
Abstract
Background
Synechocystis sp. PCC 6803 is a photosynthetic bacterium that has been genetically modified to produce industrially relevant chemicals, yet efflux mechanisms have not been well elucidated. These photosynthetic organisms live in environments that are often nutrient limited; therefore, the genome of these organisms encodes far fewer proteins used for efflux of chemicals when compared to members of the Enterobacteriaceae family. Understanding efflux mechanisms can lead to a greater efficiency of chemical production within the cyanobacterial cell.
Results
Both sll0180 and slr2131 genes that encode the Sll0180 and Slr2131 proteins, respectively, were removed from Synechocystis sp. PCC 6803 and SD277, a high fatty acid-producing Synechocystis-based strain, to test the hypothesis that Sll0180 and Slr2131 contribute to the efflux of chemicals out of Synechocystis sp. PCC 6803 and SD277. The mutant Synechocystis sp. PCC 6803 and SD277 strains with either sll0180 or slr2131 removed from the chromosome had significantly decreased half maximal inhibitory concentrations to various antibiotics. The free fatty acid (FFA) concentration of the SD277 mutant strains increased intracellularly yet decreased extracellularly indicating that Sll0180 and Slr2131 have a role in FFA efflux. E. coli wild-type gene acrA (a homolog to sll0180) was added on a plasmid to the respective mutant strains lacking the sll0180 gene. Similarly, the E. coli wild-type gene acrB (a homolog to slr2131) was added to the respective mutant strains lacking the slr2131 gene. The tolerance to chloramphenicol of each mutant strain containing the wild-type E. coli gene was restored when compared to the parent stains. The extracellular FFA concentration of SD277 Δslr2131 with E. coli acrB increased significantly compared to both SD277 and SD277 Δslr2131.
Conclusions
Two proteins involved in the transportation of antibiotics and FFAs out of the Synechocystis sp. PCC 6803 cell were identified. In an effort to alleviate costs associated with mechanically or chemically separating the cells from the FFAs, the combination of genome editing of SD277 and the addition of exogenous transport gene increased extracellular concentrations of FFAs. This understanding of active transportation is critical to improving the production efficiency for all industrially relevant chemicals produced in Synechocystis sp. PCC 6803.
Similar content being viewed by others


Background
Synechocystis sp. PCC 6803 was isolated in 1968 from a freshwater lake [1] and was the first phototrophic organism whose whole genome was completely sequenced [2]. Prior to sequencing, Synechocystis sp. PCC 6803 was shown to be naturally transformable [3] and later, able uptake plasmid DNA through conjugation [4]. The combination of these findings led to the use of Synechocystis sp. PCC 6803 as a model organism for investigating a variety of fields of study including the process of photosynthesis and the use of cyanobacterial cells to produce biofuels. However, there remain gaps of knowledge as to the function of many proteins in the Synechocystis sp. PCC 6803 cell.
Genetically modified Synechocystis sp. PCC 6803 strains produce useful chemicals such as ethanol, acetone, free fatty acids (FFAs), and 3-hydroxybutyrate [5,6,7,8]. However, there is limited understanding as to how the chemicals are transported out of the cell. Much of the chemical transportation research throughout the last 15 years focused on the process by which chemicals are transported into Synechocystis sp. PCC 6803 cells. Some examples include the Fut series of proteins that are responsible for uptake of iron from the environment [9,10,11] and the Pst proteins that are responsible for phosphate uptake [12, 13]. While uptake of nutrients is paramount to understanding ecological survival of cyanobacteria with respect to algal blooms that create anoxic or toxin-filled waterways, understanding the native chemical efflux mechanisms is vital for using the cyanobacteria to produce industrially relevant chemicals. Once the industrially relevant chemicals are synthesized inside the cyanobacterial cell, the chemicals need to be expelled for two reasons: first, altered intracellular chemical concentrations can cause chemical or protein synthesis to cease or inhibit cell growth and second, the cells do not have to be lysed if the chemicals are in the supernatant, saving time and money. By promoting the secretion of the industrially relevant chemicals, the cyanobacteria can not only survive, they can also grow continuously if the chemical can be purified from the extracellular environment. In an effort to understand a mechanism of efflux of Synechocystis sp. PCC 6803, the function of a Escherichia coli TolC homolog, Slr1270, has been identified [14, 15].
In Escherichia coli, the TolC outer membrane duct protein was originally identified to provide tolerance to colicin [16]. TolC was later shown to require other proteins to transport the intracellular substrates to the extracellular environment, providing tolerance to numerous antibiotics, dyes, and detergents [17]. The proteins necessary for the efflux of substrates using the E. coli TolC outer membrane duct are characterized into one of two groups: multidrug efflux pumps or membrane fusion proteins (MFPs). One example of a multidrug efflux pump is AcrB which works together with AcrA, an MFP, to export chemicals from within the E. coli cell out through the TolC duct [18, 19]. AcrB is responsible for transporting intracellular chemicals through the inner membrane, to the periplasmic opening of the TolC duct, while two AcrA proteins stabilize the opening of the TolC duct at the periplasmic end and bind to the homotrimer of AcrB [20,21,22]. The TolC duct allows for the substrates from the multidrug efflux pump to reach the extracellular environment passively [23] (Fig. 1). There are numerous other closely related multidrug efflux pumps and MFPs that are synthesized by E. coli, including EmrAB that has been shown to have overlapping function alongside AcrAB with respect to the efflux of FFAs; both AcrAB and EmrAB must be removed from E. coli to significantly decrease the efflux of FFAs [24]. All efflux pump systems possess the ability to actively extrude substrates from the cytoplasm through the lipid bilayer using the antiporter mechanism in which a proton is exchanged for the substrate in the multidrug efflux pump. Without any one of these three proteins, the tolerance to toxic chemicals that are substrates of the multidrug efflux system significantly decreases and resistance concomitantly increases in E. coli cells.

Schematic of the AcrAB-TolC multidrug efflux system as established in E. coli. The blue cylinder represents the TolC exit duct. The green trapezoid with two tails represents two AcrA adaptor proteins. The three red rectangles sans corners represent the AcrB homotrimer transporter protein
The TolC homolog in Synechocystis sp. PCC 6803, Slr1270, possesses a similar function to that of TolC in E. coli [14, 15]. Without Slr1270, the cyanobacteria possess a decreased tolerance to various antibiotics. In addition, Synechocystis sp. PCC 6803 that lacks a functional Slr1270 has a reduced ability to form biofilms and secrete outer membrane vesicles; the mutant strain also has a missing S-layer [15]. Slr1270 is thus predicted to be one component of the multidrug efflux mechanism for efflux of chemicals from Synechocystis sp. PCC 6803. There exist numerous putative proteins in Synechocystis sp. PCC 6803 that have been described as MFPs and multidrug efflux pumps [25,26,27,28]. However, there is a limited understanding of the multidrug efflux pump(s) and MFP(s) of Synechocystis sp. PCC 6803 that function together with Slr1270 to transport chemicals out of the cell. Evidence suggests that one multidrug efflux pump is Slr2131, while Sll0180 is one of the described MFPs [25, 28]. AcrA is created from 397 amino acids while Sll0180 is composed of 501 amino acids. When comparing the homology of the two proteins, the first 131 amino acids of Sll0180 share an 18% identity to AcrA and, similarly, nearly the last 262 amino acids of Sll0180 share a 19% identity to AcrA (Sll0180 amino acids 500 and 501 were not included in this identity analysis) [29]. AcrB share far more protein sequence homology to Slr2131 relative to the comparison between AcrA and Sll0180. Slr2131 has a protein sequence length of 1061 amino acids, while AcrB has 1049 amino acids. Amino acid 2 through 1017 of Slr2131 watched matched with the a 1016 amino acid length central region of AcrB for a 41% identity with no gaps in the alignment [29], indicating that the vast majority of the two proteins sequences share protein binding sites and transmembrane domains that a paramount to the functioning of each pump. Slr2131 contributes to the secretion of chloramphenicol (Cm) and ethidium bromide (EtBr) as evidenced by a decrease in tolerance to Cm and EtBr in Synechocystis sp. PCC 6803 with a portion of the slr2131 gene deleted [30]. A sll0180 mutant Synechocystis sp. PCC 6803 strain was previously shown to have a greater susceptibility to sodium dodecyl sulfate (SDS) and lacks the S-layer [30]. The research featured in this article provides further insight into the hypothesis that Slr2131 and Sll0180 are members of the multidrug efflux pump family and MFP family, respectively, and may function together with Slr1270 to transport toxic chemicals out of the cell. However, the roles of Slr2131 and Sll0180 may not be limited to the efflux of antibiotics and dyes; they may also have a role in efflux of FFAs. The homologous AcrA and AcrB proteins in conjunction with TolC in E. coli are responsible for efflux of FFAs in E. coli [24]. If the native mechanisms of efflux in Synechocystis sp. PCC 6803 are better understood this can lead to genetically modified cyanobacterial cells with a more efficient process for FFA production and the ensuing purification from the extracellular media.
