemic lupus erythematosus. Int J Clin Exp Med. 2015;8:20472–80.
Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–72.
Lindau D, Mussard J, Rabsteyn A, Ribon M, Kotter I, Igney A, Adema GJ, Boissier MC, Rammensee HG, Decker P. TLR9 independent interferon alpha production by neutrophils on NETosis in response to circulating chromatin, a key lupus autoantigen. Ann Rheum Dis. 2014;73:2199–207.
Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, Wright T, Punaro M, Palucka K, Guiducci C, Banchereau J, Pascual V. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. 2016;213:697–713.
Monteith AJ, Kang S, Scott E, Hillman K, Rajfur Z, Jacobson K, Costello MJ, Vilen BJ. Defects in lysosomal maturation facilitate the activation of innate sensors in systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2016;113:E2142–51.
Ronnblom L, Pascual V. The innate immune system in SLE: type I interferons and dendritic cells. Lupus. 2008;17:394–9.
Migliorini A, Anders HJ. A novel pathogenetic concept-antiviral immunity in lupus nephritis. Nat Rev Nephrol. 2012;8:183–9.
Santiago-Raber ML, Baudino L, Izui S. Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun. 2009;33(3-4):231–8.
Chang NH, Li TT, Kim JJ, Landolt-Marticorena C, Fortin PR, Gladman DD, Urowitz MB, Wither JE. Interferon-alpha induces altered transitional B cell signaling and function in systemic lupus erythematosus. J Autoimmun. 2015;58:100–10.
Rodriguez-Pla A, Patel P, Maecker HT, Rossello-Urgell J, Baldwin N, Bennett L, Cantrell V, Baisch J, Punaro M, Gotte A, Nassi L, Wright T, Palucka AK, Banchereau J, Pascual V. IFN priming is necessary but not sufficient to turn on a migratory dendritic cell program in lupus monocytes. J Immunol. 2014;192:5586–98.
Cai X, Xu Y, Kim YM, Loureiro J, Huang Q. PIKfyve, a class III lipid kinase, is required for TLR-induced type I IFN production via modulation of ATF3. J Immunol. 2014;192:3383–9.
Bezalel S, Guri KM, Elbirt D, Asher I, Sthoeger ZM. Type I interferon signature in systemic lupus erythematosus. Isr Med Assoc J. 2014;16:246–9.
Petri M, Singh S, Tesfasyone H, Dedrick R, Fry K, Lal P, Williams G, Bauer J, Gregersen P, Behrens T, Baechler E. Longitudinal expression of type I interferon responsive genes in systemic lupus erythematosus. Lupus. 2009;18:980–9.
Patole PS, Zecher D, Pawar RD, Grone HJ, Schlondorff D, Anders HJ. G-rich DNA suppresses systemic lupus. J Am Soc Nephrol. 2005;16:3273–80.
Bossaller L, Christ A, Pelka K, Nundel K, Chiang PI, Pang C, Mishra N, Busto P, Bonegio RG, Schmidt RE, Latz E, Marshak-Rothstein A. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in Pristane-induced murine lupus. J Immunol. 2016;197(4):1044–53.
Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–31.
Fukui R, Saitoh S, Kanno A, Onji M, Shibata T, Ito A, Onji M, Matsumoto M, Akira S, Yoshida N, Miyake K. Unc93B1 restricts systemic lethal inflammation by orchestrating toll-like receptor 7 and 9 trafficking. Immunity. 2011;35:69–81.
Fukui R, Saitoh S, Matsumoto F, Kozuka-Hata H, Oyama M, Tabeta K, Beutler B, Miyake K. Unc93B1 biases toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–50.
Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, Hudkins KL, Alpers CE, Liggitt D, Rawlings DJ. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. 2014;192:4525–32.
Capolunghi F, Rosado MM, Cascioli S, Girolami E, Bordasco S, Vivarelli M, Ruggiero B, Cortis E, Insalaco A, Fanto N, Gallo G, Nucera E, Loiarro M, Sette C, De Santis R, Carsetti R, Ruggiero V. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatology (Oxford). 2010;49:2281–9.
Chauhan SK, Singh VV, Rai R, Rai M, Rai G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J Clin Immunol. 2013;33:954–64.
