
Abstract
Background
Determining the grade and molecular marker status of intramedullary gliomas is important for assessing treatment outcomes and prognosis. Invasive biopsy for pathology usually carries a high risk of tissue damage, especially to the spinal cord, and there are currently no non-invasive strategies to identify the pathological type of intramedullary gliomas. Therefore, this study aimed to develop a non-invasive machine learning model to assist doctors in identifying the intramedullary glioma grade and mutation status of molecular markers.
Methods
A total of 461 patients from two institutions were included, and their sagittal (SAG) and transverse (TRA) T2-weighted magnetic resonance imaging scans and clinical data were acquired preoperatively. We employed a transformer-based deep learning model to automatically segment lesions in the SAG and TRA phases and extract their radiomics features. Different feature representations were fed into the proposed neural networks and compared with those of other mainstream models.
Results
The dice similarity coefficients of the Swin transformer in the SAG and TRA phases were 0.8697 and 0.8738, respectively. The results demonstrated that the best performance was obtained in our proposed neural networks based on multimodal fusion (SAG-TRA-clinical) features. In the external validation cohort, the areas under the receiver operating characteristic curve for graded (WHO I–II or WHO III–IV), alpha thalassemia/mental retardation syndrome X-linked (ATRX) status, and tumor protein p53 (P53) status prediction tasks were 0.8431, 0.7622, and 0.7954, respectively.
Conclusions
This study reports a novel machine learning strategy that, for the first time, is based on multimodal features to predict the ATRX and P53 mutation status and grades of intramedullary gliomas. The generalized application of these models could non-invasively provide more tumor-specific pathological information for determining the treatment and prognosis of intramedullary gliomas.
Similar content being viewed by others


Background
Intramedullary gliomas (IMGs) are the most common primary spinal cord neoplasms, accounting for approximately 80% of spinal cord tumors and 2–4% of central nervous system tumors [1]. The tumor grade and genetic and histological characteristics of gliomas are considered to affect their prognosis and response to treatment. The fifth edition of the WHO Classification of Tumors of the Central Nervous System (WHO CNS5) promotes the application of molecular characteristics for diagnosis and grading. In general, pathological examination and immunohistochemistry (IHC) during surgery or biopsy are required to analyze tumor molecular biomarkers [2, 3]. Without pathological guidance, therapeutic options for patients who are unsuitable for surgery or those who choose nonsurgical therapy may be limited. Although relatively safe, these invasive examinations can damage the normal brain or spinal cord tissue. Owing to the highly dense neural structure of the spinal cord, any minor injury may cause permanent damage to bodily function [4]. Hence, biopsy is not suitable for IMGs, and thus, the demand for alternative non-invasive approaches that can offer genetic and histological evidence of IMGs has surged [5].
Accurate classification of IMG preoperatively is crucial for physicians to develop an appropriate treatment plan. Preoperative magnetic resonance imaging (MRI) remains the most widely utilized and efficient technique for detecting spinal cord lesions in clinical practice. Increasing evidence has revealed the feasibility of using MRI to predict the type of glioma and molecular biomarkers, such as IDH1, Ki67, and H3-K27M, via machine learning or deep learning method [6,7,8]. However, only a limited number of studies have attempted to predict markers and classification of IMGs based on MRI owing to the lack of sufficient training in such rare tumors [9, 10].
Although IMGs and brain gliomas are both derived from glial cells, the molecular characteristics of primary IMGs are significantly different from those of brain gliomas [11, 12]. As summarized in the WHO CNS5, the mutation-site genotypes of genes such as isocitrate dehydrogenase (IDH), tumor protein p53 (P53), and alpha thalassemia/mental retardation syndrome X-linked (ATRX) are important indicators for the classification of gliomas [13]. Despite the commonly mentioned IDH mutations in brain gliomas, IDH mutations are rare in IMGs. The absence of IDH mutations in the spine cannot be used to distinguish between grade I and II diffuse astrocytomas. Moreover, IDH IHC would be ineffective because most IMGs lack the conventional IDH1 p.R132H mutation [12]. Hence, determining the mutation status of ATRX and P53 in IMGs is especially important.
Methods
Aim
The present study aimed to develop a non-invasive preoperative method for predicting IMG grade and mutational status of molecular markers. Towards this goal, we retrospectively analyzed the preoperative MRI scans of patients with IMGs whose ATRX and P53 mutation statuses were tested by IHC. In the experimental design, multiple feature representations and mainstream machine learning models were compared to explore the superior predictive effect. After exploring the superior feature representation of novel machine learning models, including WHO-Mind, ATRX-Mind, and P53-Mind, methods were developed to accurately classify the glioma grade and predict the ATRX and P53 mutation statuses of IMGs. Additionally, rigorous external validation was performed to verify the generalizability of the proposed method.
Patient demographic characteristics and data acquisition
This retrospective study was approved by the ethical committees of Beijing Tsinghua Changgung Hospital (hospital 1) and The First Affiliated Hospital of Zhengzhou University (hospital 2) and was conducted in accordance with the principles of the Declaration of Helsinki. The requirement for obtaining informed consent was waived.