The research featured in this article looks to further the understanding of both Slr2131 and Sll0180 in respect to their ability to promote the tolerance of Synechocystis sp. PCC 6803 to various antibiotics. SD100 is a single colony isolate of wild-type Synechocystis sp. PCC 6803 [31] and is the parental strain for all of the strains featured in this research. To begin the research, each of the SD100 genes, slr2131 and sll0180, that encode the Slr2131 and Sll0180 proteins, respectively, were removed with replacement deletions from both SD100 and SD277, a strain derived from SD100 which contains numerous genetic modifications for the promotion of FFA production [8]. Previous researchers have hypothesized that SD277 uses the flip-flop method of diffusion of FFAs across the membrane [8], in which FFAs quickly diffuse across the lipid bilayer [32]. But we contend that there exists an active mechanism for FFA transportation to the extracellular environment, so the intracellular and extracellular FFA concentrations of SD100, SD277, and the deletion mutant strains were measured to identify that Slr2131 and Sll1080 are responsible for the secretion of FFAs. Then the mutations found in the deletion mutant strains derived from SD100 and SD277 were complemented with the homologous wild-type genes from E. coli, acrB or acrA, to counteract the deleterious effects of the native gene removals. The addition of the genes encoding the E. coli AcrB and AcrA proteins affects the secretion of FFAs and the tolerance to antibiotics in the SD100 and SD277 mutant strains. By further the understanding of active efflux in Synechocystis sp. PCC 6803, the cost and time associated with using the cyanobacteria for the manufacturing of chemicals can be reduced.
Results
Sixteen strains based on either SD100 or SD277, the high FFA synthesizing strain, were created to investigate the roles of putative transporters in SD100 and SD277 and the ability to express homologous E. coli genes encoding the AcrA and AcrB proteins in both SD100 and SD277. A summary of all of the strains and the plasmids used to obtain the following results are provided (Table 1) along with the primers designed for the purposes of this research (Table 2).
The removal of slr2131 or sll0180 generally decreases tolerance to antibiotics
After deleting either slr2131 or sll0180 from both SD100 and SD277, the maximal half inhibitory concentration (IC50) was determined when each strain was exposed to the antibiotics ampicillin (Amp), chloramphenicol (Cm), or Erythromycin (Ery) at concentrations between 10 ng/mL and 100 mg/mL (Table 3). SD100 Δslr2131 possessed significant decreases in tolerance to Cm and Ery at 74 and 85%, respectively, when compared to the wild-type SD100. However, when SD100 Δslr2131 was treated with Amp, there was no significant difference when compared to wild-type SD100. SD277 Δslr2131 had significant decreases in tolerance to Amp, Cm, and Ery of 36, 61, and 60%, respectively when compared to SD277. SD100 Δsll0180 possessed significant decreases in tolerance when treated with Amp, Cm, and Ery of 37, 37, and 52%, respectively, when compared to SD100. SD277 Δsll0180 possessed significant decreases in tolerance to Amp, Cm, and Ery of 71, 77, and 92%, respectively, when compared to SD277.
The addition of the E. coli acrB gene to the cyanobacterial mutants lacking slr2131 and the addition of the E. coli acrA gene to mutants lacking sll0180 restored tolerance to chloramphenicol
SD100 Δslr2131 possessed a 74% decrease in tolerance to Cm when compared to SD100, but when the E. coli acrB gene was introduced on an expression vector (pacrB) into the deletion mutant, the resulting strain, SD100 Δslr2131 pacrB, no difference of tolerance to Cm when compared to SD100 indicating that the original tolerance was restored (Table 3). SD277 Δslr2131 pacrB and SD277 Δsll0180 pSacrA each had a significant increase in tolerance to Cm compared to each’s respective mutant parent strain, while each have an equivalent IC50 to Cm when compared SD277. SD100 Δsll0180 pSacrA had an increased tolerance to Cm with a significant 106% increase when compared to SD100; SD100 Δsll0180 pSacrA also possessed a significant 228% increase in tolerance to Cm when compared to SD100 Δsll0180. The addition of E. coli acrB to SD100 Δslr2131 or SD277 Δslr2131 did not restore the tolerance to Amp and Ery to equivalent concentrations observed in each mutant strain’s respective parent strain, SD100 and SD277. The addition of E. coli acrA to SD100 Δsll0180 or SD277 Δsll0180 did not restore the tolerance to Amp or Ery to equivalent concentrations observed in each mutant strain’s respective parent strain, SD100 and SD277. Outside of the restoration of tolerance to Cm, there does not exist any evidence that the addition of E. coli acrA or acrB compensate for the loss of tolerance due to the deletion of native sll0180 or slr2131 from SD100 and SD277.
The removal of either slr2131 or sll0180 from SD277 and SD100 modifies the extracellular and intracellular FFA concentrations
The total concentrations of extracellular FFA of SD277 Δslr2131 and SD277 Δsll0180 were significantly less than SD277 with differences of 19 and 65% respectively (Fig. 2a). These were proportional changes in concentrations from each of the 4 measured FFAs. The intracellular FFA concentrations of SD277 Δslr2131 and SD277 Δsll0180 both had increases in total concentration when compared to SD277, with differences of 62 and 17% respectively (Fig. 2b); only the 62% increase was significant. Both SD100 Δslr2131 and SD100 Δsl10180 possessed significant increases in extracellular FFA concentrations when compared to SD100, but while statistically significant, the actual values of the decreases were between 3 and 4 mg/L of total FFA concentrations (Fig. 2c). These values represent one log level less than the amount difference values of the SD277 mutant strains compared to SD277. There were no significant differences when comparing the intracellular FFA concentrations of SD100 to SD100 Δslr2131 and SD100 Δsll0180 (Fig. 2d).

FFA concentration of SD277, mutant, and complementation derivatives (a) extracellularly and (b) intracellularly. FFA concentration of SD100 and its mutant and complementation derivatives (c) extracellularly and (d) intracellularly in which * is p < 0.05, ** is p < 0.005, *** is p < 0.0005, and **** is p < 0.0001. Each color represents an average concentration of a saturated fatty acid: green represents lauric acid, orange represents myristic acid, blue represents palmitic acid, and purple represents stearic acid
The addition of the E. coli acrB to the mutant SD100 and SD277 strains lacking slr2131 and the addition of the E. coli acrA gene to mutant strains lacking sll0180 modified the extracellular and intracellular FFA concentrations
SD277 Δslr2131 pacrB, possessed a significant increase in the concentration of extracellular FFAs when compared to SD277 and SD277 Δslr2131 with an increase of 26 and 55% respectively, while the intracellular FFA concentration of SD277 Δslr2131 pacrB was no different to that of SD277 Δslr2131 (Fig. 2a-b). When E. coli acrA was added to SD277 Δsll0180, hereafter referred to as SD277 Δsll0180 pSacrA, the extracellular FFA concentration was the same as SD277 Δsll0180, maintaining a significantly lower concentration of FFAs when compared to SD277, while the intracellular concentration of SD277 Δsll0180 pSacrA was significantly lower than SD277 Δsll0180 (Fig. 2a-b). The addition of the E. coli acrB gene to SD100 Δslr2131 (SD100 Δslr2131 pacrB) did not change in the extracellular or intracellular FFA concentrations of the strain when compared to SD100 Δslr2131 (Fig. 2c-d). The addition of E. coli acrA to SD100 Δsll0180, now denoted as SD100 Δsll0180 pSacrA, significantly lowered the concentration of the extracellular FFAs when compared to SD100 Δsll0180, but still maintained a significantly higher concentration of FFA when compared to SD100 (Fig. 2c). Again, the changes in concentrations of the SD100 derivative strains were under the 4 mg/L, a log level lower than the changes observed in the corresponding SD277 and SD277 derivative strains. There were no significant differences among the concentrations of intracellular FFAs when comparing SD100 Δsll0180 pSacrA and SD100 Δslr2131 pacrB to SD100 or the SD100 derivative mutant strains (Fig. 2d).
