
Abstract
Background
KRAS gene is the most common type of mutation reported in colorectal cancer (CRC). KRAS mutation-mediated regulation of immunophenotype and immune pathways in CRC remains to be elucidated.
Methods
535 CRC patients were used to compare the expression of immune-related genes (IRGs) and the abundance of tumor-infiltrating immune cells (TIICs) in the tumor microenvironment between KRAS-mutant and KRAS wild-type CRC patients. An independent dataset included 566 cases of CRC and an in-house RNA sequencing dataset were served as validation sets. An in-house dataset consisting of 335 CRC patients were used to analyze systemic immune and inflammatory state in the presence of KRAS mutation. An immue risk (Imm-R) model consist of IRG and TIICs for prognostic prediction in KRAS-mutant CRC patients was established and validated.
Results
NF-κB and T-cell receptor signaling pathways were significantly inhibited in KRAS-mutant CRC patients. Regulatory T cells (Tregs) was increased while macrophage M1 and activated CD4 memory T cell was decreased in KRAS-mutant CRC. Prognosis correlated with enhanced Tregs, macrophage M1 and activated CD4 memory T cell and was validated. Serum levels of hypersensitive C-reactive protein (hs-CRP), CRP, and IgM were significantly decreased in KRAS-mutant compared to KRAS wild-type CRC patients. An immune risk model composed of VGF, RLN3, CT45A1 and TIICs signature classified CRC patients with distinct clinical outcomes.
Conclusions
KRAS mutation in CRC was associated with suppressed immune pathways and immune infiltration. The aberrant immune pathways and immune cells help to understand the tumor immune microenvironments in KRAS-mutant CRC patients.
Similar content being viewed by others


Background
Colorectal cancer (CRC) remains a major cause of cancer-related mortality worldwide despite advancements in tumor screening, early diagnosis, and curative resection. Currently, radical resection is the sole reliable method of cure for CRC. At the diagnostic stage, 20–25% of CRC patients show evidence of metastatic disease with no scope for radical surgery [1]. Subsequent to curative resection, the recurrence rate of metastasis in patients is approximately 70%, of which 50% are fatal [2]. Survival in patients with untreated metastatic CRC is around six months. Treatment regimens combining cytotoxic chemotherapy and biological agents improved overall survival of patients with metastatic CRC by more than two years [3]. The advent of immunotherapy further advanced the scope of prolonging survival in cancer patients. Immune checkpoint blockade therapy has shown promising therapeutic results in patients with advanced malignant tumor, such as non-small cell lung cancer, melanoma, renal cell carcinoma, and mismatch repair-deficient tumors [4, 5]. Immune checkpoint blockade therapy has been beneficial in microsatellite instability-high CRC patients [6], which account for 15% of all CRCs [7]. However, since the majority of CRC patients are microsatellite-stable, researching the immune microenvironment and identifying potential immunotherapeutic targets are important in improving the effectiveness of immunotherapy in these patients.
In CRC, the common canonical gain-of-function mutation is the oncogenic mutation of Kirsten rat sarcoma viral oncogene homolog (KRAS) that encodes GTPases, namely, KRAS4A and KRAS4B [8]. RAS, a key molecule of the mitogen-activated protein kinase (MAPK) signaling pathway, is activated by the binding of ligands such as the vascular endothelial growth factor (VEGF) to receptor tyrosine kinases (RTKs). RAS exists in two states, the active (GTP, guanosine triphosphate) or non-active-forms (GDP, guanosine diphosphate). Transition between the two states is responsible for the signal transduction crucial for cell growth and differentiation [9]. RAS mutations lead to persistent activation of multiple downstream effectors resulting in the induction of malignant transformation [10]. The prevalence of KRAS mutations in CRC patients is approximately 30–50% and is associated with poor prognosis and metastasis [11]. Clinical significance of KRAS mutation is proved by its use as a biomarker of EGFR-TKI resistance and its application in identifying suitable patients for anti-epidermal growth factor receptor (EGFR) therapies [12].
Current research evidence suggests a significant influence of KRAS mutation in tumor immunity. An unsupervised hierarchical clustering analysis of immune genes/signatures named the Co-ordinate Immune Response Cluster (CIRC), comprising 28 genes, revealed a relatively high proportion of patients with KRAS mutation in the cluster associated with low inhibitory molecule expression [13, 14]. In addition, immunophenotyping of colon tumors from mice indicated an association between Kras mutation and an immunosuppressive microenvironment characterized by decreased T-cell infiltration and increased infiltration of myeloid-derived suppressor cells (MDSCs) [15]. However, the immune landscape and altered expression of immune-related genes in CRC patients with KRAS mutation have not been fully elucidated.
The present study systematically depicts the immune landscape, profiles immune-related genes (IRGs), and compares systemic immune markers between KRAS-mutant and KRAS wild-type CRC patients based on TCGA, GEO and in-house dataset. Our results indicate an association of KRAS mutation with local and systemic immunosuppression in CRC. In addition, an immue risk (Imm-R) model was established, which was associated with immune infiltration and prognosis in CRC patients.
Materials and methods
Clinical specimens
In the present study, 8 cases of CRC samples including 3 cased of KRAS-mutant CRC samples and 5 cased of KRAS wild-type CRC samples were obtained from patients at the Guangxi Medical University Cancer Hospital. The samples were subjected to RNA sequencing. All of the patients were pathologically diagnosed as CRC without chemotherapy or radiotherapy before the collection of the tissues. Written informed consents were obtained from all patients. The study was approved by the Ethics and Human Subject Committee of Guangxi Medical University Cancer Hospital. All experiments and methods were performed according to relevant guidelines and regulations formulated by the Guangxi Medical University.
RNA-seq analysis
Total RNA was extracted using Trizol reagent (Invitrogen). The construction of RNA-seq library was based on the protocol of the IlluminaTruSeq RNA Sample Preparation Kit (illumina). Finally, RNA-seq analysis was performed by GENE + company (Beijing, China) using Illumina HiSeqX Ten platforms. After quality control and trimming adaptor, reads were mapped onto human genome GRCh38. RNA-seq data have been deposited in the China National Center for Bioinformation (ID: PRJCA003751).
Data acquisition and processing
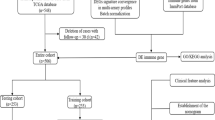
High-throughput RNA sequencing and somatic mutation data (VarScan2 Variant Aggregation and Masking) related to colon and rectal adenocarcinoma available in The Cancer Genome Atlas (TCGA) were downloaded from the GDC Data Portal (https://portal.gdc.cancer.gov/) on July 25, 2019. Totally, 535 patients with complete somatic mutation data were included in this study. Of 535 CRC patients, 528 had high-throughput RNA sequencing data. Gene symbols were annotated based on GRCh38.91. Mutation Annotation Format (MAF) files of colon and rectal adenocarcinoma were merged together to obtain somatic mutation data. Visualization and summarization of somatic mutation data was achieved using the Maftools R package [16].The corresponding clinical information of the patients was simultaneously downloaded. The GSE39582 dataset from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) repository was downloaded, which included gene expression profile of 566 cases, and served as an independent validation dataset. Gene IDs were transformed using the clusterProfiler R package [17].