We collected the records of patients from two hospitals (hospital 1: January 2015 to April 2021 and hospital 2: February 2017 to October 2021). The patient inclusion criteria were preoperative MRI examination, pathological diagnosis of IMGs according to the WHO CNS5, and IHC-confirmed ATRX and P53 mutation status. Patients who had undergone biopsy, chemotherapy, and radiotherapy and those with IMG metastases from brain glioma were excluded. Following the initial patient screening, MRI data were evaluated, and the exclusion criteria were as follows: absence of sagittal (SAG) and transverse (TRA) T2-weighted imaging (T2WI) scans and artifacts in the MRI image.
Data on clinical baseline characteristics, including age, sex, smoking, alcohol consumption, medical history with accompanying diseases, time of onset, preoperative McCormick score, and radiological features of tumor-related lesions, were collected. Moreover, all radiological features, including tumor number, axis ratio (tumor/spinal cord), bleeding, cysts, spinal cord cavity, edema, atrophy, and malformation, were independently assessed by three neuroradiologists. All of these features were not obtainable by radiomic feature extraction. Two experienced pathologists performed the pathological evaluation of all gliomas using the WHO CNS5 for glioma grading and classification [13]. We classified glioma grades as low grade (WHO I–II, benign) and high grade (WHO III–IV, malignant), and the mutation status of ATRX and P53 was determined by IHC.
Patients recruited from hospital 1 were included in the primary cohort (n = 332) and were randomly assigned to the training set and internal validation set after fivefold cross-validation according to the ATRX/P53 mutation status and tumor grade. Patients from hospital 2 were included in the independent external validation cohort (n = 129). A detailed flow diagram of the patient selection process is illustrated in Fig. 1.

Flow diagram showing the patient selection protocol and inclusion and exclusion criteria. ATRX, alpha thalassemia/mental retardation syndrome X-linked; IHC, immunohistochemistry; P53, tumor protein p53; SAG, sagittal; TRA, transverse; WI, weighted imaging
MRI acquisition and region of interest segmentation
MR data were obtained employing a 3.0-T MR scanner (Discovery MR750; GE Healthcare, Milwaukee, WI) in hospital 1 and either Siemens (Verio, Prisma, or Skyra; Siemens Healthcare, Erlangen, Germany) or Philips (Ingenia, Philips Medical System, Best, The Netherlands) MR systems in hospital 2. An overview of the parameters used to acquire MRI is given in Additional file 1, which shows that scans are obtained by scanners from different vendors employing various acquisition settings. Identifying the borders of low-grade gliomas is difficult on T1-weighted and contrast-enhanced sequences; thus, the T2-weighted (T2W) sequence is well accepted for identifying abnormal hyperintense signals representing gliomas, and tumor regions of interest (ROIs) were delineated on SAG and TRA T2WI separately [14].
Lesions were delineated on both SAG and TRA T2WI by two experienced neuroradiologists using the ITK-SNAP software (www.itksnap.org). Abnormal hyperintense signals on T2W MR images were regarded as tumor regions, and the signals of cerebrospinal fluid, spinal cord cavity, and edema were avoided. If the ROI was controversial, a senior neuroradiologist performed the final determination. T1-weighted images with and without gadolinium enhancement, short T1 inversion recovery, and fluid attenuation inversion recovery images were collected for most patients to aid in identifying tumors. Only T2WI was used in subsequent studies. Finally, to reduce the heterogeneity between different scanners, we resampled the T2W images and the corresponding ROI mask into a uniform voxel size of 0.5 × 0.5 × 5 mm across all patients for radiomic extraction.
Lesion segmentation using deep learning
After preprocessing, deep learning models were proposed to segment lesions, which greatly improved future work efficiency. This work selected a deep learning model method based on the transformer architecture (Swin transformer) because of its superiority in multiple domains [15, 16]. The Swin transformer adopts a hierarchical design containing a total of four stages: each stage decreases the resolution of the input feature map and expands the perceptual field layer by layer, similar to a convolutional neural network. In addition, it is designed with patch embedding, which cuts MRI scans into patches and embeds them into the model. Although this architecture has performed well in several tasks [17, 18], its use for segmenting IMGs has not been reported.
Both SAG and TRA images of the primary cohort were employed for the training, internal, and external validation cohorts for testing. The input channel was two dimensional, that is, all MRI sequences were converted into slices and then batch inputted, and the final output results were reconstructed in 3D. Transfer learning was adopted for training, and the pre-trained model was swin_transformer_base_patch4_window7_224_imagenet_1k. The backbone of the model was SwinTransformer_base_patch4_window7_224.
Feature extraction and selection
Radiomic features were extracted using the PyRadiomics module written in Python 3.7 in accordance with a previous study [19]. Both the SAG and TRA images extracted the following six feature classes: first-order statistics, shape, gray-level run-length matrix, gray-level size zone matrix, neighboring gray-tone difference matrix, and the gray-level dependence matrix. These feature classes used six built-in filters (wavelet, square, square root, logarithm, exponential, and gradient filters). All features were named by connecting the MRI type, the image type from which the feature was extracted, the feature class, and the feature name separated by an underscore. For example, TRA_original_glrlm_RunEntropy is a feature extracted from the TRA T2WI sequence, original image, and glrlm feature class, and the feature name is RunEntropy.
