
Abstract
Background
Acute fibrinous and organizing pneumonia (AFOP) is a rare clinicopathological condition. Studies in the literature have reported that AFOP may be associated with respiratory infections, such as respiratory syncytial virus, influenza virus, Pneumocystis jirovecii, Penicillium citrinum, and Chlamydia infections. However, AFOP associated with Legionella infection has not been reported previously. Here, we report a case of a patient with AFOP secondary to Sjögren’s syndrome and Legionella infection.
Case presentation
A 47-year-old man was admitted to the hospital because of fever, expectoration, and shortness of breath. Lung imaging showed irregular patchy consolidation. A diagnosis of Legionella pneumonia was initially considered on the basis of the patient’s history of exposure to soil before disease onset, signs of extrapulmonary involvement, and a positive Legionella urine antigen test result. However, the patient’s symptoms and lung imaging did not improve after treatment with levofloxacin, moxifloxacin, and tigecycline for Legionella infection. In addition, Sjögren’s syndrome was diagnosed on the basis of clinical manifestations and immunological indicators. Pathological changes associated with AFOP were confirmed from the results of ultrasound-guided percutaneous lung biopsy. The patient’s clinical symptoms improved rapidly after a short course of low-dose corticosteroid therapy, and lung imaging showed significant improvement.
Conclusions
The possibility of secondary AFOP should be considered when Legionella pneumonia does not improve after standard antibiotic therapy. Lung biopsy and histopathological examination are important for the adjustment of treatment strategy. Our case also highlights the importance of screening for autoimmune diseases in patients with AFOP.
Similar content being viewed by others


Background
Acute fibrinous and organizing pneumonia (AFOP) is a recently recognized rare clinicopathological condition. The histopathological characteristics of AFOP include large amounts of fibrin deposition in the alveolar cavities and changes associated with organizing pneumonia (OP) [1]. One type of AFOP has an acute onset and progresses rapidly, leading to multiple organ failure and death, with a clinical course similar to that of diffuse alveolar damage (DAD). The other type of AFOP has a subacute onset and a clinical course similar to that of cryptogenic organizing pneumonia (COP) [2]. The causes of AFOP are unclear. The onset may be related to infection, autoimmune disease, adverse drug reactions, and environmental exposures [3]. Pathogens reported to be associated with AFOP include Haemophilus influenzae, Acinetobacter baumannii, Chlamydia, respiratory syncytial virus, influenza virus A/H1N1, human immunodeficiency virus, and Pneumocystis jirovecii, Penicillium citrinum et al. [1, 4,5,6,7,8,9]. To our knowledge, there have been no reports of AFOP associated with Legionella infection. To improve the clinical understanding of AFOP, we report a case of a man with Sjögren’s syndrome and Legionella infection in which the lung pathology was consistent with that of AFOP.
Case presentation
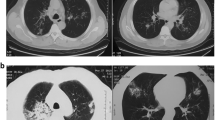
A 47-year-old Chinese male farmer, with history of contact with soil during farm work 2 days before the onset of illness, was admitted to our hospital with a 2-week history of a high fever, productive cough with purulent sputum, shortness of breath on exertion, and nausea and vomiting. Chest computed tomography (CT) performed at a local hospital before admission to our hospital had shown irregular patchy opacities in the lower lobe of the left lung (Fig. 1a). He had been treated with levofloxacin for 4 days and then switched to moxifloxacin and meropenem for 10 days at the local hospital. However, as his fever, expectoration, and shortness of breath had not improved, he was transferred to our hospital for further investigation and treatment.

