
Abstract
Plasma free fatty acids levels are increased in subjects with obesity and type 2 diabetes, playing detrimental roles in the pathogenesis of atherosclerosis and cardiovascular diseases. Increasing evidence showing that dysfunction of the vascular endothelium, the inner lining of the blood vessels, is the key player in the pathogenesis of atherosclerosis. In this review, we aimed to summarize the roles and the underlying mechanisms using the evidence collected from clinical and experimental studies about free fatty acid-mediated endothelial dysfunction. Because of the multifaceted roles of plasma free fatty acids in mediating endothelial dysfunction, elevated free fatty acid level is now considered as an important link in the onset of endothelial dysfunction due to metabolic syndromes such as diabetes and obesity. Free fatty acid-mediated endothelial dysfunction involves several mechanisms including impaired insulin signaling and nitric oxide production, oxidative stress, inflammation and the activation of the renin-angiotensin system and apoptosis in the endothelial cells. Therefore, targeting the signaling pathways involved in free fatty acid-induced endothelial dysfunction could serve as a preventive approach to protect against the occurrence of endothelial dysfunction and the subsequent complications such as atherosclerosis.
Similar content being viewed by others


Background
Cardiovascular diseases (CVDs), particularly coronary heart disease (CHD), account for the major causes of mortality worldwide [1]. The presence of atherosclerosis is a very common characteristic in patients with CHD [1], and that endothelial dysfunction (ED) is suggested as one of the early events in the pathogenesis of atherosclerotic progression. The vascular endothelium is a tightly regulated organ that forms a vast interface between the blood and neighboring tissues, and is consisted of a monolayer of endothelial cells (ECs). It regulates a wide range of functions, including the maintenance of the balance between vasodilation and vasoconstriction, the maintenance of thrombosis and hemostasis, and other inflammatory responses in order to regulate the normal functioning in the circulatory system. However, various harmful stimuli such as oxidative stress and inflammation can alter the normal endothelium functioning and lead to the onset of ED. ED can be expressed as an imbalanced ratio of vasodilation to vasoconstriction induced by the endothelium, and such impairment in vasodilation is mainly due to the diminished release of nitric oxide (NO), the most important vasodilatory agent that is released by the ECs. Although impaired NO production is the main characteristic feature of ED, the production and/or utilization of other vasodilatory agents such as prostacyclin (PGI2) and bradykinin are also compromised in the context of ED.
Free fatty acids (FFAs), or non-esterified fatty acids (NEFAs), are well-known risk factors of CVDs [2], and are closely related to the events of metabolic syndromes (MetS), such as obesity and type 2 diabetes mellitus (T2DM) [3]. Recent studies have shown that FFAs not only are the major causes of insulin resistance [4, 5], but they are also responsible for inducing inflammatory events in the tissues targeted by insulin, such as ECs, liver and skeletal muscle [3, 6]. Therefore, elevated FFAs in blood are considered as an important link between insulin resistance, inflammation, obesity, T2DM and hypertension (HTN) [3]. The impairment of insulin-mediated glucose uptake is correlated to the circulating FFA levels, and such resistance to insulin might be due to FFA-mediated inactivation of phosphoinositide 3-kinase (PI3K) [7]. Interestingly, cancer-related up-regulation of FFAs was also reported in several types of cancer in many previous studies [8,9,10,11].
In addition, a growing body of evidence has suggested the important role of FFAs in mediating ED. Specifically, insulin resistance, oxidative stress, and inflammatory burdens account for the substantial causes of FFA-induced ED [12,13,14]. Generally, diabetes and other metabolic states could give rise to elevated FFAs, which in turn impose a direct effect on transcription factors that trigger inflammation and oxidative stress in the endothelium [15]. Furthermore, FFAs also facilitate apoptosis/necroptosis of the ECs [16,17,18], and mediate many deleterious effects on endothelial progenitor cells (EPCs) [19].
Biology of the endothelium and endothelial dysfunction
Distribution, structure, and functions of the endothelium
The endothelium is a composition of a monolayer of ECs which lines on the inner surface of the vascular lumen between flowing blood and vascular smooth muscle cells (VSMCs) [20]. The endothelium has a thickness of ≤1 μm and covers a surface area of 4000 m2 throughout the whole circulation [21]. ECs are multifunctional; they are responsible for a wide range of vital functions, including the maintenance of vascular tone, blood fluidity and permeability [21, 22]. The endothelium is also responsible for the regulation of inflammatory responses and signals for the regulation of hemostasis/thrombosis, fibrinolysis and angiogenesis [20, 21, 23]. Thus, impairment of the endothelial functions is suggested to play deleterious roles in the development of several diseases, including inflammatory angiitis syndrome, thrombotic embolism, disseminated intravascular coagulation (DIC) disorder, and neovascularization, tumor progression and diabetic retinopathy [23].
ECs regulate various pathophysiological properties by synthesizing and secreting various molecules relative of the blood and/or to the extracellular matrix [24]. For example, endothelium-secreted molecules such as angiotensin II (Ang II), endothelin-1 (ET-1), thromboxane A2 (TXA2), and prostacyclin H2 participate in vasoconstriction, whereas, molecules such as NO, bradykinin, and hyperpolarizing factor contribute to vasodilation that helps maintaining a balance between the vasoconstriction and vasodilation [20]. The fine balance between these secreted molecules is critical for a proper functioning of the endothelium, and an imbalance of these molecules may contribute to failure in vascular auto-regulation, and influence the structural and functional integrity of the circulation [24].
Importance and regulation of nitric oxide (NO) in endothelial function
After the discovery of prostacyclin (or prostaglandin I2, PGI2) and endothelium derived relaxing factor (EDRF), NO was identified as the third endothelium regulator for vascular auto-regulation. In the endothelium, NO is synthesized from L-arginine, a semi-essential amino acid, by the endothelial nitric oxide synthase (eNOS), and L-citrulline is the by-product of this pathway [25]. The biosynthesis of NO also requires many co-factors, such as nicotinamide adenine dinucleotide phosphate (NADPH), flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), tetrahydrobiopterin (BH4), and calmodulin [25]. Following its synthesis, NO diffuses across the EC membranes and enters the VSMCs where it activates guanylate cyclase (GC). Activation of GC further gives rise to the intracellular cyclic guanosine-3′,5-monophosphate (cGMP), which, as a second messenger, mediates the biological actions of NO, including the control of vascular tone and platelet aggregation [25,26,27]. Shear stress is a crucial factor for the activation of eNOS under physiological circumstances; other signaling molecules such as bradykinin, adenosine, vascular endothelial growth factor (VEGF), and serotonin can also lead to the activation of eNOS [28].
Endothelial dysfunction: Pathogenesis and mechanisms
ED can be described as diminished production and/or availability of NO, and an imbalance between the endothelium-derived vasodilators and vasoconstrictors. It is an event that accounts for the risk of CVDs and precedes the development of atherosclerosis [20, 29]. Among the relevant mechanisms of ED pathogenesis, proposed in previous studies, oxidative stress and inflammation account for the majority of them [12, 13, 29,30,31]. In particular, the inactivation of NO by oxidative enzyme systems such as NADPH oxidase, xanthine oxidase (XO), cyclooxygenases (COX), lipoxygenases (LOX), myeloperoxidases (MPO), cytochrome P450 monooxygenase, uncoupled NOS, and peroxidases is one of the critical mechanisms that leads to ED through an elevated level of superoxide anion (O2 −) [31,32,33]. NADPH oxidase acts as an important source of O2 − that gives rise to vascular oxidative stress [31, 32], and approaches that could lower NADPH oxidase might have reversing effects on ED [14].
Also, eNOS uncoupling is another mechanism of ED that leads to increased O2 − synthesis instead of NO synthesis, in a scenario where BH4 is inadequate; restoration of BH4 can reduce uncoupled eNOS and reverse oxidative stress-induced ED [31, 34, 35]. Oxidative stress-induced ED is a major concern observed in patients with CVDs and MetS [30, 33, 36]. Many studies have suggested a role of inflammation in ED that underlies the pathogenesis of CVDs, obesity and T2DM. Elevated levels of pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α), interleukin-1beta (IL-1β), interleukin-6 (IL-6), and interferon gamma (IFN-γ) were observed in age-related ED, both in rodents and humans [37, 38], mainly via the activation of the NF-κB (nuclear factor-kappa B) pathway [37, 39, 40]. NF-κB, with the association of IκK, controls the global pro-inflammatory response in ECs [41], and acts as an effective “transducer for feed forward activation” of oxidative stress and inflammatory signaling, subsequently leading to ED by means of increased NADPH oxidase-mediated ROS upregulation, and upregulation of the pro-inflammatory cytokines such as IL-6 and TNF-α [42, 43]. Therefore, NF-κB is considered as a potential therapeutic target for the control and prevention of arterial aging and atherosclerosis [41, 42].
In addition, ED can also be induced by many factors such as dietary intake, drugs, and aging. For example, high-fat diet (HFD) induces a downregulation of the endothelial AMPK-PI3K-Akt-eNOS pathway which in turn causes ED; such downregulation of the AMPK-PI3K-Akt-eNOS pathway correlates with increased plasma levels of FFAs and TG, and an impaired glucose utilization [44]. Besides, uric acid (UA), which is a final product derived from purine metabolism pathway, can induce ED via the high mobility group box chromosomal protein 1/receptor for advanced glycation end products (HMGB1/RAGE axis). UA inhibits eNOS expression and subsequent NO production in human umbilical vein ECs (HUVECs), by increasing intracellular HMGB1 expression and extracellular secretion of the protein. UA also upregulates RAGE expression and other inflammatory cytokines, possibly via the activation of NF-κB transcription factor [45]. A role of HMGB1 in inducement of ED, triggered by FFAs, has also been shown in a previous study [46], which will be further discussed later in Section 4.3; other mechanisms by which FFAs can mediate ED have been discussed in Section 4. Drug-induced generation of ROS and inflammation also play a major role in the onset of ED. Several classes of drugs, including anti-cancer drugs, immunosuppressive drugs, anti-retroviral drugs, and others, have been known to induce ED. For example, doxorubicin (DOX) induces ED in a VEGF-dependent manner, and targeting VEGF rescues DOX-induced ED [47, 48]. Alteration in endothelial markers such as PGH2, TXA2, NO, ET-1, thrombomodulin and von Willebrand factor (vWF) are seen as aging progresses. Notably, NO production is reduced in elderly people which has also been seen in aged animals through a downregulation of eNOS. On the other hand, ET-1 expression is also increased with aging; ET-1 inhibits acetylcholine (ACh)-dependent platelet inhibition in the endothelium and is a promoter of vWF expression. vWF is negative regulator of NO and a well-known marker of ED [49]. More recently, Medin, a common amyloidogenic protein in humans, especially in older individuals, has been shown to induce ED. In ex vivo human adipose and leptomeningeal arterioles, the protein could decrease NO production, increase peroxynitrite, and superoxide production and enhance the expression of the proinflammatory markers such as IL-6 and IL-8 as well as the NF-κB transcription factor. These effects were shown to be mediated through RAGE and inhibition of RAGE by its specific inhibitor, FPS-ZM1, could reverse all such changes [50].