For every patient, 1960 radiomic features were extracted from SAG and TRA T2WI data, and all radiomic features were z transformed for data standardization. To avoid interobserver variations during manual segmentation, we calculated the intraclass correlation coefficient (ICC) for each feature, and only those with high stability (ICC > 0.8) were included in the analysis [20]. Then, the selected stable features were tested using the independent samples t-test or the Mann–Whitney U test to select potential important features. Features that did not meet the criteria for either of the aforementioned tests were excluded. This study adopted the least absolute shrinkage and selection operator (LASSO) on the training cohort to screen significant features with non-zero coefficients that can differentiate ATRX and P53 mutation status or glioma grade separately. For the three outcomes of ATRX, P53, and tumor grade, we used LASSO to select features in the TRA, SAG, and TRA + SAG groups, respectively. The aforementioned calculation methods are available in PyRadiomics 2.2.0 documentation [21].
Prediction model construction
In this study, three types of deep neural networks (DNNs) were constructed based on the data characteristics of the prediction tasks: WHO-Mind, ATRX-Mind, and P53-Mind. WHO-Mind has one input layer, four hidden layers, and one output layer. Both ATRX-Mind and P53-Mind have one input layer, three hidden layers, and one output layer. For each DNN, the ReLU and Adam were selected as the activation function and the solver for weight optimization, respectively. In addition, the initial learning rates and batch sizes were set to 0.01 and 64 in all models, respectively. The model architecture and the key hyperparameters are summarized in Additional file 2. However, the unbalanced nature of the data included in this study posed a challenge for model training, as data category imbalance may lead to severe overfitting. Therefore, after dividing the dataset, the Synthetic Minority Oversampling Technique (SMOTE) was adopted for the divided training set to achieve data augmentation. Its basic idea is to generate new synthetic samples by interpolating between minority class samples, thereby balancing the class distribution, and thus improving the performance of the classifier.
Specifically, this algorithm analyzes the minority class samples and artificially synthesizes new samples based on the minority class samples to add to the dataset, thereby solving the problem of model overfitting. The algorithm first randomly selects a sample from the minority class (sample A) and identifies its k nearest neighbors within the minority class. Subsequently, a neighbor (sample B) is randomly chosen from these k neighbors. The new synthetic sample is created on the line segment between sample A and sample B. This process is repeated as needed until a sufficient number of synthetic minority class samples are generated to achieve the desired class balance. It is worth noting that SMOTE solely creates new samples within the feature space without generating new class labels. Synthetic samples inherit the class label of their parent samples (i.e., sample A and sample B). To implement SMOTE in Python 3.7, we utilized the imbalanced-learn library, applying the SMOTE algorithm to the training data in order to balance the class distribution.
Additionally, considering that some mainstream models that have been developed obtained good performance in related fields [22,23,24], extreme Gradient Boosting (XGBoost), Gradient Boosting Decision Tree (LightGBM), and random forest (RF) were also trained and tested for comparison. The parameters of the above models were optimized during the training process using the GridSearchCV tool in Scikit-learn 1.1.1. The modeling was performed in the Python 3.7 programming environment, and the core library involved was Scikit-learn 1.1.1.
Experimental setup for the prediction models
The overall process of this study is illustrated in Fig. 2. Three tasks (WHO tumor grade, ATRX mutation status prediction, and P53 mutation status prediction) were included, and each was used to build four machine learning models. Moreover, six different feature representations were compared to explore the most superior model input: SAG radiomics, SAG radiomics with clinical baseline, TRA radiomics, TRA radiomics with clinical baseline, SAG + TRA radiomics, and SAG + TRA radiomics with clinical baseline features. In the primary cohort, fivefold cross-validation was employed to identify the best models. Data were randomly divided into five equal parts: one was selected for internal validation, and the rest were trained. This process was repeated five times. The best-performing models were tested using an external validation cohort to verify the generalization ability. This work was completed in Windows 10 OS and involved computing devices with a CPU AMD Ryzen 7 5800H (16 GB RAM) and GPU GeForce RTX ™ 3090 (24 GB RAM).

Illustration of the study process. Stage I includes raw image acquisition (a), manual ROI segmentation (b), and auto ROI segmentation (c). Stage II includes feature extraction and selection. From both SAG and TRA images, radiomic features, including first-order statistical, shape, texture, and wavelet features, are extracted (d). All extracted features are screened out by ICC to select stable features (e). Informative features are then selected using LASSO (f). Stage III includes model construction and validation. Selected clinical and radiomic features are entered into the deep neural networks to predict the different tasks (g), and model performance is further tested in the external validation cohort (h). ATRX, alpha thalassemia/mental retardation syndrome X-linked; ICC, intraclass correlation coefficient; LASSO, least absolute shrinkage and selection operator; P53, tumor protein p53; ROI, region of interest; SAG, sagittal; TRA, transverse
Statistical analysis
When representing continuous variables, differences were assessed using Student’s t-test or the Mann–Whitney U test, as appropriate, and data are represented as median with interquartile range. Specifically, because the fivefold cross-validation was employed in the primary cohort, we also calculated the means and 95% confidence intervals (CIs) for the evaluation metrics of the models. We adopted the chi-square test to evaluate the differences in categorical variables, and the results are presented as the number of events and relative frequency (%). The performance of all models was evaluated according to accuracy (Acc), sensitivity (Sens), specificity (Spec), F1 scores, and receiver operating characteristic (ROC) curves. A dice similarity coefficient (DSC) was adopted to evaluate the performance of the networks for IMG segmentation. All statistical analyses were performed employing R software (version 3.6.3, R Foundation for Statistical Computing, Vienna, Austria). Statistical significance was defined as p ≤ 0.05.
