
Abstract
Background
Interstitial 4q deletions are rare chromosomal alterations. Most of the previously reported deletions involving the 4q13.3 region are large chromosomal alterations with a common loss of band 4q21 resulting in marked growth restriction, severe intellectual disability, and absent or severely delayed speech. A microdeletion of 4q13.3 hasn’t been previously reported. We discuss the involvement of genes and the observed phenotype, comparing it with that of previously reported patients.
Case presentation
We report on a 4q13.3 microdeletion detected in three affected individuals of a Lithuanian family. The clinical features of two affected children and their affected mother are very similar and include short stature, congenital heart defect, skeletal anomalies, minor facial anomalies, delayed puberty, and intellectual disability. Whole genome SNP microarray analysis of one child revealed an interstitial 4q13.3 microdeletion, 1.56 Mb in size. FISH analysis confirmed the deletion in the proband and identified the same deletion in her affected sib and mother, while it was not detected in a healthy sib. Deletion includes ADAMTS3, ANKRD17, COX18, GC, and NPFFR2 protein-coding genes.
Conclusions
Our findings suggest that 4q13.3 microdeletion is a cause of a recognizable phenotype of three affected individuals. The detected microdeletion is the smallest interstitial deletion in 4q13. We highlight ADAMTS3, ANKRD17 and RNU4ATAC9P as candidate genes for intellectual disability, growth retardation and congenital heart defect.
Similar content being viewed by others

Background
Structural chromosome alterations can lead to various clinical features, the most common being intellectual disability and congenital anomalies [1, 2]. Deletion of a short (p) arm of chromosome 4, causing Wolf-Hirschhorn syndrome characterized by intellectual disability, the Greek warrior helmet appearance of the nose and forehead, growth delay and seizures [3], is well described in the literature, while deletions involving the long (q) arm of chromosome 4 are poorly investigated. Not many patients with deletions of the long arm of chromosome 4 are described in the literature [4,5,6,7,8,9]. Moreover, even fewer patients have been reported with small alterations in one particular region of chromosome 4 instead of large deletions or duplications. The use of chromosomal arrays allows to detect submicroscopic alterations and subsequently through genotype- phenotype correlations and gene function analysis create the possibility to identify candidate genes, responsible for specific clinical features.
We present three patients with a 4q13.3 deletion in an attempt to characterize clinical manifestation of this rare chromosomal alteration.
Case presentation
Clinical findings
We report on three affected individuals in two generations of the Lithuanian family: two daughters, aged 19 years and 14 years, and their mother aged 43 years (Fig. 1a).

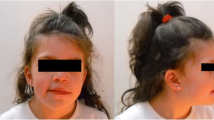
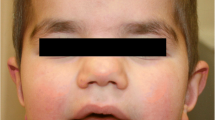
a. The genealogy of the family. The black symbol denotes affected individuals. b. Side facial view of patient 1 at age of 14 years. c. Side facial view of patient 2 at age of 9 years. d-e. Chest X-ray of patient 2 at age of 10 years showes “S” shaped II° scoliosis of the thoracolumbar spine
Patient 1 (DECIPHER 321792)
The patient is a 19-year-old female, the first child of non-consanguineous Lithuanian parents (Fig. 1a, individual II-1; Fig. 1b). She was born at the 41st-42nd gestational week by cesarean delivery due to signs of fetal asphyxia. Her birth weight was 2700 g (<3rd centile), her length was 48 cm (3rd –10th centile), and Apgar scores at 1, 5 and 10 min were 8, 9 and 10. Psychomotor developmental delay was noticed from infancy. According to the medical documentation, the girl’s height was below 3rd centile (130 cm and 138 cm at age of 15 and 16 years, respectively), weight was also below 3rd centile (28 kg and 36 kg at age of 15 and 16 years, respectively). At the age of 19 years, her head circumference was 51 cm (<3rd centile), her height was 147 cm (<3rd centile), her weight was 45 kg (<3rd centile), and her weight/height dependence was 75th–90th centile. She was found to have mild intellectual disability, microcephaly, micrognathia, upslanted palpebral fissures, coarse facial features, short neck, wide chest, spine deformation, and delayed puberty (according to Tanner B2, Ph2). An echocardiogram revealed an abnormality of the aortic valve and atrial septal defect with atrial septal aneurysm. No endocrine pathology was found, and bone age corresponded to chronological age. A chest X-ray showed S-shaped scoliosis of the thoracolumbar spine and no deformation of the ribs. No abnormalities were detected in a neck X-ray. An audiogram showed a mild neurosensory hearing impairment. Abdominal and renal ultrasound and brain MRI were normal. The patient’s IQ was measured at age 19 using the Wechsler Adult Intelligence Scale-III [10]: total IQ 56, verbal IQ 56, and non-verbal IQ 54.
