
Abstract
Background
Recent studies suggest that classical coronary risk factors play a significant role in the pathogenesis of coronary artery disease. Our study aims to explore the interaction of circRNA with classical coronary risk factors in coronary atherosclerotic disease.
Method
Combined analysis of RNA sequencing results from coronary segments and peripheral blood mononuclear cells of patients with coronary atherosclerotic disease was employed to identify critical circRNAs. Competing endogenous RNA networks were constructed by miRanda-3.3a and TargetScan7.0. The relative expression quantity of circRNA in peripheral blood mononuclear cells was determined by qRT-PCR in a large cohort including 256 patients and 49 controls. Spearman’s correlation test, receiver operating characteristic curve analysis, multivariable logistic regression analysis, one-way analysis of variance, and crossover analysis were performed.
Results
A total of 34 circRNAs were entered into our study, hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 were selected for further investigation. A circRNA-miRNA-mRNA network is composed of 20 microRNAs and 66 mRNAs. The expression of hsa_circRPRD1A (P = 0.004) and hsa_circHERPUD2 (P = 0.003) were significantly down-regulated in patients with coronary artery disease compared to controls. The area under the curve of hsa_circRPRD1A and hsa_circHERPUD2 is 0.689 and 0.662, respectively. Univariate and multivariable logistic regression analyses identified hsa_circRPRD1A (OR = 0.613, 95%CI:0.380–0.987, P = 0.044) as a protective factor for coronary artery disease. Based on the additive model, crossover analysis demonstrated that there was an antagonistic interaction between the expression of hsa_circHERPUD2 and alcohol consumption in subjects with coronary artery disease.
Conclusion
Our findings imply that hsa_circRPRD1A and hsa_circHERPUD2 could be used as biomarkers for the diagnosis of coronary artery disease and provide epidemiological support for the interactions between circRNAs and classical coronary risk factors.
Similar content being viewed by others


Introduction
Coronary artery disease (CAD), also known as ischemic heart disease, results from myocardial ischemia and hypoxia, which are associated with coronary artery atherosclerosis [1]. Myocardial infarction, arrhythmias, and even death can result from CAD if it is not diagnosed and treated in a timely manner. As stress myocardial perfusion imaging, coronary artery computed tomography angiography, and coronary angiography have developed, so has the diagnosis of CAD. However, in most cases, these examinations will not be considered, unless patients suffer from clinical symptoms. Unfortunately, approximately 1/4 of patients who experience myocardial infarction do so without any prior clinical symptoms [2]. Therefore, it is crucial to actively explore methods for early diagnosis of CAD.
Previous research has established that atherosclerosis is the result of plenty of complex factors, including the cells of the arterial wall, blood components, extracellular matrix, hemodynamic environment, immunity, and genetics [3]. Blood components such as monocytes, platelets, and low-density lipoprotein are all involved in the development of atherosclerosis. Peripheral blood mononuclear cells (PBMCs), as the name implies, are cells with a single nucleus in the peripheral blood that comprise both lymphocytes and monocytes. Monocytes play a critical role both in the development of atherosclerotic plaques and in the process of regression as members of the innate immune system [4]. Apart from that, a number of risk factors including age, blood pressure, blood lipids, smoking and obesity are also significant contributors to the onset of CAD [5, 6].
A unique form of non-coding RNA known as circular RNA (circRNA), which has a closed-loop structure and is not affected by RNA exonucleases [7]. Attempts have been made to explore the expression and function of circRNAs in CAD with microarray [8]. It should be noted, however, that such an exposition is unsatisfactory since microarray is not capable of recognizing novel circRNAs and only a limited number of circRNAs have been validated further. Research on circular RNAs has gradually intensified as high-throughput RNA sequencing (RNA-seq) and circular RNA-specific computational biology has been developed rapidly [9]. Therefore, we carried out a high-throughput RNA sequencing analysis in our prior study in order to determine the expression profile of circRNAs in CAD [10]. So far, however, there has been scanty discussion about the interaction effect of circRNAs and classical coronary risk factors on CAD.
Furthermore, this paper seeks to investigate the association between CAD and differentially expressed circRNAs in PBMCs. To achieve this purpose, a large cohort of individuals was analyzed using quantitative real-time polymerase chain reaction (qRT-PCR). Moreover, spearman correlation analysis, multivariable logistic regression analyses, as well as receiver operating characteristics analysis were conducted to evaluate the diagnostic value of circRNAs. In addition, the interaction effect of circRNAs and classical coronary risk factors is determined by crossover analysis. We will gain a better understanding of the pathogenesis of atherosclerosis by focusing on the association between circRNAs and CAD. In addition, it will also help us explore the interaction effect of circRNAs and classical coronary risk factors on CAD.
Methods
Study population
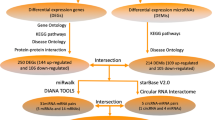
Our study population was divided into three groups in order to begin this process. Firstly, we obtained the epicardial coronary arteries from the autopsy specimen of an 81-year-old man who died from a heart attack. The autopsy was carried out by the Department of Human Anatomy in Nanjing Medical University. The preparation of coronary artery segments is described in detail in our previous study [11]. And then, Secondly, in order to investigate the relationship between circRNAs in PBMCs and CAD, transcriptome high throughput sequencing was conducted on 5 CAD patients and 5 healthy controls [12]. Finally, the validation group consisted of 256 individuals in the CAD group and 49 individuals in the control group. All patients were recruited at the First Affiliated Hospital of Nanjing Medical University. Figure 1 illustrates the flow chart of the present study.