Identifying the roles of E. coli genes and expression vectors have on FFA efflux and IC50
To identify what role the E. coli acrA and acrB genes have in SD277 and SD100, pacrA and pacrB were added to SD100 and SD277 individually. In addition, the expression vectors lacking these two E. coli genes, pKan and pSpec, were each added individually to identify if the plasmid has a role in chemical efflux. When either pacrA or pacrB was added to SD277, the resulting strains, SD277 pacrA and SD277 pacrB, had a significant decrease in the FFA concentrations extracellularly when compared to SD277 (Fig. 3a). This was the same case with respect to the strains resulting from the addition of pKan and pSpec to SD277, SD277 pKan and SD277 pSpec (Fig. 3a). With respect to intracellular FFA concentrations, when pacrA, pacrB, or pKan were added individually to SD277 none of the resulting strains had a significant difference when compared to SD277 (Fig. 3b). However, when pSpec was added to SD277, the SD277 pSpec strain had a significant decrease of FFA concentration intracellularly (Fig. 3b). SD100 pacrA and SD100 pSpec each had a significantly higher concentration of FFA extracellularly while SD100 pacrB and SD100 pKan did not vary significantly when compared to SD100 (Fig. 3c). When the concentrations of intracellular FFAs were measured, none of the SD100 based strains with the added plasmids were significantly different when compared to SD100 (Fig. 3d).

FFA concentration of SD277 and its derivatives (a) extracellularly and (b) intracellularly. FFA concentration of SD100 and its derivatives (c) extracellularly and (d) intracellularly in which * is p < 0.05, ** is p < 0.005, *** is p < 0.0005, and **** is p < 0.0001. Each color represents an average concentration of a saturated fatty acid: green represents lauric acid, orange represents myristic acid, blue represents palmitic acid, and purple represents stearic acid
With respect to the IC50 of the resulting strain when any of the four plasmids (pacrA, pacrB, pKan, pSpec) were added to SD100 or SD277, the plasmids largely either had an effect to decrease or provided no change to the IC50 relative to the parent strain (Table 4). The one exception was SD277 pKan when treated with Ery; this strain had a slight, yet significant increase in IC50 when compared to SD277.
mRNA and growth rates
To ensure that the E. coli genes were expressing the DNA that was present in the constitutively expressing promoters located on the plasmids, the mRNA was purified, and cDNA was created using reverse-transcriptase PCR (RT-PCR). In each case in which the E. coli gene was added to a cyanobacterial strain, a corresponding cDNA sequence was identified using gel electrophoresis, with none of the E. coli genes identified in parent, mutant, or vector control strains (Additional file 1: Figure S1A). Each cyanobacterial strain was also tested for the presence of petB mRNA, a constitutively expressed cyanobacterial gene. mRNA of petB was identified in each cyanobacterial sample (Additional file 1: Figure S1B). A conventional PCR was performed using the identical primers used for the RT-PCR to verify that there was no genomic cyanobacterial DNA contamination detected in the mRNA samples after the DNase treatment (Data not shown) (Table 2).
To understand the growth pattern of the cyanobacteria, each strain was grown for 168 h in triplicate beginning at an OD730 of 0.05. There were no differences in any of the growth phases among any strain presented in this research when compared to its respective parent strain, either SD100 or SD277 (Additional file 1: Figure S2A-D).
Discussion
Considering the numerous advancements using genetically modified bacterial strains based on Synechocystis sp. PCC 6803 for production of biofuels and other industrially relevant chemicals, there is a limited understanding regarding the transportation of these chemicals out of the cyanobacterial cells. This current research looked to corroborate the hypothesis that Synechocystis sp. PCC 6803 genes slr2131 and sll0180 encode homologous proteins to that of E. coli AcrB and AcrA, respectively. To do this, this research presents three hypotheses. The first is that the removal of slr2131 or sll0180 from both SD100 and SD277 will inhibit the cell’s ability to tolerate antibiotics and decrease the concentrations of extracellular FFAs while increasing intracellular FFA concentrations. Second, adding the homologous E. coli acrA and acrB genes to the SD100 and SD277 sll0180 and slr2131 mutant strains will restore the lost tolerance to Amp, Cm, and Ery; additionally, extracellular FFA concentrations will be increased. And, the final hypothesis addressed was that adding the vectors containing the E. coli acrA or acrB genes to SD100 and SD277, will increase the tolerance to Amp, Cm, and Ery, and increase the extracellular FFA concentrations.
This research showed that removing sll0180 and slr2131 from SD100 and SD277 significantly decreased tolerance to Amp, Cm, and Ery. Both SD100 and SD277 rely on the encoded proteins, Sll0180 and Slr2131, for efflux of these chemicals and without either Sll0180 or Slr2131, the tolerance decreases. Previous research performed by Gonçalves et al. (2018) supports the hypothesis that Sll0180 is necessary for native efflux in that the removal of sll0180 from Synechocystis sp. PCC 6803 caused a significant inhibition of growth due to the presence of Cm [30]. However, Gonçalves et al. (2018) did not observe a significant inhibition of growth associated with the removal of slr2131 from Synechocystis sp. PCC 6803 due to the presence of Cm, as shown in the currently presented research. With respect to the sll0180 and slr2131 mutants in Synechocystis sp. PCC 6803, Gonçalves et al. (2018) did also associate a significant growth inhibition when treated with 0.0025% SDS [30], providing further support that both of the encoded proteins confer part of an efflux mechanism. In addition, Synechocystis sp. PCC 6803 does not appear to have another efflux mechanism capable of substituting for the efflux roles that Sll0180 or Slr2131 proteins confer to the cyanobacteria. The E. coli AcrB multidrug efflux pump has other homologous pumps encoded by the E. coli genome, AcrD and AcrF, that have some identical substrates to AcrB [33]. However, Synechocystis sp. PCC 6803 does not appear to have this redundancy as indicated by the significant decrease in tolerance to Amp, Cm, and Ery, when slr2131 was removed from the Synechocystis sp. PCC 6803 chromosomes. If another protein existed that possessed a similar to either Sll0180 or Slr2131 then there would likely not be a significant decrease in the tolerance to the various chemicals of all of the mutant strains; additionally, the extracellular FFA concentration of the two SD277 mutant strains would also not be decreased significantly. The idea of redundant proteins used in transportation of chemicals is explored subsequently with respect to the E. coli EmrAB and AcrAB multidrug efflux pumps.
When either of the native slr2131 or sll0180 genes were removed from the high FFA producing SD277 strain, the results validate the hypothesis that both of the conferred proteins, Slr2131 and Sll0180, contribute to the secretion of FFAs from within the SD277 cell to the extracellular environment. When either gene was deleted, the extracellular FFA concentrations decreased significantly, while the FFA concentrations within the cell increased. This parallels the results observed when acrAB was removed concurrently with emrAB, a distinct multidrug efflux system, from E. coli. Lennen et al. (2013) caused a significant drop in the extracellular FFA concentration in E. coli that was genetically engineered to produce FFAs at concentrations 450% higher than wild-type E. coli [24]. Removing the acrAB operon alone from the high FFA producing E. coli strain did not create a strain that had a significantly lower extracellular FFA concentration than the high FFA producing E. coli strain with native acrAB present. FFAs are substrates of both E. coli emrAB and acrAB [34]. Conversely in the cyanobacterial strain SD227, because the removal of just a single gene, e.g. slr2131, from SD277 caused a significant decrease extracellular FFA concentration, there is no apparent homologous protein to Slr2131 in SD277 that can transport FFAs out of the cell in a similar manner to the complementary FFA efflux function conferred by the E. coli EmrB multidrug efflux pump when the high FFA producing E. coli strain does not have AcrB. Synechocystis sp. PCC 6803 has approximately one-third of the total transporters that E. coli does, approximately 100 compared to 300 total transporters may contribute to the lack of pumps with overlapping function in the cyanobacteria [35]. 5% of the E. coli genome encodes approximately 79 ATP-Binding Cassette (ABC) transporters [36]. This is compared to the 54 putative and identified ABC transporters of Synechocystis species [37]. This large discrepancy in the number of transporters is likely to do the dramatically different environments that E. coli and Synechocystis sp. PCC 6803 evolved throughout the millennia. But genetically modifying SD100 and SD277 can alter the results of millennia of evolution to assist in the production of precursors to biofuels.
