
Abstract
Background
The present study aims to investigate the complete long non-coding RNA (lncRNA) and messenger RNA (mRNA) expression profiles in Intracranial aneurysm (IA) patients and controls by RNA sequencing, which reveals the lncRNA with predictive value for IA risk.
Methods
The comprehensive lncRNA and mRNA expression profiles were detected by RNA-Seq in human IA walls and superficial temporal arteries (STAs), followed by bioinformatics analyses, such as GO analysis, KEGG pathway analysis, and CNC network construction. Subsequently, qRT-PCR was used to profile the expression levels of selected lncRNA (lncRNA ENST000000576153, lncRNA ENST00000607042, lncRNA ENST00000471220, lncRNA ENST00000478738, lncRNA MALAT1, lncRNA ENST00000508090 and lncRNA ENST00000579688) in 30 (small) or 130 (large) peripheral blood leukocytes, respectively. Multivariate logistic regression was utilized to analyze the effects of lncRNA on IA. Receiver operating characteristic (ROC) curve was further drawn to explore the value of lncRNA in predicting IA.
Results
Totally 900 up-regulated and 293 down-regulated lncRNAs, as well as 1297 up-regulated and 831 down-regulated mRNAs were discovered in sequencing. Enrichment analyses revealed that they were actively involved in immune/inflammatory response and cell adhesion/extracellular matrix. Co-expression analysis and further enrichment analyses showed that five candidate lncRNAs might participate in IA’s inflammatory response. Besides, after controlling other conventional risk factors, multivariate logistic regression analysis disclosed that low expression of lncRNA ENST00000607042, lncRNA ENST00000471220, lncRNA ENST00000478738, lncRNA MALAT1 in peripheral blood leukocytes were independent risk factors for IA. LncRNA ENST00000607042 has superior diagnostic value for IA.
Conclusions
This study reveals the complete lncRNAs expression profiles in IA. The inflammatory response was closely related to IA. Besides, lncRNA ENST00000607042 might be a novel biomarker for IA risk.
Similar content being viewed by others


Background
Intracranial aneurysm (IA) is a localized lesion of the cerebral arteries that affect 2 to 3% of the general population [1]. IA rupture can cause aneurysmal subarachnoid hemorrhage (aSAH), which leads to a mortality rate of 30–40% and poor postoperative recovery of patients who survive it [2,3,4]. Researchers have identified several risk factors for IA’s development, including family history of IA, smoking, alcohol, family history of hypertension, older age and female [5,6,7]. But the genetic pathology of IA remains unclear.
Long non-coding RNAs (lncRNAs) are non-coding RNAs that are > 200 nucleotides in length and widely distributed in the nucleus and cytoplasm, especially the nucleus [8]. Multiple studies have revealed that lncRNA can be involved in the inflammatory process, vascular smooth muscle cells (SMCs) phenotypic transition, endothelial cell function regulation and lipid metabolism [9,10,11,12], which play essential roles in IA. Therefore, these results indicate that lncRNA may be associated with the pathogenesis of IA.
Fewer studies investigate the relationship between lncRNA and IA [13, 14]. In this study, we explored the differential expression of lncRNAs and messenger RNAs (mRNAs) between IA patients and controls based on sequencing technology. The results indicated that several lncRNAs were closely involved in the pathogenesis of IA. Five candidate lncRNA were further validated by subsequent large-sample peripheral blood PCR experiments in IA and healthy controls. Association and predictive value of lncRNAs and IA were evaluated using receiver operating characteristic curve (ROC) and multivariate logistic regression analysis. In summary, the study aims to reveal the complete lncRNA expression profiles in IA, and to further explore the predictive value of lncRNA for IA.
Methods
Patients and samples selection for RNA sequencing
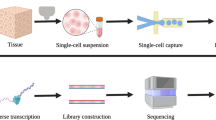
A total of 4 ruptured IA walls (one is an anterior communicating artery aneurysm, and the other three are middle cerebral artery aneurysms) and 4 paired superficial temporal arteries were collected from patients at the Second Affiliated Hospital of Fujian Medical University between December 2017 to March 2018. Their demographic characteristics are shown in supplementary Table 1 (Table S1), Fig. 1 is the illustrative figure of the study. The peripheral blood of the above patients was also collected. Patients were eligible when: 1. Diagnosis of ruptured IA (by CTA, MRA or DSA). 2. Age older than 18 years. 3. No major diseases such as malignant tumors and arteriovenous malformations. 4. Patient data is complete. The exclusion criteria were as follows: 1. The cause of subarachnoid hemorrhage is unknown. 2. Malignant tumor. 3. Other cerebral arteriovenous malformations or cerebral arteriovenous fistulas. 4. Aneurysms caused by trauma or other factors. The corresponding control participants were diagnosed as free of IA who were attending a regular health check-up, including clinical examinations (CTA, MRA or DSA) and medical history assessments. Our research protocol meets the Helsinki declaration and the plan was approved by the Fujian Medical University Ethics Committee.