The flow chart of the present study. Integrated analysis between RNA sequencing results both in coronary artery segments and peripheral blood mononuclear cells was employed to figure out hub circRNA. 4 circRNAs was picked up by sanger sequencing from 34 circRNAs. Further investigation found out the expression of hsa_circRPRD1A and hsa_circHERPUD2 were significantly down-regulated in patients with CAD compared to controls, eventually
Our study was carried out in accordance with the ethical guidelines of the First Affiliated Hospital of Nanjing Medical University’s ethical committee. We obtained written informed permission from all patients or their families according to the Declaration of Helsinki.
Ascertainment of CAD
Whether coronary artery atherosclerosis occurs depends on the degree of luminal stenosis caused by arterial atherosclerosis. Coronary artery atherosclerosis is classified into four grades based on the cross-section of the stenosis that is the most severe: grade I, luminal stenosis area less than 25%; grade II, luminal stenosis area of 26–50%; grade III, 51–75%; and grade IV, 76–100% [13]. Generally, with the exception of coronary artery spasms, grade I-II atherosclerosis normally does not significantly reduce arterial blood flow or have a direct impact on CAD. Stenosis of grade III or above is closely related to CAD. Consequently, patients with at least one major coronary artery with stenosis greater than 50% were selected as the patient group, whereas individuals with all major coronary arteries with stenosis less than 50% were considered as the control group [14]. Patients with severe bacterial/viral/fungal infections, congenital heart abnormalities, cerebrovascular illnesses, cardiomyopathy, malignancy, severe hepatic and renal insufficiency, and a history of percutaneous coronary intervention were excluded from our study.
Covariates
Baseline data including routine blood and blood biochemical tests were collected from the patient’s medical records. An interview-style questionnaire was used at baseline to collect information regarding smoking and alcohol consumption. Smoking is characterized as a person who has smoked continuously or cumulatively for 6 months or more in his lifetime. Further questions were asked about the frequency of their drinking (number of months, number of times per month), the type of alcoholic beverage they consumed (beer, liquor, wine, rice wine) and the amount of alcohol consumed per session. Lastly, individuals who consumed alcohol at least once a week during the past year and drank at least 12 g of alcohol per drink were considered to be alcohol consumers [15, 16]. The criteria for hypertension were an average systolic blood pressure greater than or equal to 140 mmHg, and/or diastolic blood pressure greater than or equal to 90 mmHg [17]. Diabetes is defined as fasting blood glucose exceeding than 7.8 mmol/l, or the use of hypoglycemic agents during the last 2 weeks [18]. Weight (kg) divided by height (m) squared was used to determine body mass index (BMI).
Peripheral blood mononuclear cells collected
A fasting venous blood sample was obtained from each participant eight hours prior to coronary arteriography, which was pretreated with regular doses of aspirin and clopidogrel. To separate and preserve the lower blood cells, all samples were anticoagulated with ethylenediaminetetraacetic acid (EDTA)-2 K and centrifuged at 3000 rpm/centrifugation for 10 min. Then, we extracted the second white cloudy cell layer from it after centrifuging at 2000 rpm/centrifugation for 20 min with an equivalent lymphocyte separation medium (TBD, Tianjin, China). Finally, resuspended PBMCs with 1ml phosphate buffer saline (Biosharp, Anhui, China) and a counting was performed.
RNA isolation
First, we extracted the total RNA from the PBMCs with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). After PBMCs were lysed in TRIzol for ten minutes at 24 °C, 200 µl chloroform was added. Then, the resulting solution was gently mixed at room temperature for ten minutes and centrifuged with 12,000 g for twenty minutes at 4 °C. Next, we meticulously aspirated the upper aqueous phase into another centrifuge tube. We precipitated with equal isopropanol with centrifuged through 12,000 g×10 min, purified with 75% ethanol through centrifuged with 7500 g×5 min, and dissolved the sediment in RNase-free water. A NanoDrop 1000 spectrophotometer (Agilent Inc. USA) was used to measure RNA concentration and mass.
Reverse transcription
To obtain complementary DNA (cDNA), we performed a reverse transcription reversed transcribe with RNA as the template in the presence of primers. Transcript one-step gDNA removal (YEASEN, Shanghai, China) was used to remove residual genomic DNA from the RNA template. And first-strand cDNA was synthesized using cDNA synthesis super-mix (YEASEN, Shanghai, China). Each stage is carried out on ice, and RNase contamination was strictly avoided throughout the procedure.
Real-time polymerase chain reaction
The cDNA, SYBR q70P1CR master mix (EnzyArtisan, Shanghai, China), the primers, and diethyl pyrocarbonate treated water were configured according to the instructions. The aforementioned mixture was centrifuged at 3000 g for three minutes before performing qRT-PCR on StepOnePlus (Applied Biosystems, USA) equipment. Each well was repeated for three times to ensure the quantification accuracy. The expression of the circRNA was calculated using the 2−ΔΔCt method, and GAPDH was employed as an internal reference gene.
Bioinformatic analyses
The microRNAs that may be in competition with our four targeted circRNAs were obtained using miRanda-3.3a [19]. We constructed a competing endogenous RNA network by predicting the target genes of the top 5 miRNAs ranked by their structure score of every circRNAs through TargetScan7.0 [20] Cytoscape 3.8.2 was used to visualized the related lncRNA, miRNA and miRNA target genes [21].