Several compounds with therapeutic value against ED, in different diseased states, have been reported in the recent years. Rosuvastatin, a lipid-lowering statin, has been shown to improve endothelial function in patients with inflammatory joint diseases, systemic sclerosis and chronic heart failure (CHF) [51,52,53]. Particularly, in the CHF patients, the drug improved the flow-mediated dilation (FMD) by inducing antioxidant effects, neovascularization and Akt phosphorylation [53]. Protective roles of pitavastatin in obesity-related ED [54], by increasing FMD and lowering triglycerides, and gliclazide in T2DM-related ED [55], by increasing FMD and EPCs, have also been reported. Pioglitazone, a peroxisome proliferator-activated receptor gamma or PPARγ agonist, was shown to prevent ischemia-induced ED in healthy subjects [56], and improve endothelial and adipose tissue dysfunction in pre-diabetic patients with coronary artery disease (CAD) [57]. In the CAD patients, the drug could improve FMD, reduce TNF-α and other inflammatory markers, and improve insulin sensitivity [57]. A synergistic protective effect of pioglitazone with quercetin, a naturally occurring plant polyphenol, on ED has also been shown on isolated rat aorta with the characteristics of T2DM [58]. A detailed meta-analysis on the effects of thiazolidinediones on the FMD has been reported elsewhere [59]. In a more recent study, it has been shown that the drug could protect EPCs by upregulating long non-coding RNA maternally expressed 3 (lncRNA MEG3) in MetS patients [60]. However, it should be noted that pioglitazone can induce heart failure in patients with underlying heart disease [61]. Efficacy of dipeptidyl peptidase IV (DPP-IV) inhibitors against ED has also been reported [62]. Efficacy of more drugs with multifaceted mechanisms in different diseased states with ED has been reviewed elsewhere [63]. Butyrate, a 4-carbon FA, when given orally in ApoE−/− mice, can lessen atherosclerotic development by reducing ROS load in ECs and macrophage migration and activation at the site of lesion; such reduction of ROS by butyrate is mediated through downregulation of the oxidative enzymes such as NADPH oxidase and iNOS in atherosclerotic lesions [64].
In addition, protective roles of many natural products, such as Traditional Chinese Medicines (TCM), have been reported to improve endothelial function. Tongxinluo, a TCM against CVDs, mediates protective role against ED in rats by means of lowering ET-1, and increasing NO [65]. Danshensu, another TCM, which is a water-extractable component of the medicinal herb Salvia miltiorrhiza, protects the endothelium in rats with hyperhomocysteinemia by modulating the abnormality in the parameters such as NO, ET-1, and other inflammatory markers induced by hyperhomocysteinemia [66]; homocysteine is a byproduct of numerous biological processes in the human body and, when elevated, it may be associated with severe atherosclerosis and thrombotic occlusions [67]. Some other types of TCM formulations with potential effects on the endothelium have been described elsewhere [68]. Other compounds having potential effects on endothelial function, which can be disturbed by FFAs, have been discussed in the later parts.
FFAs and their role in diseases
FFAs and their receptors
Fatty acids (FAs) are carboxylic acids with long aliphatic chains containing a methyl group at one end, while a carboxylic group at the other end. Depending on the presence of double bonds, they are classified into saturated fatty acids or SFAs with no double bonds, monounsaturated fatty acids or MUFAs with only one double bond and polyunsaturated fatty acids or PUFAs with at least two double bonds [69]. SFAs are primarily derived from animal and dairy products, coconut and palm oils, whereas unsaturated FAs (UFAs) such as MUFAs and PUFAs are found in olive oil, nuts and in some fatty fishes [69].
FAs, depending upon their amino acid chain lengths, can also be divided into three types: short-, medium-, and long-chain FAs (SCFAs, MCFAs, and LCFAs, respectively) [70]. SCFAs contain no more than 6 carbons, while MCFAs contain 6–12 carbons, and LCFAs have more than 12 carbons. Saturated FAs such as acetic acid (C2:0), propionic acid (C3:0), butyric acid (C4:0), valeric acid (C5:0) and caproic acid (C6:0) are examples of SCFAs, while caprylate (C8:0), capric acid (C10:0) and lauric acid (C12:0) are MCFAs; other SFAs such as myristic acid (C14:0), palmitic acid or PA (C16:0) and stearic acid or SA (C18:0), and all types of unsaturated FAs, including MUFAs and PUFAs, fall under the category of LCFAs [71].
The FFA sensing receptors (FFARs) belong to the G-protein or guanine nucleotide-binding protein coupled receptors (GPCRs) family [72]. Several GPCRs, including GPR40 (FFAR1), GPR43 (FFAR2), GPR41 (FFAR3) and GPR120 (FFAR4), have been found to be activated by extracellular FFAs [70, 73]. While SCFAs bind to FFAR2 and FFAR3, the other two subtypes, MCFAs and LCFAs, bind to FFAR1 and FFAR4. The endogenous ligand potency (EC50) of FFA2 and FFA3 for SCFAs is 0.1–1.0 mM, while that of FFA1 and FFA4 for LCFAs is 1.0–30 μM [73]. Detailed physiological and pathophysiological roles of these receptors have been described elsewhere [72, 73].
FFAs and CVDs
SFAs are largely responsible for CVDs, while UFAs are unlikely to cause CVDs, and rather UFAs are mostly found to be protective against CVDs. SFAs increase low-density lipoprotein (LDL), which is a major risk factor for CVDs [74]. Studies suggest that LCFAs bear greater risk for CHD than that by SCFAs or MCFAs [74]. The most common LCFAs occurring in western diets are myristic acid (14:0), PA (16:0) and SA (18:0). Previous plasma metabolomic studies have confirmed PA as a strong contributing factor to the development of atherosclerosis [75]. Several recent in vivo and in vitro studies have revealed the mechanisms by which PA contributes the pathogenesis of CVDs. A very recent study has shown that PA is a promoter of inflammatory responses and cellular senescence in cardiac fibroblasts which it mediates via the activation of toll-like receptor 4 (TLR4) and NLRP3 inflammasome, increasing mitochondrial ROS load and mitochondrial dysfunction, and functionality loss of the cardiac fibroblasts [76]. Another study has shown that PA could mediate apoptosis of the VSMCs by inducing the TLR4 pathway and ROS generation [77]. The specific roles and mechanisms of FFAs, in particular through mediation of ED, in the development of CVDs have been discussed in the later parts of this article (Section 4).
FFAs and insulin resistance: Role of oxidative stress and inflammation
FAs possess different physiological roles - structurally they contribute to the constituents of the membrane lipids, including phospholipids and glycolipids, whereas, functionally they are important as fuel molecules [78]. Although they are important sources of energy, particularly during a fasting condition, abnormalities in FA metabolism may contribute to the pathogenesis of MetS [78], and may bear risks for developing atherosclerosis [79]. In obesity, high levels of plasma FFAs are seen because of several reasons, such as release of more FFAs by enlarged adipose tissue mass and that the FFAs clearance may also be compromised in obesity [3, 80]. In turn, higher levels of FFAs inhibit the anti-lipolytic action of insulin, which further increases the rate of FFAs release into the circulation [81]. Clinical studies have shown that elevated level of FFAs leads to an insulin-resistant state, and that lowering of FFAs can be beneficial to insulin-stimulated glucose uptake [3]. Several mechanisms underlie FFA-induced insulin resistance such as intracellular accumulation of triglycerides (TG) and diacylglycerol (DAG), activation of serine/threonine kinases, reduced tyrosine phosphorylation of the insulin receptor substrate 1/2 (IRS 1/2) and impairment of the IRS/phosphoinositol 3 kinase (PI3K) pathway, involved in insulin signaling [3, 80]. By inducing an insulin-resistant state in all major insulin target organs, including the ECs, liver, and skeletal muscles [6, 80], FFAs contribute to the progression of T2DM, HTN, dyslipidemia and nonalcoholic fatty liver disease (NAFLD) [80]. T2DM, which is manifested by a chronic insulin-resistant state with progressive decline in functional status of β-cells, is often associated with hypertriglyceridemia or increased plasma concentrations FFAs [78].
FFAs are significant sources of reactive oxygen species (ROS), which lead to the event of oxidative stress. Not only in vascular cells [82], but also in other cells types, such as hepatocytes [83], and immune cells [84], FFAs lead to the generation of ROS, mainly through the activation of NADPH oxidase via protein kinase C (PKC) [82, 84]. A role of PKC is also seen in FFA-induced inflammation; FFA-mediated inflammation also relates to the IKK/NF-κB inflammatory signaling and leads to the activation of TNF-α, IL1-β, and IL-6, and increased plasma levels of the monocyte chemotactic protein-1 (MCP-1) [80]. All these inflammatory components play a role in chronic inflammation that might cause insulin resistance in the ECs [85, 86].
Interestingly, FFAs are also modulators of the NLRP3 inflammasome in the context of T2DM and obesity [87]. Inflammasomes act as both innate immune system receptors and sensors, and regulate a number of activities such as the activation of caspase-1 and inducement of inflammation; NLRP3 is the best characterized inflammasome [87] that can correlate to a number of human diseases, including atherosclerosis, MetS and neurodegenerative diseases [88]. Palmitate or PA has been shown to activate NLRP3, which in turn can enhance ROS generation in macrophages and subsequently weaken the AMPK signaling, which is a negative regulator ROS generation and inflammation [89]. Using a mouse liver cell line, it was also shown that palmitate is an inducer of IL-1β, which could suppress the insulin-induced Akt phosphorylation, suggesting the development of insulin resistance by FFA through the mediation of ROS and inflammatory signaling via the NLPR3 inflammasome.