When E. coli acrA was added to SD100 and SD277 strains without sll0180 or E. coli acrB was added to SD100 and SD277 strains without slr2131, the only consistent restoration of tolerance when compared to the parent strains was to Cm. Tolerance to Ery and Amp was inconsistent across the four complementation strains with examples that had decreased, increased, and statistically identical IC50 relative to the corresponding mutant strain. But none of the complementation strains had IC50 statistically the same or greater than the parent strain when treated with increasing concentration of Ery and Amp. Cm, Amp, and Ery are all established substrates of AcrB [18, 38, 39], with the E. coli tolerance to each chemical usually decreasing especially if other multidrug efflux systems are removed concurrently. So, it’s not clear why Cm is readily exported in Synechocystis-derivative strains while the other 2 antibiotics are not when the E. coli genes are expressed. However, Cm is a polar compound while Amp and Ery are not. Cm contains two N-O polar bonds in a bent conformation. There are 11 polar residues in E. coli AcrB that contribute to binding of substrates [40], which may allow for Cm to be bound preferentially to the nonpolar Ery and Amp.
The export of chemicals was not limited to antibiotics; when acrB was expressed in SD277 Δslr2131 mutants, FFA concentration was significantly increased extracellularly compared to both SD277 and SD277 Δslr2131. SD277 Δslr2131 pacrB had a greater intracellular FFA concentration than SD277 but statistically equivalent to SD277 Δslr2131. We expected the intracellular FFA concentration of SD277 Δslr2131 pacrB to have a statistically significant decrease relative to SD277 Δslr2131 as the addition of pacrB led to an increase in extracellular FFAs, but we instead observed a decreasing trend in intracellular FFA concentration. This is one example of unexpected results between extracellular and intracellular FFA concentrations. This may be attributed to CO2 limitations in the production of FFAs. If we had increased the CO2 concentration in the aeration of the samples, these trends may have had significant differences as the total FFA production would have increased [41]. However, in this research we determined the process for moving FFAs out of the cell is not a result of diffusion; the extracellular and intracellular concentrations may not be directly proportional. Furthermore, AcrB is able to transport FFAs out of the cell in Synechocystis-based strains. AcrB likely does not act alone and may use Slr1270 as a partner to transport the FFA out of the cell. Slr1270 has previously been shown to have similar physical characteristics to TolC when slr1270 is placed in E. coli [14]. Furthermore, when E. coli acrB is added to SD277, there is no increased extracellular FFA concentration in the resulting SD277 pacrB strain when compared to SD277. The native SD277 Slr2131 protein may be competitive with E. coli AcrB protein synthesized in SD277 for outer membrane ducts, such as Slr1270. Once Slr2131 is removed, constitutively synthesized E. coli AcrB continuously transports FFAs out of the SD277 Δslr2131 pacrB cells; although, it is not clear if AcrB needs an MFP to facilitate the transportation of FFA out of the SD277 Δslr2131 pacrB strain. The FFAs located intracellularly of SD277 Δslr2131 pacrB remain significantly higher than SD277 and SD277 Δslr2131, so the addition of pacrB to SD277 Δslr2131 does not alleviate the increased intracellular FFA concentration observed in SD277 Δslr2131. But the SD277 Δslr2131 pacrB cells tolerate the elevated concentrations as their growth rates do not vary significantly from SD277 (Additional file 1: Figure S2A). There are a few genetic manipulations to SD100 to modify the cell membrane structure including the removal of sll1951 and slr1710 that led to the creation SD277. Sll1951, the protein encoded by sll1951, is necessary for the native structure of the S-layer of the cyanobacteria to be present while slr1710 encodes a penicillin binding protein necessary for the peptidoglycan layer assembly [42]; these two modification to the cell membrane were an effort to allow FFAs to flow out of the cell with less encumbrance. The addition of E. coli AcrB to SD277 Δslr2131 increases the extracellular FFA concentration relative to SD277, indicating that the strategy of active transportation of FFA out of the cyanobacterial cell is a more efficient strategy than eliminating cell membrane proteins to move FFA out of the cell.
The addition of acrA to SD277 Δsll1080 did not provide any increase in FFA concentration extracellularly. E. coli AcrA may just not effectively recognize binding sites on the Slr1270 outer membrane duct or there may exist differences in the membrane structure of these cyanobacteria that may not be ideal or even necessitate AcrA. When aligning the 397 amino acids of AcrA and the 501 amino acids of Sll0180, there exists a gap from amino acids 132 to 237 of Sll0180, bearing virtually no homology to AcrA. When identifying possible protein binding regions in these 105 nonhomologous amino acids of Sll0180, there are two are predicted at sites 132 and 183. AcrA does not possess this central region and therefore these two predicted protein binding sites that are present in Sll0180. The lack of this central region may affect the ability of AcrA to bind to a protein within the membranes of Synechocystis sp. PCC 6803. In addition, previous research has supported the hypothesis that the periplasmic end of the Slr1270 outer membrane duct in Synechocystis sp. PCC 6803 remains open without the MFP, which is not the case for E. coli TolC lacking an MFP. Without E. coli AcrA or a different MFP, the periplasmic end of E. coli TolC collapses, while Slr1270 maintains its open at both ends of the duct without an MFP [14]. Based on the results of the sll0180 mutant strains, the Sll0180 MFP is necessary for a native efflux mechanism, but the resulting loss of function cannot be compensated through the addition of E. coli AcrA in SD100 or SD277 sll0180 mutant strains. The compounding differences between Sll0180 and AcrA including the amino acid sequence lengths (501 compared to 397), nonhomologous protein binding sites, and the different roles each may have in relation to the outer membrane duct may all lead to the inability for E. coli AcrA to restore the loss of function demonstrated in sll0180 mutant SD100 and SD277 strains with respect to FFA secretion and tolerance to Amp, Cm, and Ery.
Finally, the four vectors, pKan, pSpec, pacrA, and pacrB were transferred into the parent strains, SD100 or SD277, to understand the role that the addition of these plasmids had on tolerance to chemicals and FFA concentration, intracellularly and extracellularly. This resulted in SD100 derivative strains with universally decreased or unchanged tolerance to the 3 chemicals tested. However, the SD277 derivative strains featured strains that that were observed to have increased, decreased, and statistically unchanged tolerances to the 3 chemicals tested when compared to SD277, making any broad conclusions about the effect from the plasmids on the tolerance of SD277 to the various chemicals difficult to corroborate with the results presented in this research. Additionally, SD277 pSpec was observed to have a significantly lower intracellular and extracellular FFA concentration than SD277, while SD277 pKan had a lower extracellular FFA concentration but statistically identical intracellular FFA concentration to SD277. The observed decrease in the intracellular FFA concentration of SD277 pSpec, which may be the cause of the observed significant decrease in the extracellular FFA concentration provided evidence contrary to the hypothesis that the addition of the control expression vector would not change the extracellular or intracellular FFA concentrations of SD277. In numerous bacteria including Bacillus subtilis and Vibrio cholerae, the inducible SOS DNA repair system is modulated, in part, by LexA that functions as a repressor. Once DNA damage occurs, ssDNA is formed which binds to RecA. This binding cleaves the LexA dimer that functions as a repressor to the SOS system, initiating DNA repair [43]. Additionally, LexA in V. cholerae has been shown to induce the SOS DNA repair response during bacterial conjugation [44]. Interestingly, Synechocystis sp. PCC 6803 possesses a lexA homolog, sll1626, and the encoded Sll1626 protein has a role in down-regulating fab (fatty acid biosynthesis) genes in Synechocystis sp. PCC 6803. The same down-regulation was observed in a modified strain of Synechocystis sp. PCC 6803 created to increase FFA synthesis [45]. The LexA homolog found in Synechocystis sp. PCC 6803, Sll1626, unlike that in E. coli and V. cholerae, is not linked to any genes involved in DNA metabolism in SD100 [46,47,48]. Sll1626 may not be involved in SOS DNA repair in Synechocystis sp. PCC 6803; however, Sll1626 still may be induced by bacterial conjugation and may be decreasing fatty acid biosynthesis in SD277 when pacrA, pacrB, pKan, or pSpec were added to a cyanobacterial strain. This downregulation of fab genes may be causing this significant decrease in extracellular and intracellular FFA concentrations observed in SD277 pSpec. A future study removing sll1626 from the strains created for this research may help illustrate a picture of Sll1626 involvement in fatty acid biosynthesis and may help explain the decrease in FFA concentrations in SD277 strains containing exogenous plasmids.
