
Abstract
Background
Lamb-Shaffer syndrome (LAMSHF, MIM 616,803) is a rare neurodevelopmental disorder due to haploinsufficiency of SOX5. Furthermore, studies about the clinical features of LAMSHF patients with same allele of c.1477C > T (p. R493*) are very limited.
Case presentation
We analyzed the phenotypes of one of our cases and two previously reported cases with c.1477C > T (p. R493*), and reviewed the correlating literature. A de novo heterozygous variation c.1477C > T (p. R493*) in SOX5 was identified in a 4 years and 2 months old boy with global development delay by trio-based whole exome sequencing. We compared our case and previously 2 cases reported with recurrent variation, the overlapping clinical features are global developmental delay or intellectual disability, language delay and scoliosis, but their other clinical characteristics are different.
Conclusions
This study suggests that the clinical features of LAMSHF patients with recurrent variations in the SOX5 gene are different. It is suggested that the LAMSHF-related SOX5 gene should be screened and included as one of the candidate genes for neurodevelopmental disorders of unknown etiology.
Similar content being viewed by others

Background
Lamb-Shaffer syndrome (LAMSHF, MIM 616,803) is a neurodevelopmental disorder caused by genetic alterations due to haploinsufficiency of the SOX5 gene (SRY-related high-mobility-group box5, MIM 604,975) [1, 3]. LAMSHF is clinically characterized by global developmental delay (GDD), intellectual disability (ID), language delay, behavioral disturbances including autistic features, and dysmorphic facial features [1,2,3]. The SOX5 gene is located on chromosome 12p12.1, and is the key gene encoding a transcription factor regulating cell fate and differentiation during neural development [1, 4]. Haploinsufficiency of SOX5 is mostly caused by deletions and point variation [1, 5, 6]. To date, only 74 LAMSHF patients have been reported worldwide [1,2,3, 6,7,8,9,10,11,12,13]. There is no specific treatment for LAMSHF. Life expectancy of individuals with LAMSHF does not to be affected. The genotype–phenotype correlations are still not clear [6]. Here, we report the genetic and clinical manifestations of a patient with LAMSHF and compare them with 2 previously reported cases with recurrent variation. To extend the clinical characteristics and genotypes of Lamb-Shaffer syndrome, related literature was also reviewed and summarized.
Case presentation
The clinical data of the patient with LAMSHF were analyzed retrospectively. The developmental level was assessed using the Griffiths Development Scales-Chinese (GDS-C), including subscales of locomotor (A), personal-social (B), language (C), eye-hand coordination (D) and performance (E). We used trio-based whole exome sequencing (WES) to analyze the genes of the case. Sanger sequencing was performed to validate the variations and determine their parental origin. The variants were classified as “pathogenic” or “likely pathogenic” or “benign” or “likely pathogenic” according to the variant interpretation guidelines of the American College of Medical Genetics and Genomics [5]. Previous cases were compared, and related literature was reviewed.
The patient was male, the first child, born full-termly, and his parents were healthy and nonconsanguineous. The birth weight was 3750 g, and the length was 52.0 cm. He was able to stand alone at 12 months old and walk without support at 16 months old. The patient visited our clinic at the age of 50 months old, and the weight was 16.0 kg ( − 0.35 SD), the length was 105.5 cm (+ 0.26 SD) and the head circumference was 50.0 cm ( − 0.20 SD). He started to vocalize for approximately 18 months, and he could only speak 2-word phrases instead of sentences at admission. He had scoliosis and constipation. His facial features included a flat nasal bridge and overlapping toes and toe contractures of the feet. Family history was negative for any neurodevelopmental disorder or genetic disorders. A detailed description of the clinical features of our case S.Y. and previously reported cases with SOX5 variants, and comparisons with other patients are shown in Table 1 and Fig. 1.

Description of the clinical features of our case S.Y. and previously reported two cases with the same allele of SOX5 c.1477C > T (p. R493 *)
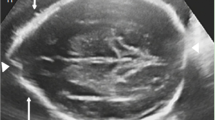
The results of Locomotor (A), Personal-social (B), Language (C), Eye-hand co-ordination (D) and Performance (E) of GDS-C were 5th pencentile, <1st pencentile, < 1th pencentile, < 1th pencentile and 2.5th pencentile, respectively. X-ray examination of the spine showed mildly scoliosis. Brain magnetic resonance imaging (MRI) showed bilateral ventricular dilatation. His thyroid function was normal. Hearing test was passed. Eye exams were normal. Electroencephalogram (EEG), cardiac sonography and abdominal sonography examinations were also normal.
