
Abstract
Background
Lamb–Shaffer syndrome (LAMSHF) is a rare neurodevelopmental disorder caused by heterozygous mutation or microdeletion involving the SOX5 gene. LAMSHF is characterize by developmental delay, intellectual disability, poor expressive speech, mild dysmorphic facial features and skeletal abnormalities.
Case presentation
We presented a case of a child with delayed psychomotor development in all areas, scoliosis, peculiar facies, and suspicion of intermittent endotropia, alteration in the alignment of one foot and difficulty in standing. These clinical features lead to genetics studies, in which a novel pathogenic variant in the SOX5 gene was detected in association with LAMSHF.
Conclusions
LAMSHF should be suspected in patients with developmental delay, speech delay, intellectual disability, behavioural disturbances, ophthalmological alterations and skeletal abnormalities. A novel pathogenic mutation in the SOX5 gene c.1627del p.(Tyr543IlefsTer14) was identified in this patient as responsible of Lamb–Shaffer syndrome. This case contributes to understanding the genetic characteristics, clinical features, and diagnosis of LAMSHF.
Similar content being viewed by others


Background
Lamb–Shaffer syndrome (LAMSHF; MIM#616803) [1, 2] is a rare neurodevelopmental disorder, described in very few patients, that is caused by heterozygous mutation or microdeletion in the SOX5 gene. Also, deletion in 12p12 region [3,4,5] or chromosomal translocation [2] containing SOX5 gene have been identified as causes of LAMSHF.
The prevalence of LAMSHF is currently unknown. According to the revised literature, LAMSHF has been only reported in less than 100 patients worldwide [6, 7], and only a small case series has been previously published [1,2,3, 8, 9].
LAMSHF is characterized by developmental delay, intellectual disability, poor expressive speech and mild dysmorphic facial features [1, 2, 5, 10, 11]. In addition, ophthalmological alterations and skeletal abnormalities [12, 13] can also be shown in these patients.
Located on chromosome 12p12.1, SOX5 gene produces at least five transcript isoforms through the expression of different promoters, alternative start sites, and alternative precursor messenger RNA (m-RNA) splicing. The longer isoform (NM_006940) encodes a protein initially named L-SOX5 (now called SOX5), which is the major isoform in the brain. SOX5 is part of the SOX [1, 14] gene family, which includes SOX2, SOX4, SOX5, SOX9, SOX10 and SRY. These genes have been described to be involved in very important processes such as sex determination, chondrogenesis, skeletogenesis and neurogenesis [15,16,17]. Most pathogenic variants of the SOX gene family are de novo (except for the SRY gene) and cause haploinsufficiency, as it happens in the case of the SOX5 gene causing LAMSHF. Only in a very few cases describing LAMSHF the mutation is inherited from parents [18].
We present a case of a child diagnosed with neurodevelopmental delay, sialorrhea and scoliosis. Trio-WES was conducted according to our hospital protocol for these patients, and a novel pathogenic, de novo mutation in SOX5 gene was identified. This case is added to the reduced number of publications previously reported regarding LAMSHF.
Case presentation and genetics findings
We reported a clinical case of a two-and-a-half-year-old male, only son of non-consanguineous parents without history of genetic disease. The mother reported a well-controlled and echographically normal pregnancy and no prior abortions. Born at 38 gestational weeks and with a normal neonatal screening, he had no episodes of hospitalization during the neonatal period.
At 4 months of age, a slight right plagiocephaly was detected at a paediatric check-up, being treated with a cranial orthosis and followed up until 10 months of age.
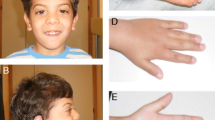
At the age of 18 months, he was evaluated by neuropaediatrics due to peculiar facies (epicanthus, broad nose, small palpebral fissures, hypertelorism, micrognathia, synophrys and big ears), delayed psychomotor development in all areas and scoliosis. He was also evaluated by ophthalmologists, who diagnosed him of marked epicanthus with suspicion of intermittent endotropia. In addition, due to an alteration in the alignment of one foot and the difficulty in standing, he was recommended to use a dynamic ankle foot orthosis (DAFO). The child was also referred to paediatric rehabilitation and neuropaediatric consultations.
Following the protocol established in our hospital for those cases in which psychomotor retardation is evident, doctors ordered both a genetic testing for Fragile X syndrome (FRAXA) [19] and a Microarray-based Comparative Genomic Hybridization (aCGH). No pathogenic findings were revealed in any case.
