
Abstract
Background
To comprehensively analyze the stemness characteristics related to prognosis and the immune microenvironment in lung adenocarcinoma (LUAD).
Methods
The OCLR machine learning method was used to calculate the stemness index (mRNAsi) of the LUAD samples. DEGs common between the low mRNAsi, normal, and high mRNAsi groups were screened and the immune-stemness genes were obtained. Then the PPI network was created and enrichment analyses were performed. Moreover, different subtypes based on immune-stemness genes associated with prognosis were identified, and the relationships between LUAD stemness and TIME variables were systematically analyzed, followed by TMB analysis.
Results
Patients in the high mRNAsi groups with poor prognosis were screened along with 144 immune-stemness genes. IL-6, FPR2, and RLN3 showed a higher degree in the PPI network. A total of 26 immune-stemness genes associated with prognosis were screened. Two clusters were obtained (cluster 1 and cluster 2). Survival analysis revealed that patients in cluster 2 had a poor prognosis. A total of 12 immune cell subpopulations exhibited significant differences between cluster 1 and cluster 2 (P < 0.05). A total of 10 immune checkpoint genes exhibited significantly higher expression in cluster 1 (P < 0.05) than in cluster 2. Further, the TMB value in cluster 2 was higher than that in cluster 1 (P < 0.05).
Conclusion
Immune-stemness genes, including L-6, FPR2, and RLN3, might play significant roles in LUAD development via cytokine–cytokine receptor interaction, neuroactive ligand‒receptor interaction, and the JAK‒STAT pathway. Immune-stemness genes were related to tumor-infiltrating immune cells, TMB, and expression of immune checkpoint gene.
Highlights
-
IL-6, FPR2, RLN3 had higher degree in PPI network.
-
The patients in cluster 1 had better prognosis.
-
Total 10 immune checkpoint genes showed higher expression in cluster 1.
-
TMB value in cluster 2 was higher than that in cluster 1.
Similar content being viewed by others


Background
Lung cancer is a malignant disease with the highest incidence rate and mortality rate across the world [1, 2]. Lung adenocarcinoma (LUAD) is a major type of lung cancer, accounting for 40–50% of all lung cancers and originates from the bronchial epithelial and mucous glands [3, 4]. Despite significant improvements in targeted and chemotherapeutic techniques, the overall survival rate of patients with LUAD is still poor [5]. Therefore, it is essential to screen markers related to LUAD prognosis.
Cancer stem cells (CSCs) have the ability of self-renewal and producing heterogeneous tumor cells [6]. However, owing to their biological characteristics and the protective effect of the tumor microenvironment (TME), CSCs are less sensitive to conventional radiotherapy and chemotherapy, a phenomenon that supports tumor recurrence and metastasis [7]. Therefore, if CSCs are not completely removed, relapse and metastasis can easily occur. Transcriptional and epigenetic disorders of cancer cells often alter the core signaling pathways—regulating the phenotype of normal stem cells—leading to carcinogenic dedifferentiation and acquisition of stemness [8]. In addition, the complex interactions between immune cells and their secreted molecules in the TME help maintain the viability and self-renewal ability of the CSCs [9]. The stemness index (mRNAsi) is an index used to describe the similarity between tumor cells and stem cells. Higher mRNAsi is related to active biological process and a higher degree of tumor dedifferentiation [10]. LUAD comprises a complex system of cancer cells, infiltrating immune cells, CSCs, and nonmalignant stromal cells. However, a synthesized understanding of the tumor immune microenvironment (TIME) and LUAD stemness is lacking.
In this study, an OCLR machine learning method was used to calculate the mRNAsi of the LUAD samples. Then, the samples were divided into low and high mRNAsi groups based on the median value of mRNAsi. The DEGs between the low mRNAsi group vs. normal group, high mRNAsi group vs. normal group, and high mRNAsi group vs. low mRNAsi group were screened, and immune-stemness genes were obtained. PPI network analysis and enrichment analysis were performed. Moreover, different subtypes based on immune-stemness genes associated with prognosis were acquired. The relationships between LUAD stemness and TIME variables were systematically evaluated, followed by tumor mutation burden (TMB) analysis. This study offers novel understanding of the stemness characteristics of LUAD and provides a theoretical basis for drug development.