Role of FFAs in inducing ED: Evidence from clinical and experimental studies
FFAs induce ED via downregulation of the AMPK/PI3K/Akt/eNOS signaling pathway
Insulin is a key mediator of NO-mediated vasodilation [90], and 5′ adenosine monophosphate-activated protein kinase (AMPK) is important in insulin signaling that serves as a target for against insulin resistance in MetS and related diseases [91, 92]. Moreover, AMPK plays a role in FA oxidation [93]. AMPK, which is a fuel-sensing enzyme, is activated by an increased AMP/ATP ratio following a number of stimuli such as exercise or its pharmacological activators, and can further phosphorylate and inactivate Acetyl-CoA carboxylases (ACC), which catalyzes the synthesis of malonyl-CoA and is important for FAs synthesis, a mechanism by which AMPK increases the oxidation of FAs [86, 93], thus serving as a target in MetS-related elevation in FAs. Not only so, but also that AMPK can modulate both oxidative stress and NF-κB-mediated inflammatory signaling [86]. FFAs downregulate insulin-mediated production of NO and peripheral blood flow via two mechanisms: 1) by reducing tyrosine phosphorylation IRS-1/2 and 2) by inhibiting the PI3K/Akt pathway, which not only controls insulin-stimulated uptake of glucose but also regulates eNOS → NO production in the ECs [80]. One study showed that increased level of FFAs in the bloodstream impairs endothelium-dependent vasodilation while the endothelium-independent vasodilation remains unaffected [94], suggesting FFAs have specific inhibitory roles on production of NO in the ECs. Another study reported that infusion of FFA in insulin-sensitive human subjects leads to a significant reduction in NO synthase flux and an impaired shear stress-induced production of NO [95]. More recently, effects of FFAs have been shown on eNOS activity by inhibiting eNOS mRNA expression in rat aortic ECs and modulating eNOS activity through possible increases in oxidative stress and inflammatory burdens [96]; a similar downregulation in insulin-mediated eNOS activity by FFAs was shown through upregulation of PTEN (phosphatase and tensin homolog) and simultaneous inhibition of Akt kinase [97]. The AMPK/PI3K/Akt/eNOS pathway relative of ED has been extensively reported, which is diminished with the increasing level of FFAs, induced by HFD [98]. Detrimental roles of the SFAs such as PA and SA were shown in porcine aortic ECs (PAECs) through the downregulation of eNOS [99]. Interestingly, even though PUFAs have a protective role against ED [100], their free circulating form could also mediate a negative action on the endothelium by decreasing NO availability and increasing ET-1 [101]. Storniolo and colleagues reported that free forms of linoleic acid (LA), which is an ω-6 PUFA, negatively regulate eNOS phosphorylation, and consequently affect the level of intracellular NO availability in ECV304 cells [102].
Protective role of several dietary constituents has been shown in FFA-induced ED via the AMPK/PI3K/Akt/eNOS pathway. For example, eicosapentaenoic acid (EPA), which is an ω-3 or n-3 PUFA, has a protective role against PA-induced ED which is mediated via activation of the AMPK/eNOS pathway; EPA also mediated its inhibitory effect on the PA-induced apoptosis of ECs and activation of apoptosis-related proteins, such as Caspase-3, p53 and Bax [18] (Table 1). Decreases in NO synthesis and increase in the level of ET-1 can be reversed by treatment with the olive oil polyphenolic compounds [102]. There are many clinical studies reporting the protective effects of EPA or olive oil constituents such as oleic acid (OA) on FMD and other endothelial markers [103,104,105], which, however, did not focus on their effects on FFA-induced ED. Although the studies by Lee et al. [18] and Storniolo et al. [102] have strong implications that EPA and olive oil polyphenols can be beneficial in FFA-induced ED, these studies were limited to the use of in vitro models only; further animal or even human studies should be carried out to support the effects of these compounds on FFA-induced ED. A role of the mitochondria-related AMPK/eNOS pathway has also been shown to alleviate ED and atherosclerosis in mice fed with HFD [106]. Salidroside (SAL), an isolated phenylpropanoid glycoside from the medicinal plant Rhodiolarosea, mitigates ED and atherosclerosis by activating the mitochondrial AMPK/PI3K/Akt/eNOS pathway and promoting NO production [106]. Moreover, L-carnitine, an important factor for FA transport/oxidation in the mitochondria, attenuates FFA-induced obesity-related ED in human subjects [107] (Table 1).
FFAs-induced ED through disruption of calcium signaling-mediated NO production/release
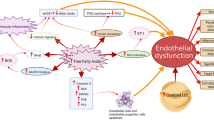
Calcium signaling plays a crucial role in endothelial function by facilitating the release of NO through activation of eNOS, which is a calcium-dependent enzyme [108]. Several receptor-operated agonists, such as adenosine 5′-triphosphate (ATP) and bradykinin facilitate breakdown of phosphatidylinositol, leading to an increase in the intracellular free calcium concentration [109]. Intracellular calcium is important for mechanosensitivity responses of ECs [110], and that shear stress-mediated release of NO largely depends on increases in cytosolic calcium levels [111, 112]. Free forms of UFAs such as oleic acid (OA) and linoleic acid (LA) diminish ATP-induced mobilization and influx of intracellular calcium in bovine aortic endothelial cells (BAECs) culture, and thus impair production/release of NO [113]. Later on, another study confirmed the deleterious effects of FFAs on endothelial calcium signaling and subsequent eNOS activity that led to diminished NO production [114] (Fig. 1).

Potential mechanisms by which FFAs induce ED. FFAs mediate ED by means of several mechanisms, which might have direct/indirect effects on NO production. For example, FFAs can mediate oxidative stress and inflammation in the endothelium which can affect insulin signaling and contribute to dysregulated NO production. Activation of the RAS by FFAs can elevate the level of ET-1, which can lead to vasoconstriction. Furthermore, FFAs can also activate the apoptotic pathways which can induces apoptosis in ECs and EPCs. Several dietary/therapeutic agents can be beneficial against FFA-induced ED through activation of the eNOS or via the inhibition of NF-κB-mediated inflammatory signaling (has been listed in Table 1)
Role of the FFAs in mediation of oxidative stress and inflammatory signaling in the endothelium
Oxidative stress-related ED is not only a critical mechanism that leads to CVDs [30], but it is also a major contributor to the pathogenesis of MetS [3]. A dose-dependent increase of ROS is seen in monocytes exposed to FFAs which leads to adhesion of the monocytes to the ECs [115] - a critical mechanism in the development of atherosclerosis at the earlier stages [116]. Interestingly, FFAs-induced increases in CV risk factors, characterized by elevated levels of endothelial markers, is seen in healthy subjects, a possible mechanism in the development of the CVDs [117]. Besides, HFD, which is a large source of FFAs, also induces oxidative stress in endothelium [118, 119]. Chinen and colleagues showed that FFA induces overexpression of NADPH and mediates oxidative stress in rats with characteristics of both obesity and T2DM [120]. Events of ER stress induced by FFAs have also been shown in ECs isolated from healthy human subjects [121]. The study has shown that intralipid infusion in healthy subjects could lead to 4.2-fold increases in the level of FFAs which was associated with a reduction of the hyperemic response in the leg without a change in FMD of the brachial artery. They also have revealed that mRNA levels of the genes ATF6 and IRE1, which are responsible for early adaptive responses to ER stress, had also been elevated in ECs; however, such changes were only adaptive rather than apoptotic.
FFAs may contribute to inflammatory states that lead to enhance endothelial permeability [122]. One of the major pathways leading to FFAs-induced ED is the activation of NF-κB, reported in many studies [123]. Intake of trans fatty acids (TFAs), consumed through foods made from partially hydrogenated vegetable oils, can activate the NF-κB pathway, leading to increased endothelial superoxide production and reduced NO production [124]. The NF-κB pathway is a major player mediating the deleterious effects of SFAs on human coronary artery ECs [125]. Surprisingly, FFAs of PUFA such as LA also may play a role in inducing inflammatory responses by increasing the levels of TNF-α, MCP-1, vascular cell adhesion molecule 1 (VCAM-1), and intercellular adhesion molecule 1 (ICAM-1) through the activation of NF-κB and activator protein 1 (AP-1) [126], and affect the release of NO [113]. Interestingly, a study has shown that IKK-β, which is an activator of NF-κB, can also diminish NO production [127] (Fig. 1). Moreover, a role of the FFAs in inducement of the NLRP3 inflammasome has been shown that could lead to an increase in the endothelial permeability [122]. In microvascular endothelial cells (MVECs), using palmitate, the authors have showed that it could activate the NLRP3 inflammasome with a resulting reduction in endothelial tight junction proteins - zonula occludens-1 and -2 (ZO-1 and ZO-2). Further exploring of the mechanisms, it had been found that FFAs mediated such effects by triggering the production of HMGB1 which might explain the early onset of endothelial injury during obesity.