Pawar RD, Patole PS, Ellwart A, Lech M, Segerer S, Schlondorff D, Anders HJ. Ligands to nucleic acid-specific toll-like receptors and the onset of lupus nephritis. J Am Soc Nephrol. 2006;17:3365–73.
Pawar RD, Patole PS, Wornle M, Anders HJ. Microbial nucleic acids pay a toll in kidney disease. Am J Physiol Renal Physiol. 2006;291:F509–16.
Imaizumi T, Aizawa T, Segawa C, Shimada M, Tsuruga K, Kawaguchi S, Matsumiya T, Yoshida H, Joh K, Tanaka H. Toll-like receptor 3 signaling contributes to the expression of a neutrophil chemoattractant, CXCL1 in human mesangial cells. Clin Exp Nephrol. 2015;19(5):761–70.
Timmerman JJ, Van Gijlswijk-Janssen DJ, Van Der Kooij SW, Van Es LA, Daha MR. Antigen-antibody complexes enhance the production of complement component C3 by human mesangial cells. J Am Soc Nephrol. 1997;8:1257–65.
Lorenz G, Lech M, Anders HJ. Toll-like receptor activation in the pathogenesis of lupus nephritis. Clin Immunol. 2017;185:86–94.
Ramaiah SK, Gunthner R, Lech M, Anders HJ. Toll-like receptor and accessory molecule mRNA expression in humans and mice as well as in murine autoimmunity, transient inflammation, and progressive fibrosis. Int J Mol Sci. 2013;14:13213–30.
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7.
Wirestam L, Schierbeck H, Skogh T, Gunnarsson I, Ottosson L, Erlandsson-Harris H, Wettero J, Sjowall C. Antibodies against high mobility group box protein-1 (HMGB1) versus other anti-nuclear antibody fine-specificities and disease activity in systemic lupus erythematosus. Arthritis Res Ther. 2015;17:338.
Qing X, Pitashny M, Thomas DB, Barrat FJ, Hogarth MP, Putterman C. Pathogenic anti-DNA antibodies modulate gene expression in mesangial cells: involvement of HMGB1 in anti-DNA antibody-induced renal injury. Immunol Lett. 2008;121:61–73.
Feng XJ, Wu C, Yang GF, Liu QJ, Liu JX, Hao J, Xing LL, Yang M, Liu SX. TLR2 plays a critical role in HMGB1-induced glomeruli cell proliferation through the FoxO1 signaling pathway in lupus nephritis. J Interferon Cytokine Res. 2016;36:258–66.
Banas MC, Banas B, Hudkins KL, Wietecha TA, Iyoda M, Bock E, Hauser P, Pippin JW, Shankland SJ, Smith KD, Stoelcker B, Liu G, Grone HJ, Kramer BK, Alpers CE. TLR4 links podocytes with the innate immune system to mediate glomerular injury. J Am Soc Nephrol. 2008;19:704–13.
Ka SM, Cheng CW, Shui HA, Wu WM, Chang DM, Lin YC, Chen A. Mesangial cells of lupus-prone mice are sensitive to chemokine production. Arthritis Res Ther. 2007;9:R67.
Lichtnekert J, Vielhauer V, Zecher D, Kulkarni OP, Clauss S, Segerer S, Hornung V, Mayadas TN, Beutler B, Akira S, Anders HJ. Trif is not required for immune complex glomerulonephritis: dying cells activate mesangial cells via Tlr2/Myd88 rather than Tlr3/Trif. Am J Physiol Renal Physiol. 2009;296:F867–74.
Pawar RD, Castrezana-Lopez L, Allam R, Kulkarni OP, Segerer S, Radomska E, Meyer TN, Schwesinger C-M, Akis N, Gröne H-J, Anders H-J. Bacterial lipopeptide triggers massive albuminuria in murine lupus nephritis by activating toll-like receptor 2 at the glomerular filtration barrier. Immunology. 2009;128:e206–21.
Imaizumi T, Aizawa T, Hayakari R, Xing F, Meng P, Tsuruga K, Matsumiya T, Yoshida H, Wang L, Tatsuta T, Tanaka H. Tumor necrosis factor-alpha synergistically enhances polyinosinic-polycytidylic acid-induced toll-like receptor 3 signaling in cultured normal human mesangial cells: possible involvement in the pathogenesis of lupus nephritis. Clin Exp Nephrol. 2015;19:75–81.