Statistical analysis
The data were analyzed by SPSS (version26.0, USA) and GraphPad Prism (version8.0, USA). Normally distributed data were presented by mean ± standard deviation (SD) determined by t-test, otherwise presented as median (interquartile range) and determined by the Wilcoxon-Mann-Whitney test. The Spearman correlation and multivariate regression were used to further identify the relationship between circRNAs expression levels and clinical characteristics in CAD patients. The receiver operating characteristic (ROC) curve analysis was carried out to evaluate the potential diagnostic value of circRNAs. Multivariate logistic regression and unconditional logistic regression were used to evaluate relationships between relative expression levels of circRNAs and CAD. We conducted crossover analyses to identify the interaction of circRNAs with classical coronary risk factors in subjects with CAD [22]. The additive interaction was evaluated using the relative excess risk owing to interaction (RERI), attributable proportion of interaction (AP), and synergy index (S). There was no additive interaction when the 95% confidence interval(95%CI) of RERI contained 0 or 95%CI of S contained 1, which demonstrate that the combined effect of circRNA and classical coronary risk factors is equal to the sum of their individual effects. For further analysis, we used one-way analysis of variance (ANOVA) and post-hoc tests to assess differences between groups. An analysis of normality and homogeneity of variance of the data was conducted using the Levene test and the Brown-Forsythe test. P < 0.05 was considered to be statistically significant.
Result
Overview of RNA sequencing results in PBMCs
The relative expression level of circRNA in CAD patients in PBMCs was determined by RNA sequencing [10]. Compared with normal controls, the expression levels of 12,905 circRNAs and 13,161 circRNAs was up-regulated and down-regulated in the CAD group, respectively (Fig. 2A). What’s more, there are 679 circRNAs and 673 circRNAs were assessed to be significantly different expressions between the two groups (P < 0.05). At the same time, our results revealed approximately 75% of circRNAs were exonic. The proportion was generally 13% at sense overlapping, approximately 10% at intronic, and less than 1% of circRNAs were classified as antisense and intergenic (Fig. 2B-C). Besides, the circRNAs locations were identified on all human chromosomes and one notable exception was that there are 29 circRNAs positioned in chrM (Fig. 2D).

Overview of circRNAs RNA sequence results in peripheral blood mononuclear cells. (A) Proportion exhibiting the number of circRNAs in the up-regulation group and down-regulation group. CircRNA with a significant difference is shown in the blue and others in the red. (B-C) Pie charts exhibit the catalog of the circRNA, including antisense, exonic, intergenic, intronic, and sense overlapping in the two groups. (D) Chromosomal distribution of all identified circRNAs. (E-F) Histogram displaying the top 20 circRNA-associated diseases of all identified circRNA indicated by circ2Trait in the up-regulation group and down-regulation group, respectively. (G) Proportion of novel circRNAs in the two groups. (H) Venn diagram exhibiting number of common circRNAs between RNA-seq in coronary artery and PBMCs.
In addition, circRNA-associated diseases were categorized by circ2Trait (http://gyanxet-beta.com/circdb/) [23]. There are 5011 up-regulated circRNAs linked to 86 different kinds of diseases and 4836 down-regulated circRNAs associated with 93 different kinds of diseases (Fig. 2E-F). Among these circRNAs, 472 circRNAs were correlated with cardiac hypertrophy. Furthermore, the results described 10,426 novel circRNAs not been mentioned in any circRNA database before (Fig. 2G).
Transcriptome profile of human coronary arteries by RNA-Seq
This research is a component of our series whose findings have already been reported [24]. CircRNAs expression was statistically calculated and normalized by TPM [25].
Integrated RNA-seq analysis
In our previous study, the circRNA profiles in PBMCs of CAD patients were identified by RNA-seq. We also characterized human coronary artery transcriptomes by RNA-seq at the same time. A total of 34 circRNAs were not only detected in coronary artery segments but also differently expressed in PBMCs of CAD patients (fold-change ≥ 2 and P < 0.05) (Fig. 2H). The characteristics of the circRNAs mentioned above are displayed in Table 1. Additionally, the expression levels of 34 circRNAs in human coronary artery segments are listed in Table 2. Then, we designed primers for circRNA junction sites. The qRT-PCR products were sequenced in order to determine the specificity of the primers used for amplification, and the results were compared with the reference sequence found in the circBase database. The Sanger sequencing results revealed that the product sequences were compatible with the database sequence, indicating that hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 could be amplified specifically by qRT-PCR (Fig. 3).

Sanger sequencing of selected candidate circRNAs. The black arrow points to the back-splice junctions. The number above the ring indicates how many bases the circRNA is made up of and every color represents an exon, respectively
Construction of competing endogenous RNA networks
It is known that circRNA regulates gene expression by competitively binding microRNAs [26, 27]. Firstly, we predicted the potential binding miRNAs of hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 by miRanda. Our findings revealed interactions between 228 miRNAs and 4 circRNAs. And then target gene prediction was performed on the top 20 miRNAs ranked by structure score, and these target genes were analyzed jointly with the mRNAs in PBMCs from CAD patients in our previous studies, and 66 target mRNAs were obtained that may be involved in the development of CAD under circRNAs regulation. As shown in Fig. 4, the rank top 5 miRNAs and the related 66 mRNAs were used to construct a circRNA-miRNA-mRNA network.

The circRNA-miRNA-mRNA interaction network. Yellow round nodes indicate four selected circRNAs. The green arrow and the blue diamond represent miRNAs and mRNAs, respectively. The lines between them indicate their interconnectedness
Validation of the circRNA expression
The quantitative real-time polymerase chain reaction was employed to validate the expression level of hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, hsa_circDHTKD1 in a large cohort. First, qRT-PCR primers for circRNA were listed in supplementary Table 1. Second, 256 CAD patients and 49 healthy controls defined by coronary angiography were included in this study. The summary statistics of the clinical characteristics are depicted in Table 3. Gender (p < 0.001), cardiac troponin T (P = 0.031), and Gensini score (p < 0.0001) were significantly different between the two groups. Finally, the relative expression levels of the four circRNAs were evaluated in PBMCs. Compared with controls, the expression levels of hsa_circRPRD1A (P = 0.004) and hsa_circHERPUD2 (P = 0.003) were significantly decreased in CAD patients. In contrast to the controls, the expression levels of hsa_circLMBR1 (P = 0.906), and hsa_circDHTKD1 (P = 0.288) presented were up-regulated in the CAD group. However, no significant difference between the two groups was evident (Fig. 5A-D).

