
Abstract
Background
Association of cigarette smoking habits with the risk of prostate cancer is still a matter of debate. This systematic review and meta-analysis aimed to assess the association between cigarette smoking and prostate cancer risk.
Methods
We conducted a systematic search on PubMed, Embase, Cochrane Library, and Web of Science without language or time restrictions on June 11, 2022. Literature search and study screening were performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement. Prospective cohort studies that assessed the association between cigarette smoking habits and the risk of prostate cancer were included. Quality assessment was conducted using the Newcastle–Ottawa Scale. We used random-effects models to obtain pooled estimates and the corresponding 95% confidence intervals.
Results
A total of 7296 publications were screened, of which 44 cohort studies were identified for qualitative analysis; 39 articles comprising 3 296 398 participants and 130 924 cases were selected for further meta-analysis. Current smoking had a significantly reduced risk of prostate cancer (RR, 0.74; 95% CI, 0.68–0.80; P < 0.001), especially in studies completed in the prostate-specific antigen screening era. Compared to former smokers, current smokers had a significant lower risk of PCa (RR, 0.70; 95% CI, 0.65–0.75; P < 0.001). Ever smoking showed no association with prostate cancer risk in overall analyses (RR, 0.96; 95% CI, 0.93–1.00; P = 0.074), but an increased risk of prostate cancer in the pre-prostate-specific antigen screening era (RR, 1.05; 95% CI, 1.00–1.10; P = 0.046) and a lower risk of prostate cancer in the prostate-specific antigen screening era (RR, 0.95; 95% CI, 0.91–0.99; P = 0.011) were observed. Former smoking did not show any association with the risk of prostate cancer.
Conclusions
The findings suggest that the lower risk of prostate cancer in smokers can probably be attributed to their poor adherence to cancer screening and the occurrence of deadly smoking-related diseases, and we should take measures to help smokers to be more compliant with early cancer screening and to quit smoking.
Trial registration
This study was registered on PROSPERO (CRD42022326464).
Similar content being viewed by others

Background
Prostate cancer (PCa) is the second most commonly diagnosed cancer and the fifth leading cause of cancer death among males, with an estimated 1.4 million new cases and 375 000 deaths worldwide in 2020, accounting for 7.3% and 3.8% of all cancers diagnosed, respectively [1]. Various endogenous and exogenous risk factors for PCa have been discussed for decades. Several factors have been identified to be associated with an increased risk of PCa, for instance, family history [2], elevated hormone levels [2], black ethnicity [2], and high alcohol consumption [3]. Conversely, several factors have been associated with a decreased risk of PCa, such as higher intake of tomatoes [4], increased coffee consumption [5] and sexual activity [6].
Smoking is a well-established risk factor for several cancers, such as lung cancer, head and neck cancer, bladder cancer, and esophageal cancer [7, 8]. However, the data on the association between smoking and PCa incidence are conflicting [9, 10]. In a meta-analysis of 24 prospective cohort studies [11], M. Huncharek showed that current smokers had no increased risk of incident PCa, but in data stratified by amount smoked, a significant elevated risk was observed, and former smokers had a higher risk of PCa in comparison with never smokers. Another meta-analysis conducted in 2014 [12] revealed an inverse association between current smoking and PCa risk, while in studies completed before the prostate-specific antigen (PSA) screening era, ever smoking was positively associated with PCa. In addition, a recent pooled study of five Swedish cohorts [13] demonstrated that former smokers and current smokers had a lower risk of PCa than never smokers, and smoking intensity was inversely associated with PCa risk, especially in the PSA screening era.