Methods
Data source and data preprocessing
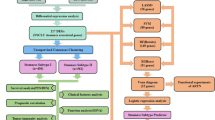
The gene expression data of 585 LUAD patients (collected using an Illumina HiSeq 2000 RNA Sequencing platform-based) were acquired from TCGA. In total, 501 LUAD tumor samples and 58 normal samples were obtained after retaining LUAD samples and normal samples with survival prognosis information. Ethical approval was obtained for accessing patient information curated on the database, and all methods were carried out in accordance with relevant application guidelines and regulations of TCGA. This study is based on open source data, so there are no ethical issues. A flowchart of this study is shown in Fig. 1.

Flow chart of this study
Evaluating the clinical significance and prognosis of mRNAsi
The mRNAsi of LUAD samples was calculated using OCLR machine learning [11] through the gelnet package in R software. Thereafter, the differences in mRNAsi values between LUAD samples and normal samples were evaluated using a t-test. In addition, the LUAD samples were categorized into low and high mRNAsi groups based on the median value of mRNAsi, and Kaplan‒Meier survival analysis was used to assess the prognosis of patients between the low and high mRNAsi groups. Fisher’s exact test (count variables such as sex) and t-test (continuous variables such as age) were used to assess the differences in clinical information between the low and high mRNAsi groups.
PPI network and enrichment analysis of immune-stemness genes
The samples were allocated into normal sample, low mRNAsi, and high mRNAsi sample groups based on the source information and mRNAsi grouping information of the samples. Thereafter, the limma package [12] of R was used to identify the DEGs between the low mRNAsi group vs. normal group, high mRNAsi group vs. normal group, and high mRNAsi group vs. low mRNAsi group with the following cutoffs: false positive discovery (FDR) < 0.05; |log2FC| > 0.5. Common DEGs were then obtained. In addition, immune-related genes were acquired from the Immport database [13]. The common DEGs were intersected with these immune-related genes and the overlapping genes were redefined as immune-stemness genes.
STRING [14] was used to analyze the interactions between immune-stemness genes encoding proteins with a PPI score > 0.7. Cytoscape [15] was used to build the PPI network. Moreover, enrichment analysis of immune-stemness genes in the PPI network was performed using DAVID [16, 17] with an FDR threshold < 0.05.
Identification of different subtypes based on immune-stemness genes related to prognosis
Immune-stemness genes in the PPI network were subjected to univariate Cox regression analysis using the survival package [18] to screen the immune-stemness genes significantly associated with survival prognosis with a threshold of P < 0.05. In addition, based on the expression of immune-stemness genes related to prognosis, the pheatmap package [19] in R software was used to conduct bidirectional hierarchical cluster analysis to identify the different subtypes using the centered Pearson correlation algorithm [20]. Subsequently, Kaplan‒Meier survival analysis was used to assess the prognosis of patients between these different subtypes. Fisher’s exact test (count variables such as sex) and t-test (continuous variables such as age) were used to evaluate the differences in clinical information between these different subtypes.
Association between different subtypes with TIME
Based on the expression profile of LUAD samples, CIBERSORT [21] was used to evaluate 22 types of tumor-infiltrating immune cells in these subtypes. The differences in the expression of numerous immune checkpoint genes were compared.
TMB analysis of different subtypes
As the biology of LUAD is different depending on the type of oncogenic driver mutations, the relations between driver mutations [Kirsten rat sarcoma viral oncogene homolog (KRAS), epidermal growth factor receptor (EGFR), anaplastic lymphoma kinase (ALK), c-Ros oncogene 1 receptor tyrosine kinase (ROS1), V-raf murine sarcoma viral oncogene homolog B1 (BRAF), mesenchymal-epithelial transition factor (MET), and RET proto-oncogene (RET)] and the stemness score were compared. In addition, based on the LUAD gene mutation data obtained from TCGA, the gene mutation frequencies of LUAD samples were evaluated. t-test was used to assess the differences in gene mutation frequency between these different subtypes. TMB is generally defined as the number of somatic coding mutations per million bases. The TMB of LUAD samples was calculated using the maftools package [22] in R, and the differences in TMB between these different subtypes were compared using Wilcox test.