WES identified a de novo heterozygous variant c.1477C > T of SOX5 and confirmed it by Sanger sequencing (Fig. 2). SOX5 c.1477C > T (p. R493*) was a nonsense variant and was classified as pathogenic. Other pathogenicity point variants previously reported are also shown as comparisons (Fig. 3).

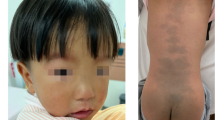
S.Y.with a de novo heterozygous SOX5 variant. a Sanger sequencing confirmation for c. 1477C > T SOX5 variant in the proband but absent in both parents. b The figure above shows flat nasal bridge. The figure below shows overlapping toes and toe contractures

Genetic location of the SOX5 point variants associated with LAMSHF identified to date. a Boxes 1 to 15, coding exons of isoform NM_006940. The variant reported in this case showed a square frame. We used bars to report the variants, and " × " represents the number of cases. The size of exon and intron is not proportional. b Distribution of amino acid changes related to the protein domains CC (coiled-coil) and HMG (high-mobility-group). Protein and domain residue boundaries are indicated underneath the schematic
Discussion and conclusions
Case S.Y. in our study was the third case with the same allele of SOX5 c.1477C > T (p. R493 *). Previously reported case 1 was a 3-year-old female with global developmental delay, hypotonia, cortical visual impairment, scoliosis, ear tubes, constipation and supernumerary nipple. The other previously reported case 2 was a 20-year-old male with intellectual disability, epilepsy and scoliosis and inability to speak [6]. The 3 cases with recurrent SOX5 variant shared similar clinical manifestations, such as global developmental delay or intellectual disability, language delay and scoliosis. Particularly, our case S.Y. exhibited flat nasal bridges, overlapping toes, toe contractures and constipation.
The common and different manifestations may suggest heterogeneous clinical phenotypes underlying the same genotype. Previous cases with other recurrent point variants of the SOX5 gene, such as p. Gln208* 2 cases, p. Arg213* 3 cases, p. Met560Val 3 cases, p. Arg571Trp 5 cases, p. Thr632Asn 2 cases also demonstrated different clinical manifestations [6, 8]. For example, 5 patients have been reported with the variant p.Arg571Trp. A total of 3/5 showed hypotonia, 1/5 showed microcephaly, and the most severe patient showed seizures, strabismus and joint hyperlaxity. The differences in the phenotypes may be explained by the additional variations associated with the same gene, however in those cases other implicated genes are not available for us. Other than point variants, patients with the same deletion alteration of SOX5 also exhibited different clinical manifestations. For instance, 2 patients with the same exon 8–10 deletion both had seizures, but only one of them showed fused vertebrae, subaortic ventricular septal defects and pulmonary stenosis [6]. Regarding the remaining 72 cases with LAMSHF, language development delay was present in almost all of the cases [1,2,3, 6,7,8,9,10,11,12,13]. Hypotonia, ophthalmic features, and scoliosis are also commonly reported [1, 6, 9]. Approximately 1/4 of reported cases showed seizures with a positive response to medication, followed by a benign course [7, 12].
The incomplete penetrance suggests that SOX5 haploinsufficiency may manifest differently in similar genetic backgrounds. One possible reason could be that some variants of SOX5 retain partial activity [14]. We hypothesize that there could be environmental influences, epigenetics, life style and genetic background of other genetic variation contributing to incomplete penetrance and variable expressivity. Longitudinal studies with larger sample sizes as well as molecular and histological studies on animal models are highly recommended for the elucidation of the pathological mechanisms of SOX5 partial activity.
Notably, all of the LAMSHF cases were caused by heterozygous variations. The majority of the cases were reported with SOX5 deletion and point variation, including 18 cases with missense variation (12 types), 13 cases with nonsense variation (9 types), and 3 cases with shift variation (3 types) [6, 7, 11, 13]. Our case was tested with the nonsense point variant c.1477C > T (p. R493 *), which will lead to premature termination and absence of the HMG domain in the translated protein.