Biological mechanisms underlying smoking and PCa risk have been studied for many years. Burning cigarettes can produce more than 7000 chemicals, and at least 70 carcinogens such as polycyclic aromatic hydrocarbons (PAHs) and cadmium [14]. Mutations or functional polymorphism in genes involved in PAH metabolism and detoxification may increase the risk of PCa [15]. The glutathione-S-transferases (GSTs) are a class of enzymes that can detoxify PAHs. The most common subtypes of GSTs in human prostate are GSTP and GSTM, which were reported to be associated with an increased risk of PCa in smokers [15, 16]. Cadmium induces prostate carcinogenesis through interaction with the androgen receptor because of its androgen-like activity, and it also enhances androgen-mediated transcriptional activity when in combination with the androgen [17]. A higher level of androgen was related to increased PCa risk [2, 18]. Smoking can increase testosterone concentrations by promoting testosterone secretion from Leydig cells or acting as an aromatase inhibitor [19]. Mutations in the p53 gene and CYP1A1 gene showed a higher risk of PCa in smokers, suggesting that smoking may have a joint effect on PCa risk when combined with susceptible genotypes [20]. Increased heme oxygenase 1 (HO-1) messenger RNA expression and upregulated HO-1 protein levels were observed in PCa cell lines DU 145 and PC3 [21], implying that HO-1 may play a role in the development of PCa for its function in promoting angiogenesis [22]. Evidence also suggested that prostatic inflammation may be involved in the development and progression of PCa [23]. Cigarette smoke augments the production of numerous pro-inflammatory cytokines, decreases the levels of anti-inflammatory cytokines, and activates macrophage and dendritic cell activity in many ways [24].
We performed this systematic review and meta-analysis to investigate the association of cigarette smoking habits with the risk of PCa. We aimed to include a larger sample of studies than previous meta-analyses and collect the latest evidence and the most comprehensive information on the association between cigarette smoking and PCa risk. Our primary objective was to assess the risk of PCa in current smokers, former smokers, and ever smokers. We hypothesized that smokers have a higher risk of PCa compared to non-smokers.
Methods
Search strategy
This systematic review and meta-analysis was performed according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement [25]. Two independent investigators (XWY and HC) searched PubMed, Embase, Cochrane Library, and Web of Science for publications from database inception to June 11, 2022. The following search terms were used: ("Prostate cancer") AND ("Cigarette" OR "Smoking" OR "Tobacco") AND ("Risk" OR "Incidence"). No language restrictions were applied. Reference lists of identified articles and relevant reviews were screened for additional studies. Details of the protocol for this systematic review were registered on PROSPERO and can be accessed by CRD42022326464.
Selection criteria
Prospective cohort studies investigating the association between cigarette smoking and PCa risk were included for analysis. The primary outcome was the risk of PCa. Those studies that provided an effect measure (i.e., a relative risk) quantifying the impact of smoking on the risk of PCa were considered for further quantitative synthesis (meta-analysis). The removal of duplicates and assessment of article eligibility were conducted independently by XWY and HC, and any disagreements were resolved by consulting the senior author (JP). Review articles, editorials, meeting abstracts, case‒control studies, cross-sectional studies, and those not on the topic were excluded.
Quality assessment
All included studies were independently assessed by XWY and HC for risk of bias using the Newcastle‒Ottawa Scale for cohort studies [26]. This scale assesses the selection of the study groups, the comparability of the groups, and the ascertainment of the outcome of interest. Studies with 7–9 scores were considered to be of high quality, those with 5–6 scores were classified as intermediate quality, and those with less than 4 scores were classified as low quality. Disagreements in the quality assessment were resolved by consulting JP.
Data extraction
Data were independently extracted by XWY and HC. All extracted variables were cross-checked to ensure their reliability. We recorded the total number of participants, PCa cases, and the mean or median follow-up time across all included studies. Relative risks (RRs) and the corresponding 95% confidence intervals (CIs) were retrieved or calculated using frequency distributions. Considering the prevalence rate of PCa in the public, we believed that the odds ratio was close to the RR [27, 28]. Hazard ratios (HRs) and RRs are different, HRs contain temporal information but RRs do not [28]. We converted HRs to RRs based on the formula provided by Shor E et al. [29], and the corresponding 95% CIs were converted using the same method. RRs and 95% CIs of ever smokers were computed by combining the results for former and current smokers when these results were not reported in the original papers. In addition, we recorded the baseline characteristics, methods, adjusted confounding factors, and other important comments to establish comparability. Discrepancies were discussed and resolved by consensus.