Validation of the circRNAs expression and identification the potential biomarker (A-D) qRT-PCR analysis of expression of 4 circRNAs in a large sample of CAD patients and healthy controls. *P < 0.05, **P < 0.001. (E-F) ROC curve analysis of hsa_circRPRD1A and hsa_circHERPUD2 discrimination of CAD patients from healthy controls
Identification of potential circRNA biomarkers
The spearman correlation analysis displayed that the expression level of hsa_circHERPUD2 was positively correlated with BMI (r = 0.152, P = 0.026) and the expression level of hsa_circLMBR1 was positively correlated with high-density lipoprotein cholesterol (r = 0.212, p < 0.001). Besides, it can be seen from Table 4 that the expression level of hsa_circRPRD1A was positively correlated with hsa_circHERPUD2 expression (r = 0.394, p < 0.001) and hsa_circDHTKD1 (r = 0.156, P = 0.046). Additionally, the expression level of hsa_circLMBR1 was also positively correlated with hsa_circDHTKD1 (r = 0.564, p < 0.001). After adjusting for a number of different clinical parameters in the multivariate linear regression model, we discovered that lip(a) (β = 0.390 P = 0.008), high-density lipoprotein cholesterol (β = 0.212, P = 0.001), and lipoprotein (a) (β = 0.134, P = 0.040) were independent factors associated with the expressions of hsa_circRPRD1A, hsa_circHERPUD2, and hsa_circLMBR1, respectively.
Diagnostic values of circRNA expressions
The results obtained from the ROC analysis of hsa_circRPRD1A and hsa_circHERPUD2 are presented in Fig. 5E-F. The area under the ROC curve (AUC) of hsa_circRPRD1A was 0.689 (95% CI:0.593–0.786, P = 0.004). A hsa_circRPRD1A expression level of 0.526 was the most appropriate cut-off value with sensitivity and specificity of 0.588 and 0.782, respectively. As for hsa_circHERPUD2, the AUC was 0.662 (95% CI:0.590–0.734, P = 0.006), and the cut-off values was 0.662 of 0.479 and 0.808 for sensitivity and specificity, respectively.
Univariate and multivariable logistic regression analysis
Multivariable logistic regression analyses were conducted to investigate risk factors and predict CAD probability. As shown in Table 5, without adjustment, hsa_circRPRD1A acts as a protective factor for coronary artery disease with an odds ratio of 0.625 (95% CI: 0.412–0.948, P = 0.027). After age, gender, BMI, smoking, alcohol consumption, diabetes mellitus, hypertension, TC, TG, Apo (a), fasting blood glucose levels, and LDL-C were adjusted, the expression of hsa_circRPRD1A (OR = 0.613, 95%CI:0.380–0.987, P = 0.044) remained significantly associated with CAD prevalence. Unfortunately, no evidence was found for multivariable logistic associations between hsa_circHERPUD2 (P = 0.837), hsa_circLMBR1 (P = 0.229), hsa_circDHTK-D1 (P = 0.441) and CAD.
Analysis of variance
The study population was classified into mild, ordinary, severe and critical groups according to the Gensini score quartile. The ANONA found no significant differences in the expression of hsa_circRPRD1A, hsa_circHERPUD2, and hsa_circLMBR1 between cases with different disease severity (Table 6). Interestingly, there were a significant difference in the expression of hsa_circDHTKD1 between the four groups (F = 3.739, P = 0.012). What’s more, the Bonferroni test was used for multiple comparisons and revealed that the expression of hsa_circDHTKD1 between ordinary cases and severe cases was significantly different (P = 0.011).
Crossover analysis of circRNA and risk factors
Based on the additive model, we analyzed the modification effect interaction between the expression of circRNA and CAD risk factors. As can be seen from Table 7, the expression of hsa_circRPRD1A is negatively interacted with smoking (S = 0.658, API = 2.395, RERI = 0.428) and hypertension (S=-2.117, API=-3.182, RERI=-1.007) in patients with CAD compared to controls. Due to the 95%CI of S containing 1, the interaction between hsa_circRPRD1A and smoking or hypertension was not considered significant. Simultaneously, the expression of hsa_circHERPUD2 negatively interacted with smoking (S = 0.682), drinking (S = 0.500), and hypertension (S=-0.130). Alcohol consumption was the only variable that significantly negative interacted with the expression of hsa_circHERPUD2 (API = 4.415, RERI = 0.707), suggesting that there was an antagonistic interaction between the expression of hsa_circHERPUD2 and alcohol consumption in subjects with CAD. Based on a further analysis of alcohol consumption, it was revealed that patients who consumed alcohol had higher levels of high-density lipoprotein cholesterol than those who did not consume alcohol (Supplementary Table 2).
Discussion
We investigated whether circRNAs have affect atherosclerosis pathogenesis and explored biomarkers for the diagnosis of CAD in our present study. To examine the relationship between differently expressed circRNAs in PBMCs with CAD, we performed qRT-PCR in large cohort populations and conducted statistical analysis. According to our results, firstly, we verified the specificity of the amplification primers of hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 by sanger sequencing. Secondly, we discovered that the expression level of hsa_circRPRD1A and hsa_circHERPUD2 were significantly down-regulated in CAD patients compared with controls. Last but not least, ROC curve analysis, multivariable logistic regression analysis along with crossover analysis observed that hsa_circRPRD1A and hsa_circHERPUD2 can function as diagnostic biomarkers and together with disease risk factors play a crucial role in the development of CAD.