Illustrative figure of the study
Study population for exploration
Totally 300 participants (150 cases and 150 controls) were enrolled and confirmed their participation by signing the consent forms based on inclusion and exclusion criteria. After corresponding the gender and age by propensity score matching (matching tolerance = 0), 130 patients and 130 controls were enrolled finally, including the blood sample. Head CTA, MRA or DSA were used to diagnosed or excluded IA. Comprehensive data were collected including age, gender, marital status, smoking, alcohol drinking, family history of stroke, tea-drinking, diets, physical exercise, SBP, DBP, TG. Alcohol drinkers and Tea drinking were defined the same as the literature [15, 16]. Active exercise participants were the individuals who exercised at least 20 min once a week. Individuals who consumed at least 20 packs of cigarettes or smoked one cigarette/day for at least 1 year were defined as smokers [17].
RNA extraction
First, peripheral blood leukocytes were isolated from 130 cases and control blood samples. Then, TRIzol reagent (Takala, USA) were used to extracted total RNA according to the kit’s instructions. RNA samples’ quality and integrity were determined. Samples with an OD260/OD280 ratio between 1.8–2.0 were accepted.
RNA sequencing
Total RNA was ribo-consumed by Ribo-ZeroTM Removal Kit (Epicentre, USA) and then treated with RNase (20 U/L) at 37 °C for an hour. Finally, RNA Cleanup magnetic beads (Geneaid, USA) was used to purify. Illumina HiSeq 4000 (Illumina, Inc.) was used for sequencing according to the manufacturer’s protocol. FastQC (v0.11.3) (The Babraham Institute, Cambridge, UK) was used to quality control. Libraries were constructed according to the standard TruSeq protocol. A fluorometer (Qubit 4.0) was then used to evaluate the database centralization while detecting its quantity by Agilent bioanalyzer 2100. The sequencing experiment’s veracity quantification was identified by Library Quantification Kit (Kapa Biosystems, USA). Subsequently, data analyses were performed in silico. At least 1.5 folds with P < 0.05 was considered a significant difference..
GO and KEGG pathway analysis
DAVID was used for GO analysis (http://www.geneongoloty.org/) and KEGG pathway analysis (http://www.genome.jp/kegg/). The meaningful annotations of genes and gene products were constructed by GO analysis. KEGG pathway analysis was used to capture the clusters of pathways involved in the molecular interaction and reaction networks. The log10 (p-value) denotes the enrichment score.
Construction of the CNC network
LncRNA and mRNA with Pearson correlation> 0.90 or < − 0.90, P < 0.05 were selected to draw the network using Cytoscape software [18]. Concentration is treated as the number of nodes associated with other nodes in the network analysis. The degree can determine the relative importance of a network [19].
qRT-PCR assay
PrimeScript RT Reagent Kit (Takara Bio Inc., Shiga, Japan) was performed to reverse transcription of quantified RNA based on the manufacturer’s instructions. The SYBR® Premix Ex Taq™ II kit (Takara Bio Inc., Shiga, Japan) was used for qRT-PCR and performed on the LightCycler 480 Real-Time PCR System (Roche, Switzerland). The 2-ΔΔCt method was used to represent the expression level of lncRNA. The internal control was β-actin. Supplementary table 2 (Table S2) listed the primer sequences.
Statistical analysis
Chi-square (χ2) test and t-test were used to analyze qualitative and quantitative data respectively. Multivariate analysis was followed. Pearson coefficient analysis was utilized to assess the co-expression network. ROC curve analysis was conducted to determine the value of lncRNA. Analyses were accomplished by using SPSS 19.0. A two-tailed P < 0.05 was considered significant.