In-house data collection
Clinical and pathological data from CRC patients hospitalized at the Guangxi Medical University Cancer Hospital (Nanning, China) between July 2013 and October 2018 were documented. The criteria for inclusion of patients in this study were as follows: (i) pathologically confirmed CRC and (ii) primary tumor resection with KRAS mutation detected based on postoperative gross specimen analysis. The exclusion criteria were: (i) exposure to prior preoperative therapy (including radiotherapy, chemotherapy, or chemoradiotherapy), (ii) with other types of cancer before or after CRC diagnosis, (iii) with known familial adenomatous polyposis or hereditary non-polyposis colorectal cancer, and (iv) diagnosed with infectious diseases or systemic stress reaction at the time of first admission to hospital.
Medical records of patients were examined to document information related to demographic and clinical characteristics such as age, sex, pathological stage, preoperative routine blood test, serum Igs, complement proteins C3 and C4, C-reactive protein (CRP), high-sensitivity C-reactive protein (hs-CRP), percentage of T lymphocytes, B lymphocytes, and natural killer (NK) cells, and KRAS mutation status. Based on the inclusion and exclusion criteria, 335 CRC patients (101 patients with KRAS mutation and 234 patients with KRAS wild-type tumors) were enrolled in the current study. The protocol of this retrospective study was approved by the Ethics and Human Subject Committee of Guangxi Medical University Cancer Hospital, and all experiments and methods met the standards of the relevant guidelines and regulations.
Generation of IRGs list
The list of IRGs was collected from the immunology database and analysis portal (ImmPort) and TISIDB [18, 19]. The ImmPort database encompasses accurately updated information related to immunology and provides a list of IRGs curated with functions and Gene Ontology terms. TISIDB is a web portal that facilitates comprehensive investigation of tumor-immune interactions and provides a list of genes associated with anti-tumor immunity reported in literature. IRGs enlisted in ImmPort and TISIDB are from different sources that complement each other. In the present study, IRGs listed in both the databases were amalgamated and the genes that were annotated by the Ensembl database were retained. Accordingly, the IRGs list in the current study was made up of 1951 genes.
Estimation of the abundance of immune cells
The tumor immune estimation resource (TIMER) is a web-accessible resource that estimates the abundance of six types of tumor-infiltrating immune cells (TIICs) (B cells, CD4 and CD8 T cells, neutrophils, macrophages, and dendritic cells) [20]. TIMER deduces the abundance of TIICs from gene expression profiles based on a deconvolution method validated by Monte Carlo simulations, orthogonal estimates from DNA methylation-based inferences, and pathological assessments. CIBERSORT is a deconvolution algorithm based on support vector regression, which uses a set of reference gene-expression values corresponding to a minimal representation for each cell type to infer cell type proportions in data from bulk tumor samples with mixed cell types [21]. CIBERSORT could sensitively and specifically discriminate 22 human immune cell phenotypes. The abundance of TIICs was analyzed using TIMER in 528 CRC patients (Seven patients without high-throughput RNA sequencing data were excluded from 535 CRC patients) enrolled in the study and then validated by CIBERSORT using the default parameters.
Construction and validation of tumor-infiltrating immune cell(TIICs)signature
The TCGA dataset served as the training set, and GSE39582 was the validation set. In the training set, we first applied the univariable survival analysis to define the prognostic value of the TIICs in patients with KRAS mutation. TIICs with significant prognostic value were first validated in the validation set. TIICs with significant prognostic value in both training set and validation set were analyzed using the multivariate Cox proportional hazards regression model, and those with a p value < 0.05 were used to construct the TIICs signature. A formula for the TIICs signature was established to predict patient survival: TIICs signature = ∑Cox coefficient of TIIC Xi × abundance of TIIC Xi. The prognostic performance of the TIICs signature was evaluated using the receiver operating characteristic (ROC) curve and area under the curve (AUC).
Identification of differentially expressed IRGs
To identify IRGs associated with KRAS mutation in CRC, the expression of 1951 IRGs between KRAS-mutant and KRAS wild-type CRC was compared using the R software package empirical analysis of digital gene expression data in R (edgeR) [22]. Trimmed mean of M-values (TMM) method was used to normalize the count data in edgeR. The threshold for filtering differentially expressed IRGs was set at a false discovery rate (FDR) of < 0.05 and a log2 fold change > 1.
Functional enrichment analysis
The pathways and biological processes affected by KRAS mutation in CRC were identified by gene set enrichment analysis (GSEA) using the clusterProfiler R package. A list of sorted genes obtained based on the fold-change of mean expression of the genes between KRAS-mutant and KRAS wild-type CRC patients represented the input file. Biological processes were evaluated using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways and gene ontology (GO).
Construction of an immune risk (Imm-R) model
The expression profile of differentially expressed IRGs between KRAS-mutant and KRAS wild-type CRC was analyzed using univariate Cox regression analysis. Input dataset was in log2 (normalized value + 1) data format. The prognostic value of differentially expressed IRGs for overall survival (OS) was defined by univariate Cox regression analysis wherein genes were regarded as significant at p < 0.05. IRGs identified as prognostic indicators by univariate Cox regression analysis were subsequently subjected to multiple Cox regression analysis. IRGs identified as independent prognostic indicators in multiple Cox regression analysis together with TIICs signature were used to construct the Imm-R model. The individual risk value was calculated by multiplying the expression value of each prognostic indicator and the cox regression coefficient. The association between the risk value and immune infiltration was analyzed by comparing the abundance of immune cells between the high- and low-risk groups.
Statistical analysis
All statistical analyses were performed using the R software (Version: 3.5.0). OS between two groups was compared using the Kaplan–Meier survival analysis and log-rank test. Spearman rank correlation test was used to evaluate the correlation between the expression of IRGs and abundance of immune cells. Results with two-sided p < 0.05 were considered statistically significant.
Results
Somatic mutation landscape of CRC patients
Somatic mutation landscape analysis was performed to analyze the status of KRAS in 535 CRC patients. Of them, 99.63% were detected to possess at least one type of gene mutation (Additional file 1: Figure S1). The most frequent mutation seen in CRC was in the adenomatous polyposis coli (APC) gene (79%) followed by the tumor protein 53 (TP53) gene (61%). KRAS mutation was the third common mutation detected in CRC with a frequency of 42%. We divided patients into two groups based on the presence or absence of KRAS mutations. The genes with the top 3 mutation frequencies (except for KRAS) in the KRAS mutant group were APC, TP53 and PIK3CA. The genes with the top 3 mutation frequencies in KRAS wild-type patients were APC, TP53 and SYNE1 (Fig. 1a). We compared the frequency of gene mutations between the two groups and found that mutation frequency of APC and PIK3CA was significantly increased in the KRAS mutant group, while mutation frequency of TP53 and ZFHX4 was significantly increased in the KRAS wild-type group (Fig. 1b). KRAS mutation-induced altered pathways were evaluated using GSEA based on the data obtained from KRAS-mutant and KRAS wild-type CRC patients. Several immune-related pathways were significantly down-regulated in KRAS-mutant compared to that of KRAS wild-type CRC patients, namely, Th1 and Th2 cell differentiation, T cell receptor signaling, and nuclear factor kappa-B (NF-κB) signaling pathways (Fig. 1c). Four pathways were significantly up regulated in KRAS-mutant compared to that of KRAS wild-type CRC patients, namely, biosynthesis of amino acids, carbon metabolism, oxidative phosphorylation and ribosome (Fig. 1c). GSEA based on biological processes and molecular function revealed inhibition of several immune-related terms in KRAS-mutant compared to that of KRAS wild-type CRC patients (Additional file 2: Figure S2). Taken together, these data indicate an association of KRAS mutation with immune-suppression in CRC. The expression of immune checkpoint molecules, such as PD-1, are a promising predictive factor for immune treatment response. We explored the association between expression of immune checkpoint molecules and KRAS mutation. Interestingly, we found that several key immune checkpoint-associated molecules (PD-L1, CTLA4 and TIM-3, all p < 0.05, Fig. 1d) were significantly downregulated in the KRAS mutant group. We speculated KRAS mutations might inhibit checkpoint molecules.