While the addition of pacrB to SD277 decreased the extracellular FFA concentration of SD277, the result appears to be due to the addition of the plasmid to the SD277 strain. But this research has established the efficacy of the addition pacrB to the SD277 Δslr2131 strain, significantly increasing the extracellular FFA relative to both SD277 and SD277 Δslr2131. This provides corroborating data to the hypothesis that the addition of the E. coli acrB gene to SD277 Δslr2131 increases the extracellular FFA concentration. This conclusion leads to the hypothesis that adding all three of the E. coli genes encoding the E. coli AcrAB-TolC multidrug efflux system to SD277 may further increase FFA concentration extracellularly. But removing the homologous Synechocystis sp. PCC 6803 genes, sll0180, slr2131, and slr1270, first from SD277 may assist in the function of the E. coli multidrug efflux pump system, considering that SD277 Δslr2131 pacrB had the highest concentration of FFA in this research. Additionally, without first understanding if bacterial conjugation modifies the expression of sll1626, the lexA homolog, which effects the resulting fab genes, E. coli gene additions should be placed in the genome. The negative phenotypes observed in this research due to the addition of the vectors may be avoided by placing the E. coli genes in the cyanobacterial chromosome.
Conclusions
In conclusion, the data presented illustrates the role of the Synechocystis sp. PCC 6803 proteins encoded by slr2131 and sll0180 in chemical efflux. Without each one of these genes present, the cyanobacteria have decreased tolerance to antibiotics. Additionally, extracellular FFA concentrations are decreased, while intracellular FFA are increased in SD277 mutant strains. E. coli homologs of slr2131 and sll0180, acrA and acrB, were expressed in SD100 and SD277 and created distinct phenotypes including the extracellular FFA concentration increase of the SD277 Δslr2131 pacrB strain, producing the highest concentration of extracellular FFA observed in this research. This research continues the understanding of chemical transportation through the cell membrane of Synechocystis sp. PCC 6803 and introduces the ability to use E. coli transport genes to alter the native efflux mechanism.
Methods
Bacterial strains, plasmids, growth conditions, and reagents
Bacterial strains and plasmids used in this study are listed in Table 1. For routine use, Synechocystis sp. PCC 6803 was inoculated on a BG-11 agar plate [49] and grown in illumination (50 μmol photons m− 2 s− 1) at 30 °C in an environmental chamber (SANYO, Osaka, Japan). Strains with the sacB-KanR cassette or pKan or plasmids derived from pKan in the cyanobacterial cell were grown with the addition of 50 μg/mL Kan. Strains with pSpec or plasmids derived from it were grown with the addition of 30 μg/mL streptomycin (Str) and spectinomcin (Spc). For FFA analysis, mRNA sequence observation, growth rate determination, and measurement of IC50, Cyanobacterial strains were also grown in BG-11 liquid media supplied with 10 mM N-[tris(hydroxymethyl) methyl]-2-aminoethanesulfonic acid (TES) NaOH (pH 8.2) in a 15 mL glass test tube 30 °C in an environmental chamber. All chemicals were purchased from Sigma Aldrich (St. Louis, MO), BD (Franklin Lakes, NJ), or Thermo Fisher Scientific (Waltham, MA). The tubes containing cyanobacteria and BG-11 liquid media was incubated with illumination and intermittent shaking for 2–4 d. Once these starter cultures reached an OD730 nm of 0.6, the samples were transferred to a 250-mL Erlenmeyer flask with aeration at 100 mL∕ min. This protocol uses TES buffer and air aeration to keep the pH around 8 at the beginning inoculation stages to minimize the lag phase. Once the culture reaches mid-log level phase, the culture was used to test the IC50. If instead the culture was used for FFA analysis, 20 mL of BG-11 liquid media without TES was added, as TES negatively affects FFA concentration [8]. This now 30 mL culture was growth to a concentration of 0.8-1 × 109 cells/mL and then subjected to FFA analysis. However, if the culture was used for mRNA analysis, 40 mL of BG-11 was added to the initial 10 mL culture to create a 50 mL culture. Once this reaches the mid-log level phase, 40 mL was removed and analyzed for the presence of specific mRNA sequences. The specific protocols for the measurements of IC50, FFA concentrations, and the presence mRNA sequences are explained in detail in subsequent sections.
To determine the growth rate of the strains, an initial OD730 of 0.05 was created in a 30 mL 250 mL Erlenmeyer flask. It was placed in the illuminated chamber and aerated at 100 mL/min, but not rotated. The OD730 was measured by spectrophotometer every 24 h for 168 h. This was performed in triplicate.
Transformation of suicide vectors containing the sacB-KanR cassette
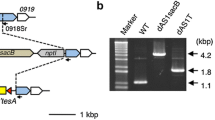
To delete the entire gene of interest, initially a suicide vector was designed so that one 700-bp sequence located immediately upstream and one 700-bp sequence located immediately downstream of sll1080 (flanking regions) were amplified from Synechocystis sp. PCC 6803 using a colony PCR and ligated to either end of the sacB-KanR cassette. This now upstream flanking region/sacB-KanR cassette/downstream flanking region nucleotide sequence was digested using restriction enzymes ligated into a restriction enzyme digested pGEM-3Z. The resulting plasmid (pΨ695) was electroporated into E. coli Top10 cells and a colony of Top10 cells containing pΨ695 was grown in liquid lysogeny broth (LB) with Miller’s modification [50] at 37 °C overnight with aeration. pΨ695 was then purified from the exponentially grown Top10 cells containing pΨ695 using the Qiagen Plasmid Miniprep Kit (Hilden, Germany) About 100 cyanobacteria cells in 10 μL BG-11 medium were mixed with 400 ng of purified pΨ695 (1–2 μL of DNA dissolved in Elution Buffer (Qiagen)) and incubated for 4 h at room temperature under ambient lighting. Then the mixtures were plated onto a filter membrane (Whatman PC MB 90MM 4 μm (Maidstone, UK)) layered on a BG-11 agar plate. After growth for 24 h on the BG-11 plate at room temperature under ambient lighting, the membrane carrying the cyanobacteria was transferred onto a BG-11 agar plate containing 50 μg∕mL Kan. Once individual colonies appear, they were picked and placed with a sterile loop onto a new BG-11 with 50 μg∕mL Kan agar plate. The cyanobacteria were moved to a BG-11 with Kan plate in order to select for colonies that contain cells that have the sacB-KanR cassette in place of the native sll0180 gene in the chromosome. This process is defined as homologous recombination in which the suicide vector has homologous regions to the native sequence of the chromosome, allowing the sacB-KanR cassette to be exchanged for sll0180. However, in order for sll0180 to be completely removed from Synechocystis sp. PCC 6803 and SD277, the gene must be removed from all copies of the chromosome; this is defined as complete segregation. In order to identify that the sacB-KanR cassette is present in place of the gene of interest in every chromosome copy, a colony PCR was performed using primers that amplify the flanking region both upstream and downstream of sll0180, A end sll1080 end F and B end sll0180 end R (Table 2). Regions with the native sll0180 are 1 kb shorter than regions with the sacB-KanR cassette when observed on 0.8% agarose DNA gel electrophoresis. If the native gene persisted, then the colony was picked using a sterile loop and plated on a new BG-11 plate containing 50 μg∕mL Kan. Again, after growth for 2–3 days in an illuminated chamber at 30 °C a colony of cyanobacteria was suspended in 3 μl of dH2O and a PCR was performed using the primers for the two flanking regions. These steps were repeated until the native gene sequence was not observed, leaving the cyanobacteria with only the sacB-KanR cassette in place of sll0180. The same protocol was performed to replace slr2131 in SD100 and SD277. The one exception is which primers were used to identify whether or not slr2131 was present in the cell since slr2131 has almost the identical nucleotide sequence length as the sacB-KanR sequence. Because of the similar sequence lengths, a new set of primers was created to amplify a 373 bp region both located with slr2131 (2131 w/in A and 2131 w/in B) (Table 2) were used to identify whether or not slr2131 was present in the cell. And finally, to ensure that the 3.2 kb nucleotide sequence of sacB-KanR was present between the flanking regions of slr2131, the primers encoding the slr2131 flanking region sequences were used. The DNA electrophoresis gels depicting the deletion confirmation are in Additional file 1: Figure S3.