Svenson J, Cunningham M, Dasgupta S, Gilkeson GS. Estrogen receptor alpha modulates mesangial cell responses to toll-like receptor ligands. Am J Med Sci. 2014;348:492–500.
Imaizumi T, Hayakari R, Matsumiya T, Yoshida H, Tsuruga K, Watanabe S, Kawaguchi S, Tanaka H. Chloroquine attenuates TLR3/IFN-beta signaling in cultured normal human mesangial cells: a possible protective effect against renal damage in lupus nephritis. Mod Rheumatol. 2017;27:1004–9.
Flur K, Allam R, Zecher D, Kulkarni OP, Lichtnekert J, Schwarz M, Beutler B, Vielhauer V, Anders HJ. Viral RNA induces type I interferon-dependent cytokine release and cell death in mesangial cells via melanoma-differentiation-associated gene-5: implications for viral infection-associated glomerulonephritis. Am J Pathol. 2009;175:2014–22.
Fairhurst AM, Xie C, Fu Y, Wang A, Boudreaux C, Zhou XJ, Cibotti R, Coyle A, Connolly JE, Wakeland EK, Mohan C. Type I interferons produced by resident renal cells may promote end-organ disease in autoantibody-mediated glomerulonephritis. J Immunol. 2009;183:6831–8.
Anders HJ, Lichtnekert J, Allam R. Interferon-alpha and -beta in kidney inflammation. Kidney Int. 2010;77:848–54.
Allam R, Lichtnekert J, Moll AG, Taubitz A, Vielhauer V, Anders HJ. Viral RNA and DNA trigger common antiviral responses in mesangial cells. J Am Soc Nephrol. 2009;20:1986–96.
Allam R, Pawar RD, Kulkarni OP, Hornung V, Hartmann G, Segerer S, Akira S, Endres S, Anders HJ. Viral 5′-triphosphate RNA and non-CpG DNA aggravate autoimmunity and lupus nephritis via distinct TLR-independent immune responses. Eur J Immunol. 2008;38:3487–98.
Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–41.
Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, Colonna M. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–91.
Fiore N, Castellano G, Blasi A, Capobianco C, Loverre A, Montinaro V, Netti S, Torres D, Manno C, Grandaliano G, Ranieri E, Schena FP, Gesualdo L. Immature myeloid and plasmacytoid dendritic cells infiltrate renal tubulointerstitium in patients with lupus nephritis. Mol Immunol. 2008;45:259–65.
Benigni A, Caroli C, Longaretti L, Gagliardini E, Zoja C, Galbusera M, Moioli D, Romagnani P, Tincani A, Andreoli L, Remuzzi G. Involvement of renal tubular toll-like receptor 9 in the development of tubulointerstitial injury in systemic lupus. Arthritis Rheum. 2007;56:1569–78.
Papadimitraki ED, Tzardi M, Bertsias G, Sotsiou E, Boumpas DT. Glomerular expression of toll-like receptor-9 in lupus nephritis but not in normal kidneys: implications for the amplification of the inflammatory response. Lupus. 2009;18:831–5.
Frieri M, Samih MA, Dzhindzhikhashvili M, Liu H, Balsam L, Rubinstein S. Toll-like receptor 9 and vascular endothelial growth factor levels in human kidneys from lupus nephritis patients. J Nephrol. 2012;25:1041–6.
Machida H, Ito S, Hirose T, Takeshita F, Oshiro H, Nakamura T, Mori M, Inayama Y, Yan K, Kobayashi N, Yokota S. Expression of toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis. Nephrol Dial Transplant. 2010;25:2530–7.
Conti F, Spinelli FR, Truglia S, Miranda F, Alessandri C, Ceccarelli F, Bombardieri M, Giannakakis K, Valesini G. Kidney expression of toll like receptors in lupus nephritis: quantification and Clinicopathological correlations. Mediat Inflamm. 2016;2016:7697592.
Kimura J, Ichii O, Miyazono K, Nakamura T, Horino T, Otsuka-Kanazawa S, Kon Y. Overexpression of toll-like receptor 8 correlates with the progression of podocyte injury in murine autoimmune glomerulonephritis. Sci Rep. 2014;4:7290.
Ehlers M, Ravetch JV. Opposing effects of toll-like receptor stimulation induce autoimmunity or tolerance. Trends Immunol. 2007;28:74–9.
Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6:823–35.