Results
Clinicodemographic patient characteristics
A total of 332 patients in hospital 1 (primary cohort) were divided into training and internal validation cohorts, and 127 patients in hospital 2 were enrolled as the independent external validation cohort. The clinical baseline features of the patients and the distribution of ATRX and P53 mutation status across the primary and external validation cohorts are presented in Additional file 3. In the primary cohort, 52 patients (15.7%) had high-grade gliomas and 280 patients (84.3%) had low-grade gliomas. The external validation cohort included 20 (58.8%) patients with high-grade gliomas and 14 (41.2%) patients with low-grade gliomas. There were significant differences in the numbers of ATRX (79/332, 23.8%; 43/127, 33.9%; p = 0.039) and P53 (72/332, 21.7%; 44/127, 34.6%; p = 0.006) mutations between the primary and external validation cohorts. Meanwhile, no significant difference was found in age, sex, onset month, accompanying diseases, smoking, alcohol consumption, or the McCormick score between the primary and external validation cohorts. Regarding the imaging features extracted by neuroradiologists, the axis ratio of the tumor in the spinal cord and the proportion of tumor bleeding differed significantly between the two hospitals.
Results of lesion segmentation
The Swin transformer model was trained using slices of SAG and TRA images and artificially labeled lesions in the primary cohort. After 50,000 iterations, the model proved to be completely convergent and had no overfitting. Therefore, this model was selected for testing at this point. In total, 20% of the samples were randomly selected from patients as the test set, and the rest were used as the training set. In the SAG phase test, the Acc and DSC of the automatic segmentation model reached 0.9929 and 0.8697, respectively. In the TRA phase test, the Acc and DSC of the automatic segmentation model were 0.9978 and 0.8738, respectively. These results demonstrated that the aforementioned deep learning model was relatively satisfactory for the segmentation of lesions in this study. Visualization of the automatic segmentation of the two phases is demonstrated in Fig. 2c.
Feature extraction and selection
For every patient, 1960 radiomic features were derived, and the ICC results showed that 1572 (80.1%) features were stable. The selected stable features in the TRA and SAG groups are listed in Additional file 4. A total of 1560, 1554, and 1517 features for the ATRX mutation status, P53 mutation status, and WHO tumor grading tasks, respectively, passed the scrutiny of the t-test or the Mann–Whitney U test. We employed fivefold cross-validation in LASSO for each prediction task to select the meaningful features. In the WHO tumor grade prediction task, 24 features were selected in SAG, 6 features in TRA, and 20 features in SAG + TRA (Additional file 5). In the ATRX mutation status prediction task, 3 features were selected in SAG, 39 features in TRA, and 5 features in SAG + TRA (Additional file 6). In the P53 mutation status prediction task, 21 features were selected in SAG, 24 features in TRA, and 57 features in SAG + TRA (Additional file 7). All features selected by LASSO are listed in Additional file 8.
Results of prediction models in the primary cohort
Four machine learning models were trained for each prediction task, and each model was fed into six feature representations. All models were parameter optimized in the experiments, which facilitated the performance comparison. The results of the WHO tumor grade prediction and ROC curves of the proposed models are shown in Fig. 3. Among the four models, WHO-Mind obtained the best average area under the ROC curve (AUC; 0.9263), with the AUC higher than that of XGBoost (0.8802), LightGBM (0.9079), and RF (0.8618), indicating the superiority of the neural network architecture in this prediction task. However, inputting radiomics features combined with clinical baseline features was superior to only inputting radiomics features, and the best-performing combination was SAG + TRA + clinical indicators. Notably, we found similar findings in performing the prediction task of ATRX and P53 mutation status, which may be due to the large variety of features covered by multimodal fusion. The results of the ATRX and P53 mutation predictions for each model are shown in Figs. 4 and 5, respectively. The proposed ATRX-Mind and P53-Mind models performed better than the compared models, where the highest AUCs were 0.9281 and 0.9173, respectively. To reflect the comprehensiveness of the evaluation of the proposed models, their Acc, Sens, Spec, and F1 scores were also calculated and summarized in Additional file 9.