The HMG domain is the core functional domain of the SOX protein family, which mediates DNA binding and bending and regulates the expression of downstream developmental genes [15]. For humans, half of the pathogenic variants of the SOX genes result in developmental disorders [14]. The SOX5 gene plays an important role in the development of the nervous system, chondrogenesis and other developmental pathways [4]. According to the amino acid sequences of the HMG domain, the SOX protein is divided into groups A-H [16]. SOX5 belongs to the SOXD group, which consists of a unique long N-terminal sequence containing a population-specific coiled-coil (CC) domain [17]. The CC domain mediates SOXD protein dimerization, thereby preferentially binding to pairs of DNA recognition sites [17].
Moreover, SOX5 can produce at least 5 transcript isoforms through promoter expression, start site alternative and precursor messenger RNA (premRNA) splicing [18]. The variant of our case was on exon 11, which leads to haploinsufficiency of 3 transcript isoforms L1-SOX5 (NM_006940), L2-SOX5 (NM_152989) and S-SOX5 (NM_178010). The isoform L1-SOX5 is composed of exons 1–15 encoding the protein. The isoform L2-SOX5 consists of a 5’ noncoding protein sequence, and exons 2–15 encode the protein. Both L1-SOX5 and L2-SOX5 are the predominant brain isoforms and play an important role in the formation and functional differentiation of the fetal brain [11, 18]. The shortest isoform S-SOX5 is composed mainly of exon 10–15 encoding protein and is testes-specific [19]. It may play an important role in the formation and function of motor cilia in the brain, lungs, testes and spermatozoa [19]. In our case, we observed developmental delay and language delay, while the effects on the lung and testis were further followed up.
In conclusion, the current study reported one LAMSHF case with the SOX5 gene variant. The case shared not only similar but also specific clinical features compared with previous cases, which helped to extend the phenotypic spectrum.
The differences in the phenotypes may be explained by the additional variations or incomplete penetrance and variable expressivity. LAMSHF may affect multiple systems, especially language and intellectual development. However, the diagnosis of LAMSHF requires genetic testing to confirm. Thus, it is suggested that the LAMSHF-related SOX5 gene should be screened and included as one of the candidate genes for neurodevelopmental disorders of unknown etiology. Systematic developmental studies with follow-up and larger sample sizes should be performed to investigate the function of SOX5 in humans and provide more evidence for clinical decisions. Future studies on LAMSHF patients with same variants iPSC derived neurons could be conducted as different phenotypic variations were observed. This could lead to better understand the disorder's molecular pathogenesis, as well as looking into the potential causes of the variation seen, including genetic modifiers and polygenic factors.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. And the genetic testing data has been uploaded to ClinVar [VCV000695081.6].
Abbreviations
- LAMSHF:
-
Lamb-Shaffer syndrome
- SOX5 :
-
SRY-related high-mobility-group box5
- GDD:
-
Global developmental delay
- ID:
-
Intellectual disability
- GDS-C:
-
Griffiths development scales-Chinese
- WES:
-
Whole exome sequencing
- NA:
-
Not available
- ASD:
-
Autism spectrum disorder
- MRI:
-
Magnetic resonance imaging
- EEG:
-
Electroencephalogram
- CC:
-
Coiled-coil
- HMG:
-
High-mobility-group
References
Lamb AN, Rosenfeld JA, Neill NJ, Talkowski ME, Blumenthal L, Girirajan S, et al. Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat. 2012;33:728–40.
Schanze I, Schanze D, Bacino CA, Douzgou S, Kerr B, Zenker M. Haploinsufficiency of SOX5, a member of the SOX (SRY-related HMGbox) family of transcription factors is a cause of intellectual disability. Eur J Med Genet. 2013;56:108–13.
Lee RW, Bodurtha J, Cohen J, Fatemi A, Batista D. Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatr Neurol. 2013;48:317–20.
Aza-Carmona M, Shears DJ, Yuste-Checa P, Barca-Tierno V, Hisado-Oliva A, Belinchon A, et al. SHOX interacts with the chondrogenic transcription factors SOX5 and SOX6 to activate the aggrecan enhancer. Hum Mol Genet. 2011;20:1547–59.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Zawerton A, Mignot C, Sigafoos A, Blackburn PR, Haseeb A, McWalter K, et al. Widening of the genetic and clinical spectrum of Lamb-Shaffer syndrome, a neurodevelopmental disorder due to SOX5 haploinsufficiency. Genet Med. 2020;22:524–37.