Protective effects of several drugs and other dietary/natural agents through suppression of inflammation and oxidative stress in FFA-induced ED have been reported both in clinical and experimental studies. For example, dihydropyridine calcium channel blockers such as nifedipine and amlodipine possess preventive effects against FFAs-induced ED, leucocyte activation and oxidative stress as evidenced by studies in human subjects; by exploring the further mechanisms, through in vitro study, the authors showed a role of NF-κB in such mediation of FFAs-induced ED, and that the protective role of the drugs were through the suppression of NF-κB p65 phosphorylation [128]. A role of Withaferin A (WA), a steroidal lactone derived from Acnistus arborescens [129], against PA-induced insulin resistance and dysfunction of the endothelium has been shown, mediated through its anti-oxidant and anti-inflammatory properties [130]. Such inhibition of the inflammatory responses by WA was through suppression of IKKβ/NF-κβ phosphorylation and decreased release of proinflammatory cytokines, such as TNF-α and IL-6. Furthermore, insulin sensitivity in the ECs was shown to be improved via the PI3K signaling while inhibiting inflammation-stimulated IRS-1 serine phosphorylation. Similar inhibitory effects on IKKβ/NF-κβ phosphorylation, and TNF-α and IL-6 upregulation, induced by PA in HUVECs, were shown to be exerted by Diosgenin, a steroidal saponin extracted from Dioscorea [131]. Diosgenin, in the same study, also reportedly improved insulin signaling by modulating serine/tyrosine phosphorylation of IRS-1 and thereby inducing insulin-mediated NO production. Cyanidin-3-O-glucoside (C3G), an anthocyanin which is abundant in human diet, was reported to provide protection to the endothelium from PA-induced toxicity via suppression of NF-κB pathways and adhesion molecules [132]. Moreover, the compound could mediate nuclear localization of Nrf2 and thereby activating its related pathways to suppress oxidative stress and increase the expression of the cytoprotective genes in HUVECs. In another study, recently reported by the same authors, showed that the protective effects of C3G in HUVECs via the modulation of PA-induced ED (suppressed eNOS expression and NO release) was related to the PI3K/Akt axis and that C3G was a direct activator of Nrf2 [133]. Protective effect of Astragalus membranaceus on FFA-induced dysfunction in ECs in an anti-NF-κB manner has also been reported [134] (Table 1).
Evidence of renin-angiotensin system activation by FFAs
The renin-angiotensin system (RAS) is a crucial regulator of the arterial blood pressure, and Ang II is known as a potent vasoconstrictor. EC membrane expresses the angiotensin converting enzyme (ACE), which is required for Ang II synthesis; Ang II causes vasoconstriction by stimulation of ET and depletion of NO, and inhibition of ACE is fruitful in boosting the NO pathway [135]. While Ang II gives rise to FFAs through downregulation of the FA oxidation pathway [136], FFAs, on the other hand, have also been shown to be activators of the RAS [137]. Mice lacking ACE (ACE−/−) show increased gene expression of enzymes related to lipolysis and FA oxidation [138], which might explain the interplay between RAS and generation of FFAs. Moreover, Ang II interferes with insulin signaling, mainly by affecting insulin-induced tyrosine phosphorylation of IRS-1 [139]. Activation of the RAS by Ang II or the activity of Ang II itself, has been implicated in the pathogenesis of ED [140, 141]. While leukocytes activation is deleterious for endothelial health, FFAs can activate leukocytes and contribute to the adhesion properties of leukocytes in an Ang II-dependent manner, leading to the onset of ED [140], and inhibition of the RAS is preventive against the FFA-induced ED in humans [141] (Fig. 1). In the latter study, Watanabe and colleagues showed that a single dose of either losartan, an Ang II receptor antagonist, or perindopril, an inhibitor of ACE, could completely prevent the FFAs-induced dysregulation of endothelium-dependent vasodilation, suggesting the blockade of RAS as an effective treatment for FFAs-induced ED. Interestingly, a more recent report studying the effect of losartan on Goto-Kakizaki (GK) rats, which mimic the symptoms of T2DM, showed that the drug could improve eNOS activity, possibly by modulating Ang II-mediated increase in phospho-IRS-1 [139].
Effects of FFAs on endothelial progenitor cells (EPCs)
EPCs participate in endothelial recovery following arterial injury, and factors such as oxidative stress contribute to dysfunction and apoptosis of the EPCs [142]. Dysfunctional EPCs are thought to be key regulators in the pathogenesis of atherosclerosis and other CVDs [143]. PA, which is the most abundant type of FFAs in the circulating blood, contributes to apoptosis of EPCs which is facilitated via the p38 and JNK/MAPKs pathways [19] (Fig. 1). Another study also depicted a deleterious role of PA on EPCs in MetS patients via regulating lncRNA MEG3 [144]. MEG3 is required for human mesenchymal stem cells (hMSCs) differentiation into ECs [145]; however, some studies showed that MEG3 may also interfere with the proliferation and angiogenesis in VECs and its expression may correlate with cardiovascular aging [146]. These data raise a conflict of interest for which the pathophysiological roles of MEG3 needs to be studied with a deeper understanding in both EPCs and ECs.
Perspectives
Targeting the AMPK/PI3K/Akt/eNOS signaling pathways: Importance of insulin and other endogenous targets
The AMPK/PI3K/Akt/eNOS signaling pathways are important for NO synthesis, and disruption of this signaling in the endothelium induces ED [98]. In the endothelium, insulin induces production of NO via the IRS-1/IRS-2 signaling. IRS1 activates the PI3K/Akt, which, in turn, phosphorylates eNOS at Ser1177, catalyzing the conversion of L-arginine to L-citrulline and NO [147]. Moreover, insulin protects the endothelium by inhibiting Caspase-mediated ECs death and inducing antioxidant enzymes, such as heme oxygenase 1 (HO-1) through the PI3K/Akt pathway [147]. However, patients with obesity or T2DM show higher levels of FFAs that lead to insulin resistance [4], which further contributes to ED, a pivotal step in the initiation and progression of atherosclerosis [148]. Thus, mitigating insulin resistance through the upregulated AMPK/PI3K/Akt/eNOS pathway seems to be a crucial therapeutic opportunity to combat FFAs-induced ED and subsequent CVD events. Additionally, a study with direct infusion of insulin in the rats showed that hyperinsulinemia could mitigate FFA-induced ED in rat aortic rings [149]; however, the direct effect of insulin infusion still remains questionable as the article is in a non-English language with unclear mechanisms.
Here, we propose that Exendin-4 and Irisin could be two novel targets against FFAs-induced ED. Exendin-4, also having its synthetic counterpart known as exenatide, a glucagon-like protein-1 (GLP-1) receptor agonist [150], increases insulin sensitivity via a PI3K-dependent mechanism [151]. Prevention of HFD-induced insulin resistance by Exendin-4 has been shown to be mediated through an increasing level of adiponectin [152]. It also mediates a direct improvement of the endothelial function via the cAMP or AMPK-eNOS pathways in isolated aortas from obese rats [153]. As FFAs have a role in downregulation of the AMPK-eNOS pathway, which is commonly observed in obesity, it can be suggested that Exendin-4 might regulate the activity of FFAs in obesity and thus improve obesity-related AMPK-eNOS pathway or endothelial function, which could be through the protective effects of Exendin-4 on FFA-induced apoptosis of the pancreatic β-cells [154, 155]. A role of Irisin in improving endothelial function via the AMPK-eNOS pathway is also noteworthy in obese mice [156] and it also has a protective role against ED and atherosclerosis in apolipoprotein E-Null (apoE(−/−)) diabetic mice [157]. A recent study reported that Irisin improves FA oxidation through the AMPK signaling pathway and glucose utilization in mouse model of T2DM [158]; in the same animal model, Irisin also reportedly inhibited hepatic gluconeogenesis via the PI3K/Akt pathway [159]. Moreover, a clinical study has suggested an antagonistic effect of Irisin on fatty acid binding protein 4 (FABP4), the FA binding protein, which is associated with an increased risk of obesity-related metabolic disorders and HTN [160]. These collective data suggest a possible protective role of Exendin-4 and Irisin on FFA-induced ED that would require further validation (Fig. 2).

Possible targets against FFAs-induced ED. The level of FFAs is elevated in obesity/T2DM which contributes to ED. For a healthy endothelium, a balance in the relative pathways should be maintained. Here, we highlight some targets that may serve as important therapeutic avenue against FFAs-induced ED. These targets include Irisin and Exendin-4, which can increase eNOS activity by several means, but their role in ED, specifically induced by FFAs, should be studied. The Nrf2/HO-1 axis, which is a modulator of oxidative stress, might also have great impact to overcome FFAs-induced ED
Nuclear factor erythroid 2-related factor 2 (Nrf2)/HO-1 pathway: A future therapeutic avenue against FFAs-induced ED?
In this part, we propose that the Nrf2/HO-1 pathway could be an important target pathway for the alleviation of FFA-induced ED. This pathway is an important regulator of oxidative stress, and its activation has been proved to be fruitful in many diseases through the modulation of oxidative stress, and inflammation [161,162,163,164,165]. Moreover, the pathway improves diet-induced cognitive deficits and fatty liver [166, 167]. Nrf2, which belongs to the subfamily of basic region leucine zipper (bZip) transcription factors, is responsible for cellular defense mechanisms against oxidative stress and is also crucial for suppression of signaling cascades relative of inflammation [168]. It plays a protective role on FFA-induced cardiotoxicity and ED [132, 169], possibly due to its direct effects on mitochondrial FA oxidation [170, 171]. On the other hand, HO-1, which also possesses anti-oxidative properties and is transcriptionally regulated by Nrf2 [172, 173], is suppressed during oxidative stress-induced EC injury [174], and inducing HO-1 in the mouse endothelium is favorable against T2DM-induced vascular injury, where HO-1 facilitates reendothelialization by increasing the number of EPCs through AMPK-mediated mechanism [175]. An anti-apoptotic role of HO-1, as well as its isoform HO-2, is found in glutamate-induced toxicity and oxidative stress in the cerebrovascular endothelium [176], where it plays a defensive role against disruption of the blood-brain barrier (BBB) and neurological deficits in stroke via the Nrf2 signaling [177]. Induction of HO-1 has been shown to improve FFA-induced ED in rat aorta by activating the AMPK/PI3K/eNOS pathway [178]. This study also showed that adiponectin, which is an anti-inflammatory protein, is induced by HO-1. Interestingly, adiponectin, which mediates its protective role in cAMP-dependent alleviation of FFA-induced ED [179], is endogenously regulated by HO-1 in obese and diabetic animal models [180, 181], suggesting an important role of HO-1 in regulation of FFA-induced endothelial toxicity through multifaceted mechanisms. Looking at the several important roles of the Nrf2/HO-1 pathway, such as suppression of oxidative stress and inflammation, improvement of insulin resistance [166], inhibition of phosphorylation of PI3K/Akt [165, 167], inhibition of NF-κB [165], as well as their individual suppressive role (independent of each other) on FFA-induced ED [132, 178], it could be endorsed that the pathway could be an avenue for future therapy for FFA-related ED (Fig. 2).