Statistical analysis
Fisher’s exact test was used to compare the differences in count variables between groups. t-test was used to compare the differences in continuous variables between groups, and the normalization was conducted before using the t-test, and non-parametric methods was used if the parameters were not normally distributed. P < 0.05 was considered significant.
Results
Evaluating the clinical significance and prognosis of mRNAsi
As shown in Fig. 2 A, the mRNAsi value corresponding to that of LUAD samples was higher than that corresponding to that of normal samples. Survival analysis revealed that the patients in the high mRNAsi group had a poor prognosis (Fig. 2B). In addition, differences in clinical information between the low and high mRNAsi groups were examined (Table 1). The results revealed that pathological T (Fig. 2 C) and pathological stage (Fig. 2D) were significantly different between the low and high mRNAsi groups (P < 0.05), and the mRNAsi value was positively correlated with pathological T and stage, which explains the poor prognosis in the high mRNAsi group.

Evaluation of the clinical significance and prognosis of mRNAsi. (A) The mRNAsi value corresponding to LUAD samples and normal samples. (B) Survival analysis of low and high mRNAsi groups. Difference in the pathological T (C) and pathological stage (D) between the low and high mRNAsi groups
PPI network enrichment analysis of immune-stemness genes
As previously mentioned, a total of 4653, 3961, and 2583 DEGs were screened from the low mRNAsi group vs. normal group, high mRNAsi group vs. normal group, and high mRNAsi group vs. low mRNAsi group, respectively, and a total of 1239 common DEGs were identified (Fig. 3 A). The expression levels of common DEGs were significantly different between samples from the high and low mRNAsi groups (Fig. 3B). In addition, 144 immune-stemness genes were identified. The PPI network of these immune-stemness genes showed that there were 109 nodes in the PPI network (Fig. 3 C), and interleukin 6 (IL-6), formyl peptide receptor-2 (FPR2), and relaxin 3 (RLN3) had a higher degree in the PPI network (Supplementary Table ); thus, L-6, FPR2, and RLN3 might play significant roles in LUAD development. Subsequently, enrichment analysis was carried out on the immune-stemness genes in the PPI network. The results showed that the 109 immune-stemness genes were involved in 42 biological processes (BPs) (Fig. 3D) and 16 Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (Fig. 3E) [23, 24].

Protein-protein interaction (PPI) network and enrichment analysis of immune-stemness genes. (A) Differentially expressed genes (DEGs) and common DEGs screened from low mRNAsi group vs. normal group, high mRNAsi group vs. normal group and high mRNAsi group vs. low mRNAsi group. (B) The expression level of common DEGs in samples from the high mRNAsi and low mRNAsi groups. (C) PPI network of these immune-stemness genes; the large nodes with higher degree. Gene ontology (GO) (D) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (E) of the immune-stemness genes in the PPI network
Identification of different subtypes based on immune-stemness genes associated with prognosis
A total of 26 immune-stemness genes associated with prognosis were screened (Supplementary Table 2). Two clusters were identified, cluster 1 and cluster 2; there were 166 and 335 samples in cluster 1 and cluster 2, respectively (Fig. 4 A). Survival analysis showed that the patients in cluster 2 had a poor prognosis (Fig. 4B). The differences in age, sex, pathologic N, pathologic T, and pathologic stage between cluster 1 and cluster 2 were significant (P < 0.05) (Fig. 4 C and Table 2).

Identification of different subtypes based on immune-stemness genes associated with prognosis. (A) Identification of the different subtypes. (B) Survival analysis cluster 1 and cluster 2. (C) Differences in age, sex, pathologic N, pathologic T, and pathologic stage between cluster 1 and cluster 2
Association between different subtypes with TIME
A total of 22 infiltrating immune subpopulations were evaluated; among them, 12 immune cell subpopulations showed significant differences between cluster 1 and cluster 2 (P < 0.05) (Fig. 5 A). A comparison of the differences in expression levels of immune checkpoint genes between cluster 1 and cluster 2 revealed that 10 immune checkpoint genes showed significantly higher expression in cluster 1 than in cluster 2 (P < 0.05) (Fig. 5B). These results provided new insights into LUAD development and immunotherapy.