Patient 2
The patient is a 14-year-old female and the third child in the family (Fig. 1, individual II-3; Fig. 1c–e). She was born at term naturally, following a normal pregnancy. Her birth weight was 2800 g (3rd centile), her length was 47 cm (3rd centile), and Apgar scores at 1 and 5 min were 9 and 10. After birth, an intrauterine infection, cystitis, urinary tract infection, CNS irritation syndrome, and bilateral subependymal cyst in the lateral ventricles of the brain were diagnosed. Psychomotor development delay was noticed from infancy; she started to walk at the age of 17 months. At the age of 8 years, she was diagnosed with hypopituitarism. According to the medical documentation, the girl’s height was below 3rd centile (123 cm and 135 cm at age of 10 and 12 years, respectively), and her weight was at 3–10 centiles (29 kg and 32 kg at age of 10 and 12 years, respectively). The patient’s psychological evaluation at age 13 yielded with full scale of 54 on Wechsler Intelligence Scale for Children (WISC-III-LT) [11], verbal IQ - 51, non-verbal IQ - 64. At the age of 14 years, her head circumference was 51.5 cm (3rd centile), her height was 141.5 cm (<3rd centile), her weight was 34 kg (<3rd centile), and her weight/height dependence was in the 25th–50th centile. She was found to have intellectual disability, irregular posture, short neck, upslanted palpebral fissures, coarse facial features, micrognathia, widely spaced nipples, and clinodactyly of the first and second digits of the foot. Deformation of the third digit of the foot was a consequence of trauma. She suffers from frequent respiratory tract infections and anemia. An echocardiogram revealed an abnormality of the aortic valve. A chest X-ray showed S-shaped second degree scoliosis of the thoracolumbar spine, thoracic kyphosis, and incomplete closure of the vertebral arches of Th8 and S1. A neck X-ray showed osteochondrosis. A brain MRI revealed cavum septi pellucidi. An inner organ ultrasound showed hypoplasia of the right kidney. Hypermetropic astigmatism and strabismus in both eyes was also diagnosed. An audiogram showed mild neurosensory hearing impairment.
Patient 3
The mother of patients II-1 and II-3 is a 43-year-old female who is a child of non-consanguineous parents (Fig. 1, individual I-2). During puberty she wore an orthopaedic corset due to a spine deformation and an orthopaedic pad for a shorter right leg. Regular menses started at age of 16 years. At the age of 40 years her head circumference was 52 cm (3rd centile), her height was 149 cm, her weight was 52 kg, and her BMI was 23. She was found to have intellectual disability, coarse facial features, hypotelorism, irregular posture, and wide chest. She suffers from frequent back pain, which spreads to both legs. She was diagnosed with arterial hypertension, second degree blood pressure increase, hypertensive cardiopathy (impaired diastolic function, moderate risk group), and a small haemodynamically insignificant atrial septal defect. An audiogram showed a neurosensory hearing impairment. An inner organ ultrasound revealed gall bladder stones. A spinal MRI performed at the age of 41 showed straightened lordosis, first degree spondylolystesis of L5, spondylolysis, first degree deforming osteochondrosis in the lower part of the thoracic spine and L1–5, second-third degree deforming osteochondrosis in the L5–S1 vertebrae, and Th12–L1 partial stenosis and L5–S1 absolute stenosis of the spinal canal. The patient’s IQ was measured at age 42 using the Wechsler Adult Intelligence Scale-III [10]: total IQ 62, verbal IQ 61, non-verbal IQ 69.
Genetic analysis
Cytogenetic analysis from a culture of peripheral blood lymphocytes was performed using conventional GTG-banding techniques at resolution of the 550-band level according to standard procedures [12].
Genomic DNA was extracted from the patients’ peripheral blood samples using phenol-chloroform extraction method. A whole genome SNP (single nucleotide polymorphism) microarray analysis was performed for the patient 1 to detect copy number variations using the HumanCytoSNP-12v2.1 BeadChip, as described [13]. The data was analyzed using GRCh37/hg19 annotation.