Results
LncRNA and mRNA expression profiles
The heatmap analysis showed that lncRNA and mRNA profiles were able to differentiate IA patients from controls (Fig. 2a,d). A total of 3342 lncRNAs were significant difference expression (FC ≥ 1.5), of which 2308 lncRNAs transcripts were up-regulated and 1034 lncRNAs were down-regulated (Fig. 2b). Further screening for statistically significant lncRNAs. 1193 lncRNAs were significantly different (FC ≥ 1.5, P < 0.05), among which 900 lncRNAs were up-regulated and 293 lncRNAs were down-regulated (Fig. 2c). And the top 10 up-regulated and top 10 down-regulated lncRNAs were displayed in Table 1. As to mRNA, totally 5338 differentially expressed mRNAs were identified (FC ≥ 1.5), in which 2856 mRNAs were obviously up-regulated while 2482 mRNAs were obviously down-regulated in IA patients compared with controls (Fig. 2e). Statistically significant mRNAs were further screened. Including 1297 up-regulated mRNA and 831 down-regulated mRNA (FC ≥ 1.5, P < 0.05), (Fig. 2f).

LncRNA and mRNA expression profiles. a, d Heatmap analysis. b, c Volcano plot and scatter plot foe lncRNA. e, f Volcano plot and scatter plot for mRNA. Red indicates relatively up-regulated RNAs, green represents relatively down-regulated RNAs, whereas black indicates RNAs with no significant difference
GO and KEGG pathway analyses
As for the up-regulated target genes, GO analysis showed that immune/inflammatory response were most of the functional terms. KEGG pathway analyses revealed that T cell receptor signaling pathway was one of the most enriched pathway (Fig. 3a-b). As for the down-regulated target genes, The most of the functional terms were related to cell adhesion and extracellular matrix. The genes were most enriched in adherens junction and lipolysis regulation in adipocytes (Fig. 3c-d).

GO and KEGG enrichment analyses. a, b GO and KEGG enrichment analyses of up-regulated genes; c, d GO and KEGG enrichment analyses of down-regulated genes
Verify the accuracy of sequencing and small sample qRT-PCR preliminary screening
Based on the lncRNA fold change, p-values and related bioinformatics analysis, three lncRNAs were selected to confirm the sequencing data (lncRNA ENST00000508090, lncRNA ENST00000607042 and lncRNA ENST000000576153). As shown in Fig. 4a-c, lncRNA ENST00000508090 and lncRNA ENST000000576153 expression in tissues were consistent with the RNA-seq results and the expression level of lncRNA ENST00000607042 in tissues was not statistically significant between the two groups. All these results confirmed the accuracy of sequencing. We also observed the expression of the above three lncRNAs in the same human peripheral blood leukocytes. As shown in Fig. 4d-f, lncRNA ENST00000607042, lncRNA ENST00000508090 and lncRNA ENST000000576153 showed the same expression trend in IA tissues and peripheral blood leukocytes.

LncRNAs expression level in different samples. a-c LncRNA expression level in tissues; d-f LncRNAs expression level in peripheral blood leukocytes. (2-ΔΔCt was used to describe the expression of lncRNA. After log 2 logarithmic transformation, t test was used to compare the difference of lncRNA between the two groups)
Next, a total of seven differentially expressed lncRNAs (lncRNA ENST000000576153, lncRNA ENST00000607042, lncRNA ENST00000471220 lncRNA ENST00000478738, lncRNA MALAT1, lncRNA ENST00000508090 and lncRNA ENST00000579688) were further selected using the same screening criteria for qRT-PCR verification among 30 cases and controls matched for age and gender. The expression level of lncRNA ENST00000607042, lncRNA ENST00000471220 and lncRNA ENST00000478738, lncRNA ENST00000508090 and lncRNA MALAT1 in peripheral blood were significantly different between the two groups (Fig. 5). Considering that the CT value of ENST00000508090 in peripheral blood leukocytes is too large, the expression is relatively low, so we did not continue to select this lncRNA for further research. Finally, we selected five lncRNAs with larger differential expression and better lncRNA expression levels for subsequent research, namely lncRNA ENST00000471220, lncRNA ENST00000607042, lncRNA ENST00000478738, MALAT1, lncRNA ENST000000576153.