CircRNAs have been discovered to play a role in the pathological of cardiovascular processes, such as myocardial infarction [28], heart failure [29], and overall atherosclerosis [30]. In addition, PBMCs are a key player in atherosclerosis plaque development. The impact of circGSAP in PBMCs on idiopathic pulmonary arterial hypertension has been examined by investigators [31]. The expression profiles of circRNAs in PBMCs from retinopathy of prematurity patients were revealed by Li et al. through microarray analysis [32]. Additionally, evidence from numerous research points to a potential link between the expression of circRNAs and the incidence and progression of rheumatoid arthritis [33, 34]. Extensive research has shown that circRNA can be considered as a potential biomarker for clinical diagnosis and treatment of CAD [35].
In this research, we found that 1352 circRNAs were significantly differentially expressed (|log2FC |≥ 2 and p < 0.05) in patients with CAD, compared with controls. As revealed by the RNA-seq results, the number of up-regulated circRNAs was similar to circRNAs exhibiting downregulation. This finding differs from Yu et al.’s study by which concluded that more down-regulated circRNAs were identified in patients with CAD [36]. This inconsistency could be attributed to the difference in sequencing instruments. We used an illumina Novaseq 6000 instrument with 150 bp paired-end reads to complete our library sequencing, while they were sequenced on an illumina HiSeq 2500 platform with 125-bp paired-end reads. In addition, a large proportion of the circRNAs were represented as exonic in our study, similar to that found by Li et al. [37]. Besides, we discovered that circRNAs were widely distributed throughout all human chromosomes, with chr1, chr2, and chr4 at the top3 of circRNA-containing chromosomes. Surprisingly, we found that there are 29 circRNAs positioned on chrM. It is possible, therefore, that these circRNAs regulate the entry of proteins into the mitochondria, thus participating in metabolic inflammatory processes such as the production of inflammatory factors [38, 39]. What’s more, there are 472 circRNAs were correlated with cardiac hypertrophy, which indicates that they may participate in protein regulation synthesis in cardiomyocytes.
Apart from that, we performed a joint analysis of RNA-seq results in human coronary artery segments and peripheral PBMCs in CAD patients. In total, there are 34 circRNAs that have attracted our attention. The sanger sequence results revealed that no junction sites were found in 30 circRNAs, contrary to some previous studies [40,41,42,43]. According to their study, hsa_circFKBP8, hsa_circLMTK2, hsa_circMRRF, and hsa_circCLASP2 were further researched. The difference may be related to the primer design, and redesigning the primers might aid in our understanding of these circRNAs.
Additionally, we constructed the circRNA-miRNA-mRNA networks for hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1, aiming to explore the possible regulation mechanism in patients with CAD. It is interesting to note that the PRKX [44] and TFRC [45] expression was downregulated in CAD patients which has already been reported. Moreover, RPS3A, BCL6, and S100B play a critical role in coronary atherosclerosis [44,45,46,47]. This finding broadly supports other studies in this area linking circRNAs with CAD, and provides evidence that hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 take part in the pathogenesis of atherosclerosis.
Given that hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 were dysregulated in patients with CAD, we sought to further investigate alterations in hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 in CAD. As expected, the expression levels of hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 were decreased in CAD patients. However, only the differential expression of hsa_circHERPUD2 was statistically significant. qRT-PCR also revealed that the hsa_circRPRD1A expression was significantly decreased in CAD patients compared with controls, which was contrary to expectations. This discrepancy could be attributed to that we had sequenced only ten samples, while we validated in 305 samples. We proceeded with post-hoc analysis for the validation group by G*Power and the power (1-β error prob) was calculated as 0.931, indicating that our experimental results are plausible. Zhang et al. validated 58 genes, 50 of which were consistent in qPCR and RNA-Seq sequencing results, suggesting that our discrepancy is acceptable [48].
To understand the diagnosis ability of hsa_circRPRD1A and hsa_circHERPUD2 for CAD, further statistical analyses were performed. Based on spearman rank correlation analysis and ROC analyses, hsa_circRPRD1A and hsa_circHERPUD2 could be potential diagnostic biomarkers for CAD. Furthermore, univariate and multivariable logistic regression analyses identified hsa_circRPRD1A as a protective factor for coronary artery disease, with the elevated expression of hsa_circRPRD1A1 and a possible decrease in CAD incidence of 37.5%. It’s interesting to note that although the expression of hsa_circDHTKD1 was not statistically different between patients with CAD and normal control subjects, ANOVA found that its expression was different between patients with ordinary and severe CAD. This indicates that although hsa_circDHTKD1 is not a suitable biomarker for diagnosing CAD, its expression still affects the progression of CAD. And it is capable of evaluating the severity of a disease based on the expression of hsa_circDHTKD1.
According to prior studies, light-to-moderate alcohol consumption was particularly protective against CAD [49, 50], which has been attributed to the connection between alcohol consumption with high-density lipoprotein cholesterol [51,52,53]. According to the additive model, crossover analysis found an antagonistic relationship between the expression of hsa_circHERPUD2 in subjects with CAD and alcohol consumption in our study. The value of RERI was 0.707, indicating that hsa_circHERPUD2 expression interacted with alcohol consumption accounted for 70.7% of the effects induced by hsa_circHERPUD2 expression and alcohol consumption. Although the expression of hsa_circHERPUD2A was not an independent protective factor for CAD, the combined effect of hsa_circHERPUD2 expression and alcohol consumption on the development of CAD is meaningful. An implication of this is the possibility that hsa_circHERPUD2 is more relevant for the development of CAD in individuals with risk factors. Nevertheless, alcohol is not recommended for preventing myocardial ischemia in our study, due to its addictive nature as well as its cancer-causing properties. This suggested that we should also be aware of the influence of classical cardiovascular risk factors in our search for molecular mechanisms of CAD.