Statistical analysis
Three authors (SZQ, XJC and YYS) performed statistical analyses using Stata software, version 16.0 (StataCorp). When both crude and adjusted RRs were provided, we used the most fully adjusted value. We calculated the pooled RRs and 95% CIs and plotted forest plots using random-effects models (DerSimonian‒Laird method) for the association of current smoking, former smoking, and ever smoking with the risk of PCa [30]. Statistical heterogeneity across the trials was assessed using the I2 statistic and the Cochran’s Q test. Values of the I2 statistic of approximately 25%, 50%, and 75% were interpreted as low, moderate, and high heterogeneity, respectively [31]. In the case of low heterogeneity, a fixed-effects model (Inverse variance method) was applied. We plotted funnel plots and used Egger’s test to examine publication bias. Additionally, a series of sensitivity analyses were performed to assess the robustness of our results. We stratified studies by reference status (never smoker, former smokers), completion year (pre-PSA screening era vs. PSA screening era), world region (North America vs. Europe vs. Asia vs. Australia), and the Newcastle‒Ottawa Scale score (≤ 6 points vs. > 6 points). We considered 1995 as a cutoff year of study completion to distinguish studies before and after the PSA screening era [12]. All tests were two-tailed, and P < 0.05 was considered statistically significant.
Results
Study population
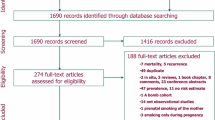
We identified 7296 citations, and after removing duplicates, 4963 citations remained for screening. After the removal of ineligible citations, we retained 60 articles that we assessed for eligibility by reading the full text; 16 of these were excluded for specific reasons. Finally, 44 studies met our inclusion criteria for qualitative synthesis and meta-analysis (Fig. 1). The number of participants and PCa cases from each selected study for systematic review ranged separately from 997 to 844 455 and 54 to 40 821, with a median of 22 677 and 382, respectively. Overall, 39 studies with 3 296 398 participants and 130 924 cases were identified for meta-analysis, and 5 studies with 91 377 participants and 1364 cases were not included in meta-analysis due to lack of information (Additional file 1). Articles were published between 1989 and 2022 and were from studies conducted in the following geographic regions: 19 from Europe (4 from the United Kingdom, 4 from Norway, 3 from Sweden, 2 from Finland, 1 from France, 1 from the Netherlands, 1 from Denmark, 1 from Lithuania, 1 from Iceland, and one from 10 European countries), 18 from North America (17 from the United States, 1 from Canada), 5 from Asia (3 from Japan, 1 from South Korea, 1 from Singapore), and 2 from Australia. The median score of quality assessment for all eligible studies was 7, with a range of 6–9 (Additional file 2).

Flow diagram of included studies
Current smoking
In total, 37 studies [6, 13, 32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66] reported the risk of current smoking on PCa, among which 6 studies [32, 35, 41, 53, 55, 63] took non-smokers as the reference and the remaining 31 studies [6, 13, 33, 34, 36,37,38,39,40, 42,43,44,45,46,47,48,49,50,51,52, 54, 56,57,58,59,60,61,62, 64,65,66] took never smokers as the reference. We defined non-smokers as never smokers plus former smokers. RRs and 95% CIs of current smokers versus non-smokers were calculated using frequency distributions in never smokers and former smokers when the risk estimates were not provided in original studies. Ten studies [34, 36, 38, 39, 42, 43, 54, 58, 59, 66] did not provide enough data on frequency distribution and were not included in analysis. Twenty-seven studies [6, 13, 32, 33, 35, 37, 40, 41, 44,45,46,47,48,49,50,51,52,53, 55,56,57, 60,61,62,63,64,65] were included to calculate the pooled RR and 95% CI. The results showed that current smoking at baseline was associated with a reduced risk of PCa (RR, 0.74; 95% CI, 0.68–0.80; P < 0.001) (Fig. 2). The I2 statistic and the Cochran’s Q test showed high heterogeneity (I2 = 90.5%; P < 0.001). Inspection of the funnel plot did not demonstrate publication bias (P = 0.231; Fig. 3).