Conclusions
ED is an early event in atherosclerosis and other CVDs. FFAs, which are elevated in blood due to metabolic defects under different diseased states, such as obesity, and T2DM, contribute to ED and subsequent events of CVDs, by means of several mechanisms such as decreased insulin signaling and NO production, impaired endothelium/insulin-dependent vasodilation, and increased oxidative stress and inflammation. Thus, early intervention of FFA-induced ED could be beneficial against the CVDs related to ED. Choosing the right lifestyle, such as cutting HFD and eating foods rich in ω-3 or ω-6 FAs or other dietary components that have proven protective role on the endothelium, could be a preventive approach against ED. However, as it is proven that the circulating forms of ω-3 or ω-6 FAs could also sometimes contribute to oxidative stress and inflammation and thus cause ED, it is not completely feasible to conclude the idea of taking more ω-3 or ω-6 FA-rich foods. Therefore, it would require a better understanding of this field and identify some better, possible targets that could be used to develop better therapeutic approaches to intervene the early events of ED-related health conditions and pave the way for a better living.
Abbreviations
- AMPK:
-
5′ adenosine monophosphate-activated protein kinase
- CVD:
-
Cardiovascular disease
- ED:
-
Endothelial dysfunction
- EPCs:
-
Endothelial progenitor cells
- FA:
-
Fatty acid
- FFA:
-
Free fatty acid
- HO-1:
-
Heme oxygenase 1
- MUFA:
-
Monounsaturated fatty acid
- MVECs:
-
Microvascular endothelial cells
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NF-κB:
-
Nuclear factor kappa B
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- Nrf2:
-
Nuclear factor erythroid 2-related factor 2
- PA:
-
Palmitic acid
- PGI2 :
-
Prostacyclin
- PI3K:
-
Phosphoinositide 3-kinase
- PUFA:
-
Polyunsaturated fatty acid
- ROS:
-
Reactive oxidative stress
- SFAs:
-
Saturated fatty acids
- T2DM:
-
type 2 diabetes mellitus
- VSMCs:
-
Vascular smooth muscle cells
References
Zapolska DD, Bryk D, Olejarz W. Trans fatty acids and atherosclerosis-effects on inflammation and endothelial function. J Nutr Food Sci. 2015;5:426. doi:10.4172/2155-9600.1000426.
Egan BM, Greene EL, Goodfriend TL. Nonesterified fatty acids in blood pressure control and cardiovascular complications. Curr Hypertens Rep. 2001;3(2):107.
Boden G. Obesity and free fatty acids. Endocrinol Metab Clin N Am. 2008;37(3):635–46.
Capurso C, Capurso A. From excess adiposity to insulin resistance: the role of free fatty acids. Vasc Pharmacol. 2012;57(2):91–7.
Delarue J, Magnan C. Free fatty acids and insulin resistance. Current Opinion in Clinical Nutrition & Metabolic Care. 2007;10(2):142–8.
Haus JM, Solomon TP, Marchetti CM, Edmison JM, Gonzalez F, Kirwan JP. Free fatty acid-induced hepatic insulin resistance is attenuated following lifestyle intervention in obese individuals with impaired glucose tolerance. The Journal of Clinical Endocrinology & Metabolism. 2010;95(1):323–7.
Steinberg H, Baron A. Vascular function, insulin resistance and fatty acids. Diabetologia. 2002;45(5):623–34.
Liu J, Mazzone PJ, Cata JP, Kurz A, Bauer M, Mascha EJ, et al. Serum free fatty acid biomarkers of lung cancer. Chest. 2014;146(3):670–9.
Zhang Y, He C, Qiu L, Wang Y, Zhang L, Qin X, et al. Serum unsaturated free fatty acids: potential biomarkers for early detection and disease progression monitoring of non-small cell lung cancer. J Cancer. 2014;5(8):706.
Lv W, Yang T. Identification of possible biomarkers for breast cancer from free fatty acid profiles determined by GC–MS and multivariate statistical analysis. Clin Biochem. 2012;45(1):127–33.
Zhang Y, He C, Qiu L, Wang Y, Qin X, Liu Y, et al. Serum unsaturated free fatty acids: a potential biomarker panel for early-stage detection of colorectal cancer. J Cancer. 2016;7(4):477.
Iantorno M, Campia U, Di Daniele N, Nistico S, Forleo G, Cardillo C, et al. Obesity, inflammation and endothelial dysfunction. J Biol Regul Homeost Agents. 2014;28(2):169–76.
Virdis A. Endothelial dysfunction in obesity: role of inflammation. High Blood Pressure & Cardiovascular Prevention. 2016;23(2):83–5.
Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, et al. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol. 2009;587(13):3271–85.
Nilsson J, Ares MP, Dichtl W. VLDL and atherosclerosis. Endothelial dysfunctions in vascular disease. 2007:85–94.
Artwohl M, Roden M, Waldhäusl W, Freudenthaler A, Baumgartner-Parzer SM. Free fatty acids trigger apoptosis and inhibit cell cycle progression in human vascular endothelial cells. FASEB J. 2004;18(1):146–8.
Khan MJ, Alam MR, Waldeck-Weiermair M, Karsten F, Groschner L, Riederer M, et al. Inhibition of autophagy rescues palmitic acid-induced necroptosis of endothelial cells. J Biol Chem. 2012;287(25):21110–20.
Lee C-H, Lee S-D, Ou H-C, Lai S-C, Cheng Y-J. Eicosapentaenoic acid protects against palmitic acid-induced endothelial dysfunction via activation of the AMPK/eNOS pathway. Int J Mol Sci. 2014;15(6):10334–49.
Jiang H, Liang C, Liu X, Jiang Q, He Z, Wu J, et al. Palmitic acid promotes endothelial progenitor cells apoptosis via p38 and JNK mitogen-activated protein kinase pathways. Atherosclerosis. 2010;210(1):71–7.
Lai WKC, Kan MY. Homocysteine-induced endothelial dysfunction. Ann Nutr Metab. 2015;67(1):1–12.
Chlopicki S. Perspectives in pharmacology of endothelium: from bench to bedside. Pharmacological reports: PR. 2015;67(4):vi.
Aird WC. Endothelial cell heterogeneity. Crit Care Med. 2003;31(4):S221–S30.
Maruyama I. Biology of endothelium. Lupus. 1998;7(2_suppl):41–3.
Jaffe EA. Cell biology of endothelial cells. Hum Pathol. 1987;18(3):234–9.
Vallance P, Chan N. Endothelial function and nitric oxide: clinical relevance. Heart. 2001;85(3):342–50.
Huang PL. Endothelial nitric oxide synthase and endothelial dysfunction. Curr Hypertens Rep. 2003;5(6):473–80.
Deanfield J, Donald A, Ferri C, Giannattasio C, Halcox J, Halligan S, et al. Endothelial function and dysfunction. Part I: methodological issues for assessment in the different vascular beds: a statement by the working group on Endothelin and endothelial factors of the European Society of Hypertension. J Hypertens. 2005;23(1):7–17.
Arrebola-Moreno AL, Laclaustra M, Kaski JC. Noninvasive assessment of endothelial function in clinical practice. Revista Española de Cardiología (English Edition). 2012;65(1):80–90.
Hadi HA, Carr CS, Al SJ. Endothelial dysfunction: cardiovascular risk factors, therapy, and outcome. Vasc Health Risk Manag. 2005;1(3):183.
Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87(10):840–4.
Donato AJ, Morgan RG, Walker AE, Lesniewski LA. Cellular and molecular biology of aging endothelial cells. J Mol Cell Cardiol. 2015;89:122–35.
Hamilton CA, McIntyre M, Brosnan MJ, Graham D, Dominiczak AF. Superoxide excess in hypertension and aging: a common cause of endothelial dysfunction? Am Heart Assoc. 2000;37(2):529–34.
Avogaro A, de Kreutzenberg SV. Mechanisms of endothelial dysfunction in obesity. Clin Chim Acta. 2005;360(1):9–26.
Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. 1997;237(2):340–4.
Eskurza I, Myerburgh LA, Kahn ZD, Seals DR. Tetrahydrobiopterin augments endothelium-dependent dilatation in sedentary but not in habitually exercising older adults. J Physiol. 2005;568(3):1057–65.
Roberts AC, Porter KE. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diab Vasc Dis Res. 2013;10(6):472–82.
Lesniewski LA, Durrant JR, Connell ML, Folian BJ, Donato AJ, Seals DR. Salicylate treatment improves age-associated vascular endothelial dysfunction: potential role of nuclear factor κB and forkhead box O phosphorylation. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences. 2011;66(4):409–18.
Wang M, Zhang J, Jiang L-Q, Spinetti G, Pintus G, Monticone R, et al. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension. 2007;50(1):219–27.
Pierce GL, Lesniewski LA, Lawson BR, Beske SD, Seals DR. Nuclear factor-κB activation contributes to vascular endothelial dysfunction via oxidative stress in overweight/obese middle-aged and older humans. Circulation. 2009;119(9):1284–92.
Walker AE, Kaplon RE, Pierce GL, Nowlan MJ, Seals DR. Prevention of age-related endothelial dysfunction by habitual aerobic exercise in healthy humans: possible role of nuclear factor κB. Clin Sci. 2014;127(11):645–54.
Kempe S, Kestler H, Lasar A, Wirth T. NF-κB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 2005;33(16):5308–19.
Donato AJ, Pierce GL, Lesniewski LA, Seals DR. Role of NFκB in age-related vascular endothelial dysfunction in humans. Aging. 2009;1(8):678.
Csiszar A, Wang M, Lakatta EG, Ungvari Z. Inflammation and endothelial dysfunction during aging: role of NF-κB. J Appl Physiol. 2008;105(4):1333–41.
Garcia-Prieto CF, Hernandez-Nuno F, Rio DD, Ruiz-Hurtado G, Aranguez I, Ruiz-Gayo M, et al. High-fat diet induces endothelial dysfunction through a down-regulation of the endothelial AMPK-PI3K-Akt-eNOS pathway. Mol Nutr Food Res. 2015;59(3):520–32.
Cai W, Duan XM, Liu Y, Yu J, Tang YL, Liu ZL, et al. Uric acid induces endothelial dysfunction by activating the HMGB1/RAGE signaling pathway. Biomed Res Int. 2017;2017:4391920.