At this point, with the aim of extending the study due to nonspecific dysmorphic features and a phenotype indistinguishable from many other inherited disorders with intellectual disability, a Trio-WES (whole exome sequencing) was performed in the patient and his biological parents.
The library for this technique was prepared using the Comprehensive Exome Panel technology (captures > 20,000 genes, > 99% coverage of the genes included in the RefSeq, CCDS and GENCODE databases > 85% of the alterations responsible for diseases of genetic origin and flanking splicing regions (5–20 bp), with a size of 41.2 Mb).
The sequencing was performed on a state-of-the-art mass sequencer, NovaSeq 6000 SystemTM (Illumina). The sequences were aligned against the reference genome (GRCh37/hg19), filtered according to specific quality criteria and analysed with DRAGENTM BioIT Platform software.
A heterozygous de novo frameshift variant c.1627del; p.(Tyr543IlefsTer14) was detected in exon 13 of SOX5 gene (NM_006940.5), (GRCh37:g.23696291delA) with a probable genotype–phenotype association (Table 1), being later confirmed by Sanger sequencing. This finding in the patient was not detected in his parents.
Not having been previously reported in clinical databases (Clinvar, PubMed, LOVD) this variant was also not included in the control population databases (1Kgenome, gnomAD). However, some nonsense and frameshift mutations in this gene (Table 2) have been described as a cause of the LAMSHF with a dominant inheritance pattern [14, 20]. These patients showed, among other clinical features, language delay from a mild to severe degree.
Therefore, it was then considered that this frameshift variant (c.1627del; p.(Tyr543IlefsTer14) changes the reading frame, generating a premature stop codon and causing a loss-of-function effect on the SOX5 protein.
According to the American College of Medical Genetics and Genomics (ACMG) variant classification guidelines [23], this variant should be considered pathogenic. There criteria supporting this state are the following: It is a null variant in a gene where LOF (Loss of function) is a known mechanism of disease (PVS1), it is absent from controls in Exome Aggregation Consortium (PS2), it does not exist in general population databases (gnomAD, 1000 G, ESP) (PM2) and the protein length changes as a result of in-frame deletions in a non-repeat region or stop-loss variants (PM4).
The demonstration of a de novo pathogenic variant in the SOX5 gene in a patient with symptomatology compatible with LAMSHF, and the evidence of other nonsense and frameshift pathogenic variants reported as cause of LAMSHF, lead us to ascertain that the new mutation c.1627delT; p.(Tyr543IlefsTer14) in the SOX5 gene, causes LAMSHF.
Other variants with low population frequency were also identified in Trio-WES (Table 3) but for them, a genotype–phenotype correlation was not possible.
Our patient was finally diagnosed with LAMSHF. He currently carries DAFO and has been performing autonomous walks and runs from 22 months of age. At the time of writing this article, the boy was two-and-a-half years old, 12.0 kg (P15) weight, 91.5 cm (P25) height and craniofacial perimeter 47.7 cm (P5). Nowadays, he is still being followed by Neuropaediatrics due to his neurodevelopmental disorder. He goes to kindergarten, eats, understands everything, points, imitates, uses propositional bisyllables and parents refer a positive evolution since the early childhood care.
Discussion
Pathogenic variants in the SOX genes have been proposed to cause developmental disorders [24]. Concretely, the SOX5 gene has been described in association with developmental delay, language and motor deficit, intellectual disability, behavioural disturbance including autistic traits, and other partially penetrating features [1] (facials features, strabismus and skeletal abnormalities).
LAMSHF should be suspected in individuals with developmental delay, speech delay, intellectual disability, behavioural disturbances, ophthalmological alterations and skeletal abnormalities. According to Zawerton's [1] language delay (97%), behavioural disturbances (49%) and seizures (19%) have been described as the most frequent clinical features in affected patients. Other symptoms were hypotonia (51%) and strabismus (27%). Skeletal abnormalities (fused vertebrae, microcephaly, thoracic kyphosis, joint hyperlaxity and tooth anomalies) have also been described in LAMSHF. These percentages will presumably change as new cases are described.
Our patient presented delayed psychomotor development in all areas, epicanthus phenotype, broad nose, small palpebral clefts, hypotonia, strabismus, scoliosis and joint laxity. All these clinical features are similar to those described in the LAMSHF. Skeletal anomalies (pectoral malformations) and epileptic seizures were not evident in the patient.
Hypertelorism, small eyes, small fingers, retromicrognathia, synophrys, large and separated ear and sialorrhea were described in our patient but not included in OMIM as clinical symptoms of LAMSHF. Thus, they should also be considered in future medical reviews of the disease.