Forest plot for the association between current smoking and prostate cancer. RR, relative risk; CI, confidence interval; PCa, prostate cancer; US, United States; UK, United Kingdom. a Rohrmann et al. [49] had two sub-populations. b RR and 95% CI were calculated using frequency distributions. c RR and 95% CI were converted from HR and corresponding 95% CI using the formula RR ≈ (1-e HR x ln (1−P0))/P0 (P0 refers to the incidence rate of PCa in the control group). d Weights were from random effects analysis

Funnel plot for publication bias in the studies investigating current smoking and prostate cancer risk. SE, standard error. Twenty-eight dots from 27 studies. P = 0.231
When performing sensitivity analyses (Additional file 3) stratified by reference status, studies using never smokers as the reference [6, 13, 33, 34, 36,37,38,39,40, 42,43,44,45,46,47,48,49,50,51,52, 54, 56,57,58,59,60,61,62, 64,65,66] showed a similar inverse association with PCa risk (RR, 0.90; 95% CI, 0.86–0.95; P < 0.001), with the heterogeneity lower than that of analysis of studies using non-smokers as the reference (I2 = 66.7%; P < 0.001). Compared to former smokers, current smokers had a significant lower risk of PCa (RR, 0.70; 95% CI, 0.65–0.75; P < 0.001) based on 21 studies [6, 13, 33, 37, 40, 44,45,46,47,48,49,50,51,52, 56, 57, 60,61,62, 64, 65]. In the pre-PSA screening era, current smoking showed a decreased risk of PCa (RR, 0.79; 95% CI, 0.64–0.98; P = 0.033) compared to non-smokers, while in the PSA screening era, the risk was significantly lower (RR, 0.72; 95% CI, 0.66–0.79; P < 0.001). When stratified by world region, studies conducted in North America, Europe, Asia, and Australia showed a negative association between current smoking and PCa risk. We also performed subgroup analyses in 21 studies with quality scores ≥ 7 [6, 13, 32, 33, 35, 40, 44,45,46, 49,50,51,52, 55,56,57, 60,61,62, 64, 65] and 6 studies with quality scores of 6 [37, 41, 47, 48, 53, 63]. Thereupon, both demonstrated a reduced risk of PCa.
Former smoking
Meta-analysis on former smoking as a risk factor for PCa was performed in 31 studies (Fig. 4) [6, 13, 33, 34, 36,37,38,39,40, 42,43,44,45,46,47,48,49,50,51,52, 54, 56,57,58,59,60,61,62, 64,65,66], and the results showed no significant association between former smoking and the risk of PCa (RR, 0.98; 95% CI, 0.95–1.02; P = 0.313). The data were heterogeneous according to the I2 statistic and the Cochran’s Q test (I2 = 61.5%; P < 0.001). Inspection of the corresponding funnel plot did not show evidence of publication bias (P = 0.431; Fig. 5). Sensitivity analyses stratified by PSA screening era, world region, and quality score also demonstrated no association between former smoking and PCa risk (Additional file 3).

Forest plot for the association between former smoking and prostate cancer. a Rohrmann et al. [49] had two sub-populations. b RR and 95% CI were calculated using frequency distributions. c RR and 95% CI were converted from HR and corresponding 95% CI using the formula RR ≈ (1-e HR x ln (1−P0))/P0 (P0 refers to the incidence rate of PCa in the control group). d Weights were from random effects analysis

Funnel plot for publication bias in the studies investigating former smoking and prostate cancer risk. Thirty-two dots from 31 studies. P = 0.431
Ever smoking
Thirty-three studies were included in the meta-analysis to assess the association of ever smoking with the risk of PCa (Fig. 6) [6, 13, 33, 34, 36,37,38,39,40, 42,43,44,45,46,47,48,49,50,51,52, 54, 56,57,58,59,60,61,62, 64,65,66,67,68]. Two of those studies [67, 68] provided RRs and 95% CIs in the original paper, and the risk estimates of the remaining 31 studies [6, 13, 33, 34, 36,37,38,39,40, 42,43,44,45,46,47,48,49,50,51,52, 54, 56,57,58,59,60,61,62, 64,65,66] were calculated by combining results for former and current smokers. Thereupon, the pooled RR and 95% CI showed no association with the risk of PCa (RR, 0.96; 95% CI, 0.93–1.00; P = 0.074), with an I2 value of 67.0% and a negative result of publication bias ((P = 0.672; Fig. 7). The association was inverse when analyzing studies completed in the PSA screening era (RR, 0.95; 95% CI, 0.91–0.99; P = 0.011), but in the pre-PSA screening era, ever smokers showed a significantly increased risk of PCa compared to never smokers (RR, 1.05; 95% CI, 1.00–1.10; P = 0.046) (Additional file 3). Four studies [50, 57, 60, 67] from Asia showed a pooled reduced risk of PCa in ever smokers (RR, 0.82; 95% CI, 0.74–0.91; P < 0.001), and studies from North America, Europe, and Australia revealed no association between ever smoking and PCa incidence. In terms of subgroup analyses stratified by quality score, the studies with a quality score ≥ 7 showed a modest negative association with PCa risk (RR, 0.96; 95% CI, 0.92–1.00; P = 0.047), while the studies with a quality score of 6 showed no association.