Wang L, Chen Y, Li X, Zhang Y, Gulbins E, Zhang Y. Enhancement of endothelial permeability by free fatty acid through lysosomal cathepsin B-mediated Nlrp3 inflammasome activation. Oncotarget. 2016;7(45):73229–41.
Rasanen M, Degerman J, Nissinen TA, Miinalainen I, Kerkela R, Siltanen A, et al. VEGF-B gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc Natl Acad Sci U S A. 2016;113(46):13144–9.
Yin Z, Zhao Y, Li H, Yan M, Zhou L, Chen C, et al. miR-320a mediates doxorubicin-induced cardiotoxicity by targeting VEGF signal pathway. Aging (Albany NY). 2016;8(1):192–207.
Sepulveda C, Palomo I, Fuentes E. Mechanisms of endothelial dysfunction during aging: predisposition to thrombosis. Mech Ageing Dev. 2017;164:91–9.
Migrino RQ, Davies H, Truran S, Karamanova N, Franco DA, Serrano G, et al. Amyloidogenic Medin induces endothelial dysfunction and vascular inflammation through the receptor for advanced Glycation Endproducts. FASEB J. 2017;31(1 Supplement):1015.19.
Ikdahl E, Hisdal J, Rollefstad S, Olsen IC, Kvien TK, Pedersen TR, et al. Rosuvastatin improves endothelial function in patients with inflammatory joint diseases, longitudinal associations with atherosclerosis and arteriosclerosis: results from the RORA-AS statin intervention study. Arthritis Res Ther. 2015;17:279.
Timar O, Szekanecz Z, Kerekes G, Vegh J, Olah AV, Nagy G, et al. Rosuvastatin improves impaired endothelial function, lowers high sensitivity CRP, complement and immuncomplex production in patients with systemic sclerosis--a prospective case-series study. Arthritis Res Ther. 2013;15(5):R105.
Erbs S, Beck EB, Linke A, Adams V, Gielen S, Krankel N, et al. High-dose rosuvastatin in chronic heart failure promotes vasculogenesis, corrects endothelial function, and improves cardiac remodeling--results from a randomized, double-blind, and placebo-controlled study. Int J Cardiol. 2011;146(1):56–63.
Nagashima H, Endo M. Pitavastatin prevents postprandial endothelial dysfunction via reduction of the serum triglyceride level in obese male subjects. Heart Vessel. 2011;26(4):428–34.
Chen LL, Yu F, Zeng TS, Liao YF, Li YM, Ding HC. Effects of gliclazide on endothelial function in patients with newly diagnosed type 2 diabetes. Eur J Pharmacol. 2011;659(2–3):296–301.
Sakatani Y, Miyoshi T, Oe H, Noda Y, Ohno Y, Nakamura K, et al. Pioglitazone prevents the endothelial dysfunction induced by ischemia and reperfusion in healthy subjects. J Cardiovasc Pharmacol. 2014;64(4):326–31.
Rizza S, Cardellini M, Porzio O, Pecchioli C, Savo A, Cardolini I, et al. Pioglitazone improves endothelial and adipose tissue dysfunction in pre-diabetic CAD subjects. Atherosclerosis. 2011;215(1):180–3.
Kunasegaran T, Mustafa MR, Achike FI, Murugan DD. Quercetin and pioglitazone synergistically reverse endothelial dysfunction in isolated aorta from fructose-streptozotocin (F-STZ)-induced diabetic rats. Eur J Pharmacol. 2017;799:160–70.
Stojanović M, Prostran M, Radenković M. Thiazolidinediones improve flow-mediated dilation: a meta-analysis of randomized clinical trials. Eur J Clin Pharmacol. 2016;72(4):385–98.
Jialal I, Fadini GP, Pollock K, Devaraj S. Circulating Levels of Endothelial Progenitor Cell Mobilizing Factors in the Metabolic Syndrome. The American Journal of Cardiology. 2010;106(11):1606-8.
Jearath V, Vashisht R, Rustagi V, Raina S, Sharma R. Pioglitazone-induced congestive heart failure and pulmonary edema in a patient with preserved ejection fraction. J Pharmacol Pharmacother. 2016;7(1):41–3.
Akoumianakis I, Antoniades C. Dipeptidyl peptidase IV inhibitors as novel regulators of vascular disease. Vasc Pharmacol. 2017;S1537-1891(17):30155–6.
Radenkovic M, Stojanovic M, Potpara T, Prostran M. Therapeutic approach in the improvement of endothelial dysfunction: the current state of the art. Biomed Res Int. 2013;2013:252158.
Aguilar EC, Santos LC, Leonel AJ, de Oliveira JS, Santos EA, Navia-Pelaez JM, et al. Oral butyrate reduces oxidative stress in atherosclerotic lesion sites by a mechanism involving NADPH oxidase down-regulation in endothelial cells. J Nutr Biochem. 2016;34:99–105.
Dai W, Wei C, Kong H, Jia Z, Han J, Zhang F, et al. Effect of the traditional Chinese medicine tongxinluo on endothelial dysfunction rats studied by using urinary metabonomics based on liquid chromatography-mass spectrometry. J Pharm Biomed Anal. 2011;56(1):86–92.
Yang RX, Huang SY, Yan FF, Lu XT, Xing YF, Liu Y, et al. Danshensu protects vascular endothelia in a rat model of hyperhomocysteinemia. Acta Pharmacol Sin. 2010;31(10):1395–400.
Brattstrom L, Wilcken DE. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr. 2000;72(2):315-23.
Xiong X, Yang X, Liu Y, Zhang Y, Wang P, Wang J. Chinese herbal formulas for treating hypertension in traditional Chinese medicine: perspective of modern science. Hypertens Res. 2013;36(7):570–9.
Vadivel V, Kunyanga CN, Biesalski HK. Health benefits of nut consumption with special reference to body weight control. Nutrition. 2012;28(11-12):1089-97.
Mielenz M. Invited review: nutrient-sensing receptors for free fatty acids and hydroxycarboxylic acids in farm animals. Animal. 2017;11(6):1008–16.
Miyamoto J, Hasegawa S, Kasubuchi M, Ichimura A, Nakajima A, Kimura I. Nutritional signaling via free fatty acid receptors. Int J Mol Sci. 2016;17(4):450.
Yonezawa T, Kurata R, Yoshida K, Murayama MA, Cui X, Hasegawa A. Free fatty acids-sensing G protein-coupled receptors in drug targeting and therapeutics. Curr Med Chem. 2013;20(31):3855–71.
Dranse HJ, Kelly ME, Hudson BD. Drugs or diet?--developing novel therapeutic strategies targeting the free fatty acid family of GPCRs. Br J Pharmacol. 2013;170(4):696–711.
Briggs MA, Petersen KS, Kris-Etherton PM. Saturated fatty acids and cardiovascular disease: replacements for saturated fat to reduce cardiovascular risk. Healthcare (Basel). 2017;5(2):E29.
Chen X, Liu L, Palacios G, Gao J, Zhang N, Li G, et al. Plasma metabolomics reveals biomarkers of the atherosclerosis. J Sep Sci. 2010;33(17–18):2776–83.
Sokolova M, Vinge LE, Alfsnes K, Olsen MB, Eide L, Kaasboll OJ, et al. Palmitate promotes inflammatory responses and cellular senescence in cardiac fibroblasts. Biochim Biophys Acta. 2017;1862(2):234–45.
Zhang Y, Xia G, Zhang Y, Liu J, Liu X, Li W, et al. Palmitate induces VSMC apoptosis via toll like receptor (TLR)4/ROS/p53 pathway. Atherosclerosis. 2017;263:74–81.
Fatehi-Hassanabad Z, Chan CB. Transcriptional regulation of lipid metabolism by fatty acids: a key determinant of pancreatic β-cell function. Nutrition & metabolism. 2005;2(1):1.
Thijssen M, Mensink R. Fatty acids and atherosclerotic risk. Atherosclerosis: Diet and Drugs: Springer. 2005:165–94.
Boden G. Free Fatty Acids, A Major Link Between Obesity, Insulin Resistance, Inflammation, and Atherosclerotic Vascular Disease. In: Fonseca VA, editor. Cardiovascular Endocrinology: Shared Pathways and Clinical Crossroads. Totowa: Humana Press; 2009. p. 61-70.
Jensen MD, Haymond MW, Rizza RA, Cryer PE, Miles J. Influence of body fat distribution on free fatty acid metabolism in obesity. J Clin Investig. 1989;83(4):1168.
Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD (P) H oxidase in cultured vascular cells. Diabetes. 2000;49(11):1939–45.
Soardo G, Donnini D, Domenis L, Catena C, De Silvestri D, Cappello D, et al. Oxidative stress is activated by free fatty acids in cultured human hepatocytes. Metab Syndr Relat Disord. 2011;9(5):397–401.
Cury-boaventura MF, Rui C. Regulation of reactive oxygen species (ROS) production by C18 fatty acids in Jurkat and Raji cells. Clin Sci. 2005;108(3):245–53.
Savage DB, Petersen KF, Shulman GI. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol Rev. 2007;87(2):507–20.
Ruderman N, Cacicedo J, Itani S, Yagihashi N, Saha A, Ye J, et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. London: Portland Press Limited; 2003.
Legrand-Poels S, Esser N, L’homme L, Scheen A, Paquot N, Piette J. Free fatty acids as modulators of the NLRP3 inflammasome in obesity/type 2 diabetes. Biochem Pharmacol. 2014;92(1):131–41.
Guo H, Callaway JB, Ting JP-Y. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677.
Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT-H, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. 2011;12(5):408–15.
Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Investig. 1994;94(3):1172.
Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123(7):2764.
Chopra I, Li H, Wang H, Webster K. Phosphorylation of the insulin receptor by AMP-activated protein kinase (AMPK) promotes ligand-independent activation of the insulin signalling pathway in rodent muscle. Diabetologia. 2012;55(3):783–94.
Wakil SJ, Abu-Elheiga LA. Fatty acid metabolism: target for metabolic syndrome. J Lipid Res. 2009;50(Supplement):S138–S43.
Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J Clin Investig. 1997;100(5):1230.