Expression level of selected lncRNAs in 30 cases and controls. a lncRNA ENST00000471220; b lncRNA ENST00000607042; c lncRNA ENST00000478738; d lncRNA MALAT1; e lncRNA ENST000000576153; f lncRNA ENST00000508090; g lncRNA ENST00000579688. (2-ΔΔCt was used to describe the expression of lncRNA. After log 2 logarithmic transformation, t test was used to compare the difference of lncRNA between the two groups)
Construction of five differential expressed lncRNAs CNC network and function prediction
To explore the potential interplay between lncRNAs and mRNAs, We next constructed the gene CNC network between candidate lncRNAs and differential expression mRNAs based on Pearson correlation> 0.90 or < − 0.90 and P < 0.05. As shown in Fig. 6, five lncRNAs and 593 mRNAs were included in this network, suggesting that a significant correlation exists between the expression profile of lncRNAs and mRNAs. Interestingly, we observed many mRNAs that may be involved in the pathogenesis of IA. Such as Eukaryotic elongation factor 2 kinase (EEF2K) [20], Scm polycomb group protein-like 4 (SCML4) [21], Matrix metalloproteinases 19 (MMP19) [22], Lymphoid enhancer factor 1 (LEF1) [23]. The further GO analyses revealed that the co-expression genes of the 5 candidate lncRNAs were related to T cell activation and leukocyte activation, etc. And KEGG enrichment analyses indicated that the genes were most enriched in the T cell receptor signaling pathway, cytokines-cytokines receptor interaction and chemokine signaling pathway. These items and pathways are closely related to the development of IA (Fig. 7).

Network of lncRNAs-mRNAs. The red nodes represent lncRNAs, and the blue nodes represent mRNAs. The dotted line indicates a negative correlation and the solid line indicates a positive correlation

Enrichment analyses for the co-expression genes of the 5 candidate lncRNAs. a GO analyses; b KEGG enrichment analyses
Large sample verification of candidate lncRNAs in peripheral blood leukocyte
A total of 130 IA patients and healthy people were collected for lncRNA qRT-PCR validation. The characteristics of 130 IA patients and controls were listed in Table 2. No significant difference was found in age, gender, marital status (all P > 0.05). People who smoke, high-salty diet were highly prone to IA. However, tea-drinking and physical exercise were protected factors for IA, consistent with current research results [24,25,26,27,28]. The expression level of lncRNA ENST00000607042, lncRNA ENST00000471220 and lncRNA ENST00000478738, lncRNA ENST00000576153 and lncRNA MALAT1 were significantly different between the 2 groups (Fig. 8a-e). The expression trend of lncRNA ENST00000607042, lncRNA ENST00000471220, lncRNA ENST00000478738 in blood differed from that in sequencing.

Expression level of selected lncRNAs in 130 cases and controls. a lncRNA ENST00000471220; b lncRNA ENST00000607042; c lncRNA ENST00000478738; d lncRNA MALAT1; e lncRNA ENST000000576153 (2-ΔΔCt was used to describe the expression of lncRNA. After log 2 logarithmic transformation, t test was used to compare the difference of lncRNA between the two groups)
ROC curves of related lncRNA for predicting IA risk
We further performed a logistic regression analysis to predict the risk of IA. General demographic characteristics (gender (0 = man,1 = woman), age (0 = < 60, 1 = ≥60), marital status (0 = Marriage,1 = Single and others)) and variables with significant differences in univariate analysis (smoking(0 = no,1 = yes), tea drinking (0 = no,1 = yes), physical exercise (0 = no,1 = yes) were included in the logistic model (Enter). The results disclosed that low expression of lncRNA ENST00000471220 (0R = 0.604, 95%CI: 0.482–0.756), lncRNA ENST00000607042 (0R = 0.077, 95%CI: 0.037–0.160), lncRNA ENST00000478738 (0R = 0.616, 95%CI: 0.497–0.764) and lncRNA MALAT1 (0R = 0.754, 95%CI: 0.597–0.951) were risk factors for IA (Table 3). ROC curves showed that the AUC of lncRNA ENST00000471220, ENST00000607042, ENST00000478738, MALAT1 were 0.689 (95%CI: 0.625–0.753), 0.958 (95%CI: 0.936–0.980), 0.714 (95%CI: 0.653–0.776) and 0.580 (95%CI: 0.510–0.650) (Fig. 9) which shows a good predictive value of lncRNA ENST00000607042 in IA patients.

The ROC curve of lncRNAs. a lncRNA ENST00000471220; b ENST00000607042; c ENST00000478738; d MALAT1
Stratified analysis of lncRNA ENST00000607042
Stratification analysis by various influencing factors revealed the statistically significant associations between lncRNA ENST00000607042 and IA risks in young and old individuals, males and females, smokers and non-smokers, alcohol drinkers and non-alcohol drinkers, high-salt diets and non-high-salt diets, tea drinkers and non-tea drinkers, physical exercise and no physical exercise (Table 4). These results show that lncRNA ENST00000607042 exhibit significant differences in various populations.