Computed tomography (CT) of the patient’s chest showing progression of the pulmonary lesions. a Chest CT performed at a local hospital prior to admission to our hospital showing irregular patchy opacities in the left lower lobe. b Chest CT performed on admission to our hospital showing extension of the patchy opacities in the left lower lobe since the previous scan, and new small patchy opacities in the right middle lobe. c Follow-up chest CT performed after 1 month of corticosteroid treatment showing almost complete resolution of the pulmonary lesions
The patient had a 7-year history of dry mouth and dry eyes which had not previously been medically evaluated or treated, but had no history of other possible autoimmune disease such as recurrent oral ulcers, Raynaud’s phenomenon, rash, or arthritis. He was a smoker with a 30 pack-year smoking history. He denied a history of pulmonary disease. He didn’t undergo any previous chest X-ray examination and reported no symptoms and signs of previous interstitial lung disease such as gradually progressive dyspnea on exertion, dry cough, fatigue of unknown origin. He also denied a history of occupational dust exposure, keeping pets and drug abuse. He had no family history of pulmonary disease and cancer.
On admission, he had a temperature of 37.6 °C, pulse of 82 beats/min, respiratory rate of 16 breaths/min, and blood pressure of 123/71 mmHg. The skin on both his legs was dry and rough, but there was no rash or bruising. There were no rales in the lungs on auscultation, and no other obvious abnormalities were noted on physical examination.
The chest CT images on admission at our hospital showed that the patchy opacities in the left lower lobe had extended since the previous chest CT, and there were new small patches in the right middle lobe (Fig. 1b). Laboratory tests revealed an elevated white blood cell count and neutrophil percentage, elevated levels of procalcitonin and C-reactive protein, an increased erythrocyte sedimentation rate, mild hyponatremia, and elevated levels of liver enzymes. The serum human immunodeficiency virus antibody was negative. No infectious agents were detected in the patient’s blood culture and sputum microbiological examination (Table 1).
Because the patient had symptoms and signs of extrapulmonary system involvement, including gastrointestinal symptoms, hyponatremia, and signs of liver damage, we could not rule out Legionella pneumonia and thus we performed a Legionella urine antigen test, which was positive. The patient was treated with moxifloxacin 0.4 g once a day and piperacillin sodium and tazobactam sodium 4.5 g three times a day for 8 days. His white blood cell count, neutrophil percentage and C-reactive protein level had decreased since admission (Table 1). However, his symptoms of fever, expectoration, and shortness of breath persisted. Thus, the antibiotic treatment was switched to intravenous tigecycline 50 mg twice a day for 5 days after an initial dose of 100 mg. However, his symptoms still did not improve. To confirm the diagnosis, we performed an ultrasound-guided percutaneous needle lung biopsy and tested for markers of autoimmune disease.
The result of a tear secretion test was positive. Salivary gland emission computed tomography dynamic imaging showed impaired uptake in both submandibular glands and parotid glands. Immunological index testing gave positive results for antinuclear antibodies, anti-SSA/Ro60, anti-SSA/Ro52, anti-SSB/La and anti-histone antibody, and the serum rheumatoid factor level was increased (Table 1). Test for autoimmune liver disease-related antibodies, antineutrophil cytoplasmic antibodies, were all negative, and the serum complement C3 and C4 levels were normal.
Lung histology showed formation of fibrin balls in the alveolar cavities accompanied by changes associated with OP (Fig. 2). No pathogens were detected in the lung tissue using metagenomic next generation sequencing (mNGS). Based on the results of the lung biopsy, antibiotics were stopped and intravenous methylprednisolone 40 mg daily was initiated. The patient’s symptoms improved rapidly, and his body temperature returned to normal on the first day of corticosteroid therapy. He was discharged home 4 days later, and continued oral prednisone 10 mg daily for 20 days followed by 5 mg daily for 5 days. At the follow-up visit one month after discharge, his chest CT showed almost complete resolution of the pulmonary lesions (Fig. 1c).