Somatic mutation landscape of colorectal cancer (CRC) patients based on KRAS status. a Somatic mutation landscape of CRC patients with (left panel) or without (right panel) KRAS mutation. Seven common mutation types were counted. b Genes with significantly different mutation frequencies between the KRAS mutant and KRAS wild-type group. c Significantly different pathways between KRAS mutant and KRAS wild-type groups. Pathways significantly down regulated in KRAS-mutant CRC patients (left) and significantly up regulated in KRAS-mutant CRC patients (right). d The expression of key immune checkpoint-associated molecules between KRAS mutant and KRAS wild-type groups
Immune landscape of CRC patients in the presence and absence of KRAS mutation
The immune landscape was successfully analyzed among 528 CRC patients (KRAS mutant: 224; KRAS wild-type: 304). We comprehensively compared the spectrum of immune cell infiltration in the presence and absence of KRAS mutation. As shown in Fig. 2a, significant variations were observed in the proportion of TIICs among different individuals. Thus, variation in the proportion of TIICs represent intrinsic characteristics that underlie individual differences. Correlation analysis revealed different subpopulations of immune cells displaying weak to moderate (correlation coefficient ranging from 0.36 to 0.75) positive correlation (Fig. 2b). The abundance of different types of immune cells was compared between the KRAS-mutant and KRAS-wild type CRC patients (Fig. 2c). The abundance of B cells (0.08 ± 0.06 vs. 0.09 ± 0.08), neutrophils (0.11 ± 0.06 vs. 0.13 ± 0.07), and macrophage (0.05 ± 0.06 vs. 0.07 ± 0.08) were significantly down-regulated in KRAS-mutant compared to that of KRAS wild-type CRC patients. Considering that immune cells have multiple subtypes, we used CIBERSORT for further typing of immune cells and validation the results of TIMER. The abundance of 22 immune cells estimated by CIBERSORT was shown in Additional file 3: Figure S3. Comparing the abundance of 22 immune cells between the KRAS-mutant and KRAS wild-type groups, we found that native B cells, neutrophils and macrophage M1 were significantly down-regulated in the KRAS-mutant group (all p < 0.05, Fig. 2d), which was a further refinement of the results of TIMER. In addition, we observed that activated CD4 memory T cell was significantly decreased but regulatory T cells (Tregs) was significantly increased in the KRAS-mutant group (Fig. 2d). Microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) CRC exhibit an active immune microenvironment due to the hyper-mutated state of the tumor cells [23]. We excluded samples with MSI-H or dMMR and re-analyzed the differences in the immune microenvironment between the two groups. Totally, 72 cases (13.63%) of CRC with MSI-H or dMMR were excluded, including 21 KRAS mutant and 51 KRAS wild-type. We found that four of the above five TIICs (native B cells, macrophage M1, activated CD4 memory T cell and Tregs), except neutrophils, had significant differences between the two groups (Additional file 4: Figure S4a). We further used in-house RNA sequencing data to compare the abundance of the above five TIICs in the KRAS-mutant and KRAS wild-type groups (KRAS-mutant groups: n = 3; KRAS wild-type groups: n = 5). Macrophage M1 was also significantly down-regulated in the KRAS-mutant group (p = 0.039, Additional file 4: Figure S4b). For several other TIICs, we observed similar trends to the above results, but the differences were not statistically significant due to the small sample size.

Immune landscape of colorectal cancer (CRC) patients in the presence and absence of KRAS mutation. a Percentage abundance of six types of tumor-infiltrating immune cells. b Correlation matrix showing abundance of six types of immune cells. Correlation coefficients displayed are expanded 100 times. c Abundance of six types of tumor-infiltrating immune cells in KRAS-mutant and KRAS wild-type CRC patients. d Differential abundance of tumor-infiltrating immune cells between the KRAS-mutant and KRAS wild-type CRC
Development and validation of the tumor-infiltrating immune cell(TIICs)signature
Given that TIICs were associated with KRAS mutations, we investigated whether TIICs were associated with survival in patients with KRAS mutations. We performed survival analysis to identify survival-associated TIICs in patients with KRAS mutation based on above differentially expressed TIICs. We observed that high abundance of macrophage M1 and activated CD4 memory T cell were associated with better prognosis while high abundance of Tregs was associated with poorer prognosis in patients with KRAS mutation (Fig. 3a). There was no significant correlation between the abundance of native B cells and neutrophils and prognosis (Fig. 3a). We validated these results using an independent validation set (GSE39582) which included the gene expression profile of 566 cases of CRC. Survival analysis likewise suggested that macrophage M1 and activated CD4 memory T cells were associated with better prognosis, whereas high abundance of Tregs was associated with poorer prognosis in patients with KRAS mutations (Fig. 3b). We further investigated the prognostic value of the above TIICs in KRAS wild-type patients. In the training set, only macrophage M1 was significantly associated with survival in KRAS wild-type patients (Additional file 5: Figure S5a, p = 0.008) and the prognostic value of macrophage M1 in KRAS wild-type patients could not validated by validation set (Additional file 5: Figure S5b, p = 0.185). These results suggested that macrophage M1, activated CD4 memory T cells and Tregs had a more robust prognostic value in the KRAS-mutant CRC. Next, we performed multivariate Cox regression analysis to identify independent prognostic TIICs and the results showed that macrophage M1, activated CD4 memory T cells and Tregs were independent prognostic TIICs for KRAS-mutant CRC (Additional file 9: Table S1). Therefore, collection of macrophage M1, activated CD4 memory T cells and Tregs were defined as the TIICs signature to predict prognosis of KRAS-mutant CRC. Based on the TIICs signature, we constructed a method to calculate scores of TIICs signature (TIICs score), which was calculated as follows: (-0.75 × abundance of macrophage M1) + (− 0.84 × abundance of activated CD4 memory T cells) + (0.93 × abundance of Tregs). The TIICs scores successfully distinguished KRAS-mutant CRC patients into high-risk or low-risk groups. Patients with high risk had significantly poorer overall survival compared with those with low risk in the TCGA dataset (Fig. 3c). The TIICs signature yielded similar results in KRAS-mutant CRC patients of validation set. Compared with those with low risk, high-risk patients had poorer OS (Fig. 3d). The AUC under ROC curve of TIICs scores for predicting OS in the training set and validation set were 0.75 and 0.68, respectively (Fig. 3e).