Conjugation to add an expression vector containing an E. coli gene
Two expression vectors were created for the currently presented research based on derivatives of RSF1010 by the authors, with each conferring resistance to either Kan or Spc and Str, named pKan and pSpec, respectively. The aphA gene from E. coli MG1655 is expressed in pKan, while the aadA gene from the E. coli R100 plasmid is expressed in pSpec. Each expression vector contains the Ptrc promoter that was used constitutively to express the E. coli gene, either acrA or acrB. E. coli acrA or acrB genes were amplified from the E. coli chromosome using isolated genomic DNA (Wizard Genomic DNA Purification Kit, Promega, Madison, WI) using the following primers: 5’ AcrA F, 3’ AcrA R, 5’ AcrB F, 3’ AcrB R (Table 2). E. coli acrA or acrB genes were individually ligated into the multiple cloning site adjacent to Ptrc of pKan and pSpec respectively, resulting in vectors named pacrA and pacrB (Table 1). Additionally, acrA was ligated into pSpec resulting in pSacrA. In order to move these expression vectors into SD277 or SD100, conjugation must be performed to alleviate the effects of native restriction enzyme digestion in the cyanobacterial cells using an established method [51, 52]. To begin the process of conjugation, two cultures of Top10 E. coli cells containing either pRL443, which mobilizes the conjugational pair, facilitating the transfer of plasmids between cells, or pRL528 (a helper plasmid) and the expression vector (pKan, pSpec, pacrA, or pacrB) were grown in 15 mL of liquid lysogeny broth (LB) using the Miller modification [50] overnight at 37 °C. Simultaneously, 10 mL of the cyanobacteria strain was grown in a liquid BG-11 culture to an OD730 of 0.6. Then, all three strains, both E. coli and cyanobacteria, underwent a washing process in which the initial sample was centrifuged for 10 min at 3000 x g. The supernatant was removed, and the sample pellet was resuspended in 10 mL of sterilized BG-11. This process was repeated two more times. After the final wash, the strains were resuspended in 5 mL of BG-11. The process of conjugation was begun by the addition of 1 mL of the cyanobacterial strain to 2 mL of the E. coli strain with pRL443 and 2 mL of the E. coli strain containing both the expression vector and pRL528. These cultures then sat for 20 min at room temperature. Then 400 μL of each of the conjugation and control groups were spread onto a nitrocellulose membrane that had been placed on BG-11 with 0.5% LB media. This sat on the lab bench overnight. The next morning the plates were put in an illuminated chamber (SANYO) at 30 °C until a light green culture of bacteria appeared on the surface of the filter (usually after a day or two). The nitrocellulose membrane was moved to BG-11 with antibiotics corresponding to the expression vector to select for cyanobacterial strains containing the desired expression vector. Individual, bright green colonies grew on the nitrocellulose membrane within a week that potentially represented successfully modified cyanobacterial cells. The presence of the E. coli gene was confirmed using colony PCR using the same primers that were initially used to amplify the corresponding nucleotide sequence from the E. coli chromosome as described in the following section.
Confirmation of gene replacement or addition using colony PCR
The colony PCR is performed by suspending the cyanobacterial cells from the colony of interest into 3 μL of dH2O in a 200 μL-PCR tube. The suspension then proceeds through a triplicated freeze/thaw cycle from -80 °C to 55 °C. Then GoTaq DNA polymerase (Promega, Madison, WI) is used in a reaction mixture with the primers explained in previous sections to amplify the targeted DNA sequence. The thermocycler temperature and time conditions were as follows: Initial denaturation for 2 min at 95oC, then the next 3 conditions were repeated 30 times of denaturation for 1 min at 95oC, annealing for 1 min at 56oC, and extension for 1.5 min at 72oC, and finally, the final extension step occurs for 7 min at 72oC.
FFA analysis
The cyanobacterial strain was grown to a concentration of 0.8-1 × 109 cells/mL and 20 mL of the culture was removed and centrifuged at 6000 x g. The supernatant was placed into a new centrifuge tube after being filtered for any cells that remained suspended in the liquid media. The cell pellet was then processed for intracellular FFA concentration analysis using a modified Folch method as described previously [53]. Briefly, 3 mL of a ratio of 2:1 of chloroform:methanol mixture was added to the pellet, which is vortexed at room temperature for 24 h at low speed. The solvent and resuspended pellet mixture is filtered using a PTFE membrane resulting in the solvent with FFAs, which was then evaporated using positive nitrogen evaporator (N-EVAP 111, Organomation, Berlin, MA) until the sample was dry. Both the dried sample from the extracted pellet and the collected supernatant from the initial centrifugation were processed for FFA concentrations by adding 10 mL of hexane and 200 μL of 3 M H3PO4. These samples were agitated on a rotator at 180 rpms for 30 min at 37 °C and then the sample was centrifuged at 6000 x g for 5 min. 5 mL of the resulting uppermost layer (hexane layer) was removed and moved to a glass tube (13 × 100 mm) and dried on a nitrogen evaporator. The dried samples were then dissolved in 1 mL of hexane and analyzed by a gas chromatograph (GC) (Shimadzu GC 2010) with a Supelco Nukol capillary column (30 m × 0.53 mm × 0.5 μm) and flame ionization detector [54]. GC operating conditions were as follows: split ratio 1∶5; inject volume 1 μL; N2 carrier gas with a constant flow rate 30 mL∕ min; H2 40 mL∕ min, air 400 mL∕ min, make up gas (nitrogen) 5 mL∕ min; injector and detector temperature 250 °C; and oven temperature was started at 100 °C and increased at a rate of 10 °C∕ min to 220 °C and held for 10 min. Each FFA compound was identified by comparing its retention time with that of a standard FFA (Sigma Aldrich, St. Louis, MO, USA). The GC was used to test for the presence of the following hydrocarbons: lauric acid, myristic acid, palmitic acid, stearic acid, pentadecanoic acid, margaric acid, 6-[(10Z)-heptadecenyl] salicylic acid, hydroxyhexadecanoic acid, and hydroxytetradecanoic acid. Only lauric, myristic, palmitic, and stearic acids were observed in consistent concentrations greater than 1 mg/mL. FFA concentrations in samples were quantified based on the area under the chromatogram peak in comparison with the standards. Each sample was run through the gas chromatograph in triplicate and the concentration of each FFA present is averaged with the three data points. Each strain underwent this entire growth, supernatant/pellet separation, and analysis process in triplicate.
Half maximal inhibitory concentration (IC50) determination
Once the cyanobacterial strains reached mid-level growth phase as described in a previous section, they were assessed for the IC50 to Amp, Cm, and Ery. A 96-well flat bottom plate (Corning Inc., Corning, NY) in which each well was filled with a 50 μL solution containing a range of concentrations of each antibiotic (Amp, Cm, or Ery) that created final concentrations between 10 ng/mL to 10 mg/mL was used to inoculate 50 μL of the cyanobacterial sample to create a final volume of 100 μL. Each cyanobacterial sample in the well of the plate was measured at an initial OD790 between 0.35 and 0.4 using a microplate reader (Biotek Synergy H1, Winooski, VT). Each 96-well plate was placed in an illuminated chamber at 30 °C on a rotator (60 rpm) for 72 h at which point the final OD790 was measured of each sample.