Marshak-Rothstein A. Tolling for autoimmunity-prime time for 7. Immunity. 2006;25:397–9.
Broad A, Kirby JA, Jones DE. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120:103–11.
Dalpke AH, Lehner MD, Hartung T, Heeg K. Differential effects of CpG-DNA in toll-like receptor-2/−4/−9 tolerance and cross-tolerance. Immunology. 2005;116:203–12.
Rui L, Vinuesa CG, Blasioli J, Goodnow CC. Resistance to CpG DNA-induced autoimmunity through tolerogenic B cell antigen receptor ERK signaling. Nat Immunol. 2003;4:594–600.
Abe M, Tokita D, Raimondi G, Thomson AW. Endotoxin modulates the capacity of CpG-activated liver myeloid DC to direct Th1-type responses. Eur J Immunol. 2006;36:2483–93.
Moseman EA, Liang X, Dawson AJ, Panoskaltsis-Mortari A, Krieg AM, Liu YJ, Blazar BR, Chen W. Human plasmacytoid dendritic cells activated by CpG oligodeoxynucleotides induce the generation of CD4+CD25+ regulatory T cells. J Immunol. 2004;173:4433–42.
Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and toll-like receptors. Immunity. 2007;27:64–75.
Eckl-Dorna J, Batista FD. BCR-mediated uptake of antigen linked to TLR9 ligand stimulates B-cell proliferation and antigen-specific plasma cell formation. Blood. 2009;113:3969–77.
Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–7.
Rookhuizen DC, DeFranco AL. Toll-like receptor 9 signaling acts on multiple elements of the germinal center to enhance antibody responses. Proc Natl Acad Sci U S A. 2014;111:E3224–33.
DeFranco AL. Germinal centers and autoimmune disease in humans and mice. Immunol Cell Biol. 2016;94:918–24.
Hou B, Saudan P, Ott G, Wheeler ML, Ji M, Kuzmich L, Lee LM, Coffman RL, Bachmann MF, DeFranco AL. Selective utilization of toll-like receptor and MyD88 signaling in B cells for enhancement of the antiviral germinal center response. Immunity. 2011;34:375–84.
Browne EP. Toll-like receptor 7 controls the anti-retroviral germinal center response. PLoS Pathog. 2011;7:e1002293.
Walsh KB, Teijaro JR, Zuniga EI, Welch MJ, Fremgen DM, Blackburn SD, von Tiehl KF, Wherry EJ, Flavell RA, Oldstone MB. Toll-like receptor 7 is required for effective adaptive immune responses that prevent persistent virus infection. Cell Host Microbe. 2012;11:643–53.
Clingan JM, Matloubian M. B cell-intrinsic TLR7 signaling is required for optimal B cell responses during chronic viral infection. J Immunol. 2013;191:810–8.
Holl EK, Shumansky KL, Borst LB, Burnette AD, Sample CJ, Ramsburg EA, Sullenger BA. Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proc Natl Acad Sci U S A. 2016;113:9728–33.
Liu Y, Anders HJ. Lupus nephritis: from pathogenesis to targets for biologic treatment. Nephron Clin Pract. 2014;128:224–31.
Anders HJ, Rovin B. A pathophysiology-based approach to the diagnosis and treatment of lupus nephritis. Kidney Int. 2016;90:493–501.
Allam R, Anders HJ. The role of innate immunity in autoimmune tissue injury. Curr Opin Rheumatol. 2008;20:538–44.
Kaiser R, Tang LF, Taylor KE, Sterba K, Nititham J, Brown EE, Edberg JC, McGwin G Jr, Alarcon GS, Ramsey-Goldman R, Reveille JD, Vila LM, Petri M, Rauch J, Miller E, Mesznik K, Kwok PY, Kimberly RP, Salmon JE, Criswell LA. A polymorphism in TLR2 is associated with arterial thrombosis in a multiethnic population of patients with systemic lupus erythematosus. Arthritis Rheumatol. 2014;66:1882–7.
Devaraju P, Gulati R, Antony PT, Mithun CB, Negi VS. Susceptibility to SLE in south Indian Tamils may be influenced by genetic selection pressure on TLR2 and TLR9 genes. Mol Immunol. 2015;64:123–6.
Laska MJ, Troldborg A, Hansen B, Stengaard-Pedersen K, Junker P, Nexo BA, Voss A. Polymorphisms within toll-like receptors are associated with systemic lupus erythematosus in a cohort of Danish females. Rheumatology (Oxford, England). 2014;53:48–55.