Simultaneously, there was no significantly interaction between the hsa_circRPRD1A1 expression and classical cardiovascular risk factors in patients with CAD. However, with a smaller sample size in the control group than in the coronary group, caution must be applied, as such imbalance may lead to overly wide confidence intervals and an unstable combined effect of hsa_circRPRD1A1 expression and classical cardiovascular risk factors. According to these data, we can still infer that the expression of hsa_circRPRD1A1 and hsa_circHERPUD2 are tightly associated with the development of CAD. The interaction effect between circRNAs and classical cardiovascular risk factors in patients with CAD cannot be ignored.
Our study adds to the accumulating evidence that the expression of circRNAs can influence the occurrence and development of CAD, possibly through the regulation of protein entry into mitochondria or ceRNA mechanisms. At the same time, hsa_circRPRD1A and hsa_circHERPUD2 were proven to work as biomarkers for the CAD diagnosis in our study. What’s more, our findings might provide epidemiological support for the independent associations of circRNAs and environment factors and the interactions between circRNAs and environment factors. There are some limitations in our study, nevertheless. Our subjects were a homogenous group of the southern Han Chinese population; hence, it is doubtful whether our results are applied to other groups. Likewise, our study only included 305 individuals, which may be too few to extend this conclusion. Thus, further studies with large populations are needed.
Conclusion
Coronary artery disease leads to the most morbidity and mortality worldwide nowadays, while most diagnosis examinations are invasive or radiation intensive and difficult to accept by patients. In order to explore potential biomarkers for early CAD diagnosis, we performed high-throughput RNA-seq and qRT-PCR. As the research has demonstrated, hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1 caught our attention and were further investigated. The circRNA-miRNA-mRNA network was then constructed based on the rank of the top 5 miRNAs and the 66 mRNAs they are related to. To validate the expression level of hsa_circRPRD1A, hsa_circHERPUD2, hsa_circLMBR1, and hsa_circDHTKD1, a qRT-PCR assay in a large cohort was performed. We can see that the expression of hsa_circRPRD1A and hsa_circHERPUD2 significantly decreased in CAD patients compared with controls. In-depth statistical analyses infer that they could be used as biomarkers for the CAD diagnosis. What’s more, crossover analysis confirmed that the interaction effect between circRNAs and environment factors on patients with CAD cannot be ignored. Our findings might provide epidemiological support for the independent association between circRNAs and classical coronary risk factors as well as their interactions. Future research will concentrate on the significant role that hsa_circRPRD1A and hsa_circHERPUD2 play in CAD pathogenesis.
Data Availability
CloudSeq Biotech (Shanghai, China) performed high throughput transcriptome sequencing. RNA libraries were constructed by NEBNext® rRNA Depletion Kit (New England Biolabs, Inc., Massachusetts, USA) and TruSeq Stranded Total RNA Library Prep Kit(Illumina, USA). Library quality control and quantification using the BioAnalyzer 2100 instrument (Agilent Technologies, USA). Library sequencing was performed on an Illumina Novaseq 6000 instrument, eventually. The datasets used and analyzed during the current study are deposited in the GEO database with the accession number GSE205255. The resources can be viewed at https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE205255 with the reviewer secure token: cpwncewgbdmrpcr.
Abbreviations
- CAD:
-
Coronary artery disease
- circRNA:
-
circular RNA
- ROC:
-
receiver operating characteristic curves
- AUC:
-
the area under receiver operating characteristic curves
- PBMCs:
-
peripheral blood mononuclear cells
- qRT-PCR:
-
quantitative real-time polymerase chain reaction
- RPRD1A:
-
regulation of nuclear pre-mRNA domain containing 1 A
- HERPUD2:
-
HERPUD family member 2
- LMBR1:
-
limb development membrane protein 1
- DHTKD1:
-
dehydrogenase E1 and transketolase domain containing 1
References
Libby P, Pasterkamp G, Crea F, Jang IK. Reassessing the Mechanisms of Acute Coronary Syndromes. Circ Res. 2019;124(1):150–60.
Dawber TR, Meadors GF, Moore FE Jr. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41(3):279–81.
Pothineni NVK, Subramany S, Kuriakose K, Shirazi LF, Romeo F, Shah PK, Mehta JL. Infections, atherosclerosis, and coronary heart disease. Eur Heart J. 2017;38(43):3195–201.
Ghattas A, Griffiths HR, Devitt A, Lip GY, Shantsila E. Monocytes in coronary artery disease and atherosclerosis: where are we now? J Am Coll Cardiol. 2013;62(17):1541–51.
Powell-Wiley TM, Poirier P, Burke LE, Despres JP, Gordon-Larsen P, Lavie CJ, Lear SA, Ndumele CE, Neeland IJ, Sanders P, et al. Obesity and Cardiovascular Disease: A Scientific Statement from the American Heart Association. Circulation. 2021;143(21):e984–e1010.
Cullen P, Schulte H, Assmann G. Smoking, lipoproteins and coronary heart disease risk. Data from the Munster Heart Study (PROCAM). Eur Heart J. 1998;19(11):1632–41.
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777.
Wang L, Shen C, Wang Y, Zou T, Zhu H, Lu X, Li L, Yang B, Chen J, Chen S, et al. Identification of circular RNA Hsa_circ_0001879 and Hsa_circ_0004104 as novel biomarkers for coronary artery disease. Atherosclerosis. 2019;286:88–96.