The fivefold cross-validation results in the WHO prediction task. a The comparison results of the proposed WHO-Mind with the mainstream machine learning models. b The receiver operating characteristic (ROC) curves of WHO-Mind when inputting features of the SAG phase (left), TRA phase (middle), and SAG combined with TRA phase (right). c The ROC curves of WHO-Mind when the features of the SAG phase-clinical baseline (left), TRA phase-clinical baseline (middle), and SAG phase-TRA phase-clinical baseline (right) are inputted. AUC, area under the curve; SAG, sagittal; TRA, transverse

The fivefold cross-validation results in the ATRX genotype prediction task. a The comparison results of the proposed ATRX-Mind with the mainstream machine learning models. b The ROC curves of ATRX-Mind when inputting features of the SAG phase (left), TRA phase (middle), and SAG combined with TRA phase (right). c The ROC curves of ATRX-Mind when the features of the SAG phase-clinical baseline (left), TRA phase-clinical baseline (middle), and SAG phase-TRA phase-clinical baseline (right) are inputted. ATRX, alpha thalassemia/mental retardation syndrome X-linked; AUC, area under the curve; ROC, receiver operating characteristic; SAG, sagittal; TRA, transverse

The fivefold cross-validation results in the P53 genotype prediction task. a The comparison results of the proposed P53-Mind with the mainstream machine learning models. b The ROC curves of P53-Mind when inputting features of the SAG phase (left), TRA phase (middle), and SAG combined with TRA phase (right). c The ROC curves of P53-Mind when the features of the SAG phase-clinical baseline (left), TRA phase-clinical baseline (middle), and SAG phase-TRA phase-clinical baseline (right) are inputted. AUC, area under the curve; P53, tumor protein p53; ROC, receiver operating characteristic; SAG, sagittal; TRA, transverse
Results of prediction models in the external test cohort
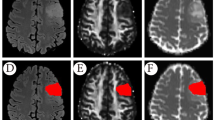
The best-performing models (i.e., WHO-Mind, ATRX-Mind, and P53-Mind) were selected for external testing. Multimodal fused feature representations were fed into each model, namely, the SAG + TRA + clinical indicators. The automatic segmentation effect of the Swin transformer is shown in Fig. 6a. This was found to segment the lesion area with relative accuracy. The performance of the models in each test task is shown in Fig. 6b. WHO-Mind reached the highest AUC (0.8431), and both WHO-Mind and ATRX-Mind obtained the highest Acc (0.8889). In addition, the ROCs of the three models were plotted to visualize their generalization performance in more detail (Fig. 6c–e). More detailed test results (e.g., Acc, Sens, Spec, and F1 scores) are displayed in Additional file 10.

Results of external validation using the proposed models. a The visualization of lesion segmentation using the automatic segmentation model in SAG and TRA phases, respectively. b The external validation results adopting the proposed models in different tasks. c–e The ROC curves for each model in the prediction tasks. ATRX, alpha thalassemia/mental retardation syndrome X-linked; ROC, receiver operating characteristic; P53, tumor protein p53; SAG, sagittal; TRA, transverse; WHO, World Health Organization
Discussion
Biopsy to obtain tumor tissue for histological examination in spinal cord tumors is dangerous and not worthwhile because of the high risk. In this study, we developed three models that can segment the IMG region automatically and predict the ATRX/P53 mutation status and the grade of IMGs based on preoperative sagittal and transverse MRI scans. The prediction models offer a non-invasive alternative for identifying the tumor grade and histopathological characteristics in patients with IMGs.
Many studies have used machine learning methods to identify the genetic and histological features of brain gliomas [6, 7]. Li et al. [25] adopted a support vector machine model to predict the genetic characteristics of ATRX. Given that their method is only effective for low-grade gliomas, the tumor grade should be known in advance, complicating the use of their method in the preoperative period. Gao et al. [7] used multiple machine learning-based radiomic approaches to predict the brain glioma grade and status of pathologic biomarkers, including Ki-67, GFAP, and S100. However, these biomarkers are interrelated and are not highly specific to gliomas, according to the WHO CNS5. In real-world clinical settings, it is not possible to accurately determine the type of glioma preoperatively. Therefore, models must address all types of gliomas, including WHO grades I–IV. As such, this study consecutively enrolled all types of spinal cord gliomas from two centers.
Many studies have focused on predicting IDH mutation and 1p/19q co-deletion status [6]. Although IDH mutation and 1p/19q co-deletion status are important biomarkers for the prognosis and survival of gliomas, we did not predict this in our study. On the one hand, IDH mutation in IMGs is not as common as in brain gliomas and cannot be used to distinguish between grade I pilocytic astrocytoma and grade II diffuse astrocytoma. On the other hand, IDH IHC would be ineffective because most IMGs lack the conventional IDH1 p.R132H mutation [12]. Furthermore, diffuse astrocytomas with IDH mutations are most likely to harbor mutations in ATRX and P53 [26], and cIMPACT-NOW Update 2 states that a definite loss of ATRX nuclear expression and/or p53 immunopositivity is sufficient for the diagnosis of astrocytoma without the need for a 1p/19q test [27]. Therefore, ATRX and P53 mutations are more useful for identifying IMGs and are also important clinical behavior indicators. Our models, focusing on ATRX and P53 mutation status, will bring significant convenience in diagnosing IMGs.
Traditional lesion segmentation is performed manually, which is time-consuming and labor-intensive. Progress in the automatic segmentation of gliomas has been reported more frequently [28,29,30,31], but only few studies have attempted to segment spinal cord tumor automatically due to the lack of sufficient training in such rare tumors [8, 10]. This study employed a deep learning strategy based on transformer architecture to segment IMGs, and relatively satisfactory segmentation results were obtained (the DSC was 0.8697 for SAG and 0.8738 for TRA). To our best knowledge, this study is the first to employ such a strategy. Although the segmentation results temporarily have a certain gap compared with manual annotation, the method significantly improves efficiency and facilitates end-to-end prediction. We will further improve this algorithm to achieve more accurate automatic segmentation.