Histopathology of the lung tissue. Fibrin balls are present in the alveolar cavities (arrows), and the alveolar septa are widened, with infiltration of chronic inflammatory cells and fibrosis. (Hematoxylin and eosin stain; magnification × 200). Picture was taken by ZEISS Microscope Model AXIO Lab A1, ZEISS camera Model Axiocam ICc 5 and ZEN software at a resolution of 96dpi, and processed in adobe photoshop 21.0.2 at a resolution of 300 dpi. No downstream processing was utilized
Discussion and conclusions
The concept of AFOP was first proposed by Beasley et al. [1] to describe a class of pathological tissue types that does not conform to the clinicopathological manifestations of DAD, OP, or eosinophilic pneumonia. AFOP is a disease that pathologically manifests as the formation of fibrin balls in alveolar cavities, accompanied by the development of OP. In terms of course and prognosis, there are two main types of AFOP: a type with an acute or fulminant course leading to respiratory failure with rapid progression to death and a type with a subacute course with a generally good prognosis. Our patient’s condition conformed to the latter type. It is still debated whether AFOP should be classified as an independent disease, but AFOP was classified as a rare histologic pattern in the 2013 edition of the classification of idiopathic interstitial pneumonias by the American Thoracic Society/European Respiratory Society [10]. The clinical manifestations of AFOP lack specificity and primarily manifest as shortness of breath, cough, and fever. AFOP may be related to numerous clinical conditions, such as infections, autoimmune diseases, adverse drug reactions, and environmental exposures (Table 2) [1, 3,4,5,6,7,8,9, 11,12,13]. There are no significant differences in the clinical and lung imaging manifestations of AFOP according to the cause. AFOP associated with different conditions exhibits a number of common clinical manifestations and the characteristics usually overlap with those of the primary disease.
Beasley et al. [1] reported that the amount of chronic inflammatory cell infiltration surrounding the fibrin balls in AFOP lesions varies and noted that AFOP may be associated with lung infection. AFOP may improve following antibiotic treatment [14], suggesting that infection may trigger the onset of AFOP. Previous reports have shown that a variety of infections may be associated with AFOP, including bacterial infections (Haemophilus influenzae, Acinetobacter baumannii), viral infections (respiratory syncytial virus, influenza virus A/H1N1, human immunodeficiency virus), and fungal infections (Pneumocystis jirovecii, Penicillium citrinum) [1, 5,6,7,8,9]. In the reported case, Legionella pneumonia was diagnosed based on a history of contact with soil before the onset; symptoms of fever, expectoration and shortness of breath; extrapulmonary manifestations (gastrointestinal symptom, liver function injury and hyponatremia), and the positive Legionella urine antigen test result. The reason that mNGS did not reveal Legionella in lung tissue was probably because the infection was already under control because of the early use of antibiotics effective against Legionella. Based on the result of the mNGS, the patient was treated with corticosteroid alone following discharge and remained in a stable condition.
AFOP and OP have some imaging and pathological manifestations that are common to both. Previous studies have found that Legionella pneumonia can cause changes associated with OP [15]. Haroon et al. [15] suggested that alveolar epithelial cell injury caused by Legionella infection and subsequent tissue repair are involved in the pathology of OP and the development of its imaging manifestations. After alveolar injury, proteins and serous fluid in the capillaries enter the alveolar cavity, where they may be involved in fibrin ball formation [14]. This may explain the AFOP manifestations in the lungs of this patient.
Legionella infection often manifests as severe pneumonia, with a mortality of 8–12% [16]. In nosocomial cases, the mortality can be as high as 34% [16]. When patients with Legionella pneumonia have poor response to treatment and their condition worsens, clinicians may consider whether the antibiotics are adequate, but rarely consider the possibility that the worsening may be due to the development of OP or AFOP as a complication. It has been reported that AFOP may have a case fatality rate of more than 50% [1]. Corticosteroids are the first-line treatment for AFOP, and some patients require corticosteroid pulse therapy. The use of corticosteroids during the acute stage of treatment for Legionella pneumonia is not recommended and corticosteroids should not be used routinely in patients with Legionella pneumonia. Therefore, in patients with Legionella pneumonia who do not respond to adequate antibiotic treatment, if the clinical features and imaging suggest AFOP, timely lung biopsy is very important to make the diagnosis of secondary AFOP, such that the treatment can be changed accordingly, leading to an improved prognosis.