Association between different subtypes with tumor immune microenvironment (TIME). (A) Differences in immune cell subpopulations between the cluster 1 and cluster 2 groups. (B) Differences in the expression levels of immune checkpoint genes between cluster 1 and cluster 2 groups. * represents P < 0.05
TMB analysis of different subtypes
As shown in Fig. 6, the stemness scores of KRAS, EGFR, ALK, ROS1, BRAF, MET, and RET showed no significant difference between the Mut and Wild groups (P > 0.05). The gene mutation frequency of LUAD samples was determined; tumor protein p53 (TP53), titin (TTN), mucin 16, cell surface associated (MUC16), etc. had a high mutation frequency (Fig. 7 A); the mutation frequency of TP53, TTN, CUB, and Sushi multiple domains 3 (CSMD3), among others, in cluster 1 and cluster 2 were significantly different (Fig. 7B). The TMB value in cluster 2 was higher than that in cluster 1 (P < 0.05) (Fig. 7 C), which is explained by the results of survival analysis.

Association between driver mutations and the stemness score

Tumor mutation burden (TMB) analysis of different subtypes. (A) Gene mutation frequency of LUAD samples. (B) Gene mutation frequency in cluster 1 and cluster 2. (C) TMB value in cluster 1 and cluster 2
Discussion
In this study, patients in the high mRNAsi group with a poor prognosis and a total of 144 immune-stemness genes were screened. There were 109 nodes in the PPI network, and enrichment analysis revealed that the 109 immune-stemness genes were involved in 42 BPs and 16 KEGG pathways. In total, 26 immune-stemness genes associated with prognosis were screened. Two clusters were identified, namely, cluster 1 and cluster 2. Survival analysis revealed that patients in cluster 2 had a poor prognosis. A total of 12 immune cell subpopulations showed significant differences between cluster 1 and cluster 2 (P < 0.05), and a total of 10 immune checkpoint genes showed significantly higher expression in cluster 1 than in cluster 2 (P < 0.05); TMB values in cluster 2 were higher than those in cluster 1.
Studies have suggested that mRNAsi might serve as an effective index for the survival, classification, and disease progression of tumor patients [25,26,27]. Huang et al. found that basal breast cancer patients have high mRNAsi values [28]. In the present study, we found that the mRNAsi value in LUAD samples was higher than that in normal samples, and survival analysis revealed that patients in the high mRNAsi group had a poor prognosis. These findings are in accordance with the results of the above studies. In addition, clinical analysis showed that the mRNAsi value was positively correlated with pathological T and stage, and patients with higher pathological T and stage had a higher mRNAsi value. These findings verify the results of survival analysis.
A total of 144 immune-stemness genes were screened in this study; IL-6, FPR2, and RLN3 had a high degree in the PPI network and therefore could be considered the hub genes. IL-6 is a pleiotropic four-helix bundle cytokine that plays various functions in the body [29]. The IL-6 family is one of the most important cytokine families involved in the process of tumorigenesis and metastasis, particularly IL-6 [30]. IL-6 promotes tumorigenesis by regulating all hallmarks of cancer and multiple signaling pathways [31]. FPR2 is a G-protein coupled receptor that plays a major role in cancer development and inflammation [32]. Several studies have shown that FPR2 stimulates tumor cell invasion and proliferation [33, 34]. For instance, Lu et al. showed that FPR2 enhances colorectal cancer progression by promoting epithelial–mesenchymal transition process [35]. Xie et al. found that FPR2 is involved in epithelial ovarian cancer progression through RhoA-mediated M2 macrophage polarization [36]. In addition, Zhang et al. found that RLN3 may be a key gene affecting the progression of hepatocellular carcinoma [37]. Liu et al. revealed that RLN3 was differentially expressed and prognostically relevant in patients with KRAS-mutant colorectal cancer [38]. KEGG pathway analysis revealed that the 109 immune-stemness genes in the PPI network were involved in 16 KEGG pathways, including cytokine−receptor interaction, neuroactive ligand−receptor interaction, and JAK−STAT signaling pathway. Cytokine receptor interaction may be the key to determining the role of inflammation in disease development [39]. The JAK−STAT signaling pathway is highly associated with many inflammatory and immune diseases [40]. Zhou et al. found that cytokine−receptor interaction and JAK−STAT signaling pathway are related to the development of glioblastoma [41]. Chen et al. showed that neuroactive ligand−receptor interaction is correlated with the occurrence of glioma [42], suggesting that these immune-stemness genes might play key roles in LUAD development.