Sharma AR, Bhattacharya M, Bhakta S, Saha A, Lee SS, Chakraborty C. Recent research progress on circular RNAs: Biogenesis, properties, functions, and therapeutic potential. Mol Ther Nucleic Acids. 2021;25:355–71.
Ji WF, Chen JX, He S, Zhou YQ, Hua L, Hou C, Zhang S, Gan XK, Wang YJ, Zhou HX, et al. Characteristics of circular RNAs expression of peripheral blood mononuclear cells in humans with coronary artery disease. Physiol Genomics. 2021;53(8):349–57.
Hou C, Gu L, Guo Y, Zhou Y, Hua L, Chen J, He S, Zhang S, Jia Q, Zhao C, et al. Association between circular RNA expression content and severity of coronary atherosclerosis in human coronary artery. J Clin Lab Anal. 2020;34(12):e23552.
Chen JX, He S, Wang YJ, Gan XK, Zhou YQ, Hua L, Hou C, Zhang S, Zhou HX, Jia EZ. Comprehensive Analysis of mRNA expression profiling and identification of potential diagnostic biomarkers in coronary artery disease. ACS Omega. 2021;6(37):24016–26.
Gensini GG. A more meaningful scoring system for determining the severity of coronary heart disease. Am J Cardiol. 1983;51(3):606.
Nikolsky E, Halabi M, Roguin A, Zdorovyak A, Gruberg L, Hir J, Grenadier E, Boulos M, Markiewicz W, Linn S, et al. Staged versus one-step approach for multivessel percutaneous coronary interventions. Am Heart J. 2002;143(6):1017–26.
Hu C, Huang C, Li J, Liu F, Huang K, Liu Z, Yang X, Liu X, Cao J, Chen S, et al. Causal associations of alcohol consumption with cardiovascular diseases and all-cause mortality among chinese males. Am J Clin Nutr. 2022;116(3):771–9.
Kloner RA, Rezkalla SH. To drink or not to drink? That is the question. Circulation. 2007;116(11):1306–17.
Bourne G. The criteria of hypertension. Practitioner. 1947;158(948):470–5.
Criteria for. Establishing the diagnosis in diabetes. Res Newsl 1957(17):360.
Enright AJ, John B, Gaul U, Tuschl T, Sander C, Marks DS. MicroRNA targets in Drosophila. Genome Biol. 2003;5(1):R1.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4.
Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Curr Protoc Bioinformatics. 2014;47:81311–24.
Wang PH, Shen HB, Chen F, Zhao JK. [Study on the significance and application of crossover analysis in assessing gene-environmental interaction]. Zhonghua Liu Xing Bing Xue Za Zhi. 2005;26(1):54–7.
Ghosal S, Das S, Sen R, Basak P, Chakrabarti J. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet. 2013;4:283.
Pan RY, Zhao CH, Yuan JX, Zhang YJ, Jin JL, Gu MF, Mao ZY, Sun HJ, Jia QW, Ji MY, et al. Circular RNA profile in coronary artery disease. Am J Transl Res. 2019;11(11):7115–25.
Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y, Zhao X, Liang C, Wang Y, Sun L, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS ONE. 2010;5(12):e15224.
Feng C, Li Y, Lin Y, Cao X, Li D, Zhang H, He X. CircRNA-associated ceRNA network reveals ErbB and Hippo signaling pathways in hypopharyngeal cancer. Int J Mol Med. 2019;43(1):127–42.
Zhao R, Li FQ, Tian LL, Shang DS, Guo Y, Zhang JR, Liu M. Comprehensive analysis of the whole coding and non-coding RNA transcriptome expression profiles and construction of the circRNA-lncRNA co-regulated ceRNA network in laryngeal squamous cell carcinoma. Funct Integr Genomics. 2019;19(1):109–21.
Geng HH, Li R, Su YM, Xiao J, Pan M, Cai XX, Ji XP. The circular RNA Cdr1as promotes myocardial infarction by mediating the regulation of miR-7a on its target genes expression. PLoS ONE. 2016;11(3):e0151753.
Shen L, Hu Y, Lou J, Yin S, Wang W, Wang Y, Xia Y, Wu W. CircRNA0044073 is upregulated in atherosclerosis and increases the proliferation and invasion of cells by targeting miR107. Mol Med Rep. 2019;19(5):3923–32.
Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, Zhou LY, Sun T, Wang M, Yu T, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37(33):2602–11.
Yuan P, Wu WH, Gong SG, Jiang R, Zhao QH, Pudasaini B, Sun YY, Li JL, Liu JM, Wang L. Impact of circGSAP in Peripheral Blood mononuclear cells on idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2021;203(12):1579–83.
Li Y, Zhou H, Huang Q, Tan W, Cai Y, Wang Z, Zou J, Li B, Yoshida S, Zhou Y. Potential biomarkers for retinopathy of prematurity identified by circular RNA profiling in peripheral blood mononuclear cells. Front Immunol. 2022;13:953812.
Zheng F, Yu X, Huang J, Dai Y. Circular RNA expression profiles of peripheral blood mononuclear cells in rheumatoid arthritis patients, based on microarray chip technology. Mol Med Rep. 2017;16(6):8029–36.
Wen J, Liu J, Zhang P, Jiang H, Xin L, Wan L, Sun Y, Huang D, Sun Y, Long Y et al. RNA-seq reveals the circular RNA and miRNA expression profile of peripheral blood mononuclear cells in patients with rheumatoid arthritis. Biosci Rep 2020, 40(4).
Yu F, Zhang Y, Wang Z, Gong W, Zhang C. Hsa_circ_0030042 regulates abnormal autophagy and protects atherosclerotic plaque stability by targeting eIF4A3. Theranostics. 2021;11(11):5404–17.