Forest plot for the association between ever smoking and prostate cancer. DM, diabetes mellitus. a Rohrmann et al. [49] had two sub-populations. b RR and 95% CI were calculated using frequency distributions or risk estimates and 95% CIs in subgroups. c RR and 95% CI were converted from HR and corresponding 95% CI using the formula RR ≈ (1-e HR x ln (1−P0))/P0 (P0 refers to the incidence rate of PCa in the control group). d Weights were from random effects analysis. e Onitilo et al. [68] had two sub-populations

Funnel plot for publication bias in the studies investigating ever smoking and prostate cancer risk. Thirty-five dots from 33 studies. P = 0.672
Studies not included in the meta-analysis
Of these 5 studies (Additional file 1) [69,70,71,72,73], 4 studies (involving 211 cases, 524 cases, 127 cases, and 129 cases) [69, 70, 72, 73] reported no significant association between cigarette smoking and the risk of PCa, 2 of which had a smoking category increment of 10 cigarette per day [69] or cigarette pack-years per 10 years [72]. The study conducted by Karlsen et al. [73] did not differentiate cigarette, cigar, cheroot, and pipe when assessing the risk of PCa in smokers, and as a result, this study could not be included in the meta-analysis. In the study conducted by Chamie et al. [71], a reduced PCa risk was reported in participants with a smoking history (with 13,144 participants and 363 cases; RR, 0.78; 95% CI, 0.72–0.85; P < 0.001).
Discussion
In this systematic review and meta-analysis, we found that current smoking was inversely associated with the risk of PCa, especially in the PSA screening era, which was inconsistent with our hypothesis but was consistent with the results of the recent studies [12, 13]. In studies using never smokers as the reference, current smoking revealed a similar negative correlation with PCa risk, accompanied by less heterogeneity. Current smokers had a lower risk of PCa compared to former smokers. Former smoking and ever smoking were not associated with PCa risk in the overall analyses. However, when stratified by completion year, ever smoking showed an increased risk of PCa in the pre-PSA screening era and a lower risk of PCa in the PSA screening era. Studies from North America, Europe, Asia, and Australia showed a similar reduced PCa risk in current smokers compared to non-smokers, whereas in ever smokers, only studies conducted in Asia demonstrated a decreased risk of PCa. There are several explanations for these results. Current smoking was believed to be associated with a lower likelihood of PSA testing [74, 75], and individuals with a smoking history were less likely to undergo prostate biopsy [62, 76]. As a consequence, the detection rate of PCa could be relatively lower among participants in the PSA screening era. The difference in the patterns of the association between ever smoking and PCa risk in Asia and other regions can be attributed to the higher proportion of studies in the PSA screening era in Asia than afterward. Additionally, the differences in race/ethnicity, socioeconomic status, educational attainment, and health literacy may also play important roles in explaining regional distinctions [77,78,79]. In a national cross-sectional survey, PSA testing was significantly higher in US-born men and older non-Hispanic White men than in foreign-born men and men from other racial categories [77]. Another study revealed that White men aged > 50 years were more likely than Black men to undergo PSA testing, and those with lower socioeconomic status were associated with less PSA testing [78]. The association of education levels with the preference for PSA screening was inconsistent [77, 79]. Johnson JA et al. [77] declared that higher educational levels were associated with higher odds of ever having had a PSA test; however, Pickles K et al. [79] announced that the preference for PSA screening was stronger in those without tertiary education and with inadequate health literacy. The age of the participants in the selected studies varied widely, and therefore, the willingness to receive PSA screening differs considerably; older people often show poorer adherence to PSA testing guidelines [77]. On the other hand, the relationship between PSA levels and smoking is still a matter of debate. According to an Italian cross-sectional study [80], PSA accuracy was reported to be lower in smokers than in nonsmokers and former smokers, suggesting that the need for PSA-based prostate biopsy can be affected to a certain extent by smoking.