Immunotherapy has become an effective treatment for cancer; immune cells are an important part of the TME and play vital roles in tumor immunotherapy [43]. In this study, two clusters were screened, including cluster 1 and cluster 2, and the survival analysis uncovered that the patients in cluster 1 had a better prognosis. A total of 12 immune cell subpopulations, including M0 macrophages, regulatory T cells (Tregs), and memory B cells, showed significant differences between cluster 1 and cluster 2. A total of 10 immune checkpoint genes, including PDCD1, CTLA4, and CD274, showed significantly higher expression in cluster 1 than in cluster 2. Among immune checkpoint therapies, PD-1/PD-L1 and CTLA-4 inhibitors have shown promising therapeutic outcomes [44]. TMB is a new biomarker for predicting the effect of immunotherapy [45, 46]. Negrao et al. showed that low TMB was a predictive factor for worse outcomes in lung cancer [47]. Zhang et al. found that high TMB levels led to poor survival outcomes in clear cell renal cell carcinoma [48]. In this study, we found that the TMB value in cluster 2 was higher than that in cluster 1, which explains why cluster 1 had a better prognosis.
This study has some limitations. First, the data analyzed in this study were all publicly available; thus, the key genes and molecular mechanisms should be explored in further experiments in the future. Second, the infiltrating immune subpopulations were evaluated using only the CIBERSORT algorithm; therefore, further tools and relevant experiments should be carried out to validate our findings. Third, large-scale prospective clinical studies are required to evaluate the obtained immune-stemness genes.
Conclusion
Immune-stemness genes, including L-6, FPR2, and RLN3, might play significant roles in LUAD development via cytokine–cytokine receptor interaction, neuroactive ligand–receptor interaction, and JAK–STAT signaling pathway. In addition, immune-stemness genes were related to tumor-infiltrating immune cells, TMB, and immune checkpoint gene expression. Therefore, this study proposed novel insights into the clinical treatment of LUAD.
Data availability
The datasets used and/or analyzed during the current study are available from TCGA (dataset ID: TCGA-LUAD.htseq_fpkm.tsv; https://gdc-hub.s3.us-east-1.amazonaws.com/download/TCGA-LUAD.htseq_fpkm.tsv.gz; Full metadata).
Abbreviations
- LUAD :
-
Lung adenocarcinoma
- CSCs :
-
Cancer stem cells
- TME :
-
Tumor microenvironment
- TIME :
-
Tumor immune microenvironment
- TMB :
-
Tumor mutation burden
- FPR2 :
-
Formyl peptide receptor-2
- RLN3 :
-
Relaxin 3
- BPs :
-
Biological processes
- TP53 :
-
Tumor protein p53
- TTN :
-
Titin
- MUC16 :
-
Mucin 16
- Tregs :
-
Regulatory T cells
References
Rachow T, Schiffl H, Lang SM. Risk of lung cancer and renin-angiotensin blockade: a concise review. J Cancer Res Clin Oncol. 2021;147(1):195–204.
Imyanitov EN, Iyevleva AG, Levchenko EV: Molecular testing and targeted therapy for non-small cell lung cancer: Current status and perspectives. Critical reviews in oncology/hematology 2021, 157:103194.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–54.
Denisenko TV, Budkevich IN, Zhivotovsky B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018;9(2):117.
Peitzsch C, Kurth I, Kunz-Schughart L, Baumann M, Dubrovska A. Discovery of the cancer stem cell related determinants of radioresistance. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2013;108(3):378–87.
Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3.
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12(2):133–43.