In this patient, the possibility of AFOP related to Sjögren’s syndrome must also be considered, as the course of corticosteroid therapy was decided on the basis of the possibility that the AFOP was secondary to Sjögren’s syndrome. Previous studies have found that the pathological types of interstitial lung lesions caused by Sjögren’s syndrome include non-specific interstitial pneumonia and usual interstitial pneumonia, with some cases manifesting as OP and lymphocytic interstitial pneumonia [17,18,19]. Chest CT imaging typically shows predominantly ground-glass opacities, linear opacities, interlobular septal thickening, cysts, reticulation, and nodules [18,19,20,21,22]. Consolidation opacities similar to those observed in community-acquired pneumonia are rare. So far, including our case, a total of 3 cases of Sjögren’s syndrome with AFOP have been reported (Table 3) [13, 23]. The lung imaging abnormalities in those two previous reported patients consisted primarily of nodules or nodular consolidation with or without ground-glass opacities. One of the patients was treated with immunosuppressive agents in addition to corticosteroids. Our patient’s lung imaging was reported to show patchy consolidation, which differed from the usual pulmonary involvement associated with Sjögren’s syndrome. Sjögren’s syndrome is a relatively indolent connective tissue disease, and not all patients with Sjögren’s syndrome require corticosteroid and/or immunosuppressive therapy. According to the European League Against Rheumatism criteria for assessment of Sjögren’s syndrome [24], if our patient’s lung changes were secondary to Sjögren’s syndrome, this would be consistent with moderate Sjögren’s syndrome, and a medium-to-high-dose corticosteroid therapy would be required, with tapering and maintenance at a low dose before discontinuation. Treatment with immunosuppressive agents may also be required in patients with AFOP secondary to autoimmune disease. Wang et al. [13] reviewed 13 cases of AFOP associated with autoimmune disease and found that most cases required immunosuppressive therapy in addition to corticosteroid therapy. However, our patient required only a short course of low-dose corticosteroid therapy, and his symptoms and imaging rapidly improved. In addition, although the patient didn’t undergo previous chest X-ray, he denied a history of pulmonary disease and lacked risk factors, symptoms and signs of interstitial lung disease. Furthermore, the patient had experienced symptoms of Sjögren’s syndrome for many years before the onset of AFOP, without respiratory symptoms, suggesting that the AFOP is most likely to have been secondary to Legionella infection rather than Sjögren’s syndrome. It is possible that Sjögren’s syndrome played a synergistic role with Legionella infection in the pathogenesis of AFOP in our patient.
In summary, we reported a case of AFOP secondary to Sjögren’s syndrome with Legionella infection. Although AFOP is a rare clinicopathological disorder, the possibility of AFOP secondary to Legionella pneumonia should be considered in patients with Legionella pneumonia who do not respond to antibiotic therapy and in patients with Sjögren’s syndrome with acute onset of respiratory symptoms, with or without infectious conditions. Pathological examination should be conducted as early as possible if patients with Legionella pneumonia do not respond to appropriate antibiotic treatment, and corticosteroids should be administered to improve prognosis. Our report also highlights the importance of screening for autoimmune diseases in patients with AFOP.
Availability of data and materials
All data and material analyzed during this study are included in this published article.
Abbreviations
- AFOP:
-
Acute fibrinous and organizing pneumonia
- COP:
-
Cryptogenic organizing pneumonia
- CT:
-
Computed tomography
- mNGS:
-
Metagenomic next generation sequencing
- OP:
-
Organizing pneumonia
References
Beasley MB, Franks TJ, Galvin JR, Gochuico B, Travis WD. Acute fibrinous and organizing pneumonia: a histological pattern of lung injury and possible variant of diffuse alveolar damage. Arch Pathol Lab Med. 2002;126(9):1064–70. https://doi.org/10.5858/2002-126-1064-AFAOP.