Discussion
Several studies have shown that dysregulation of lncRNA expression Related to a variety of diseases such as cancer, diabetes, cardiovascular and cerebrovascular diseases [29,30,31,32]. We found many differentially expressed lncRNAs and mRNAs by sequencing. GO, KEGG pathway analyses and a CNC network were constructed to elucidate the functions and possible mechanisms of differentially expressed genes. In addition, the lncRNA ENST00000607042 was found to have superior diagnostic value of IA patients.
Notably, we found that lncRNAs could be involved in immune/inflammatory response, cell adhesion and extracellular matrix by bioinformatics analyses, which we considered crucial to the pathogenesis of IA. The possible explanations for the results might be that: for the inflammatory response, studies have demonstrated that lncRNAs are key factors in inflammation-related diseases, including IA [33,34,35,36]. Recent studies have shown that the accumulation of lipids in aneurysms wall is related to the remodeling and destruction of aneurysm wall. The destruction of the aneurysm wall is related to Hemoxygenaze-1 secreted by certain inflammatory cells [37, 38]. For cell adhesion and extracellular matrix, one of the important biological processes that have been proven to be the pathogenesis of IA [39]. Extracellular matrix (ECM) serves important functions to cell adhesion, several cell adhesion molecules, such as Versican (VCAN) and Polyclonal Antibody to Integrin Alpha 10 (ITGA10), play a role in maintaining the function of ECM. The remodeling of ECM contributes significantly to the structure and integrity of intracranial arteries [40].
Most lncRNA functions are still poorly understood [41]. Therefore, the lncRNA-mRNA co-expression network was constructed based on 5 candidate lncRNAs and differentially expressed mRNA from sequencing. There are many lncRNAs-mRNAs pairs in the co-expression network that may be involved in the mechanism of IA, such as lncRNA ENST00000471220-EEF2K, lncRNA ENST00000607042-SCML4, lncRNA ENST00000576153-MMP19, lncRNA ENST00000471220-LEF1. The further GO and KEGG enrichment analyses indicated that the genes were most enriched in the T cell receptor signaling pathway, cytokines-cytokines receptor interaction and chemokine signaling pathway. Inflammation is driven by a network comprising cytokines, chemokines, their target receptors and leukocytes [42], indicating that the selected lncRNAs were closely related to the IA development. Because the inflammatory response plays an important role in IA [43].
Five candidate lncRNAs were finally selected and validated in 130 IA patients and 130 controls by qRT-PCR assay. The expression level of lncRNA ENST00000607042, lncRNA ENST00000471220, lncRNA ENST00000478738 and lncRNA MALAT1 in IA patient were lower than that of the controls (all P < 0.05). While the expression level of lncRNA ENST00000476153 was higher than the controls. It is worth noting that the expression levels of lncRNA ENST00000607042, lncRNA ENST00000471220, lncRNA ENST00000478738 in peripheral blood leukocytes are contradictory to sequencing. We consider it for the following reasons: (1) this may be related to the heterogeneous expression of lncRNA and the chemotaxis of central inflammation [44]. (2) The sample size used for sequencing is relatively small, and the result of false positives is inevitable. Further functional experiments are needed to confirm this speculation.
Multivariate regression analysis revealed that low expression of lncRNA ENST00000471220, lncRNA ENST00000607042, lncRNA ENST00000478738 and lncRNA MALAT1 were the risk factor of IA. The further AUC curve of lncRNA ENST00000607042 shows superior predictive value for IA, which indicated that lncRNA ENST00000607042 might serve as a biomarker for IA diagnosis and treatment. Further stratified analysis results show that lncRNA ENST00000607042 exhibit significant differences in various populations. Especially, We focus on the co-expression relationship pairs associated with lncRNA ENST00000607042 to explore the potential biological functions in IA. We found that SCML4 is associated with endothelial dysfunction and vascular remodeling [21]. CXCR4, another mRNA co-expressed with lncRNA ENST00000607042, has been reported to be likely to regulate chemotaxis and adhesion through the CXCR4–SDF-1 pathway, which is closely related to cardiac and macrovascular development [45]. All these results suggested that lncRNA ENST00000607042 may be involved in the development of IA.