Steinberg HO, Paradisi G, Hook G, Crowder K, Cronin J, Baron AD. Free fatty acid elevation impairs insulin-mediated vasodilation and nitric oxide production. Diabetes. 2000;49(7):1231–8.
Yu Y, Li H, Zhang X. Effects of free fatty acids on nitric oxide synthase activity and mRNA expression in endothelial cell of SD rat aorta. Sichuan da xue xue bao Yi xue ban. Journal of Sichuan University Medical science edition. 2008;39(2):193–6.
Wang XL, Zhang L, Youker K, Zhang M-X, Wang J, LeMaire SA, et al. Free fatty acids inhibit insulin signaling–stimulated endothelial nitric oxide synthase activation through upregulating PTEN or inhibiting Akt kinase. Diabetes. 2006;55(8):2301–10.
García-Prieto CF, Hernández-Nuño F, Rio DD, Ruiz-Hurtado G, Aránguez I, Ruiz-Gayo M, et al. High-fat diet induces endothelial dysfunction through a down-regulation of the endothelial AMPK–PI3K–Akt–eNOS pathway. Mol Nutr Food Res. 2015;59(3):520–32.
Van Vickle GD. The effect of free fatty acids on endothelial cells: University of Missouri--Columbia; 2005.
Das U. Long-chain polyunsaturated fatty acids interact with nitric oxide, superoxide anion, and transforming growth factor-beta to prevent human essential hypertension. Eur J Clin Nutr. 2004;58(2):195.
Mitjavila M, Storniolo C, Moreno J. Olive oil polyphenols protect endothelial dysfunction induced by high glucose and free fatty acids by modulating nitric oxide and endothelin-1. Atherosclerosis. 2014;235(2):e110–e1.
Storniolo CE, Roselló-Catafau J, Pintó X, Mitjavila MT, Moreno JJ. Polyphenol fraction of extra virgin olive oil protects against endothelial dysfunction induced by high glucose and free fatty acids through modulation of nitric oxide and endothelin-1. Redox Biol. 2014;2:971–7.
Stirban A, Nandrean S, Götting C, Tamler R, Pop A, Negrean M, et al. Effects of n–3 fatty acids on macro-and microvascular function in subjects with type 2 diabetes mellitus. Am J Clin Nutr. 2010;91(3):808–13.
Sawada T, Tsubata H, Hashimoto N, Takabe M, Miyata T, Aoki K, et al. Effects of 6-month eicosapentaenoic acid treatment on postprandial hyperglycemia, hyperlipidemia, insulin secretion ability, and concomitant endothelial dysfunction among newly-diagnosed impaired glucose metabolism patients with coronary artery disease. An open label, single blinded, prospective randomized controlled trial. Cardiovasc Diabetol. 2016;15(1):121.
Schwingshackl L, Christoph M, Hoffmann G. Effects of olive oil on markers of inflammation and endothelial function-a systematic review and meta-analysis. Nutrients. 2015;7(9):7651–75.
Xing S-S, Yang X-Y, Zheng T, Li W-J, Wu D, Chi J-Y, et al. Salidroside improves endothelial function and alleviates atherosclerosis by activating a mitochondria-related AMPK/PI3K/Akt/eNOS pathway. Vasc Pharmacol. 2015;72:141–52.
Shankar SS, Mirzamohammadi B, Walsh JP, Steinberg HO. L-Carnitine may attenuate free fatty acid-induced endothelial dysfunction. Ann N Y Acad Sci. 2004;1033(1):189–97.
Binienda ZK, Sarkar S, Silva-Ramirez S, Gonzalez C. Role of free fatty acids in physiological conditions and mitochondrial dysfunction. Food Nutr Sci. 2013;4(09):6.
Lückhoff A, Pohl U, Mülsch A, Busse R. Differential role of extra-and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. Br J Pharmacol. 1988;95(1):189–96.
Demer L, Wortham C, Dirksen E, Sanderson M. Mechanical stimulation induces intercellular calcium signaling in bovine aortic endothelial cells. Am J Phys Heart Circ Phys. 1993;264(6):H2094–H102.
Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Phys Cell Phys. 2003;285(3):C499–508.
Nauli SM, Kawanabe Y, Kaminski JJ, Pearce WJ, Ingber DE, Zhou J. Endothelial cilia are fluid shear sensors that regulate calcium signaling and nitric oxide production through polycystin-1. Circulation. 2008;117(9):1161–71.
Kuroda R, Hirata K-I, Kawashima S, Yokoyama M. Unsaturated free fatty acids inhibit ca^ 2^+ mobilization and NO release in endothelial cells. Kobe J Med Sci. 2001;47(5):211–20.
Esenabhalu VE, Schaeffer G, Graier WF. Free fatty acid overload attenuates Ca2+ signaling and NO production in endothelial cells. Antioxid Redox Signal. 2003;5(2):147–53.
Zhang W-Y, Schwartz E, Wang Y, Attrep J, Li Z, Reaven P. Elevated concentrations of nonesterified fatty acids increase monocyte expression of CD11b and adhesion to endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26(3):514–9.
Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends in cardiovascular medicine. 2008;18(6):228–32.
Mathew M, Tay E, Cusi K. Elevated plasma free fatty acids increase cardiovascular risk by inducing plasma biomarkers of endothelial activation, myeloperoxidase and PAI-1 in healthy subjects. Cardiovasc Diabetol. 2010;9(1):9.
Williams MJ, Sutherland WH, McCormick MP, de Jong SA, Walker RJ, Wilkins GT. Impaired endothelial function following a meal rich in used cooking fat. J Am Coll Cardiol. 1999;33(4):1050–5.
Edirisinghe I, Burton-Freeman BM. Age associated endothelial dysfunction: role of oxidative stress, inflammation and western diet. Nutrition and Aging. 2014;2(4):197–211.
Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, et al. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007;148(1):160–5.
Tampakakis E, Tabit CE, Holbrook M, Linder EA, Berk BD, Frame AA, et al. Intravenous lipid infusion induces endoplasmic reticulum stress in endothelial cells and blood mononuclear cells of healthy adults. J Am Heart Assoc. 2016;5(1):e002574.
Li X, Wang L, Nguyen T, Zhang Y. Enhanced endothelial permeability by free fatty acid due to lysosomal cathepsin B-mediated activation of Nlrp3 inflammasome. The FASEB Journal. 2016;30(1 Supplement):1198.2.
Li H, Li H, Bao Y, Zhang X, Yu Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. Int J Cardiol. 2011;152(2):218–24.
Iwata NG, Pham M, Rizzo NO, Cheng AM, Maloney E, Kim F. Trans fatty acids induce vascular inflammation and reduce vascular nitric oxide production in endothelial cells. PLoS One. 2011;6(12):e29600.
Staiger K, Staiger H, Weigert C, Haas C, Häring H-U, Kellerer M. Saturated, but not unsaturated, fatty acids induce apoptosis of human coronary artery endothelial cells via nuclear factor-κB activation. Diabetes. 2006;55(11):3121–6.
Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. 2002;75(1):119–25.
Kim F, Tysseling KA, Rice J, Pham M, Haji L, Gallis BM, et al. Free fatty acid impairment of nitric oxide production in endothelial cells is mediated by IKKβ. Arterioscler Thromb Vasc Biol. 2005;25(5):989–94.
Yasu T, Kobayashi M, Mutoh A, Yamakawa K, Momomura S-I, Ueda S. Dihydropyridine calcium channel blockers inhibit non-esterified-fatty-acid-induced endothelial and rheological dysfunction. Clin Sci. 2013;125(5):247–55.
Kupchan SM, Anderson WK, Bollinger P, Doskotch RW, Smith RM, Saenz-Renauld JA, et al. Tumor inhibitors. XXXIX. Active principles of Acnistur arborescens. Isolation and structural and spectral studies of withaferin a and withacnistin. The Journal of organic chemistry. 1969;34(12):3858–66.
Batumalaie K, Amin MA, Murugan DD, Sattar MZA, Abdullah NA. Withaferin a protects against palmitic acid-induced endothelial insulin resistance and dysfunction through suppression of oxidative stress and inflammation. Sci Rep. 2016;6:27236.
Liu K, Zhao W, Gao X, Huang F, Kou J, Liu B. Diosgenin ameliorates palmitate-induced endothelial dysfunction and insulin resistance via blocking IKKβ and IRS-1 pathways. Atherosclerosis. 2012;223(2):350–8.
Fratantonio D, Speciale A, Ferrari D, Cristani M, Saija A, Cimino F. Palmitate-induced endothelial dysfunction is attenuated by cyanidin-3-O-glucoside through modulation of Nrf2/Bach1 and NF-κB pathways. Toxicol Lett. 2015;239(3):152–60.
Fratantonio D, Cimino F, Molonia MS, Ferrari D, Saija A, Virgili F, et al. Cyanidin-3-O-glucoside ameliorates palmitate-induced insulin resistance by modulating IRS-1 phosphorylation and release of endothelial derived vasoactive factors. Biochim Biophys Acta. 2017;1862(3):351–7.
Wang Y, Yu Y. Protective effects of Astragalus Membranaceus on free fatty acid-induced vascular endothelial cell dysfunction. Sichuan da xue xue bao Yi xue ban= Journal of Sichuan University Medical science edition. 2011;42(1):48–51.
Lüscher TF. Endothelial dysfunction: the role and impact of the renin-angiotensin system. Heart. 2000;84(suppl 1):i20–i2.
Pellieux C, Montessuit C, Papageorgiou I, Lerch R. Angiotensin II downregulates the fatty acid oxidation pathway in adult rat cardiomyocytes via release of tumour necrosis factor-α. Cardiovasc Res. 2009;82(2):341–50.
Sun J, Luo J, Ruan Y, Xiu L, Fang B, Zhang H, et al. Free fatty acids activate renin-angiotensin system in 3T3-L1 adipocytes through nuclear factor-kappa B pathway. Journal of diabetes research. 2015;2016:1587594.
Jayasooriya AP, Mathai ML, Walker LL, Begg DP, Denton DA, Cameron-Smith D, et al. Mice lacking angiotensin-converting enzyme have increased energy expenditure, with reduced fat mass and improved glucose clearance. Proc Natl Acad Sci. 2008;105(18):6531–6.