FISH analyses were carried out on blood lymphocytes culture using commercial probes BACs ̶ RP11-373 J21 (orange) from 4q13.3 region and RP11-118 N21 (green) from 4p15.2 region which served as control probe (Illumina, San Diego, CA, USA). All FISH procedures were followed according to the manufacturer’s protocol. Fluorescent signals on metaphases and interphase nuclei were analysed using a Nikon 80i fluorescent microscope with CytoVision version 3.6 (Applied Imaging, UK).
Genetic findings
Normal female karyotypes at 550 band resolution were observed in the affected sibs (patients 1 and 2). SNP oligonucleotide microarray analysis of the patient 1 revealed an interstitial 1.56 Mb deletion, arr[hg19] 4q13.3(72,647,749_74,208,199)×1 (Fig. 2). FISH analysis with RP11-373 J21 probe which overlaps part of ADAMTS3 gene confirmed the deletion in the Patient 1 and identified the deletion at 4q13.3 in her affected sib (patient 2) and mother (patient 3). The deletion was not detected in healthy sib (II-2).

1.56 Mb deletion, arr[hg19] 4q13.3(72,647,749_74,208,199)×1 detected by SNP oligonucleotide microarray analysis and a schematic view of the genes. Horizontal white bars below represent deletions
Discussion and conclusions
We have provided a clinical and molecular characterization of a previously unreported 4q13.3 microdeletion, 1.5 Mb in size, detected in three affected individuals of a Lithuanian family. All patients presented with short stature, congenital heart defect, skeletal anomalies, minor facial anomalies, delayed puberty, and intellectual disability.
Most of the previously reported deletions involving the 4q13.3 region are large chromosomal alterations with a common loss of band 4q21, resulting in marked growth restriction, severe intellectual disability, and absent or severely delayed speech [5,6,7,8,9, 14]. The deletion detected in our patients is unique. There are no low copy repeats or other specific DNA elements flanking deleted region, therefore deletion can be caused either by nonhomologous end joining or microhomology-mediated replication-dependent recombination. Only four unrelated patients with overlapping 4q13.3 monosomy smaller than 10 Mb and not involving the 4q21 region have been reported in literature [8] or recorded in the DECIPHER database (271,532, 4688, and 355,915) [14] (Table 1). Quintela I et al. [8] have reported a de novo 6.85 Mb deletion at 4q13.2-q13.3 in the patient with psychomotor developmental delay, mild intellectual disability, behavioral disorder, short stature and facial anomalies, including a triangular craniofacial profile with a broad forehead, narrow and slightly upslanted palpebral fissures, a broad nasal tip and a prominent narrow chin with a dimple in its medial part [8]. Neurodevelopmental abnormalities including intellectual disability, delayed speech and language development, or autism were common for all patients with an interstitial deletion in 4q13.3. Short stature was found in the DECIPHER patient 4688 and the patient reported by Quintela I et al. [8, 14]. Additionally, the phenotype of DECIPHER patient 4688 is remarkable for strabismus and skeletal abnormalities which also manifested in our patients. The size of the common region in our patients and DECIPHER patients 271532, 4688, and 355915 is 905 kb. It includes three genes (ADAMTS3, ANKRD17, COX18) and four pseudogenes (HNRNPA1P67, RNU4ATAC9P, RNU6ATAC5P, HMGA1P2).
The ADAMTS3 gene encodes an enzyme belonging to the metalloproteases family, which is responsible for procollagen I and II processing in various tissues [15]. It is highly expressed in cartilage formation during embryogenesis and after birth. Studies have shown that Adamts3 was also expressed in developing mouse connective tissues, especially tendon and bone [16]. ADAMTS2 mutations in humans lead to characteristic craniofacial changes and decreased growth [16]. Therefore it could be associated with such clinical features presented in our patients as skeletal anomalies and short stature. The same study discovered Adamts3 expression in several regions of developing mouse brains, including the cerebral cortex [16].
ANKRD17 encodes an ubiquitously expressed protein that was found to be essential to vascular integrity during embryogenesis. Ankrd17-deficient mice developed various heart defects and haemorrhages and a decrease in vascular smooth muscle cells leading to death [17]. Deletion of this gene could be related to the various heart defects described in our patients. ANKRD17 is also thought to play an essential role in DNA replication [18]. Study suggests that it is involved in DNA pre-replication complex formation as well as remodelling and transcribing of chromatin [18]. A loss of or decrease in ANKRD17 blocks DNA replication and inhibits cell cycle progression [18]. However, more studies are required to determine ANKRD17 protein function more precisely.