Hu W, Wu S, Zhang Y, Sigdel KR, Lin Y, Zhong H. Association between toll-like receptor 4 polymorphisms and systemic lupus erythematosus susceptibility: a Meta-analysis. Biomed Res Int. 2016;2016:7842587.
Bogaczewicz A, Sobow T, Bogaczewicz J, Kaleta B, Sysa-Jedrzejowska A, Robak E, Lukaszkiewicz J, Dariusz S, Wozniacka A. Toll-like receptor 4 gene polymorphism 1196 C/T does not influence the risk of neuropsychiatric systemic lupus erythematosus in polish population--a preliminary report. Lupus. 2013;22:1504–8.
Wen SJ, Wu FY, Fang L, Liu H, Zheng WJ, Lin YK. Associations of genetic polymorphisms of TLR5, TLR9 and transduction molecules in MyD88 signaling pathway with systemic lupus erythematosus in Zhuang and Han ethnics of Guangxi province, China. Int J Clin Exp Med. 2015;8:7872–80.
Wang Y, Ewart D, Crabtree JN, Yamamoto A, Baechler EC, Fazeli P, Peterson EJ. PTPN22 variant R620W is associated with reduced toll-like receptor 7-induced type I interferon in systemic lupus erythematosus. Arthritis Rheumatol. 2015;67:2403–14.
Enevold C, Nielsen CH, Jacobsen RS, Hermansen ML, Molbo D, Avlund K, Bendtzen K, Jacobsen S. Single nucleotide polymorphisms in genes encoding toll-like receptors 7, 8 and 9 in Danish patients with systemic lupus erythematosus. Mol Biol Rep. 2014;41:5755–63.
Deng Y, Zhao J, Sakurai D, Kaufman KM, Edberg JC, Kimberly RP, Kamen DL, Gilkeson GS, Jacob CO, Scofield RH, Langefeld CD, Kelly JA, Ramsey-Goldman R, Petri MA, Reveille JD, Vila LM, Alarcon GS, Vyse TJ, Pons-Estel BA, Freedman BI, Gaffney PM, Sivils KM, James JA, Gregersen PK, Anaya JM, Niewold TB, Merrill JT, Criswell LA, Stevens AM, Boackle SA, Cantor RM, Chen W, Grossman JM, Hahn BH, Harley JB, Alarcomicronn-Riquelme ME, Brown EE, Tsao BP. MicroRNA-3148 modulates allelic expression of toll-like receptor 7 variant associated with systemic lupus erythematosus. PLoS Genet. 2013;9:e1003336.
Hu LY, Cheng Z, Zhang B, Yin Q, Zhu XW, Zhao PP, Han MY, Wang XB, Zheng HF. Associations between PTPN22 and TLR9 polymorphisms and systemic lupus erythematosus: a comprehensive meta-analysis. Arch Dermatol Res. 2017;309:461–77.
Yusuf JH, Kaliyaperumal D, Jayaraman M, Ramanathan G, Devaraju P. Genetic selection pressure in TLR9 gene may enforce risk for SLE in Indian Tamils. Lupus. 2017;26:307–10.
Shahin RM, El Khateeb E, Khalifa RH, El Refai RM. Contribution of toll-like receptor 9 gene single-nucleotide polymorphism to systemic lupus erythematosus in Egyptian patients. Immunol Investig. 2016;45:235–42.
Panda AK, Pattanaik SS, Tripathy R, Das BK. TLR-9 promoter polymorphisms (T-1237C and T-1486C) are not associated with systemic lupus erythematosus: a case control study and meta-analysis. Hum Immunol. 2013;74:1672–8.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (AN372/24–1) and by the European Union’s Horizon 2020 research and innovation program under grant agreement No. 668036 (RELENT). The views expressed here are the responsibility of the author(s) only. The EU Commission takes no responsibility for any use made of the information set out.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Financial disclosure and COI statements
HJA received consultancy fees from AstraZeneca.
Author information
Authors and Affiliations
Contributions
SKD and HJA drafted, read, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Devarapu, S.K., Anders, HJ. Toll-like receptors in lupus nephritis. J Biomed Sci 25, 35 (2018). https://doi.org/10.1186/s12929-018-0436-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-018-0436-2