Development and validation of the tumor-infiltrating immune cell signature. a Univariate survival analysis identified survival-associated TIICs in patients with KRAS mutation based on training set. b Univariate survival analysis identified survival-associated TIICs in patients with KRAS mutation based on validation set. c Kaplan–Meier curves for patients with high- and low TIICs score in the training set. d Kaplan–Meier curves for patients with high- and low TIICs score in the validation set. e ROC curves for measuring the predictive value of the TIICs score in the training set and validation set
Differentially expressed IRGs in KRAS-mutant and KRAS wile-type CRC patients
The expression of IRGs was compared to explore the immune molecular characteristics of CRC patients in the presence and absence of KRAS mutation. Among the 1951 IRGs, the edgeR algorithm identified 73 differentially expressed IRGs, of which 24 were up regulated and 49 down regulated in CRC with KRAS mutation (Additional file 6: Figure S6a). The pathways and biological processes influenced by differentially expressed IRGs were explored using enrichment analysis. The humoral immune response was the most significantly enriched pathway related to biological processes (Additional file 6: Figure S6b). Neuroactive ligand − receptor interaction, cytokine − cytokine receptor interaction, and Ras signaling pathway were the first three most significant pathways related to KEGG (Additional file 6: Figure S6c). Molecular functions enrichment analysis identified receptor ligand activity as the most frequent molecular function (Additional file 6: Figure S6d). Thus, these results indicate an association of a majority of differentially expressed IRGs with signal transduction. Results of protein–protein interaction (PPI) network analysis performed based on differentially expressed IRGs detected 133 edges and identified albumin (ALB), glucagon (GCG), leptin (LEP), insulin-like growth factor 2 (IGF2), CRP, and (pro-platelet basic protein) PPBP as core genes from the networks (Additional file 6: Figure S6e).
Development and validation of the immune risk (Imm-R) model by intergrating TIICs and IRGs
Immune-related genes (IRGs) were reported to orchestrate tumor-associated immune responses. The integration of the TIICs signature and IRGs signature may enable more comprehensive assessment of immune status and more precise prognostic prediction. Preliminary screening of survival-associated IRGs using univariate Cox regression analysis revealed significant association of 13 out of 73 differentially expressed IRGs with OS (Fig. 4a). Importantly, majority of the survival-associated IRGs (12 out of 13) were identified as risk factors for poor prognosis. Further, independent prognostic factors were identified by multivariate COX regression analysis. Of the 13 genes subjected to multivariate COX regression model, three independent prognostic factors were identified, namely, VGF, relaxin 3 (RLN3), and cancer/testis antigen family 45 member A1 (CT45A1). The immune risk (Imm-R) model was constructed by intergrating TIICs signature and IRGs using multivariate COX regression. VGF, RLN3, CT45A1 and TIICs signature were all independent prognostic factors for KRAS-mutant CRC (all p < 0.05, Fig. 4b). In the Imm-R model, an immune risk score (Imm-R score) was generated using the formula: Imm-R score = (0.165 × VGF) + (0.453 × RLN3) + (0.203 × CT45A1) + (0.372 × TIICs signature). The Imm-R model could effectively distinguish KRAS-mutant CRC patients with discrete clinical outcomes (Fig. 4c). Patients at high-risk had significantly shorter survival compared to those at low-risk (p = 0.0013). We further validated the Imm-R model in KRAS-mutant CRC patients in validation set. The Imm-R model was powerful to distinguish KRAS-mutant CRC patients with good or bad prognosis. Patients with high-risk had significantly shorter OS compared with those with low risk in the validation set (p = 0.0096, Fig. 4d). The AUC under ROC curve of Imm-R model for predicting OS in the training set and validation set were 0.76 and 0.68, respectively (Fig. 4e). Nomogram is a user‑friendly graphical regression model with excellent applicability in clinical settings [24, 25]. To improve the usability of the Imm-R model, we constructed a nomogram to depict the Imm-R model better (Fig. 4f). The nomogram included above four features, and a point for each feature was assigned based on the scale on the top. The total score was defined as the sum of the points of the eight variables. By drawing a perpendicular line from the total point axis to the two-outcome axis, estimated three- and five-year OS probabilities could be obtained. To assess the goodness-of-fit of the nomogram, we compared the predicted three- and five-year survival probabilities to the actual three- and five-year survival probabilities using calibration plots (Fig. 4g, h). The calibration curve revealed good concordance between the predicted and observed probabilities in both of the three- and five-year survival probabilities. These results proved that the Imm-R model had very appropriate calibration.

Development and validation of the immune risk (Imm-R) model. a Differentially expressed IRGs related with overall survival in univariate Cox regression analysis. P values are indicated by color scale on the side. Horizontal bars represent 95% confidence intervals. b Independent prognostic factors in multivariate COX regression analysis. P values are indicated by color scale on the side. Horizontal bars represent 95% confidence intervals. c Kaplan–Meier curves for patients with high- and low Imm-R score in the training set. d Kaplan–Meier curves for patients with high- and low Imm-R score in the validation set. e ROC curves for measuring the predictive value of the Imm-R model in the training set and validation set. f The nomogram of Imm-R model for predicting the three- and five-year survival probabilities. Points are assigned for four features. The score for each feature was calculated by drawing a line upward to the 'Points' line, and the sum of the four scores was 'Total Points'. The total points on the bottom scales correspond to the predicted three- and five-year survival. g The calibration plot of the nomogram predicting three-year survival. The x-axis is the nomogram-predicted survival and the y-axis is the actual survival. The reference line is 45° and indicates perfect calibration. h The calibration plot of the nomogram predicting five-year survival
Association between immune infiltration and Imm-R model
The relationship between immune infiltration and Imm-R model was investigated by comparing the abundance of the 22 types of immune cells between the low- and high-risk CRC patients. The abundance of native B cells, Tregs, macrophage M0, activated mast cells were significantly increased in patients with high risk, while CD8 + T cells, activated CD4 memory T cell, follicular helper T cells, macrophage M1 and M2, resting dendritic cells and esoinophils were significantly decreased in patients with high risk (Fig. 5a). To explore the underlying biological mechanisms of the Imm-R model, we performed Gene Set Enrichment Analysis (GSEA). The results observed that several pathways related to cancer or metabolism were significantly activated in those high-risk patients, included Basal cell carcinoma, Wnt signaling pathway, melanogenesis and Taurine and hypotaurine metabolism, reflecting the active tumor metabolism in high-risk patients (Fig. 5b). Inversely, several pathways related to immune, such as chemokine signaling pathway, NF-kappa B signaling pathway and T cell recepter signaling pathway, were significantly down-regulated in high-risk patients, suggesting immunosuppression in high-risk patients (Fig. 5c). We further investigated the association between the expression of IRGs and TIICs. The IRGs significantly associated with macrophage M1 and activated CD4 memory T cell were summarized in Additional file 7: Figure S7. Interestingly, we found that FGF6 was significantly associated with the three TIICs mentioned above, which indicated that FGF6 might play an important role in the tumor microenvironment of CRC patients with KRAS mutation.