Molecular genetic testing approaches depends on hospital protocols and laboratory possibilities. In our patient, with intellectual disability and nonspecific dysmorphic features, the performance of a single-gene testing or multigene panel was intended to discard other inherited disorders with the same clinical manifestations and from which the patient’s phenotype was indistinguishable. Therefore, a Trio-WES was conducted on the patient and their parents.
Variants in the SOX5 gene have been described in association with the LAMSHF, which can be detected by array-CGH [3, 5] or WES [10]. The majority are nonsense and missense variants [1]. According to the inheritance pattern, it has been demonstrated that most patients with LAMSHF present a de novo mutation. Here, we reported a frameshift pathogenic variant de novo (loss-of-function) in exon 13 of SOX5 gene.
Conclusion
LAMSHF is a rare genetic neurodevelopmental disorder that should be suspected in individuals with developmental delay, speech delay, intellectual disability, behavioural disturbances, ophthalmological alterations and skeletal abnormalities. Trio-WES is the technique with the best results for the diagnostic approach of these patients and so, it should be performed initially. In our patient, the presence of a pathogenic de novo variant in SOX5 together with clinical symptoms allowed us to diagnose LAMSHF. This case is added to the reduced number of publications regarding LAMSHF and contributes to understanding the genetic characteristics, clinical features, and diagnosis of this syndrome.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- LAMSHF:
-
Lamb–Shaffer syndrome
- NGS:
-
Next-generation sequencing
- WES:
-
Whole-exome sequencing
- Array-CGH:
-
Array comparative genomic hybridization
- m-RNA:
-
Messenger RNA
- DAFO:
-
Dynamic ankle foot orthosis
- FRAXA:
-
Fragile X syndrome
- NMD:
-
Nonsense Mediated Decay
- ACMG:
-
American College of Medical Genetics and Genomics
- XLD:
-
X-linked dominant
- AR:
-
Autosomal recessive
- AD:
-
Autosomal dominant
- SNV:
-
Single nucleotide variant
- NA:
-
Not applicable
- NAv:
-
Not available
References
Zawerton A, Mignot C, Sigafoos A et al (2020) Widening of the genetic and clinical spectrum of Lamb–Shaffer syndrome, a neurodevelopmental disorder due to SOX5 haploinsufficiency. Genet Med 22:524–537. https://doi.org/10.1038/s41436-019-0657-0
Fukushi D, Yamada K, Suzuki K, Inaba M, Nomura N, Suzuki Y, Katoh K, Mizuno S, Wakamatsu N (2018) Clinical and genetic characterization of a patient with SOX5 haploinsufficiency caused by a de novo balanced reciprocal translocation. Gene 655:65–70. https://doi.org/10.1016/j.gene.2018.02.049
Lamb AN, Rosenfeld JA, Neill NJ et al (2012) Haploinsufficiency of SOX5 at 12p12.1 is associated with developmental delays with prominent language delay, behavior problems, and mild dysmorphic features. Hum Mutat 33:728–740. https://doi.org/10.1002/humu.22037
Arroyo-Carrera I, de Zaldívar-Tristancho MS, Martín-Fernández R, Hernández-Martín R, López-Lafuente A, Rodríguez-Revenga L (2015) Microdeletion 12p12 involving SOX5 gene: a new syndrome with developmental delay. Rev Neurol 60:453–456. https://doi.org/10.33588/rn.6010.2014569. (Spanish)
Lee RWY, Bodurtha J, Cohen J, Fatemi A, Batista D (2013) Deletion 12p12 involving SOX5 in two children with developmental delay and dysmorphic features. Pediatr Neurol 48:317–320. https://doi.org/10.1016/j.pediatrneurol.2012.12.013
Rhead S, Weerapperuma N, Robertson C (2020) G67(P) a rare diagnosis of Lamb–Shaffer syndrome in twins with Klinefelter’s. Arch Dis Child 105:A20–A21. https://doi.org/10.1136/archdischild-2020-rcpch.50
Orphanet Journal of Rare Diseases (2022) https://www.orpha.net/consor/cgi-bin/index.php. Accessed 2 Aug 2022
Schanze I, Schanze D, Bacino CA, Douzgou S, Kerr B, Zenker M (2013) Haploinsufficiency of SOX5, a member of the SOX (SRY-related HMG-box) family of transcription factors is a cause of intellectual disability. Eur J Med Genet 56:108–113. https://doi.org/10.1016/j.ejmg.2012.11.001