Wu YC, Tang SJ, Sun GH, Sun KH. CXCR7 mediates TGFβ1-promoted EMT and tumor-initiating features in lung cancer. Oncogene. 2016;35(16):2123–32.
Bai KH, He SY, Shu LL, Wang WD, Lin SY, Zhang QY, Li L, Cheng L, Dai YJ. Identification of cancer stem cell characteristics in liver hepatocellular carcinoma by WGCNA analysis of transcriptome stemness index. Cancer Med. 2020;9(12):4290–8.
Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, Kamińska B, Huelsken J, Omberg L, Gevaert O, et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell. 2018;173(2):338–54.e315.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47–7.
Huang R, Mao M, Lu Y, Yu Q, Liao L. A novel immune-related genes prognosis biomarker for melanoma: associated with tumor microenvironment. Aging. 2020;12(8):6966.
Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44.
Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13.
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P. The STRING database in 2017: quality-controlled protein–protein association networks, made broadly accessible. Nucleic acids research 2016:gkw937.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Wang P, Wang Y, Hang B, Zou X, Mao J-H. A novel gene expression-based prognostic scoring system to predict survival in gastric cancer. Oncotarget. 2016;7(34):55343.
Wang L, Cao C, Ma Q, Zeng Q, Wang H, Cheng Z, Zhu G, Qi J, Ma H, Nian H. RNA-seq analyses of multiple meristems of soybean: novel and alternative transcripts, evolutionary and functional implications. BMC Plant Biol. 2014;14(1):1–19.
Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95(25):14863–8.
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling tumor infiltrating immune cells with CIBERSORT. In: Cancer systems biology. edn.: Springer; 2018. pp. 243–59.
Zhang C, Li Z, Qi F, Hu X, Luo J. Exploration of the relationships between tumor mutation burden with immune infiltrates in clear cell renal cell carcinoma. Annals of translational medicine 2019, 7(22).
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545-d551.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein science: a publication of the Protein Society. 2019;28(11):1947–51.
Zhang Y, Tseng JT, Lien IC, Li F, Wu W, Li H. mRNAsi Index: Machine Learning in Mining Lung Adenocarcinoma Stem Cell Biomarkers. Genes 2020, 11(3).
Pan S, Zhan Y, Chen X, Wu B, Liu B. Identification of Biomarkers for Controlling Cancer Stem Cell Characteristics in Bladder Cancer by Network Analysis of Transcriptome Data Stemness Indices. Front Oncol. 2019;9:613.
Wang C, Qin S, Pan W, Shi X, Gao H, Jin P, Xia X, Ma F. mRNAsi-related genes can effectively distinguish hepatocellular carcinoma into new molecular subtypes. Comput Struct Biotechnol J. 2022;20:2928–41.
Huang K, Wu Y, Xie Y, Huang L, Liu H: Analyzing mRNAsi-Related Genes Identifies Novel Prognostic Markers and Potential Drug Combination for Patients with Basal Breast Cancer. Disease markers 2021, 2021:4731349.
Schmidt-Arras D, Rose-John S. IL-6 pathway in the liver: From physiopathology to therapy. J Hepatol. 2016;64(6):1403–15.
Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26(1):54–74.
Kumari N, Dwarakanath BS, Das A, Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2016;37(9):11553–72.
Li Y, Ye D. Molecular biology for formyl peptide receptors in human diseases. J Mol Med. 2013;91(7):781–9.
Hou XL, Ji CD, Tang J, Wang YX, Xiang DF, Li HQ, Liu WW, Wang JX, Yan HZ, Wang Y, et al. FPR2 promotes invasion and metastasis of gastric cancer cells and predicts the prognosis of patients. Sci Rep. 2017;7(1):3153.
Xie X, Yang M, Ding Y, Yu L, Chen J. Formyl peptide receptor 2 expression predicts poor prognosis and promotes invasion and metastasis in epithelial ovarian cancer. Oncol Rep. 2017;38(6):3297–308.
Lu J, Zhao J, Jia C, Zhou L, Cai Y, Ni J, Ma J, Zheng M, Lu A. FPR2 enhances colorectal cancer progression by promoting EMT process. Neoplasma. 2019;66(5):785–91.