Kim JY, Doo KW, Jang HJ. Acute fibrinous and organizing pneumonia: Imaging features, pathologic correlation, and brief literature review (✰). Radiol Case Rep. 2018;13(4):867–70. https://doi.org/10.1016/j.radcr.2018.04.028.
Akhtar A, Ul AZ. Acute fibrinous and organizing pneumonia masquerading as a lower respiratory tract infection: a case report and review of the literature. BMC Res Notes. 2015;8:38. https://doi.org/10.1186/s13104-015-0984-4.
Ribera A, Llatjós R, Casanova A, Santin M. Chlamydia pneumoniae infection associated to acute fibrinous and organizing pneumonia. Enferm Infecc Microbiol Clin. 2011;29(8):632–4. https://doi.org/10.1016/j.eimc.2011.01.018.
Cincotta DR, Sebire NJ, Lim E, Peters MJ. Fatal acute fibrinous and organizing pneumonia in an infant: The histopathologic variability of acute respiratory distress syndrome. Pediatr Crit Care Med. 2007;8(4):378–82. https://doi.org/10.1097/01.PCC.0000269375.10806.60.
Otto C, Huzly D, Kemna L, Hüttel A, Benk C, Rieg S, et al. Acute fibrinous and organizing pneumonia associated with influenza A/H1N1 pneumonia after lung transplantation. BMC Pulm Med. 2013;13:30. https://doi.org/10.1186/1471-2466-13-30.
Rapaka V, Hussain MA, Niazi M, Diaz-Fuentes G. Severe acute fibrinous and organizing pneumonia causing acute respiratory distress syndrome and shock. J Bronchol Interv Pulmonol. 2011;18(3):269–73. https://doi.org/10.1097/LBR.0b013e318222a4f2.
Heo JY, Song JY, Noh JY, Yong HS, Cheong HJ, Kim WJ. Acute fibrinous and organizing pneumonia in a patient with HIV infection and Pneumocystis jiroveci pneumonia. Respirology. 2010;15(8):1259–61. https://doi.org/10.1111/j.1440-1843.2010.01845.x.
Zhao J, Shi Y, Yuan D, Shi Q, Wang W, Su X. A case report of fungal infection associated acute fibrinous and organizing pneumonitis. BMC Pulm Med. 2020;20(1):98. https://doi.org/10.1186/s12890-020-1145-7.
Travis WD, Costabel U, Hansell DM, King TE Jr, Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48. https://doi.org/10.1164/rccm.201308-1483ST.
Arnaud D, Surani Z, Vakil A, Varon J, Surani S. Acute fibrinous and organizing pneumonia: a case report and review of the literature. Am J Case Rep. 2017;18:1242–6. https://doi.org/10.12659/ajcr.905627.
Yamamoto M, Murata K, Kiriu T, Kouzai Y, Takamori M. Acute fibrinous and organizing pneumonia with myelodysplastic syndrome: corticosteroid monotherapy led to successful ventilator weaning. Intern Med. 2016;55(21):3155–9. https://doi.org/10.2169/internalmedicine.55.6864.
Wang Y, Zhao S, Du G, Ma S, Lin Q, Lin J, et al. Acute fibrinous and organizing pneumonia as initial presentation of primary Sjogren’s syndrome: a case report and literature review. Clin Rheumatol. 2018;37(7):2001–5. https://doi.org/10.1007/s10067-018-4128-9.
Jabbour R, Kumar H, Alvi S, Nannaka VB, Niazi M, Patel M, et al. An unusual presentation of acute fibrinous and organizing pneumonia. Am J Case Rep. 2017;18:532–6. https://doi.org/10.12659/ajcr.903539.