Association between immune infiltration and Imm-R model. a Abundance of immune cells in low- and high-risk CRC patients. b Pathways significantly enriched in CRC patients with high-risk. c Pathways significantly enriched in CRC patients with low-risk
Systemic immune and inflammatory state in CRC patients in the presence of KRAS mutation
The systemic impact of aberrant immune infiltration in localized tumor tissue was evaluated by comparing the level of immune cells, Igs, blood platelets, and acute phase proteins, such as CRP and hs-CRP in the blood of 335 CRC patients (101 patients tested positive for KRAS-mutation and 234 had KRAS wild-type). The clinical information related to the enrolled patients is listed in Table 1. The mean age of patients was 59 years. The proportion of patients with distant metastasis was significantly higher in KRAS wild-type patients compared to those harboring KRAS mutation (p = 0.02). Age, sex, tumor location, and TNM (T describes the size of the tumor and any spread of cancer into nearby tissue; N describes spread of cancer to nearby lymph nodes; and M describes metastasis) stage were similar between the two groups (p > 0.05). The systemic levels of hs-CRP, CRP, and IgM were significantly lower in KRAS-mutant compared to that of the KRAS wild-type patients (Additional file 8: Figure S8). The number of leukocyte, neutrophils, lymphocytes, blood platelets, lgM, lgA, C3, C4, helper T lymphocytes, inhibitory T lymphocytes, NK cells and B lymphocytes were similar between the KRAS-mutant and KRAS wild-type patients (p > 0.05).
Discussion
The benefits of immunotherapy have received immense research interest because of the impressive long-lasting response seen in several solid tumors [26]. In CRC, immune response and survival benefit are limited to mismatch-repair-deficient and microsatellite instability-high (dMMR–MSI-H) CRC patients, who account for only a small percentage of CRC patients. Thus, a deeper understanding of the immune landscape and identification of novel immunotherapeutic targets are needed. The present study systematically depicted the immune landscape and identified aberrant IRGs in KRAS-mutant and KRAS wild-type CRC patients. IRGs prognostic signature-based stratification effectively classified CRC patients into high- and low-risk groups with significantly evident differences in immune infiltration. This study provides a conceptual framework to understand the nature of immune infiltration in CRC in the context of KRAS mutation. This understanding might help interpret the probable responses to immunotherapy and treatment strategies designed to treat KRAS-mutant CRC patients.
KRAS mutation has been associated with immunosuppression in CRC. The presence of RAS mutation in CRC has been shown to down-regulate the IFNγ pathway, result in restricted CD8 + T cell activation [8, 27]. Immune checkpoint blockers, such as anti-PD-1 and anti-PD-L1 antibodies block the interaction between PD-1 and PD-L1, enhancing T cell activation that results in cytotoxic killing of tumor cells. However, inhibition of PD-L1 in KRAS-mutant CRC failed to bring about the desired result [28]. Several studies have explored the mechanism of immunosuppression and have provided insights to explain the mechanism of resistance to immunotherapy in KRAS-mutant CRC patients. Mutant KRAS inhibits the expression of interferon regulatory factor 2 (IRF2), a key transcription factor required for the activation of IFN-mediated responses [15]. Overexpression of IRF2 enhances sensitivity of KRAS-mutant CRC cells to anti-PD-1 therapy [15]. However, comprehensive analysis of aberrant IRGs and pathways associated with KRAS mutation in CRC still needs to be elucidated. Our study indicates down regulation of several immune and inflammatory pathways, such as NF-κB and T-cell receptor signaling pathways in KRAS-mutant CRC patients. The NF-κB signaling pathway is an important component of innate and adaptive immunity [29]. In innate immunity, upon activation of pattern recognition receptors (PRRs), NF-κB is crucial for the secretion of cytokines and for the production of perforin and IFN-γ in NK cells [30]. In adaptive immunity, the NF-κB signaling pathway is essential for the differentiation of B and T lymphocytes, and for the production of survival and maturation factors [31]. Inhibition of the NF-κB signaling pathway in the tumor microenvironment is a novel therapeutic target in immunotherapy. NF-κB-activating receptors are potential targets for combating the anti-inflammatory and regulatory effects of infiltrating regulatory T cells (Tregs) and functions as an important supportive therapy for checkpoint inhibitors [31]. In addition, metabolic pathways such as biosynthesis of amino acids and carbon metabolism were activated in KRAS-mutant CRC. Metabolic reprogramming, in which increased utilization of glucose and glutamine to support rapid growth is a hallmark of most cancers [32]. KRAS-driven metabolic rewiring occurs by up-regulating rate-limiting enzymes involved in amino acid, fatty acid, or nucleotide biosynthesis [33]. Targeting abnormal metabolic pathways may offer novel therapeutic strategies for the treatment of KRAS mutant CRC.
In CRC, KRAS mutation is associated with aberrant immune infiltration [34]. Our study found a significant decrease in the abundance of native B cells, neutrophils and macrophage M1, activated CD4 memory T cell in KRAS-mutant CRC. The the abundance of Tregs was significantly in KRAS-mutant CRC. A naive B cell is a B cell that has not been exposed to an antigen [35]. The antitumor activity of B cells is largely facilitated through IgG-mediated antigen presentation and activation of anti-tumor T cell responses [36]. In vivo, allogeneic IgG triggered a significantly more potent anti-tumor immune response than syngeneic IgG [37]. However, the functional role and mechanism of native B cells in tumor immunology remains unknown. Neutrophils are the first line of defense against pathogens. In the tumor microenvironment, tumor-associated neutrophils (TAN) exhibit a dual role in the form of N1 (tumor-suppressive) and N2 (tumor-promoting) phenotypes depending on the stage of disease progression [38]. Anti-tumor neutrophils activated by tumor cells bind to tumor cells, secrete cytotoxic mediators such as hydrogen peroxide (H2O2), and induce tumor cell apoptosis [39]. Interaction between neutrophils and T cells is essential to raise an appropriate anti-tumor immune response [40]. Neutrophils present antigens and provide accessory signals required for T cell activation [41]. Macrophages play a dual role in tumor immunity [42]. Of the two subtypes, M1 macrophages are differentiated from monocytes when exposed to Th1-type cytokines while M2 macrophages are differentiated under the influence of macrophage colony-stimulating factor (M-CSF), prostaglandin F (PGF) and vitamin D3 [43]. The M1 and M2 macrophages have distinct functions. M1 macrophages secrete higher levels of interleukin (IL)-12 and lower level of IL-10, and thereby contribute to the anti-tumor immune response. M2 macrophages produce immuno-suppressive cytokines such as IL-10, transforming growth factor-beta (TGF-β) and VEGF, resulting in the suppression of the immune surveillance system [42]. In the tumor microenvironment, T cells play a prominent role compared to B cells in cancer immunotherapy [36]. Activated CD4 memory T cell derived from CD4 memory T cells stimulated again by antigen [44]. Activated CD4 memory T cell undergo rapid expansion, eliciting a more effective and rapid immune response than the primary immune response [45]. The persistence of antitumor immunotherapy is related to the number of CD4 memory T cell [46]. Treg play a major role in orchestrating immunomodulation during CRC [47]. Treg cells can inhibit an anti-tumor specific immune response in patients with CRC and is associated with tumor progression during CRC [48]. Aberrant immune infiltration seen in KRAS-mutant CRC provides a promising ground for improving the response rate of immunotherapy. Further research is warranted to elucidate the interaction between immune and tumor cells to provide new targets for immunotherapy.