The role of the COX18 gene and pseudogenes within the deletion is still poorly understood. Pseudogenes may be transcribed into RNA and can be processed into short interfering RNAs that regulate coding genes. It is known that the RNU4ATAC9P pseudogene and its parental gene RNU4ATAC could form a regulatory pair that can influence each other [19]. Mutations in the RNU4ATAC cause the autosomal recessive Roifman Syndrome (MIM#616651), which is characterized by growth retardation, cognitive delay, spondyloepiphyseal dysplasia, and antibody deficiency [20]. Still, the association of genes within the deletion to human disorders remains to be elucidated.
Our report presents detailed molecular and phenotypic characteristics of three affected individuals providing new data supporting future genotype-phenotype studies and identification of candidate genes possibly responsible for specific clinical features. The detected microdeletion in affected individuals of the presented family is the smallest interstitial deletion in 4q13. We highlight ADAMTS3, ANKRD17 and RNU4ATAC9P as candidate genes for intellectual disability, growth retardation and a congenital heart defect. Further contributions of genotype-phenotype descriptions are required for the delineation of the complete spectrum of findings that may be seen associated with microdeletions in 4q13.3 region and clarifying the role of each of the deleted genes.
Availability of data and materials
The datasets generated and analyzed during the current study are available in the Gene Expression Omnibus repository, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE147730.
Abbreviations
- ADAMTS3 :
-
ADAM Metallopeptidase With Thrombospondin Type 1 Motif 3
- ADAMTS2 :
-
ADAM Metallopeptidase With Thrombospondin Type 1 Motif 2
- ANKRD17:
-
Ankyrin Repeat Domain 17
- COX18 :
-
Cytochrome C Oxidase Assembly Factor COX18
- GC :
-
Group-specific component
- HNRNPA1P67 :
-
Heterogeneous Nuclear Ribonucleoprotein A1 Pseudogene 67
- MGA1P2 :
-
High Mobility Group AT-Hook 1 Pseudogene 2
- NPFFR2 :
-
Neuropeptide FF Receptor 2
- p:
-
Short arm of chromosome
- q:
-
Long arm of chromosome
- RNU4ATAC :
-
RNA, U4atac Small Nuclear (U12-Dependent Splicing)
- RNU6ATAC5P :
-
RNA, U6atac Small Nuclear 5, Pseudogene
- RNU4ATAC9P :
-
RNA, U4atac Small Nuclear 9, Pseudogene
- SNP:
-
Single nucleotide polymorphism
References
Preiksaitiene E, Molytė A, Kasnauskiene J, Ciuladaite Z, Utkus A, Patsalis PC, Kučinskas V. Considering specific clinical features as evidence of pathogenic copy number variants. J Appl Genet. 2014;55(2):189–96.
Vissers LELM, de Vries BBA, Veltman JA. Genomic microarrays in mental retardation: from copy number variation to gene, from research to diagnosis. J Med Genet. 2010;47(5):289.
Zollino M, Murdolo M, Marangi G, Pecile V, Galasso C, Mazzanti L, Neri G. On the nosology and pathogenesis of Wolf–Hirschhorn syndrome: genotype–phenotype correlation analysis of 80 patients and literature review. In: American Journal of Medical Genetics Part C: Seminars in Medical Genetics: 2008: Wiley Online Library; 2008. p. 257–69. https://onlinelibrary.wiley.com/doi/abs/10.1002/ajmg.c.30190.
Bonnet C, Andrieux J, Béri-Dexheimer M, Leheup B, Boute O, Manouvrier S, Delobel B, Copin H, Receveur A, Mathieu M, et al. Microdeletion at chromosome 4q21 defines a new emerging syndrome with marked growth restriction, mental retardation and absent or severely delayed speech. J Med Genet. 2010;47(6):377.
Hemati P, du Souich C, Boerkoel CF. 4q12–4q21. 21 deletion genotype–phenotype correlation and the absence of piebaldism in presence of KIT haploinsufficiency. Am J Med Genet A. 2015;167(1):231–7.
Lipska BS, Brzeskwiniewicz M, Wierzba J, Morzuchi L, Piotrowski A, Limon J. 8.6Mb interstitial deletion of chromosome 4q13.3q21.23 in a boy with cognitive impairment, short stature, hearing loss, skeletal abnormalities and facial dysmorphism. Genet Couns. 2011;22(4):353–63.