Haroon A, Higa F, Hibiya K, Haranaga S, Yara S, Tateyama M, et al. Organizing pneumonia pattern in the follow-up CT of Legionella-infected patients. J Infect Chemother. 2011;17(4):493–8. https://doi.org/10.1007/s10156-010-0205-y.
Phin N, Parry-Ford F, Harrison T, Stagg HR, Zhang N, Kumar K, et al. Epidemiology and clinical management of Legionnaires’ disease. Lancet Infect Dis. 2014;14(10):1011–21. https://doi.org/10.1016/S1473-3099(14)70713-3.
Reina D, Roig Vilaseca D, Torrente-Segarra V, Cerda D, Castellvi I, Diaz Torne C, et al. Sjogren’s syndrome-associated interstitial lung disease: a multicenter study. Reumatol Clin. 2016;12(4):201–5. https://doi.org/10.1016/j.reuma.2015.09.003.
Luppi F, Sebastiani M, Silva M, Sverzellati N, Cavazza A, Salvarani C, et al. Interstitial lung disease in Sjögren’s syndrome: a clinical review. Clin Exp Rheumatol. 2020;38 Suppl 126(4):291–300.
Luppi F, Sebastiani M, Sverzellati N, Cavazza A, Salvarani C, Manfredi A. Lung complications of Sjogren syndrome. Eur Respir Rev. 2020;29(157):200021. https://doi.org/10.1183/16000617.0021-2020.
Flament T, Bigot A, Chaigne B, Henique H, Diot E, Marchand-Adam S. Pulmonary manifestations of Sjogren’s syndrome. Eur Respir Rev. 2016;25(140):110–23. https://doi.org/10.1183/16000617.0011-2016.
Lin W, Xin Z, Zhang J, Liu N, Ren X, Liu M, et al. Interstitial lung disease in Primary Sjögren’s syndrome. BMC Pulm Med. 2022;22(1):73. https://doi.org/10.1186/s12890-022-01868-5.
Dong X, Zhou J, Guo X, Li Y, Xu Y, Fu Q, et al. A retrospective analysis of distinguishing features of chest HRCT and clinical manifestation in primary Sjögren’s syndrome-related interstitial lung disease in a Chinese population. Clin Rheumatol. 2018;37(11):2981–8. https://doi.org/10.1007/s10067-018-4289-6.
Fasanya A, Gandhi V, DiCarlo C, Thirumala R. Acute fibrinous and organizing pneumonia in a patient with Sjogren’s syndrome. Respir Med Case Rep. 2016;20:28–30. https://doi.org/10.1016/j.rmcr.2016.11.010.
Ramos-Casals M, Brito-Zeron P, Bombardieri S, Bootsma H, De Vita S, Dorner T, et al. EULAR recommendations for the management of Sjogren’s syndrome with topical and systemic therapies. Ann Rheum Dis. 2020;79(1):3–18. https://doi.org/10.1136/annrheumdis-2019-216114.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the National Key R&D Program of China (No. 2017YFC1309702) and the National Natural Science Foundation of China (No. 81170009). The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
YL collected the data and participated in manuscript writing. YC conceived this study and participated in manuscript writing. WZ, WC, XY, LZ and YC critically revised the manuscript and provided guidance. WZ, LZ and YC critically treated the patient. WC and XY performed the histological analysis. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
As this is a case report, not a research study, ethical approval was not required. All procedures performed were in accordance with the ethical standards of the institutional research committee and with the Helsinki Declaration (as revised in 2013).
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and the accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lu, Y., Zheng, W., Cao, W. et al. Acute fibrinous and organizing pneumonia in a patient with Sjögren’s syndrome and Legionella pneumonia: a case report and literature review. BMC Pulm Med 22, 205 (2022). https://doi.org/10.1186/s12890-022-01997-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-022-01997-x