IRGs and TIICs can predict the prognosis of patients with CRC. Down-regulated M1 and up-regulated M2 macrophages are associated with poor prognosis in CRC [49]. A prognostic immunoscore model based on immune cells was established to predict OS in CRC patients [50]. However, the study was focused either on immune infiltration or on IRGs in CRC. The current study systematically analysed variation in immune infiltration and IRGs in KRAS-mutant CRC and established a prognostic model by intergrating TIICs and IRGs to determine the immune status of patients. The present model shows prognostic biomarkers that could be used to categorize patients to help improve the effectiveness of immunotherapy. The prognostic model consist of three IRGs, namely VGF, RLN3 and CT45A1. VGF is a neuroendocrine polypeptide secreted by neuroendocrine cells and functions to enhance neuronal growth and to prevent apoptosis [51]. VGF-expression influences the mechanism involved in counter regulating the decrease in functionality of T lymphocytes [52]. However, the functional role and mechanism of VGF in CRC remains unknown. RLN3 encodes relaxin-3, a peptide hormone belonging to the insulin superfamily [53]. RLN3 play an important role in the regulation of energy homeostasis and appetite [53]. Recent studies substantiate the role of RLN3 in development and tumorigenesis. Relaxins promote tumor growth and metastatic colonization in brain [54]. RLN3 is implicated in the prognosis of hepatocellular carcinoma (HCC) [55]. In phylogenetics, CT45A1 belongs to a new family of genes. CT45A1 and is aberrantly overexpressed in various types of cancer [56]. Overexpression of CT45A1 advances epithelial-mesenchymal transition, and enhances cell stemness, tumorigenesis, invasion, and metastasis. CT45-derived human leukocyte antigen (HLA) class I peptides efficiently activated patient-derived cytotoxic T cells and promoted tumor cell killing, indicating its potential as an immunotherapeutic target [57].
Cancer is a systematic disease in which the progression is driven not only by the underlying genetic alteration but also by complex systemic processes [58,59,60]. Interaction between the host and tumor plays an important role in cancer progression [61]. The cancerous state releases an abundance of proinflammatory cytokines into the circulation, resulting in systemic inflammation. Antigens are then expressed on the surface of tumor cells induce high levels of antibody and T cell response [62]. Aberrant inflammatory and immune responses are common in poorly differentiated and advanced CRC and are associated with a less favorable outcome [63]. Thus, immune recognition and inflammatory mechanism in cancer does not always result in protective immune response [64]. In the present study, relatively lower levels of systemic inflammation (low CRP and hs-CRP) and lgM were seen in KRAS-mutant compared to that of KRAS wild-type CRC patients. Of note, high serum CRP is associated with poor prognosis. Serum CRP levels also correlate with PD-L1 expression. Patients with lower serum CRP have a longer median time with regard to failure of immunotherapy compared to that of patients with higher levels of serum CRP [65]. IgM is released following initial contact with potential pathogens and is the first line of adaptive immune response [66]. Tumor-directed IgM antibodies directed against tumor-specific variants are promising agents for anti-tumor therapy [67]. In the present study, lower abundance of B cells in KRAS-mutant CRC patients corresponded with lower levels of serum IgM, indicating diminished immune infiltration related to inhibited antigen recognition and presentation. The use of serum CRP and IgM levels as predictable biomarker of immunotherapy in KRAS-mutant CRC patients remains to be elucidated.
Although the present findings provide new insights into the impact of KRAS mutation on the tumor microenvironment of CRC, there are limitations to our work. First, we identified three immune-related genes (VGF, RLN3 and CT45A1) which were differentially expressed and prognostically relevant in patients with KRAS mutations. But whether these three genes function exclusively through immune mechanisms in CRC remains undetermined. Second, the small sample size of the in-house RNA sequencing data used in this study may increase the probability of type II error. Third, the above findings may require further molecular biology experiments to confirm and clarify the underlying mechanisms.
Conclusions
In summary, this study systematically revealed the immune landscape and profiled the IRGs in KRAS-mutant and KRAS wild-type CRC patients. Mutant KRAS in CRC was associated with suppressed immune pathways and immune infiltration. An Imm-R model by intergrating TIICs and IRGs was established to determine the immune status, and for prognostic prediction in KRAS-mutant CRC patients. This study thus provides a conceptual framework to understand the tumor immune microenvironments of CRC in the context of KRAS mutation and treatment strategies designed to treat KRAS-mutant CRC patients.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request. All data analysed during this study are included in this published article (Additional files:1:8).
Abbreviations
- CRC:
-
Colorectal cancer
- IRGs:
-
Immune-related genes
- hs-CRP:
-
Hypersensitive C-reactive protein
- KRAS:
-
Kirsten rat sarcoma viral oncogene homolog
- MAPK:
-
Mitogen-activated protein kinase
- VEGF:
-
Vascular endothelial growth factor
- RTKs:
-
Receptor tyrosine kinases
- GTP:
-
Guanosine triphosphate
- GDP:
-
Guanosine diphosphatase
- EGFR:
-
Epidermal growth factor receptor
- CIRC:
-
Co-ordinate Immune Response Cluster
- MDSCs:
-
Myeloid-derived suppressor cells
- TCGA:
-
The Cancer Genome Atlas
- MAF:
-
Mutation Annotation Format
- GEO:
-
Gene Expression Omnibus
- CRP:
-
C-reactive protein
- NK:
-
Natural killer
- TIMER:
-
Tumor immune estimation resource
- TMM:
-
Trimmed mean of M-values
- FDR:
-
False discovery rate
- GSEA:
-
Gene set enrichment analysis
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- GO:
-
Gene ontology
- OS:
-
Overall survival
- PI:
-
Prognosis index
- APC:
-
Adenomatous polyposis coli
- TP53:
-
Tumor protein 53
- NF-κB:
-
Nuclear factor kappa-B
- PPI:
-
Protein–protein interaction
- ALB:
-
Albumin
- GCG:
-
Glucagon
- LEP:
-
Leptin
- IGF2:
-
Insulin-like growth factor 2
- PPBP:
-
Pro-platelet basic protein
- RLN3:
-
Relaxin 3
- CT45A1:
-
Cancer/testis antigen family 45 member A1
- TP63:
-
Tumor protein P63
- TNM:
-
T describes the size of the tumor and any spread of cancer into nearby tissue
- N:
-
Describes spread of cancer to nearby lymph nodes
- M:
-
Describes metastasi
- dMMR–MSI-H:
-
Mismatch-repair-deficient and microsatellite instability-high
- IRF2:
-
Interferon regulatory factor 2
- PRRs:
-
Pattern recognition receptors
- Tregs:
-
Regulatory T cells
- TAN:
-
Tumor-associated neutrophils
- H2O2:
-
Hydrogen peroxide
- M-CSF:
-
Macrophage colony-stimulating factor
- PGF:
-
Prostaglandin F
- IL:
-
Interleukin
- TGF-β:
-
Transforming growth factor-beta
- HCC:
-
Hepatocellular carcinoma
- HLA:
-
Human leukocyte antigen
References
Shibutani M, Maeda K, Nagahara H, Fukuoka T, Iseki Y, Matsutani S, Kashiwagi S, Tanaka H, Hirakawa K, Ohira M. Tumor-infiltrating lymphocytes predict the chemotherapeutic outcomes in patients with Stage IV colorectal cancer. Vivo. 2018;32:151–8.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Ciombor KK, Wu C, Goldberg RM. Recent therapeutic advances in the treatment of colorectal cancer. Annu Rev Med. 2015;66:83–95.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–20.