Recent literature also reported the relationship between lncRNAs and IA through microarray analysis [14]. We all found many different lncRNAs, and functional analysis suggested that lncRNAs were involved mainly in regulating immune/inflammatory processes/pathways. Compared with this report, our main advantages are listed as follows. First, in addition to predicting the function of lncRNA based on the results of bioinformatics, a large sample qRT-PCR verification was conducted to ensure the reliability of the screening results; moreover, we also included other adjust common risk factors to determine further the lncRNA that may be related to IA, and draw the ROC curve to explore the value of lncRNA in predicting IA, the research content is more abundant.
There were some limitations to the current study. Firstly, the sample size used for sequencing is relatively small because it is difficult to obtain IA tissues. Secondly, The results of this study are mostly based on bioinformatics predictions, and further experiments are needed to clarify the specific role in IA. Thirdly, although we discovered good predictive value of lncRNA ENST00000607042 and related inflammatory response in IA such as chemotaxis and adhesion, The detailed mechanism of lncRNA ENST00000607042 in the development and progression of IA remains to be further explored in our future work.
Conclusions
In summary, our study facilitates comprehensive understanding of lncRNA expression profiles in IA and reveals the close relationship between inflammatory response and IA. Moreover, the low expression of lncRNA ENST00000471220, lncRNA ENST00000607042, lncRNA ENST00000478738 and lncRNA MALAT1 were independent risk factors for IA. LncRNA ENST00000607042 has superior diagnostic value which might be served as novel biomarkers for IA risk.
Availability of data and materials
All the relevant data and Additional file 2 are all available. The datasets generated and analysed during the current study are available in the [GEO: GSE158558] repository, [http://www.ncbi.nlm.nih.gov/geo/]”.
Abbreviations
- IA:
-
Intracranial aneurysm
- LncRNA:
-
Long non-coding RNA
- mRNA:
-
Messenger RNA
- STAs:
-
Superficial temporal arteries
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- CNC:
-
Coding non-coding
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- ROC curve:
-
Receiver operating characteristic curve
- aSAH:
-
Aneurysmal subarachnoid hemorrhage
- SMCs:
-
vascular smooth muscle cells
- BP:
-
Biological processes
- CC:
-
Cellular components
- MF:
-
Molecular functions
- EEF2K:
-
Eukaryotic elongation factor 2 kinase
- SCML4:
-
Scm polycomb group protein-like 4
- MMP19:
-
Matrix metalloproteinases 19
- LEF1:
-
Lymphoid enhancer factor 1
References
Vlak MH, Algra A, Brandenburg R, Rinkel GJ. Prevalence of unruptured intracranial aneurysms, with emphasis on sex, age, comorbidity, country, and time period: a systematic review and meta-analysis. Lancet Neurol. 2011;10(7):626–36.
Greving JP, Wermer MJ, Brown RD, Morita A, Juvela S, Yonekura M, et al. Development of the PHASES score for prediction of risk of rupture of intracranial aneurysms: a pooled analysis of six prospective cohort studies. Lancet Neurol. 2014;13(1):59–66.
Wiebers DO, Whisnant JP, Huston J, et al. Unruptured intracranial aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet (London, England). 2003;362(9378):103–10.
Connolly ES, Rabinstein AA, Carhuapoma JR, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/american Stroke Association. Stroke. 2012;43(6):1711–37.
Hu S, Yu N, Li Y, Hao Z, Liu Z, Li MH. A meta-analysis of risk factors for the formation of de novo intracranial aneurysms. Neurosurgery. 2019;85(4):454–65.
Kim T, Lee H, Ahn S, Kwon OK, Bang JS, Hwang G, et al. Incidence and risk factors of intracranial aneurysm: a national cohort study in Korea. Int J Stroke. 2016;11(8):917–27.
Caranci F, Briganti F, Cirillo L, Leonardi M, Muto M. Epidemiology and genetics of intracranial aneurysms. Eur J Radiol. 2013;82(10):1598–605.
Yu B, Wang S. Angio-lncRs: LncRNAs that regulate angiogenesis and vascular disease. Theranostics. 2018;8(13):3654–75.
Song CL, Wang JP, Xue X, Liu N, Zhang XH, Zhao Z, et al. Effect of circular ANRIL on the inflammatory response of vascular endothelial cells in a rat model of coronary atherosclerosis. Cell Physiol Biochem. 2017;42(3):1202–12.
Lino Cardenas CL, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B, et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun. 2018;9(1):1009.