Rosenfeld JA, Ballif BC, Torchia BS, Sahoo T, Ravnan JB, Schultz R, Lamb A, Bejjani BA, Shaffer LG (2010) Copy number variations associated with autism spectrum disorders contribute to a spectrum of neurodevelopmental disorders. Genet Med 12:694–702. https://doi.org/10.1097/GIM.0b013e3181f0c5f3
Innella G, Greco D, Carli D, Magini P, Giorgio E, Galesi O, Ferrero GB, Romano C, Brusco A, Graziano C (2021) Clinical spectrum and follow-up in six individuals with Lamb-Shaffer syndrome (SOX5). Am J Med Genet Part A 185:608–613. https://doi.org/10.1002/ajmg.a.62001
Gkirgkinoudis A, Tatsi C, DeWard SJ, Friedman B, Faucz FR, Stratakis CA (2020) A SOX5 gene variant as a possible contributor to short stature. Endocrinol Diabetes Metab Case Rep. https://doi.org/10.1530/EDM-20-0133
Kawasaki K, Kawasaki M, Watanabe M et al (2015) Expression of Sox genes in tooth development. Int J Dev Biol 59:471–478. https://doi.org/10.1387/ijdb.150192ao
Lefebvre V (2019) Roles and regulation of SOX transcription factors in skeletogenesis. Curr Top Dev Biol 133:171–193. https://doi.org/10.1016/bs.ctdb.2019.01.007
Nesbitt A, Bhoj EJ, McDonald Gibson K et al (2015) Exome sequencing expands the mechanism of SOX5-associated intellectual disability: a case presentation with review of sox-related disorders. Am J Med Genet A 167:2548–2554. https://doi.org/10.1002/ajmg.a.37221
Stevanovic M, Drakulic D, Lazic A, Ninkovic DS, Schwirtlich M, Mojsin M (2021) SOX transcription factors as important regulators of neuronal and glial differentiation during nervous system development and adult neurogenesis. Front Mol Neurosci. https://doi.org/10.3389/fnmol.2021.654031
Jiang T, Hou C-C, She Z-Y, Yang W-X (2013) The SOX gene family: function and regulation in testis determination and male fertility maintenance. Mol Biol Rep 40:2187–2194. https://doi.org/10.1007/s11033-012-2279-3
Kamachi Y, Kondoh H (2013) Sox proteins: regulators of cell fate specification and differentiation. Development 140:4129–4144. https://doi.org/10.1242/dev.091793
Zech M, Poustka K, Boesch S, Berutti R, Strom TM, Grisold W, Poewe W, Winkelmann J (2017) SOX5-null heterozygous mutation in a family with adult-onset hyperkinesia and behavioral abnormalities. Case Rep Genet 2017:1–6. https://doi.org/10.1155/2017/2721615
Garber KB, Visootsak J, Warren ST (2008) Fragile X syndrome. Eur J Hum Genet 16:666–672. https://doi.org/10.1038/ejhg.2008.61
Online Mendelian Inheritance in Man (OMIM) (2022) https://www.omim.org/. Accessed 2 Aug 2022
National Center for Biotechnology Information (NCBI) (2022) ClinVar database. https://www.ncbi.nlm.nih.gov/clinvar/. Accessed 11 Oct 2022
Leiden Open Variation Database (LOVD) (2022) https://www.lovd.nl/. Accessed 11 Oct 2022
Ellard S, Baple EL, Berry I et al (2019) ACGS best practice guidelines for variant classification. In: Association for clinical genomic science. British Society for Genetic Medicine. London. https://www.leedsth.nhs.uk/assets/Genetics-Laboratory/86fa75f316/ACGS-variant-classification-guidelines-2019.pdf. Accessed 2 Aug 2022
Angelozzi M, Lefebvre V (2019) SOXopathies: growing family of developmental disorders due to SOX mutations. Trends Genet 35:658–671. https://doi.org/10.1016/j.tig.2019.06.003
Acknowledgements
To the patient and his parents.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial or not for profit sectors.
Author information
Authors and Affiliations
Contributions
JCA and RGT drafted and critically reviewed the manuscript. JLPS was the principal physician in the patient’s case. ECA, ESR, NGR and SIA contributed to data collection, literature search and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cuenca Alcocel, J., Criado Álamo, E., Salvador-Rupérez, E. et al. A novel mutation in the SOX5 gene c.1627del; p.(Tyr543IlefsTer14) is associated to Lamb–Shaffer syndrome: a case report. Egypt J Med Hum Genet 24, 32 (2023). https://doi.org/10.1186/s43042-023-00395-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43042-023-00395-0