Another possible explanation is that smoking is the leading risk factor for death among males [81]. Smokers may die from smoking attributable diseases including cancers, cardiovascular diseases, and respiratory diseases before their diagnosis of PCa. The majority of cases of lung cancer [7], head and neck cancer [82], approximately 50% of bladder cancer cases [83], and 49% of esophageal squamous cell carcinoma cases [84] are caused by cigarette smoking. Furthermore, smoking was reported to cause nearly 90% of lung cancer deaths [7] and showed significant associations with poor survival in patients with head and neck cancer [85]. Moreover, the detection of asymptomatic PCa can be frequently ignored when focusing on a more aggressive cancer. In addition, smoking increases the risk for stroke and coronary heart disease by 2 to 4 times, and stroke and coronary heart disease are considered to be the leading causes of death in the United States [8], and most of these deaths are caused by smoking [86]. Smoking can also cause chronic obstructive pulmonary disease (COPD), increasing 12 to 13-fold risk of dying from COPD than nonsmokers [8], and nearly 80% of deaths from COPD can be ascribed to smoking [86].
Our study found an increased risk of PCa among ever smokers in the pre-PSA screening era, indicating that it is necessary to promote smoking cessation as early as possible. Nearly one in five deaths are caused by cigarette smoking in the United States, leading to more than 480 000 deaths each year [8]. Continued tobacco use has been shown to limit the effectiveness of major cancer treatments, increase the risk of treatment-related complications and the development of secondary cancers, and lower cancer survival rates and the quality of life of patients [7]. In patients with PCa, smokers at the time of PCa diagnosis are associated with more aggressive characteristics, and the risk of experiencing biochemical recurrence, distant metastasis, cancer-specific mortality, and overall mortality is much higher [9, 10, 12, 87, 88]. Nicotine-induced chronic prostatic inflammation [23, 89], aberrant CpG methylations of adenomatous polyposis coli and glutathione S-transferase pi are the potential biological mechanisms responsible for these [90]. Although the effect of smoking cessation on PCa progression remains unclear, the negative impact of smoking has suggested to be maintained as long as 10 years after smoking cessation [10]. Additionally, active smoking is associated with adverse reproductive health outcomes, type 2 diabetes mellitus, and rheumatoid arthritis, harming nearly every organ of the body and resulting in significant economic costs for smokers, their families, and society [7].
Much progress has been made in promoting smoking cessation in recent decades. However, it is far from sufficient. In 2018, 13.7% of all adults (34.2 million people) in the United States were reported as current cigarette smokers [91]. Of them, 55.1% had made an attempt to quit in the past 12 months, but only 7.5% achieved success. Overcoming both physical nicotine dependence and long-standing rewarding behavior is a huge challenge, and most individuals relapse within 3 months after quitting smoking [92]. Evidence has indicated that the combination of behavioral and pharmacological interventions produces the largest cessation effects [7, 8, 92]. Nevertheless, fewer than one-half of tobacco users were offered cessation treatment according to a survey of oncology providers [93], and the inability to get patients to quit and patient resistance to treatment are two dominant barriers to cessation intervention. A brief intervention may be more acceptable and sustainable to help smokers quit smoking, according to a randomized clinical trial performed at emergency departments in Hong Kong [94]. Quitlines are good alternatives to interventions for both patients and clinicians because of their convenience and specialization, and their roles in improving smoking cessation rates have been confirmed [95]. For smokers with time constraints, internet-based self-help materials such as the website smokefred.gov and newer smartphone applications have also shown benefits in promoting smoking cessation and can serve as good alternatives [96, 97].