Additionally, model performance was compared using different combinations of SAG images, TRA images, and clinical features. The combination of SAG and TRA features exhibited a better discriminatory ability than either feature alone, probably because SAG and TRA features can form some complementary properties. Most IMGs are confined to the spinal cord and grow along the long axis of the spinal cord. SAG images can show the greatest anatomical detail and other tumor-related lesions, such as the spinal cord cavity and edema. However, only a few scanning slices are obtained from SAG T2WI, typically 11 slices, which is insufficient for constructing 3D tumor models. TRA T2WI scans usually have smaller slice spacing, have the advantage of accurately constructing a 3D tumor ROI, and may allow for more accurate delineation of the tumor contour during segmentation. Additionally, previous research has confirmed that clinical features such as age and sex are related to glioma type [32, 33]. Wu et al. [34] developed a nomogram strategy to adopt clinical features in radiomics-based prediction models. Fifteen clinical baseline features, including seven baseline features and eight radiological signs that are difficult to represent with the radiomic approach, selected by univariate regression and prior clinical experience were adopted in our models. Although the SAG + TRA radiomics input had already achieved satisfactory performance, the model showed better differentiation ability when we added clinical features.
It is important to emphasize that the three proposed neural networks showed superior performance to the mainstream ensemble learning models in all prediction tasks, which was encouraging. This may be related to the models’ architecture. Specifically, the neural networks proposed in this study underwent extensive pre-experimentation, including setting the number of layers and the number of neurons. External tests were also performed to verify their generalizability, and the results were satisfactory. For the WHO tumor grade, ATRX mutation status, and P53 mutation status prediction tasks, the proposed model obtained AUCs of 0.8431, 0.7622, and 0.7954, respectively, in the external validation set.
Our study has some limitations. First, we selected only the commonly used ATRX and P53 biomarkers for IMGs from a broad range of biomarkers. The detection of other important biomarkers in IMGs, such as Ki-67 and H3 K27M, may also benefit from an automatic machine learning approach. Second, our models should input more patient characteristics in the future to allow for a more comprehensive assessment of patient conditions. For example, our models still need input with respect to some radiological characteristics, such as the spinal cord cavity, edema, and atrophy, which require a priori knowledge assessment by radiologists. Multiple imaging inputs, including normal spinal cord and tumor-related lesions, may help to extract all radiological characteristics automatically. Third, although our data originated from two institutes with one for external validation, a larger dataset from multiple institutes is needed to balance the heterogeneity in different MR-acquiring machines. Finally, there is a degree of imbalance in the proportion of data of different types (e.g., different grades or mutation states) in this study. The dataset should be balanced in the future.
Conclusions
We developed the first machine learning strategy to predict IMG grade and ATRX and P53 mutation status while simultaneously automatically segmenting the tumor. Neural networks based on multimodal fusion (i.e., the SAG phase-TRA phase-clinical baseline) were shown to have the best prediction performance. Although further validation must be performed in more institutes before widespread clinical application, we believe this research is a significant attempt at the automatic diagnosis of IMGs.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- Acc:
-
Accuracy
- ATRX:
-
Alpha thalassemia/mental retardation syndrome X-linked
- DNN:
-
Deep neural network
- DSC:
-
Dice similarity coefficient
- ICC:
-
Intraclass correlation coefficient
- IDH:
-
Isocitrate dehydrogenase
- IHC:
-
Immunohistochemistry
- IMG:
-
Intramedullary glioma
- LASSO:
-
Least absolute shrinkage and selection operator
- LightGBM:
-
Gradient Boosting Decision Tree
- MRI:
-
Magnetic resonance imaging
- P53:
-
Tumor protein p53
- RF:
-
Random forest
- ROC:
-
Receiver operating characteristic
- ROI:
-
Region of interest
- SAG:
-
Sagittal
- T2WI:
-
T2-weighted imaging
- TRA:
-
Transverse
- XGBoost:
-
Extreme Gradient Boosting
References
Bruneau M, Riva M, Brotchi J. Primary intramedullary tumors. In: Van De Kelft E, editor. Surgery of the spine and spinal cord: a neurosurgical approach. Cham: Springer International Publishing; 2016. p. 699–716.
Jackson RJ, Fuller GN, Abi-Said D, Lang FF, Gokaslan ZL, Shi WM, et al. Limitations of stereotactic biopsy in the initial management of gliomas. Neuro Oncol. 2001;3:193–200.
Kristensen BW, Priesterbach-Ackley LP, Petersen JK, Wesseling P. Molecular pathology of tumors of the central nervous system. Ann Oncol. 2019;30:1265–78.
Chen CC, Hsu PW, Erich Wu TW, Lee ST, Chang CN, Wei KC, et al. Stereotactic brain biopsy: single center retrospective analysis of complications. Clin Neurol Neurosurg. 2009;111:835–9.
Zhou M, Scott J, Chaudhury B, Hall L, Goldgof D, Yeom KW, et al. Radiomics in brain tumor: image assessment, quantitative feature descriptors, and machine-learning approaches. AJNR Am J Neuroradiol. 2018;39:208–16.
Van Der Voort SR, Incekara F, Wijnenga MMJ, Kapsas G, Gahrmann R, Schouten JW, et al. Combined molecular subtyping, grading, and segmentation of glioma using multi-task deep learning. Neuro Oncol. 2023;25:279–89.
Gao M, Huang S, Pan X, Liao X, Yang R, Liu J. Machine learning-based radiomics predicting tumor grades and expression of multiple pathologic biomarkers in gliomas. Front Oncol. 2020;10:1676.
Liu J, Chen F, Pan C, Zhu M, Zhang X, Zhang L, et al. A cascaded deep convolutional neural network for joint segmentation and genotype prediction of brainstem gliomas. IEEE Trans Biomed Eng. 2018;65:1943–52.