Innella G, Greco D, Carli D, Magini P, Giorgio E, Galesi O, et al. Clinical spectrum and follow-up in six individuals with Lamb-Shaffer syndrome (SOX5). Am J Med Genet A. 2021;185:608–13.
Chérot E, Keren B, Dubourg C, Carré W, Fradin M, Lavillaureix A, et al. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: experience of 2 clinical units and 216 patients. Clin Genet. 2018;93:567–76.
Quintela I, Barros F, Lago-Leston R, Castro-Gago M, Carracedo A, Eiris J, et al. A maternally inherited 16p13.11-p12.3 duplication concomitant with a de novo SOX5 deletion in a male patient with global developmental delay, disruptive and obsessive behaviors and minor dysmorphic features. Am J Med Genet A. 2015;167:1315–22.
Nesbitt A, Bhoj EJ, McDonald Gibson K, Yu Z, Denenberg E, Sarmady M, et al. Exome sequencing expands the mechanism of SOX5-associated intellectual disability: a case presentation with review of sox-related disorders. Am J Med Genet A. 2015;167A:2548–54.
Fukushi D, Yamada K, Suzuki K, Inaba M, Nomura N, Suzuki Y, et al. Clinical and genetic characterization of a patient with SOX5 haploinsufficiency caused by a de novo balanced reciprocal translocation. Gene. 2018;655:65–70.
Tumienė B, Maver A, Writzl K, Hodžić A, Čuturilo G, Kuzmanić-Šamija R, et al. Diagnostic exome sequencing of syndromic epilepsy patients in clinical practice. Clin Genet. 2018;93:1057–62.
Cao J, Li J, Zhang Y, Niu H, Zhou Y, Li Z, et al. Variant analysis of SOX5 gene in a Lamb-Shaffer syndrome family. Chin J Med Genet. 2021;38:765–7.
Angelozzi M, Lefebvre V. SOXopathies: growing family of developmental disorders due to SOX mutations. Trends Genet. 2019;35:658–71.
Kamachi Y, Kondoh H. Sox proteins: regulators of cell fate specification and differentiation. Development. 2013;140:4129–44.
Schepers GE, Teasdale RD, Koopman P. Twenty pairs of sox: extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev Cell. 2002;3:167–70.
Lefebvre V. The SoxD transcription factors-SOX5, Sox6, and Sox13-are key cell fate modulators. Int J Biochem Cell B. 2010;42:429–32.
Ikeda T, Zhang J, Chano T, Mabuchi A, Fukuda A, Kawaguchi H, et al. Identification and characterization of the human long form of SOX5 (L-SOX5) gene. Gene. 2002;298:59–68.
Kiselak EA, Shen X, Song J, Gude DR, Wang J, Brody SL, et al. Transcriptional regulation of an axonemal central apparatus gene, sperm-associated antigen 6, by a SRY-related high mobility group transcription factor, S-SOX5. J Biol Chem. 2010;285:30496–505.
Acknowledgements
We thank the patient and family members at all stages of this work.
Funding
This research was funded by the Key Subject Construction Project of Shanghai Municipal Health Commission, Grant number shslczdzk02903, the Young Clinical Scientist Program of Children's Hospital of Fudan University, Grant number 2022LCKXJ0, and by the National Science Foundation of China, Grant numbers 81701496 and 82171540. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
QX, GZ and PD conceived and conducted the study; QX, GZ, HL, PL, XP and LH identified the patients and carried out the clinical characterizations; GZ and PD carried out the molecular genetics studies; CH contributed WES analysis; GZ, PD and QX wrote the manuscript; QX, DL, CH, HL, PL, XP, LH and XX reviewed and edited manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the Children’s Hospital of Fudan University (protocol code 2014125). The parents of the patient have read and signed the informed consent form to participate.
Consent for publication
Written informed consent for publication of identifying images or other personal or clinical details was obtained from both of the parents.
Competing interests
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported. And there is also no conflict of interest for each of the funding bodies in our study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, Gq., Dong, P., Li, Dy. et al. Clinical characterization of Lamb-Shaffer syndrome: a case report and literature review. BMC Med Genomics 16, 22 (2023). https://doi.org/10.1186/s12920-023-01448-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01448-4