RT-PCR analysis of mRNA
A 40 mL culture of a cyanobacterial strain is grown to mid-log level phase as explained previously, which was then centrifuged at 6000 x g for 10 min to pellet the cells. The supernatant was removed, and the remaining pelleted cells were resuspended in 500 μL of BG-11 media and 1 mL of RNAprotect Cell Reagent (Qiagen). Each sample was stored at -20 °C until all samples were ready for RNA isolation. For RNA extraction, the Qiagen RNeasy Kit was used. Briefly, the stored samples were thawed and pelleted using a centrifuge at a force of 6000 x g for 10 min. The supernatant was removed, and the pellet was resuspended in 700 μL of RLT Buffer (Qiagen) and transferred to a 2 mL locking lid microcentrifuge tube (Eppendorf, Hamburg, Germany) containing 0.2 g of 0.25 mm-diameter acid washed glass beads (Sartorius Stedim, Göttingen, Germany). The mixture was placed in a Bullet Blender Storm 24 homogenizer (Next Advance, Troy, NY) and homogenized for 5 min at the maximum speed. The remaining extraction steps were followed exactly as Qiagen recommended using the RNeasy Protect Bacterial Mini Kit. The mRNA concentration of each sample was quantified using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Each mRNA sample (1 μg) was treated with one unit of RQ1 RNase-free DNase (Promega) according to the manufacturer’s instructions. To create the cDNA, 250 ng of RNA was transcribed using the Qiagen One-Step RT-PCR Kit in a final reaction volume of 50 μL and then placed in the thermal cycler using the manufacturer’s recommendations, using the primers previously published to identify mRNA expression of E. coli genes acrA and acrB [55] (Table 2). Separately, a set of primers from previously published work was used to identify petB mRNA (Table 2), a constitutively expressed Synechocystis sp. PCC 6803 gene [56], ensuring that mRNA was isolated from every cyanobacterial strain, even in those that did not possess any E. coli genes.
To ensure there was no DNA contamination in the mRNA samples after the DNase treatment, each mRNA sample underwent a conventional PCR using GoTaq polymerase, appropriate reagents, and the corresponding primers to the gene of interest (acrA or acrB).
Statistics
Statistical analyses were performed using the GraphPad Prism 5 software package (GraphPad Software, San Diego, CA). A non-linear regression curve using the log (chemical concentration) versus the cell concentration in 4-parameter variable slope model was used to identify the IC50 as well as the 95% confidence intervals of that concentration. To compare the total FFA concentration of strains, a Mann-Whitney test was used. To determine the difference of growth rate of strains, a one-way ANOVA was used in which each time point was considered a matched result to be compared. Differences were considered significant at a P value of < 0.05. *p < 0.05, **p < 0.005, ***p < 0.0005, **** < 0.0001.
Change history
13 February 2019
Following publication of the original article [1], the author reported that the gene/protein names of slr2131 and sll0180 were swapped in the Discussion section. The details of the correction are mentioned below:
Abbreviations
- ABC:
-
ATP-binding cassette
- Amp:
-
Ampicillin
- Cm:
-
Chloramphenicol
- FFA:
-
Free fatty acid
- GC:
-
Gas chromatograph
- IC50 :
-
Half maximal inhibitory concentration
- Kan:
-
Kanamycin
- LB:
-
Lysogeny Broth
- MFP:
-
Membrane fusion protein
- OD:
-
Optical Density
- PCR:
-
Polymerase Chain Reaction
- RT-PCR:
-
Reverse transcriptase-polymerase chain reaction
- SDS:
-
Sodium dodecyl sulfate
- Spc:
-
Spectinomcin
- Str:
-
Streptomycin
- TES:
-
N-[tris(hydroxymethyl) methyl]-2-aminoethanesulfonic acid
References
Stanier RY, Kunisawa R, Mandel M, Cohen-Bazire G. Purification and properties of unicellular blue-green algae (order Chroococcales). Bacteriol Rev. 1971;35:171–205.
Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp strain PCC 6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996;3:109–36.
Grigorieva G, Shestakov S. Transformation in the cyanobacterium Synechocystis sp. 6803. FEMS Microbiol Lett. 1982;13:367–70.
Marraccini P, Bulteau S, Cassier-Chauvat C, Mermet-Bouvier P, Chauvat F. A conjugative plasmid vector for promoter analysis in several cyanobacteria of the genera Synechococcus and Synechocystis. Plant Mol Biol. 1993;23:905–9.
Gao Z, Zhao H, Li Z, Tan X. Lu X. photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energy Environ Sci. 2012;5:9857–65.
Zhou J, Zhang H, Zhang Y, Li Y, Ma Y. Designing and creating a modularized synthetic pathway in cyanobacterium Synechocystis enables production of acetone from carbon dioxide. Metab Eng. 2012;14:394–400.
Wang B, Pugh S, Nielsen DR, Zhang W, Meldrum DR. Engineering cyanobacteria for photosynthetic production of 3-hydroxybutyrate directly from CO2. Metab Eng. 2013;16:68–77.
Liu X, Sheng J, Curtiss IIIR. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci. 2011;108:6899–904.
Katoh H, Hagino N, Ogawa T. Iron-binding activity of FutA1 aubunit of an ABC-type iron transporter in the cyanobacterium Synechocystis sp strain PCC 6803. Plant Cell Physiol. 2001;42:823–7.
Koropatkin N, Randich AM, Bhattacharyya-Pakrasi M, Pakrasi HB, Smith TJ. The structure of the iron-binding protein, FutA1, from Synechocystis 6803. J Biol Chem. 2007;282:27468–77.
Badarau A, Firbank SJ, Waldron KJ, Yanagisawa S, Robinson NJ, Banfield MJ, et al. FutA2 is a ferric binding protein from Synechocystis PCC 6803. J Biol Chem. 2008;283:12520–7.
Suzuki S, Ferjani A, Suzuki I, Murata N. The SphS-SphR two component system is the exclusive sensor for the induction of gene expression in response to phosphate limitation in Synechocystis. J Biol Chem. 2004;279:13234–40.
Pitt FD, Mazard S, Humphreys L, Scanlan DJ. Functional characterization of Synechocystis sp strain PCC 6803 Pst1 and Pst2 gene clusters reveals a novel strategy for phosphate uptake in a freshwater cyanobacterium. J Bacteriol. 2010;192:3512–23.
Agarwal R, Zakharov S, Hasan SS, Ryan CM, Whitelegge JP, Cramer WA. Structure–function of cyanobacterial outer-membrane protein, Slr1270: homolog of Escherichia coli drug export/colicin import protein, TolC. FEBS Lett. 2014;588:3793–801.
Oliveira P, Martins NM, Santos M, Pinto F, Büttel Z, Couto NAS, et al. The versatile TolC-like Slr1270 in the cyanobacterium Synechocystis sp PCC 6803. Environ Microbiol. 2016;18:486–502.
de Zwaig RN, Luria SE. Genetics and physiology of colicin-tolerant mutants of Escherichia coli. J Bacteriol. 1967;94:1112–23.
Nakamura H. Gene-controlled resistance to acriflavine and other basic dyes in Escherichia coli. J Bacteriol. 1965;90:8–14.
Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Genes acrA and acrB encode a stress-induced efflux system of Escherichia coli. Mol Microbiol. 1995;16:45–55.
Ma D, Cook DN, Alberti M, Pon NG, Nikaido H, Hearst JE. Molecular cloning and characterization of acrA and acrE genes of Escherichia coli. J Bacteriol. 1993;175:6299–313.
Murakami S, Nakashima R, Yamashita E, Yamaguchi A. Crystal structure of bacterial multi-drug efflux transporter AcrB. Nihon Kessho Gakkaishi. 2003;45:256–61.
Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature. 2006;443:173–9.
Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, et al. Structure of the AcrAB–TolC multidrug efflux pump. Nature. 2014;509:512–5.
Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–9.
Lennen RM, Politz MG, Kruziki MA, Pfleger BF. Identification of transport proteins involved in free fatty acid efflux in Escherichia coli. J Bacteriol. 2013;195:135–44.
Huang F, Parmryd I, Nilsson F, Persson AL, Pakrasi HB, Andersson B, et al. Proteomics of Synechocystis sp strain PCC 6803: identification of plasma membrane proteins. Mol Cell Proteomics. 2002;1:956–66.
Huang F, Hedman E, Funk C, Kieselbach T, Schröder WP, Norling B. Isolation of outer membrane of Synechocystis sp. PCC 6803 and its proteomic characterization. Mol Cell Proteomics. 2004;3:586–95.
Pisareva T, Shumskaya M, Maddalo G, Ilag L, Norling B. Proteomics of Synechocystis sp PCC 6803: identification of novel integral plasma membrane proteins. FEBS J. 2007;274:791–804.