Pang B, Chai RC, Zhang YW, Chang YZ, Liu W-H, Jia W-Q, et al. A comprehensive model including preoperative peripheral blood inflammatory markers for prediction of the prognosis of diffuse spinal cord astrocytoma following surgery. Eur Spine J. 2021;30(10):2857–66.
Lemay A, Gros C, Zhuo Z, Zhang J, Duan Y, Cohen-Adad J, et al. Automatic multiclass intramedullary spinal cord tumor segmentation on MRI with deep learning. Neuroimage Clin. 2021;31:102766.
Teng YD, Abd-El-Barr M, Wang L, Hajiali H, Wu L, Zafonte RD. Spinal cord astrocytomas: progresses in experimental and clinical investigations for developing recovery neurobiology-based novel therapies. Exp Neurol. 2019;311:135–47.
Lebrun L, Meléndez B, Blanchard O, De Nève N, Van Campenhout C, Lelotte J, et al. Clinical, radiological and molecular characterization of intramedullary astrocytomas. Acta Neuropathol Commun. 2020;8:128.
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 2021;23:1231–51.
Kinoshita M, Sakai M, Arita H, Shofuda T, Chiba Y, Kagawa N, et al. Introduction of high throughput magnetic resonance T2-weighted image texture analysis for WHO grade 2 and 3 gliomas. PLoS ONE. 2016;11:e0164268.
Wu J, Xu Q, Shen Y, Chen W, Xu K, Qi XR. Swin transformer improves the IDH mutation status prediction of gliomas free of MRI-based tumor segmentation. J Clin Med. 2022;11:4325.
Yan S, Wang C, Chen W, Lyu J. Swin transformer-based GAN for multi-modal medical image translation. Front Oncol. 2022;12:942511.
Islam MN, Hasan M, Hossain MK, Alam MGR, Uddin MZ, Soylu A. Vision transformer and explainable transfer learning models for auto detection of kidney cyst, stone and tumor from CT-radiography. Sci Rep. 2022;12:11440.
Pan X, Gao X, Wang H, Zhang W, Mu Y, He X. Temporal-based Swin transformer network for workflow recognition of surgical video. Int J Comput Assist Radiol Surg. 2023;18:139–47.
Ma C, Zhang Y, Niyazi T, Wei J, Guocai G, Liu J, et al. Radiomics for predicting hematoma expansion in patients with hypertensive intraparenchymal hematomas. Eur J Radiol. 2019;115:10–5.
Aerts HJ, Velazquez ER, Leijenaar RT, Parmar C, Grossmann P, Carvalho S, et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat Commun. 2014;5:4006.
Van Griethuysen JJM, Fedorov A, Parmar C, Hosny A, Aucoin N, Narayan V, et al. Computational radiomics system to decode the radiographic phenotype. Cancer Res. 2017;77:e104–7.
Sakai Y, Yang C, Kihira S, Tsankova N, Khan F, Hormigo A, et al. MRI radiomic features to predict IDH1 mutation status in gliomas: a machine learning approach using gradient tree boosting. Int J Mol Sci. 2020;21:8004.
Le NQK, Hung TNK, Do DT, Lam LHT, Dang LH, Huynh TT, et al. Radiomics-based machine learning model for efficiently classifying transcriptome subtypes in glioblastoma patients from MRI. Comput Biol Med. 2021;132:104320.
Yan Z, Wang J, Dong Q, Zhu L, Lin W, Jiang X. XGBoost algorithm and logistic regression to predict the postoperative 5-year outcome in patients with glioma. Ann Transl Med. 2022;10:860.
Li Y, Liu X, Qian Z, Sun Z, Xu K, Wang K, et al. Genotype prediction of ATRX mutation in lower-grade gliomas using an MRI radiomics signature. Eur Radiol. 2018;28:2960–8.
Cancer Genome Atlas Research Network, Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372:2481–98.
Louis DN, Giannini C, Capper D, Paulus W, Figarella-Branger D, Lopes MB, et al. cIMPACT-NOW update 2: diagnostic clarifications for diffuse midline glioma, H3 K27M-mutant and diffuse astrocytoma/anaplastic astrocytoma, IDH-mutant. Acta Neuropathol. 2018;135:639–42.
Li SD, Liu JW, Song ZJ. Brain tumor segmentation based on region of interest-aided localization and segmentation U-Net. Int J Mach Learn Cybern. 2022;13:2435–45.
Khorasani A, Kafieh R, Saboori M, Tavakoli MB. Glioma segmentation with DWI weighted images, conventional anatomical images, and post-contrast enhancement magnetic resonance imaging images by U-Net. Phys Eng Sci Med. 2022;45:925–34.
Yu YM, Du ZF, Yuan CX, Li J. Exploration of CT images based on the BN-U-net-W network segmentation algorithm in glioma surgery. Contrast Media Mol Imaging. 2022;2022:4476412.
Kihira S, Mei XY, Mahmoudi K, Liu Z, Dogra S, Belani P, et al. U-Net based segmentation and characterization of gliomas. Cancers (Basel). 2022;14:4457.
Claus EB, Cannataro VL, Gaffney SG, Townsend JP. Environmental and sex-specific molecular signatures of glioma causation. Neuro Oncol. 2022;24:29–36.