Strengths and limitations
The key strength of this systematic review is that the study comprised a total of 44 prospective cohort studies, 39 of which were included in the meta-analysis, with the largest number of participants and PCa cases to date. Furthermore, we included all the data on current smoking, former smoking, and ever smoking in the analysis without date and language restrictions, which means that the study provides the latest evidence and the most comprehensive information on the association between cigarette smoking and risk of PCa. We assessed the quality of each selected study using the Newcastle‒Ottawa Scale for cohort studies, and the median score was 7 and the lowest score was 6, suggesting that the quality of the included studies can be guaranteed. Other strengths include applying independent literature search, quality assessment, and data extraction by two investigators; conducting several sensitivity analyses; and using Egger’s test to examine publication bias.There are some limitations of our study. Most of the information on smoking habits was obtained from self-administered questionnaires, and the definitions of current smokers and former smokers were not completely the same between different studies. Some participants may have changed their smoking habits after baseline investigations, but repeated assessment of smoking exposure was absent in primary studies. We calculated RRs and the corresponding 95% CIs using frequency distributions without adjusting confounding factors when risk estimates were not reported. We focused on the impact of cigarette smoking on the risk of PCa; second-hand cigarette smoke and the use of other tobacco products (cigars, smokeless tobacco, e-cigarettes, pipes, etc.) that have showed increased risk of many cancers in numerous studies [98, 99] were not discussed. Alcohol consumption showed a significant dose–response relationship with PCa risk in several studies [3, 100], and were often used concurrently with cigarette smoking [101], but we didn’t analyze the effect of concurrent use of cigarette and alcohol on risk of PCa due to lack of information on alcohol consumption in the included studies. High heterogeneity was showed by the I2 statistics and the Cochran’s Q test, and the difference in adjusted confounding factors may be one of the reasons. We have included multivariate results as much as possible to reduce the bias, and there was no indication of publication bias. Dividing studies into pre-PSA screening era and PSA screening era based on publication year (1995 as the cut-off) may produce bias because many of the cohorts published and categorized into the PSA screening era extended into the pre-PSA screening era. Another limitation is that we failed to calculate the impact of quantitative cigarette consumption on the PCa risk due to a lack of data. However, we have to point out that the meta-regression conducted by Islami et al. [12] was methodologically wrong as including multiple data points from a single study with the same control group counts the effect of that control group multiple times (i.e., unit-of-analysis error).
Conclusions
To the best of our knowledge, this systematic review and meta-analysis contained the largest sample of prospective cohort studies, the latest evidence and the most comprehensive information on the association between cigarette smoking habits and the risk of PCa. The smokers’ poor adherence to cancer screening and the occurrence of smoking-related aggressive cancers as well as cardiovascular, pulmonary, and several other deadly diseases may explain the negative association. Regional distinctions can be attributed to the difference of participants in age, ethnicity, socioeconomic status, and educational levels. In addition, a correct methodology is important, the choice of different effect models should base on the heterogeneity and characteristics of enrolled studies. However, it is difficult to conclude a positive association between cigarette smoking and PCa risk as we hypothesized due to these affecting factors. We should focus on taking measures to help smokers to be more compliant with early cancer screening and to quit smoking.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- PCa:
-
Prostate cancer
- PSA:
-
Prostate-specific antigen
- PAHs:
-
Polycyclic aromatic hydrocarbons
- GSTs:
-
Glutathione-S-transferases
- HO-1:
-
Heme oxygenase 1
- PRISMA:
-
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
- Cig/d:
-
Cigarettes per day
- Pk-yr:
-
Pack-year
- Yr:
-
Year
- CI:
-
Confidence interval
- RR:
-
Relative risk
- FU:
-
Follow-up
- NR:
-
Not reported
- US:
-
United States
- BP:
-
Blood pressure
- BMI:
-
Body mass index
- UK:
-
United Kingdom
- DM:
-
Diabetes mellitus
- NS:
-
Non-significant
- COPD:
-
Chronic obstructive pulmonary disease
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Bostwick DG, Burke HB, Djakiew D, Euling S, Ho SM, Landolph J, et al. Human prostate cancer risk factors. Cancer. 2004;101(10 Suppl):2371–490.
Dickerman BA, Markt SC, Koskenvuo M, Pukkala E, Mucci LA, Kaprio J. Alcohol intake, drinking patterns, and prostate cancer risk and mortality: a 30-year prospective cohort study of Finnish twins. Cancer Causes Control. 2016;27(9):1049–58.
Rowles JL 3rd, Ranard KM, Applegate CC, Jeon S, An R, Erdman JW Jr. Processed and raw tomato consumption and risk of prostate cancer: a systematic review and dose-response meta-analysis. Prostate Cancer Prostatic Dis. 2018;21(3):319–36.
Chen X, Zhao Y, Tao Z, Wang K. Coffee consumption and risk of prostate cancer: a systematic review and meta-analysis. BMJ Open. 2021;11(2): e038902.
Perez-Cornago A, Key TJ, Allen NE, Fensom GK, Bradbury KE, Martin RM, et al. Prospective investigation of risk factors for prostate cancer in the UK Biobank cohort study. Br J Cancer. 2017;117(10):1562–71.
Karam-Hage M, Cinciripini PM, Gritz ER. Tobacco use and cessation for cancer survivors: an overview for clinicians. CA Cancer J Clin. 2014;64(4):272–90.
U.S. Department of Health and Human Services. Smoking cessation. A report of the surgeon general. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2020. (Accessed 30 Sep 2022).