Xie X, He J, Wang Q, Liu Y, Chen W, Shi K. FPR2 participates in epithelial ovarian cancer (EOC) progression through RhoA-mediated M2 macrophage polarization. J ovarian Res. 2021;14(1):177.
Zhang Y, Wang S, Xiao J, Zhou H. Bioinformatics analysis to identify the key genes affecting the progression and prognosis of hepatocellular carcinoma. Bioscience reports 2019, 39(2).
Liu J, Huang X, Liu H, Wei C, Ru H, Qin H, Lai H, Meng Y, Wu G, Xie W, et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J translational Med. 2021;19(1):27.
Bass WT, Buescher ES, Hair PS, White LE, Welch JC, Burke BL. Proinflammatory cytokine-receptor interaction model improves the predictability of cerebral white matter injury in preterm infants. Am J Perinatol. 2008;25(4):211–8.
Xin P, Xu X, Deng C, Liu S, Wang Y, Zhou X, Ma H, Wei D, Sun S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int Immunopharmacol. 2020;80:106210.
Zhou L, Tang H, Wang F, Chen L, Ou S, Wu T, Xu J, Guo K. Bioinformatics analyses of significant genes, related pathways and candidate prognostic biomarkers in glioblastoma. Mol Med Rep. 2018;18(5):4185–96.
Chen J, Wang Z, Wang W, Ren S, Xue J, Zhong L, Jiang T, Wei H, Zhang C. SYT16 is a prognostic biomarker and correlated with immune infiltrates in glioma: A study based on TCGA data. Int Immunopharmacol. 2020;84:106490.
Steven A, Fisher SA, Robinson BW. Immunotherapy for lung cancer. Respirol (Carlton Vic). 2016;21(5):821–33.
Darvin P, Toor SM, Sasidharan Nair V, Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp Mol Med. 2018;50(12):1–11.
Park SE, Park K, Lee E, Kim JY, Ahn JS, Im YH, Lee C, Jung H, Cho SY, Park WY, et al. Clinical implication of tumor mutational burden in patients with HER2-positive refractory metastatic breast cancer. Oncoimmunology. 2018;7(8):e1466768.
Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, et al. Tumor Mutational Burden and Efficacy of Nivolumab Monotherapy and in Combination with Ipilimumab in Small-Cell Lung Cancer. Cancer Cell. 2019;35(2):329.
Negrao MV, Lam VK, Reuben A, Rubin ML, Landry LL, Roarty EB, Rinsurongkawong W, Lewis J, Roth JA, Swisher SG, et al. PD-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations Are Stronger Predictors of Benefit from Immune Checkpoint Blockade than HLA Class I Genotype in Non-Small Cell Lung Cancer. J Thorac oncology: official publication Int Association Study Lung Cancer. 2019;14(6):1021–31.
Zhang C, Li Z, Qi F, Hu X, Luo J. Exploration of the relationships between tumor mutation burden with immune infiltrates in clear cell renal cell carcinoma. Ann Transl Med. 2019;7(22):648.
Acknowledgements
None.
Funding
This work was supported by the Health Science and Technology Talent Training Project and Technical Center Construction Project of Kunming City (Program No. 2020-SW (Technology)-01-18), Scientific Research Fund Project of Department of Education, Yunnan province (Program No. 2021J0361), and Health Research Project of Kunming Health Commission (Program No. 2020-04-02-111).
Author information
Authors and Affiliations
Contributions
HW conceived and designed the research, YW was involved in the acquisition of data. WL and XZ carried out the analysis and interpretation of data. ZY were involved in designing the study and performed the statistical analysis. HW, KW, and JD conceived the study, participated in its design and coordination, and helped draft the manuscript and revision of manuscript for important intellectual content. RC helped to revision of manuscript for important intellectual content. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable. All methods were carried out in accordance with the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Han Wang and Ying Wang should be regard as co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, H., Wang, Y., Luo, W. et al. Integrative stemness characteristics associated with prognosis and the immune microenvironment in lung adenocarcinoma. BMC Pulm Med 22, 463 (2022). https://doi.org/10.1186/s12890-022-02184-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-022-02184-8