Robert C, Ribas A, Schachter J, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil CM, Lotem M, et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 2019;20:1239–51.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91.
Carr PR, Alwers E, Bienert S, Weberpals J, Kloor M, Brenner H, Hoffmeister M. Lifestyle factors and risk of sporadic colorectal cancer by microsatellite instability status: a systematic review and meta-analyses. Ann Oncol. 2018;29:825–34.
Lal N, White BS, Goussous G, Pickles O, Mason MJ, Beggs AD, Taniere P, Willcox BE, Guinney J, Middleton GW. KRAS mutation and consensus molecular subtypes 2 and 3 are independently associated with reduced immune infiltration and reactivity in colorectal cancer. Clin Cancer Res. 2018;24:224–33.
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov. 2014;13:828–51.
Serna-Blasco R, Sanz-Alvarez M, Aguilera O, Garcia-Foncillas J. Targeting the RAS-dependent chemoresistance: the Warburg connection. Semin Cancer Biol. 2019;54:80–90.
Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, et al. Kirsten ras mutations in patients with colorectal cancer: the “RASCAL II” study. Br J Cancer. 2001;85:692–6.
Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Janne PA, Januario T, Johnson DH, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900–9.
Lal N, Beggs AD, Willcox BE, Middleton GW. An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Oncoimmunology. 2015;4:e976052.
Mo X, Huang X, Feng Y, Wei C, Liu H, Ru H, Qin H, Lai H, Wu G, Xie W, et al. Immune infiltration and immune gene signature predict the response to fluoropyrimidine-based chemotherapy in colorectal cancer patients. OncoImmunology. 2020;9:1832347.
Liao W, Overman MJ, Boutin AT, Shang X, Zhao D, Dey P, Li J, Wang G, Lan Z, Li J, et al. KRAS-IRF2 axis drives immune suppression and immune therapy resistance in colorectal cancer. Cancer Cell. 2019;35(559–572):e557.
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Bhattacharya S, Andorf S, Gomes L, Dunn P, Schaefer H, Pontius J, Berger P, Desborough V, Smith T, Campbell J, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234–9.
Ru B, Wong CN, Tong Y, Yi Zhong J, Wa Zhong SS, Wu WC, Chu KC, Wong CY, Lau CY, Chen I, et al. TISIDB: an integrated repository portal for tumor-immune system interactions. Bioinformatics. 2019. https://doi.org/10.1093/bioinformatics/btz210.
Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, Jiang P, Shen H, Aster JC, Rodig S, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174.
Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21:938–45.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40.
Asaoka Y, Ijichi H, Koike K. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 1979;2015:373.
Huang X, Liu J, Wu G, Chen S, Pc FJ, Xie W, Tang W. Development and validation of a nomogram for preoperative prediction of perineural invasion in colorectal cancer. Med Sci Monit. 2019;25:1709–17.
Liu J, Huang X, Yang W, Li C, Li Z, Zhang C, Chen S, Wu G, Xie W, Wei C, et al. Nomogram for predicting overall survival in stage II-III colorectal cancer. Cancer Med. 2020. https://doi.org/10.1002/cam4.2896.
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16:361–75.
Governa V, Trella E, Mele V, Tornillo L, Amicarella F, Cremonesi E, Muraro MG, Xu H, Droeser R, Daster SR, et al. The interplay between neutrophils and CD8(+) T cells improves survival in human colorectal cancer. Clin Cancer Res. 2017;23:3847–58.
Bendell JC, Bang Y-J, Chee CE, Ryan DP, McRee AJ, Chow LQ, Desai J, Wongchenko M, Yan Y, Pitcher B, et al. A phase Ib study of safety and clinical activity of atezolizumab (A) and cobimetinib (C) in patients (pts) with metastatic colorectal cancer (mCRC). J Clin Oncol. 2018;36:560–560.
Pires BRB, Silva R, Ferreira GM. NF-kappaB: two sides of the same coin. Genes. 2018. https://doi.org/10.3390/genes9010024.
Tato CM, Mason N, Artis D, Shapira S, Caamano JC, Bream JH, Liou HC, Hunter CA. Opposing roles of NF-kappaB family members in the regulation of NK cell proliferation and production of IFN-gamma. Int Immunol. 2006;18:505–13.
Feng B, Cheng S, Pear WS, Liou HC. NF-kB inhibitor blocks B cell development at two checkpoints. Med Immunol. 2004;3:1.
Hutton JE, Wang X, Zimmerman LJ, Slebos RJ, Trenary IA, Young JD, Li M, Liebler DC. Oncogenic KRAS and BRAF drive metabolic reprogramming in colorectal cancer. Mol Cell Proteomics. 2016;15:2924–38.
Pupo E, Avanzato D, Middonti E, Bussolino F, Lanzetti L. KRAS-driven metabolic rewiring reveals novel actionable targets in cancer. Front Oncol. 2019;9:848.
Chatani PD, Yang JC. Mutated RAS: Targeting the “Untargetable” with T-cells. Clin Cancer Res. 2019. https://doi.org/10.1158/1078-0432.CCR-19-2138.
Heath WR, Kato Y, Steiner TM, Caminschi I. Antigen presentation by dendritic cells for B cell activation. Curr Opin Immunol. 2019;58:44–52.
Wang JZ, Zhang YH, Guo XH, Zhang HY, Zhang Y. The double-edge role of B cells in mediating antitumor T-cell immunity: Pharmacological strategies for cancer immunotherapy. Int Immunopharmacol. 2016;36:73–85.
Carmi Y, Spitzer MH, Linde IL, Burt BM, Prestwood TR, Perlman N, Davidson MG, Kenkel JA, Segal E, Pusapati GV, et al. Allogeneic IgG combined with dendritic cell stimuli induce antitumour T-cell immunity. Nature. 2015;521:99–104.
Hinshaw DC, Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79:4557–66.
Gershkovitz M, Caspi Y, Fainsod-Levi T, Katz B, Michaeli J, Khawaled S, Lev S, Polyansky L, Shaul ME, Sionov RV, et al. TRPM2 mediates neutrophil killing of disseminated tumor cells. Cancer Res. 2018;78:2680–90.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, Worthen GS, Albelda SM. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16:183–94.
Radsak M, Iking-Konert C, Stegmaier S, Andrassy K, Hansch GM. Polymorphonuclear neutrophils as accessory cells for T-cell activation: major histocompatibility complex class II restricted antigen-dependent induction of T-cell proliferation. Immunology. 2000;101:521–30.
Tamura R, Tanaka T, Yamamoto Y, Akasaki Y, Sasaki H. Dual role of macrophage in tumor immunity. Immunotherapy. 2018;10:899–909.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel). 2016. https://doi.org/10.3390/cancers8030036.