Michalik KM, You X, Manavski Y, Hamblin MH, Yin KJ. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel GrowthNovelty and significance. Circ Res. 2014;114(9):1389–97.
Muret K, Désert C, Lagoutte L, Boutin M, Gondret F, Zerjal T, et al. Long noncoding RNAs in lipid metabolism: literature review and conservation analysis across species. BMC Genomics. 2019;20(1):882.
Wu C, Song H, Wang Y, Gao L, Cai Y, Cheng Q, et al. Long non-coding RNA TCONS_00000200 as a non-invasive biomarker in patients with intracranial aneurysm. Biosci Rep. 2019;39(11):BSR20182224.
Li H, Yue H, Hao Y, Li H, Wang S, Yu L, et al. Expression profile of long noncoding RNAs in human cerebral aneurysms: a microarray analysis. J Neurosurg. 2017;127:1055–62.
Kaluza J, Harris HR, Linden A, Wolk A. Alcohol consumption and risk of chronic obstructive pulmonary disease: a prospective cohort study of men. Am J Epidemiol. 2019;188(5):907–16.
Tian T, Lv J, Jin G, Yu C, Guo Y, Bian Z, et al. Tea consumption and risk of stroke in Chinese adults: a prospective cohort study of 0.5 million men and women. Am J Clin Nutr. 2020;111(1):197–206.
Sun Y, Chen R, Lin S, Xie X, Ye H, Zheng F, et al. Association of circular RNAs and environmental risk factors with coronary heart disease. BMC Cardiovasc Disord. 2019;19(1):223.
Kohl M, Wiese S, Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303.
Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet. 2004;5(2):101–13.
Xu B, Zhang Y, Du XF, Li J, Zi HX, Bu JW, et al. Neurons secrete miR-132-containing exosomes to regulate brain vascular integrity. Cell Res. 2017;27(7):882–97.
Li Y, Wang DW, Chen Y, Chen C, Guo J, Zhang S, et al. Genome-wide association and functional studies identify SCML4 and THSD7A as novel susceptibility genes for coronary artery disease. Arterioscler Thromb Vasc Biol. 2018;38(4):964–75.
Jackson V, Olsson T, Kurtovic S, Folkersen L, Paloschi V, Wågsäter D, et al. Matrix metalloproteinase 14 and 19 expression is associated with thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2012;144(2):459–66.
Ruigrok YM, Rinkel GJ, van’t Slot R, Wolfs M, Tang S, Wijmenga C. Evidence in favor of the contribution of genes involved in the maintenance of the extracellular matrix of the arterial wall to the development of intracranial aneurysms. Hum Mol Genet. 2006;15(22):3361–8.
Chalouhi N, Ali MS, Starke RM, Jabbour PM, Tjoumakaris SI, Gonzalez LF, et al. Cigarette smoke and inflammation: role in cerebral aneurysm formation and rupture. Mediat Inflamm. 2012;2012(6):271582.
Jiang W, Ye L, Yang Y, Wang P, Pan W, Du J, et al. TRPP2 associates with STIM1 to regulate cerebral vasoconstriction and enhance high salt intake-induced hypertensive cerebrovascular spasm. Hypertens Res. 2019;42(12):1894–904.
Fraser ML, Mok GS, Lee AH. Green tea and stroke prevention: emerging evidence. Complement Ther Med. 2007;15(1):46–53.
China Kadoorie Biobank Study Collaborative Group. Association of physical activity with risk of major cardiovascular diseases in Chinese men and women. JAMA Cardiol. 2017;2(12):1349–58.
Hamer M, O'Donovan G, Stamatakis E. Association between physical activity and sub-types of cardiovascular disease death causes in a general population cohort. Eur J Epidemiol. 2019;34(5):483–7.
Shan K, Liu C, Liu BH, Chen X, Dong R, Liu X, et al. Circular non-coding RNA HIPK3 mediates retinal vascular dysfunction in diabetes mellitus. Circulation. 2017;136(17):1629–42.
Xiao H, Zhang F, Zou Y, Li J, Liu Y, Huang W. The function and mechanism of long non-coding RNA-ATB in cancers. Front Physiol. 2018;9:321.
Kaur H, Sarmah D, Saraf J, Vats K, Kalia K, Borah A, et al. Noncoding RNAs in ischemic stroke: time to translate. Ann N Y Acad Sci. 2018;1421(1):19–36.