Hyare H, Rice L, Thust S, Nachev P, Jha A, Milic M, et al. Modelling MR and clinical features in grade II/III astrocytomas to predict IDH mutation status. Eur J Radiol. 2019;114:120–7.
Wu S, Zhang X, Rui W, Sheng Y, Yu Y, Zhang Y, et al. A nomogram strategy for identifying the subclassification of IDH mutation and ATRX expression loss in lower-grade gliomas. Eur Radiol. 2022;32:3187–98.
Acknowledgements
Not applicable.
Funding
Tsinghua Precision Medicine Foundation [2022ZLB003, 10001020618]; National Key Research and Development Program of China [2021YFE0204700]; Provincial and ministerial co-construction project of Henan Medical Science and Technology Research Plan [SB201901007]; and Beijing Natural Science Foundation project [7212007].
Author information
Authors and Affiliations
Contributions
Conceiving the study and design: C.M., Y.W., and G.W. Expert radiologist reads: L.J., Y.L., D.L., W.M., and K.Y. Collection and curation of the clinical datasets: D.S., C.G., Z.M., and H.Z. Surgery/biopsy performance: G. W. and F. G. Image processing and programming: C.M., Y.W., P.X., and Y.Z. Data analysis and interpretation: C.M., Y.W., and Y.Z. Drafting of the manuscript: C.M., Y.W., and G.W. Critical analysis and manuscript revision: all authors. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The Ethical Committees of Beijing Tsinghua Changgung Hospital (16149–0110) and The First Affiliated Hospital of Zhengzhou University (2021-KY-0156–002) approved this study, and the requirement to obtain informed consent was waived.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
MRI parameters per manufacturer. Three MRI scanners, namely, GE, Siemens and Philips, were used in this study. Each machine has different scanning parameters for sagittal and transverse T2-weighted images of the cervical, thoracic, and lumbar vertebrae. This table lists the important MRI scanning parameters before image preprocessing.
Additional file 2.
Architecture of the proposed models and optimized key hyperparameter settings.
Additional file 3.
Clinical characteristics of the patients and distribution of the ATRX and P53 mutation status across the primary and external validation cohorts. ATRX, alpha thalassemia/mental retardation syndrome X-linked; IQR, interquartile range; P53, tumor protein p53; WHO, World Health Organization.
Additional file 4.
Selected stable features in TRA and SAG images. To avoid interobserver variations during manual segmentation, we calculated the intraclass correlation coefficientfor each feature, and only those with high stabilitywere included in the analysis. The above table shows the proportion of stable features in each feature class extracted from TRA and SAG images. GLDM, gray-level dependence matrix; GLRLM, gray-level run-length matrix; GLSZM, gray-level size zone matrix; NGTDM, neighboring gray-tone difference matrix; SAG, sagittal; TRA, transverse.
Additional file 5.
Feature selection with LASSO in the WHO grading task.Feature selection in the SAG group. Twenty-four features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG group. Each colored line represents the coefficient of each feature.Feature selection in the TRA group. Six features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the TRA group.Feature selection in the SAG+TRA group. Twenty features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG+TRA group. LASSO, least absolute shrinkage and selection operator; SAG, sagittal; TRA, transverse; WHO, World Health Organization.
Additional file 6.
Feature selection with LASSO in the ATRX task.Feature selection in the SAG group. Three features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG group. Each colored line represents the coefficient of each feature.Feature selection in the TRA group. Thirty-nine features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the TRA group.Feature selection in the SAG+TRA group. Five features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG+TRA group. ATRX, alpha thalassemia/mental retardation syndrome X-linked, LASSO, least absolute shrinkage and selection operator; SAG, sagittal; TRA, transverse.
Additional file 7.
Feature selection with LASSO in the P53 task.Feature selection in the SAG group. Twenty-one features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG group. Each colored line represents the coefficient of each feature.Feature selection in the TRA group. Twenty-four features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the TRA group.Feature selection in the SAG+TRA group. Fifty-seven features with non-zero coefficients were selected using the minimum criteria.LASSO coefficient profiles of the features in the SAG+TRA group. LASSO, least absolute shrinkage and selection operator; P53, tumor protein p53; SAG, sagittal; TRA, transverse.
Additional file 8.
Selected radiomics features in each prediction task. In the WHO tumor grade prediction task, 24 features in SAG, 6 features in TRA, and 20 features in SAG+TRA were selected. In the ATRX prediction task, 3 features in SAG, 39 features in TRA, and 5 features in SAG+TRA were selected. In the P53 prediction task, 21 features in SAG, 24 features in TRA, and 57 features in SAG+TRA were selected. SAG, sagittal; TRA, transverse.
Additional file 9.
Detailed results of the 5-fold cross-validation for the proposed models using multimodal fusion features in the primary cohort. Acc: accuracy, Sens: sensitivity, Spec: specificity. The above results are expressed using the mean of 5 experiments and the corresponding 95% confidence interval.
Additional file 10.
Detailed test results in the external test cohort. Acc: accuracy, Sens: sensitivity, Spec: specificity.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ma, C., Wang, L., Song, D. et al. Multimodal-based machine learning strategy for accurate and non-invasive prediction of intramedullary glioma grade and mutation status of molecular markers: a retrospective study. BMC Med 21, 198 (2023). https://doi.org/10.1186/s12916-023-02898-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-023-02898-4