Nemoto S, Kobayashi T, Taguchi K, Matsumoto T, Kamata K. Losartan improves aortic endothelium-dependent relaxation via proline-rich tyrosine kinase 2/Src/Akt pathway in type 2 diabetic Goto-Kakizaki rats. Am J Phys Heart Circ Phys. 2011;301(6):H2383–H94.
Azekoshi Y, Yasu T, Watanabe S, Tagawa T, Abe S, Yamakawa K, et al. Free fatty acid causes leukocyte activation and resultant endothelial dysfunction through enhanced angiotensin II production in mononuclear and polymorphonuclear cells. Hypertension. 2010;56(1):136–42.
Watanabe S, Tagawa T, Yamakawa K, Shimabukuro M, Ueda S. Inhibition of the renin-angiotensin system prevents free fatty acid–induced acute endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2005;25(11):2376–80.
Sorrentino SA, Bahlmann FH, Besler C, Müller M, Schulz S, Kirchhoff N, et al. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus. Circulation. 2007;116(2):163–73.
Werner N, Nickenig G. Influence of cardiovascular risk factors on endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2006;26(2):257–66.
Liu H, Wang Q, Zhang Y, Qi D, Li M, Guo W, et al. Pioglitazone up-regulates long non-coding RNA MEG3 to protect endothelial progenitor cells via increasing HDAC7 expression in metabolic syndrome. Biomed Pharmacother. 2016;78:101–9.
Huang N, Zhang R, Shi D, Li D, He H, Zhou H, et al. LncRNA MEG3 is involved in endothelial differentiation of human bone marrow-derived Mesenchymal stem cells. 2nd international conference on architectural, civil and hydraulics engineering (ICACHE 2016). 2016.
Boon RA, Hofmann P, Michalik KM, Lozano-Vidal N, Berghäuser D, Fischer A, et al. Long noncoding RNA Meg3 controls endothelial cell aging and function. J Am Coll Cardiol. 2016;68(23):2589.
Arce-Esquivel AA, Bunker AK, Mikus CR, Laughlin MH. Insulin resistance and endothelial dysfunction: macro and microangiopathy. Type 2 diabetes: InTech; 2013.
Imrie H, Abbas A, Kearney M. Insulin resistance, lipotoxicity and endothelial dysfunction. Biochimica et Biophysica Acta (BBA)-molecular and cell biology of. Lipids. 2010;1801(3):320–6.
Yu Y, Zhu J, Wu Y, Baron A. Insulin mitigates the effect of free fatty acid to cause endothelial dysfunction in rat aortic rings. Zhonghua Yi Xue Za Zhi. 2002;82(6):422–5.
Ding X, Saxena NK, Lin S, Gupta N, Anania FA. Exendin-4, a glucagon-like protein-1 (GLP-1) receptor agonist, reverses hepatic steatosis in ob/ob mice. Hepatology. 2006;43(1):173–81.
Idris I, Patiag D, Gray S, Donnelly R. Exendin-4 increases insulin sensitivity via a PI-3-kinase-dependent mechanism: contrasting effects of GLP-1. Biochem Pharmacol. 2002;63(5):993–6.
Li L, Yang G, Li Q, Tan X, Liu H, Tang Y, et al. Exenatide prevents fat-induced insulin resistance and raises adiponectin expression and plasma levels. Diabetes Obes Metab. 2008;10(10):921–30.
Han L, Yu Y, Sun X, Wang B. Exendin-4 directly improves endothelial dysfunction in isolated aortas from obese rats through the cAMP or AMPK–eNOS pathways. Diabetes Res Clin Pract. 2012;97(3):453–60.
Liu L, Wang Y, Wang L, Lin Y, Liu X, Liu X, et al. Exendin-4 protects murine pancreatic β-cells from free fatty acid-induced apoptosis through PI-3K signaling. Endocr Res. 2013;38(1):40–7.
Tews D, Werner U, Eckel J. Enhanced protection against cytokine-and fatty acid-induced apoptosis in pancreatic beta cells by combined treatment with glucagon-like peptide-1 receptor agonists and insulin analogues. Horm Metab Res. 2008;40(03):172–80.
Han F, Zhang S, Hou N, Wang D, Sun X. Irisin improves endothelial function in obese mice through the AMPK-eNOS pathway. Am J Phys Heart Circ Phys. 2015;309(9):H1501–H8.
Lu J, Xiang G, Liu M, Mei W, Xiang L, Dong J. Irisin protects against endothelial injury and ameliorates atherosclerosis in apolipoprotein E-null diabetic mice. Atherosclerosis. 2015;243(2):438–48.
Xin C, Liu J, Zhang J, Zhu D, Wang H, Xiong L, et al. Irisin improves fatty acid oxidation and glucose utilization in type 2 diabetes by regulating the AMPK signaling pathway. Int J Obes. 2016;40(3):443.
Liu T-Y, Shi C-X, Gao R, Sun H-J, Xiong X-Q, Ding L, et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin Sci. 2015;129(10):839–50.
Zhang S, Yang L, Chen P, Jin H, Xie X, Yang M, et al. Circulating Adipocyte fatty acid binding protein (FABP4) levels are associated with Irisin in the middle-aged general Chinese population. PLoS One. 2016;11(1):e0146605.
Ye F, Li X, Li L, Yuan J, Chen J. T-BHQ provides protection against lead neurotoxicity via Nrf2/HO-1 pathway. Oxidative Med Cell Longev. 2015;2016:2075915.
Wang C, Wang P, Zeng W, Li W. Tetramethylpyrazine improves the recovery of spinal cord injury via Akt/Nrf2/HO-1 pathway. Bioorg Med Chem Lett. 2016;26(4):1287–91.
Lou J, Cao G, Li R, Liu J, Dong Z, Xu L. β-Caryophyllene attenuates focal cerebral ischemia-reperfusion injury by Nrf2/HO-1 pathway in rats. Neurochem Res. 2016;41(6):1291–304.
Hu T, Wei G, Xi M, Yan J, Wu X, Wang Y, et al. Synergistic cardioprotective effects of Danshensu and hydroxysafflor yellow a against myocardial ischemia-reperfusion injury are mediated through the Akt/Nrf2/HO-1 pathway. Int J Mol Med. 2016;38(1):83–94.
Yan T, Yu X, Sun X, Meng D, Jia J-M. A new steroidal saponin, furotrilliumoside from Trillium Tschonoskii inhibits lipopolysaccharide-induced inflammation in Raw264. 7 cells by targeting PI3K/Akt, MARK and Nrf2/HO-1 pathways. Fitoterapia. 2016;115:37–45.
Li H, Qin T, Li M, Ma S. Thymol improves high-fat diet-induced cognitive deficits in mice via ameliorating brain insulin resistance and upregulating NRF2/HO-1 pathway. Metab Brain Dis. 2017;32(2):385–93.
Li W, Ma F, Zhang L, Huang Y, Li X, Zhang A, et al. S-Propargyl-cysteine exerts a novel protective effect on Methionine and Choline deficient diet-induced fatty liver via Akt/Nrf2/HO-1 pathway. Oxidative Med Cell Longev. 2016;2016:4690857.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26.
Zeng C, Zhong P, Zhao Y, Kanchana K, Zhang Y, Khan ZA, et al. Curcumin protects hearts from FFA-induced injury by activating Nrf2 and inactivating NF-κB both in vitro and in vivo. J Mol Cell Cardiol. 2015;79:1–12.
Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J. 2014;457(3):415–24.
Dinkova-Kostova AT, Abramov AY. The emerging role of Nrf2 in mitochondrial function. Free Radic Biol Med. 2015;88:179–88.
Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103(11):1232–40.
Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275(21):16023–9.
Yachie A, Niida Y, Wada T, Igarashi N, Kaneda H, Toma T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J Clin Investig. 1999;103(1):129.
Li FY, Lam KS, Tse H-F, Chen C, Wang Y, Vanhoutte PM, et al. Endothelium-selective activation of AMP-activated protein kinase prevents diabetes-induced impairment in vascular function and re-endothelialization via induction of heme oxygenase-1 in mice. Circulation. 2012:CIRCULATIONAHA. 112.108159.
Parfenova H, Basuroy S, Bhattacharya S, Tcheranova D, Qu Y, Regan RF, et al. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: contributions of HO-1 and HO-2 to cytoprotection. Am J Phys Cell Phys. 2006;290(5):C1399–C410.
Alfieri A, Srivastava S, Siow RC, Cash D, Modo M, Duchen MR, et al. Sulforaphane preconditioning of the Nrf2/HO-1 defense pathway protects the cerebral vasculature against blood–brain barrier disruption and neurological deficits in stroke. Free Radic Biol Med. 2013;65:1012–22.
Han F, Hui Z, Zhang S, Hou N, Wang Y, Sun X. Induction of haemeoxygenase-1 improves FFA-induced endothelial dysfunction in rat aorta. Cell Physiol Biochem. 2015;35(3):1230–40.
Wang B, Yu Y, Han L. Adiponectin improves endothelial dysfunction caused by elevated FFAs levels, partially through cAMP-dependent pathway. Diabetes Res Clin Pract. 2012;97(1):119–24.
Li M, Kim DH, Tsenovoy PL, Peterson SJ, Rezzani R, Rodella LF, et al. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes. 2008;57(6):1526–35.
Kim DH, Burgess AP, Li M, Tsenovoy PL, Addabbo F, McClung JA, et al. Heme oxygenase-mediated increases in adiponectin decrease fat content and inflammatory cytokines tumor necrosis factor-α and interleukin-6 in Zucker rats and reduce adipogenesis in human mesenchymal stem cells. J Pharmacol Exp Ther. 2008;325(3):833–40.
Acknowledgements
Not applicable.
Funding
The authors gratefully acknowledge the financial support from the Griffith University and Hong Kong Polytechnic University Collaborative Research Grants Scheme.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
AG, LG, AT, PMS, and CWKL organized the content for this manuscript. AG and CWKL collected relevant information and prepared the draft. CWKL, LG, and AT drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ghosh, A., Gao, L., Thakur, A. et al. Role of free fatty acids in endothelial dysfunction. J Biomed Sci 24, 50 (2017). https://doi.org/10.1186/s12929-017-0357-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-017-0357-5