Rosenblum MD, Way SS, Abbas AK. Regulatory T cell memory. Nat Rev Immunol. 2016;16:90–101.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6.
Olguín JE, Medina-Andrade I, Rodríguez T, Rodríguez-Sosa M, Terrazas LI. Relevance of regulatory T Cells during colorectal cancer development. Cancers (Basel). 2020. https://doi.org/10.3390/cancers12071888.
Ling ZA, Zhang LJ, Ye ZH, Dang YW, Chen G, Li RL, Zeng JJ. Immunohistochemical distribution of FOXP3+ regulatory T cells in colorectal cancer patients. Int J Clin Exp Pathol. 2018;11:1841–54.
Xiong Y, Wang K. Profiles of immune infiltration in colorectal cancer and their clinical significant: A gene expression-based study. Cancer Med. 2018. https://doi.org/10.1002/cam4.1745.
Peng D, Wang L, Li H, Cai C, Tan Y, Xu B, Le H. An immune infiltration signature to predict the overall survival of patients with colon cancer. IUBMB Life. 2019. https://doi.org/10.1002/iub.2124.
Severini C, Ciotti MT, Biondini L, Quaresima S, Rinaldi AM, Levi A, Frank C, Possenti R. TLQP-21, a neuroendocrine VGF-derived peptide, prevents cerebellar granule cells death induced by serum and potassium deprivation. J Neurochem. 2008;104:534–44.
Busse S, Steiner J, Micheel J, Dobrowolny H, Mawrin C, Krause TJ, Adamaszek M, Bogerts B, Bommhardt U, Hartig R, Busse M. Age-related increase of VGF-expression in T lymphocytes. Aging (Albany NY). 2014;6:440–53.
Nair VB, Samuel CS, Separovic F, Hossain MA, Wade JD. Human relaxin-2: historical perspectives and role in cancer biology. Amino Acids. 2012;43:1131–40.
Binder C, Chuang E, Habla C, Bleckmann A, Schulz M, Bathgate R, Einspanier A. Relaxins enhance growth of spontaneous murine breast cancers as well as metastatic colonization of the brain. Clin Exp Metastasis. 2014;31:57–65.
Zhang Y, Wang S, Xiao J, Zhou H. Bioinformatics analysis to identify the key genes affecting the progression and prognosis of hepatocellular carcinoma. Biosci Rep. 2019. https://doi.org/10.1042/BSR20181845.
Shang B, Gao A, Pan Y, Zhang G, Tu J, Zhou Y, Yang P, Cao Z, Wei Q, Ding Y, et al. CT45A1 acts as a new proto-oncogene to trigger tumorigenesis and cancer metastasis. Cell Death Dis. 2014;5:e1285.
Coscia F, Lengyel E, Duraiswamy J, Ashcroft B, Bassani-Sternberg M, Wierer M, Johnson A, Wroblewski K, Montag A, Yamada SD, et al. Multi-level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell. 2018;175(159–170):e116.
McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–27.
Huang X, Liu J, Mo X, Liu H, Wei C, Huang L, Chen J, Tian C, Meng Y, Wu G, et al. Systematic profiling of alternative splicing events and splicing factors in left- and right-sided colon cancer. Aging. 2019. https://doi.org/10.18632/aging.102319.
Zhou X, Huang X, Liang S, Tang S, Wu S, Huang T, Mo Z, Wang Q. Identifying miRNA and gene modules of colon cancer associated with pathological stage by weighted gene co-expression network analysis. Oncotargets Ther. 2018;11:2815–30.
McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010;28:4022–8.
Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117.
Park JH, Watt DG, Roxburgh CS, Horgan PG, McMillan DC. Colorectal cancer, systemic inflammation, and outcome: staging the tumor and staging the host. Ann Surg. 2016;263:326–36.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70.
Akamine T, Takada K, Toyokawa G, Kinoshita F, Matsubara T, Kozuma Y, Haratake N, Takamori S, Hirai F, Tagawa T, et al. Association of preoperative serum CRP with PD-L1 expression in 508 patients with non-small cell lung cancer: A comprehensive analysis of systemic inflammatory markers. Surg Oncol. 2018;27:88–94.
Gronwall C, Vas J, Silverman GJ. Protective roles of natural IgM antibodies. Front Immunol. 2012;3:66.
Rasche L, Duell J, Castro IC, Dubljevic V, Chatterjee M, Knop S, Hensel F, Rosenwald A, Einsele H, Topp MS, Brandlein S. GRP78-directed immunotherapy in relapsed or refractory multiple myeloma - results from a phase 1 trial with the monoclonal immunoglobulin M antibody PAT-SM6. Haematologica. 2015;100:377–84.
Acknowledgements
The authors thank Lingxu Huang, Chao Tian, Jianhong Chen, and Zhaoting Bu for their contribution towards collection of clinical data.
Funding
This study was funded by the Guangxi Science and Technology Department Project (Guike AB16380202); the National Natural Science Foundation of China (81973533), 2019 Guangxi University High-level Innovation Team and the Project of Outstanding Scholars Program, and Guangxi Science and Technology Project(2019AC03004), Guangxi Clinical Research Center for Colorectal Cancer (Guike: AD19245197), Guangxi Science and Technology Research Project (Guike AB18221086).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: WT, XH, JL; performed the data collection: JL, XH, CW, HL, GW, HR, HQ, HL, WX, XM, WT, YM; analyzed the data: XH, JL, GW, HL, GW, HR, HQ; contributed reagents/materials/analysis tools: JL, XH, GW, CW, HR, HQ, HL, YM, XM, WT; contributed to the writing of the manuscript: JL, XH, CW, HL, YM, GW, WT; revision of the manuscript: JL, XH, CJ, YZ; all authors reviewed the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The protocol of this study was approved by the Ethics and Human Subject Committee of Guangxi Medical University Cancer Hospital (ID: LW2018039), and all experiments and methods met the standards of the relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Figure S1. Somatic mutation landscape of colorectal cancer (CRC) patients.
Additional file 2:
Figure S2. Gene set enrichment analysis for gene ontology terms.
Additional file 3: Figure S3.
The abundance of 22 immune cells estimated by CIBERSORT between the KRAS-mutant and KRAS wild-type groups.
Additional file 4:
Figure S4. Differential abundance of tumor-infiltrating immune cells between the KRAS-mutant and KRAS wild-type CRC.
Additional file 5:
Figure S5. a Univariate survival analysis of TIICs in KRAS wild-type patients based on training set. b Univariate survival analysis of TIICs in KRAS wild-type patients based on validation set.
Additional file 6:
Figure S6. Differentially expressed immune-related genes (IRGs) in CRC in the presence and absence of KRAS mutation.
Additional file 7:
Figure S7. The association between the expression of IRGs and TIICs.
Additional file 8: Figure S8.
Systemic immune and inflammatory state in colorectal cancer (CRC) with Kirsten rat sarcoma viral oncogene homolog (KRAS) mutation.
Additional file 9: Table S1.
Multivariate analyses of prognostic tumor-infiltrating immune cells in patients with KRAS-mutation
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, J., Huang, X., Liu, H. et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J Transl Med 19, 27 (2021). https://doi.org/10.1186/s12967-020-02638-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02638-9