Yu F, Tie Y, Zhang Y, Wang Z, Yu L, Zhong L, Zhang C. Circular RNA expression profiles and bioinformatic analysis in coronary heart disease. Epigenomics. 2020;12(5):439–54.
Li Q, Wang Y, An Y, Wang J, Gao Y. The Particular expression profiles of circular RNA in Peripheral blood of myocardial infarction patients by RNA sequencing. Front Cardiovasc Med. 2022;9:810257.
Liu X, Wang X, Li J, Hu S, Deng Y, Yin H, Bao X, Zhang QC, Wang G, Wang B, et al. Identification of mecciRNAs and their roles in the mitochondrial entry of proteins. Sci China Life Sci. 2020;63(10):1429–49.
Zhao Q, Liu J, Deng H, Ma R, Liao JY, Liang H, Hu J, Li J, Guo Z, Cai J, et al. Targeting Mitochondria-Located circRNA SCAR alleviates NASH via reducing mROS output. Cell. 2020;183(1):76–93e22.
Wang S, Tang D, Wang W, Yang Y, Wu X, Wang L, Wang D. circLMTK2 acts as a sponge of mir-150-5p and promotes proliferation and metastasis in gastric cancer. Mol Cancer. 2019;18(1):162.
Jin G, Wang Q, Hu X, Li X, Pei X, Xu E, Li M. Profiling and functional analysis of differentially expressed circular RNAs in high glucose-induced human umbilical vein endothelial cells. FEBS Open Bio. 2019;9(9):1640–51.
Chen H, Li Y. Circular RNA hsa_circ_0000915 promotes propranolol resistance of hemangioma stem cells in infantile haemangiomas. Hum Genomics. 2022;16(1):43.
Ma C, Wang X, Yang F, Zang Y, Liu J, Wang X, Xu X, Li W, Jia J, Liu Z. Circular RNA hsa_circ_0004872 inhibits gastric cancer progression via the miR-224/Smad4/ADAR1 successive regulatory circuit. Mol Cancer. 2020;19(1):157.
Long F, Wang L, Yang L, Ji Z, Hu Y. Screening hub genes in coronary artery disease based on integrated analysis. Cardiol J. 2018;25(3):403–11.
Bonanni A, d’Aiello A, Pedicino D, Di Sario M, Vinci R, Ponzo M, Ciampi P, Lo Curto D, Conte C, Cribari F et al. Molecular Hallmarks of Ischemia with non-obstructive coronary arteries: the “INOCA versus Obstructive CCS” challenge. J Clin Med 2022, 11(6).
Chen H, Wang Y, Sun B, Bao X, Tang Y, Huang F, Zhu S, Xu J. Negative correlation between endoglin levels and coronary atherosclerosis. Lipids Health Dis. 2021;20(1):127.
Zhang X, Cheng M, Tong F, Su X. Association between RAGE variants and the susceptibility to atherosclerotic lesions in chinese Han population. Exp Ther Med. 2019;17(3):2019–30.
Zhang W, Chen J, Keyhani NO, Zhang Z, Li S, Xia Y. Comparative transcriptomic analysis of immune responses of the migratory locust, Locusta migratoria, to challenge by the fungal insect pathogen, Metarhizium acridum. BMC Genomics. 2015;16:867.
Leong DP, Smyth A, Teo KK, McKee M, Rangarajan S, Pais P, Liu L, Anand SS, Yusuf S, Investigators I. Patterns of alcohol consumption and myocardial infarction risk: observations from 52 countries in the INTERHEART case-control study. Circulation. 2014;130(5):390–8.
Lv J, Yu C, Guo Y, Bian Z, Yang L, Chen Y, Tang X, Zhang W, Qian Y, Huang Y, et al. Adherence to healthy Lifestyle and Cardiovascular Diseases in the Chinese Population. J Am Coll Cardiol. 2017;69(9):1116–25.
Rimm EB, Williams P, Fosher K, Criqui M, Stampfer MJ. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319(7224):1523–8.
Mukamal KJ, Jensen MK, Gronbaek M, Stampfer MJ, Manson JE, Pischon T, Rimm EB. Drinking frequency, mediating biomarkers, and risk of myocardial infarction in women and men. Circulation. 2005;112(10):1406–13.
Linn S, Carroll M, Johnson C, Fulwood R, Kalsbeek W, Briefel R. High-density lipoprotein cholesterol and alcohol consumption in US white and black adults: data from NHANES II. Am J Public Health. 1993;83(6):811–6.
Acknowledgements
Not applicable.
Funding
This study received support from the National Natural Science Foundation of China (Grants 81170180, 30400173, 30971257, and 81970302) and the Natural Science Foundation of Tibet Autonomous Region (Grants XZ202101ZR0068G).
Author information
Authors and Affiliations
Contributions
Conceptualization, QZ; Data curation, CCL and XKG; Formal analysis, SH and HXZ; Funding acquisition, EZJ; Investigation, QWJ; Methodology, YHF; Project administration, SH; Resources, XKG; Software, RLJ; Supervision, XMC and EJZ; Validation, YJW; Writing – original draft, SH. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The study was performed according to the ethics committee of Nanjing Medical University and the First Affiliated Hospital of Nanjing Medical University. In accordance with the Declaration of Helsinki, written informed consent is obtained from all patients or their families. The study was approved by the ethics committee of Nanjing Medical University and the First Affiliated Hospital of Nanjing Medical University.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, S., Fu, Y., Li, C. et al. Interaction between the expression of hsa_circRPRD1A and hsa_circHERPUD2 and classical coronary risk factors promotes the development of coronary artery disease. BMC Med Genomics 16, 131 (2023). https://doi.org/10.1186/s12920-023-01540-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01540-9