Pisareva T, Kwon J, Oh J, Kim S, Ge C, Wieslander Å, et al. Model for membrane organization and protein sorting in the cyanobacterium Synechocystis sp PCC 6803 inferred from proteomics and multivariate sequence analyses. J Proteome Res. 2011;10:3617–31.
Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res 2004;32 Web Server issue:W321–6.
Gonçalves CF, Pacheco CC, Tamagnini P, Oliveira P. Identification of inner membrane translocase components of TolC-mediated secretion in the cyanobacterium Synechocystis sp PCC 6803. Environ Microbiol. 2018;20:2354–69.
Liu X, Curtiss R. Nickel-inducible lysis system in Synechocystis sp PCC 6803. Proc Natl Acad Sci. 2009;106:21550–4.
Hamilton JA. New insights into the roles of proteins and lipids in membrane transport of fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2007;77:355–61.
Elkins CA, Nikaido H. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominately by two large periplasmic loops. J Bacteriol. 2002;184:6490–8.
Nikaido H. Multidrug efflux pumps of gram-negative bacteria. J Bacteriol. 1996;178:5853–9.
Paulsen IT, Chen J, Nelson KE, Saier, Jr. MH. Comparative genomics of microbial drug efflux systems. J Mol Microbiol Biotechnol 2001;3:145–150.
Linton KJ, Higgins CF. The Escherichia coli ATP-binding cassette (ABC) proteins. Mol Microbiol. 1998;28:5–13.
Tomii K, Kanehisa M. A comparative analysis of ABC transporters in complete microbial genomes. Genome Res. 1998;8:1048–59.
Okusu H, Ma D, Nikaido H. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (mar) mutants. J Bacteriol. 1996;178:306–8.
Nishino K, Yamaguchi A. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol. 2001;183:5803–12.
Vargiu AV, Nikaido H. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci. 2012;109:20637–42.
Cuellar-Bermudez SP, Romero-Ogawa MA, Vannela R, Lai YS, Rittmann BE, Parra-Saldivar R. Effects of light intensity and carbon dioxide on lipids and fatty acids produced by Synechocystis sp. PCC 6803 during continuous flow Algal Res. 2015;12:10–6.
Trautner C, Vermaas WFJ. The sll1951 gene encodes the surface layer protein of Synechocystis sp strain PCC 6803. J Bacteriol. 2013;195:5370–80.
Lenhart J, Schroeder J, Walsh B, Simmons L. DNA repair and genome maintenance in Bacillus subtilis. Microbiol Mol Biol Rev. 2012;76:530–64.
Baharoglu Z, Bikard D, Mazel D. Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet. 2010;6:e1001165.
Kizawa A, Kawahara A, Takashima K, Takimura Y, Nishiyama Y, Hihara Y. The LexA transcription factor regulates fatty acid biosynthetic genes in the cyanobacterium Synechocystis sp PCC 6803. Plant J. 2017;92:189–98.
Domain F, Houot L, Chauvat F, Cassier-Chauvat C. Function and regulation of the cyanobacterial genes lexA, recA, and ruvB: LexA is critical to the survival of cells facing inorganic carbon starvation. Mol Microbiol. 2004;53:65–80.
Patterson-Fortin LM, Colvin KR, Owttrim GWA. LexA-related protein regulates redox-sensitive expression of the cyanobacterial RNA helicase. crhR Nucleic Acids Res. 2006;34:3446–54.
Oliveira P, Lindblad P. Novel insights into the regulation of LexA in the cyanobacterium Synechocystis sp strain PCC 6803. J Bacteriol. 2011;193:3804–14.
Allen MM, Stanier RY. Selective isolation of blue-green algae from water and soil. Microbiology. 1968;51:203–9.
Miller J. Experiments in molecular genetics / Jeffrey H. Miller. 3rd, illustrated edition. Cold Spring Harbor. Laboratory. 1972.
Elhai J, Wolk CP. Conjugal transfer of DNA to cyanobacteria. In: Methods in enzymology: Academic Press; 1988. p. 747–54. https://doi.org/10.1016/0076-6879(88)67086-8.
Elhai J, Vepritskiy A, Muro-Pastor AM, Flores E, Wolk CP. Reduction of conjugal transfer efficiency by three restriction activities of Anabaena sp strain PCC 7120. J Bacteriol. 1997;179:1998–2005.
Sheng J, Vannela R, Rittmann BE. Evaluation of methods to extract and quantify lipids from Synechocystis PCC 6803. Bioresour Technol. 2011;102:1697–703.
Lalman JA, Bagley DM. Extracting long-chain fatty acids from a fermentation medium. J Am Oil Chem Soc. 2004;81:105–10.
Swick MC, Morgan-Linnell SK, Carlson KM, Zechiedrich L. Expression of multidrug efflux pump genes acrAB-tolC, mdfA, and norE in Escherichia coli clinical isolates as a function of fluoroquinolone and multidrug resistance. Antimicrob Agents Chemother. 2011;55:921–4.
Pinto F, Pacheco CC, Ferreira D, Moradas-Ferreira P, Tamagnini P. Selection of suitable reference genes for RT-qPCR analyses in cyanobacteria. PLoS One. 2012;7:e34983.
Acknowledgements
We would like to thank Dr. Susan Bellefleur and Alexandria Voigt Bellefleur for their help editing the manuscript.
Funding
This research was supported at Arizona State University by funding available to Roy Curtiss and by a University of Florida startup grant for Prof. Roy Curtiss, III.
Availability of data and materials
The datasets used and analyzed for the current study are available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
MPAB, SYW, and RCIII designed this study. MPAB and SYW created primers, plasmids, and strains. MPAB performed all of the experiments and created all of the figures and tables. MPAB, SYW, and RCIII analyzed all of the data. MPAB, SYW, and RCIII wrote and edited the article. MPAB, SYW, and RCIII read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
RCIII is a founder, part owner and member of the Board of Directors of Curtiss Healthcare, Inc., a start-up biotech company in Gainesville, FL to develop and market vaccines against diseases of farm animals. The company has exclusive licenses to inventions and patents pertaining to vaccine technologies filed as a faculty member at Washington University in St. Louis, Arizona State University and the University of Florida. While RCIII with collaborators invented two US patents on production and recovery of biofuels from cyanobacteria, these inventions were assigned to ASU and RCIII is no longer pursuing this line of research. MPAB and SYW have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Figure S1. Electrophoresis Gel displaying the DNA fragments created from RT-PCR of (A) E. coli genes acrA and acrB and (B) of the Synechocystis sp. PCC6803 petB gene. Figure S2. Optical density measurements at 730 nm wavelength every 24 h for 168 h of (A) SD277 and its mutant and complementation derivatives, (B) SD100 and its mutant and complementation derivatives, (C) SD277 and its plasmid addition derivatives, and (D) SD100 and its plasmid addition derivatives. The culture conditions were as follows: illumination was 50 μmol photons m− 2 s− 1, temperature was 30 °C, aeration of filtered air was pumped at a rate of 100 mL/min, and CO2 concentration was the normal atmospheric concentration. Figure S3. Electrophoresis gel displaying the DNA fragments created from PCR of using primers of either end fragments of sll0180 in (A) SD100, SD100 ∆sll0180, SD100 ∆sll0180 pSacrA and (B) SD277, SD277 ∆sll0180, SD277 ∆sll0180 pSacrA. Electrophoresis gel displaying the DNA fragments created from PCR using primers of either end fragments of slr2131 in (C) SD100, SD100 ∆slr2131, SD100 ∆slr2131 pacrB, SD277, SD277 ∆slr2131, and SD277 ∆slr2131 pacrB. Electrophoresis gel displaying the DNA fragments created from PCR of using primers of a region within slr2131 in (D) SD100, SD100 ∆slr2131, SD100 ∆slr2131 pacrB, SD277, SD277 ∆slr2131, and SD277 ∆slr2131 pacrB. (DOCX 6501 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Bellefleur, M.P.A., Wanda, SY. & Curtiss, R. Characterizing active transportation mechanisms for free fatty acids and antibiotics in Synechocystis sp. PCC 6803. BMC Biotechnol 19, 5 (2019). https://doi.org/10.1186/s12896-019-0500-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-019-0500-3