Chen T, Zhang C, Liu Y, Zhao Y, Lin D, Hu Y, et al. A gastric cancer LncRNAs model for MSI and survival prediction based on support vector machine. BMC Genomics. 2019;20(1):846.
Zhang Q, Chao TC, Patil VS, Qin Y, Tiwari SK, Chiou J, et al. The long noncoding RNA regulates inflammatory gene expression. EMBO J. 2019;38(8):1–18.
Li H, Wang W, Zhang L, Lan Q, Wang J, Cao Y, et al. Identification of a long noncoding RNA-associated competing endogenous RNA network in intracranial aneurysm. World neurosurg. 2017;97:684–92.
Ma X, Zhao X, Wang K, Tang X, Guo J, Mi M, et al. Identification and analysis of long non-coding RNAs that are involved in inflammatory process in response to transmissible gastroenteritis virus infection. BMC Genomics. 2019;20(1):806.
Gruszka W, Zbroszczyk M, Komenda J, Gruszczyńska K, Baron J. The role of inflammation and potential pharmacological therapy in intracranial aneurysms. Neurol Neurochir Pol. 2018;52(6):662–9.
Laaksamo E, Tulamo R, Liiman A, Baumann M, Friedlander RM, Hernesniemi J, et al. Oxidative stress is associated with cell death, wall degeneration, and increased risk of rupture of intracranial aneurysm wall. Neurosurgery. 2013;72(1):109–17.
Ollikainen E, Tulamo R, Lehti S, Lee-Rueckert M, Hernesniemi J, Niemelä M, et al. Smooth muscle cell foam cell formation, apolipoproteins, and ABCA1 in intracranial aneurysms: implications for lipid accumulation as a promoter of aneurysm wall rupture. J Neuropathol Exp Neurol. 2016;75(7):689–99.
Jia W, Yu L, Huang X, Wang Y, Zhao J. Comparative proteome analysis of saccular intracranial aneurysms with iTRAQ quantitative proteomics. J Proteome. 2016;130:120–8.
Penn DL, Witte SR, Komotar RJ, Sander Connolly E Jr. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci. 2014;21(1):28–32.
Zhao F, Qu Y, Liu J, Liu H, Zhang L, Feng Y, et al. Microarray profiling and co-expression network analysis of lncRNAs and mRNAs in neonatal rats following hypoxic-ischemic brain damage. Sci Rep. 2015;5(3):13850.
Bhattacharya S, Kawamura A. Using evasins to target the chemokine network in inflammation. Adv Protein Chem Struct Biol. 2020;119:1–38.
Signorelli F, Sela S, Gesualdo L, Chevrel S, Tollet F, Pailler-Mattei C, et al. Hemodynamic stress, inflammation, and intracranial aneurysm development and rupture: a systematic review. World Neurosurg. 2018;15:234–44.
Huang Q, Huang QY, Sun Y, Wu S. High-throughput data reveals novel circular RNAs via competitive endogenous RNA networks associated with human intracranial aneurysms. Med Sci Monit. 2019;25:4819–30.
Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cellderived factor (SDF-1) in the developing and mature central nervous system. Glia. 2013;42(2):139–48.
Acknowledgments
The authors thank the participants and participating physicians from the Second Affiliated Hospital of Fujian Medical University, China, as well as investigators and staff for making this research possible.
Funding
This research was funded by Joint Funds for the Innovation of Science and Technology, Fujian Province (2018Y9089), the Natural Science Foundation of Fujian Province (2019 J01315), and the Professor Development Fund Project of Fujian Medical University (JS15002). The funder had no role in the study design, data collection, analysis, decision to publish, or manuscript preparation.
Author information
Authors and Affiliations
Contributions
SW and HL contributed to the study design and helped revise the manuscript; YS, YW involved in writing and in a manuscript review. YS, QR, LY, SH conducted data analysis and interpretation of data; QR, LY, SH, XX, YC acquisition of data. All authors contributed to the critical revision of the final manuscript and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The current study was approved by the Biomedical Research Ethics Committee of Fujian Medical University. Written Informed consent was obtained from all individuals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Clinical characteristics of included patients.
Additional file 2: Table S2.
List of the primers used for qRT-PCR experiments.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, Y., Wen, Y., Ruan, Q. et al. Exploring the association of long noncoding RNA expression profiles with intracranial aneurysms, based on sequencing and related bioinformatics analysis. BMC Med Genomics 13, 147 (2020). https://doi.org/10.1186/s12920-020-00805-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-020-00805-x