Nowaczyk MJ, Teshima IE, Siegel-Bartelt J, Clarke JT. Deletion 4q21/4q22 syndrome: two patients with de novo 4q21.3q23 and 4q13.2q23 deletions. Am J Med Genet. 1997;69(4):400–5.
Quintela I, Barros F, Fernandez-Prieto M, Martinez-Regueiro R, Castro-Gago M, Carracedo A, Gomez-Lado C, Eiris J. Interstitial microdeletions including the chromosome band 4q13. 2 and the UBA6 gene as possible causes of intellectual disability and behavior disorder. Am J Med Genet A. 2015;167(12):3113–20.
Shimada S, Okamoto N, Nomura S, Fukui M, Shimakawa S, Sangu N, Shimojima K, Osawa M, Yamamoto T. Microdeletions of 5.5 Mb (4q13. 2–q13. 3) and 4.1 Mb (7p15. 3–p21. 1) associated with a saethre–chotzen-like phenotype, severe intellectual disability, and autism. Am J Med Genet A. 2013;161(8):2078–83.
Drozdick LW, Wahlstrom D, Zhu J, Weiss LG. The Wechsler adult intelligence scale—fourth edition and the Wechsler memory scale—fourth edition; 2012.
Sattler JM. Assessment of children's intelligence: Saunders Philadelphia; 1974.
Wegner R-D. Diagnostic cytogenetics: Springer Science & Business Media; 2013.
Aleksiūnienė B, Preiksaitiene E, Morkūnienė A, Ambrozaitytė L, Utkus A. A de novo 1q22q23.1 interstitial microdeletion in a girl with intellectual disability and multiple congenital anomalies including congenital heart defect. Cytogen Genome Res. 2018;154(1):6–11.
Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. DECIPHER: database of chromosomal imbalance and phenotype in humans using Ensembl resources. Am J Hum Genet. 2009;84(4):524–33.
Apte SS. A Disintegrin-like and Metalloprotease (Reprolysin-type) with Thrombospondin type 1 motif (ADAMTS) superfamily: functions and mechanisms. J Biol Chem. 2009;284(46):31493–7.
Le Goff C, Somerville RP, Kesteloot F, Powell K, Birk DE, Colige AC, Apte SS. Regulation of procollagen amino-propeptide processing during mouse embryogenesis by specialization of homologous ADAMTS proteases: insights on collagen biosynthesis and dermatosparaxis. Development. 2006;133(8):1587–96.
Hou S-C, Chan L-W, Chou Y-C, Su C-Y, Chen X, Shih Y-L, Tsai P-C, Shen CKJ, Yan Y-T. Ankrd17, an ubiquitously expressed ankyrin factor, is essential for the vascular integrity during embryogenesis. FEBS Lett. 2009;583(17):2765–71.
Deng M, Li F, Ballif BA, Li S, Chen X, Guo L, Ye X. Identification and functional analysis of a novel Cyclin E/Cdk2 substrate Ankrd17. J Biol Chem. 2009;284(12):7875–88.
An Y, Furber KL, Ji S. Pseudogenes regulate parental gene expression via ce RNA network. J Cell Mol Med. 2017;21(1):185–92.
Merico D, Roifman M, Braunschweig U, Yuen RKC, Alexandrova R, Bates A, Reid B, Nalpathamkalam T, Wang Z, Thiruvahindrapuram B, et al. Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman syndrome by disrupting minor intron splicing. Nat Commun. 2015;6:8718.
Acknowledgements
We are very thankful to the family for taking part in this study.
Funding
This work was funded by the Research Council of Lithuania [No. S-MIP-17-19/LSS-150000-1179, Ingenes project]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
EP provided genetic counseling to the patients. ZM, along with EV, drafted the manuscript. BA and ZM carried out the molecular cytogenetic analysis. EP and AU substantively revised the work and gave the final approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consents were obtained from the participants and the parents of the child for genetic investigation. The study was approved by the Vilnius Regional Biomedical Research Ethics Committee of Lithuania.
Consent for publication
The family provided the consent for the case report to be published. Written informed consents regarding photographs for publication have been obtained from the participant and the parents of the child.
Competing interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maldžienė, Ž., Vaitėnienė, E.M., Aleksiūnienė, B. et al. A case report of familial 4q13.3 microdeletion in three individuals with syndromic intellectual disability. BMC Med Genomics 13, 63 (2020). https://doi.org/10.1186/s12920-020-0711-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-0711-4