De Nunzio C, Andriole GL, Thompson IM Jr, Freedland SJ. Smoking and prostate cancer: a systematic review. Eur Urol Focus. 2015;1(1):28–38.
Brookman-May SD, Campi R, Henríquez JDS, Klatte T, Langenhuijsen JF, Brausi M, et al. Latest evidence on the impact of smoking, sports, and sexual activity as modifiable lifestyle risk factors for prostate cancer incidence, recurrence, and progression: a systematic review of the literature by the European Association of Urology Section of Oncological Urology (ESOU). Eur Urol Focus. 2019;5(5):756–87.
Huncharek M, Haddock KS, Reid R, Kupelnick B. Smoking as a risk factor for prostate cancer: a meta-analysis of 24 prospective cohort studies. Am J Public Health. 2010;100(4):693–701.
Islami F, Moreira DM, Boffetta P, Freedland SJ. A systematic review and meta-analysis of tobacco use and prostate cancer mortality and incidence in prospective cohort studies. Eur Urol. 2014;66(6):1054–64.
Jochems SHJ, Fritz J, Häggström C, Järvholm B, Stattin P, Stocks T. Smoking and risk of prostate cancer and prostate cancer death: a pooled study. Eur Urol. 2022;3:S0302-2838(22)01804-8.
Centers for Disease Control and Prevention (US); National Center for Chronic Disease Prevention and Health Promotion (US); Office on Smoking and Health (US). How tobacco smoke causes disease: the biology and behavioral basis for smoking-attributable disease: a report of the surgeon general. Atlanta: Centers for Disease Control and Prevention (US); 2010.
Nock NL, Tang D, Rundle A, Neslund-Dudas C, Savera AT, Bock CH, et al. Associations between smoking, polymorphisms in polycyclic aromatic hydrocarbon (PAH) metabolism and conjugation genes and PAH-DNA adducts in prostate tumors differ by race. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1236–45.
Mao GE, Morris G, Lu QY, Cao W, Reuter VE, Cordon-Cardo C, et al. Glutathione S-transferase P1 Ile105Val polymorphism, cigarette smoking and prostate cancer. Cancer Detect Prev. 2004;28(5):368–74.
Ye J, Wang S, Barger M, Castranova V, Shi X. Activation of androgen response element by cadmium: a potential mechanism for a carcinogenic effect of cadmium in the prostate. J Environ Pathol Toxicol Oncol. 2000;19(3):275–80.
Watts EL, Appleby PN, Perez-Cornago A, Bueno-de-Mesquita HB, Chan JM, Chen C, et al. Low free testosterone and prostate cancer risk: a collaborative analysis of 20 prospective studies. Eur Urol. 2018;74(5):585–94.
Shiels MS, Rohrmann S, Menke A, Selvin E, Crespo CJ, Rifai N, et al. Association of cigarette smoking, alcohol consumption, and physical activity with sex steroid hormone levels in US men. Cancer Causes Control. 2009;20(6):877–86.
Quiñones LA, Irarrázabal CE, Rojas CR, Orellana CE, Acevedo C, Huidobro C, et al. Joint effect among p53, CYP1A1, GSTM1 polymorphism combinations and smoking on prostate cancer risk: an exploratory genotype-environment interaction study. Asian J Androl. 2006;8(3):349–55.
Birrane G, Li H, Yang S, Tachado SD, Seng S. Cigarette smoke induces nuclear translocation of heme oxygenase 1 (HO-1) in prostate cancer cells: nuclear HO-1 promotes vascular endothelial growth factor secretion. Int J Oncol. 2013;42(6):1919–28.
Miyake M, Fujimoto K, Anai S, Ohnishi S, Kuwada M, Nakai Y, et al. Heme oxygenase-1 promotes angiogenesis in urothelial carcinoma of the urinary bladder. Oncol Rep. 2011;25(3):653–60.
De Nunzio C, Kramer G, Marberger M, Montironi R, Nelson W, et al. The controversial relationship between benign prostatic hyperplasia and prostate cancer: the role of inflammation. Eur Urol. 2011;60(1):106–17.
Arnson Y, Shoenfeld Y, Amital H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J Autoimmun. 2010;34(3):J258–65.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372: n71.
Lo CK, Mertz D, Loeb M. Newcastle-Ottawa Scale: comparing reviewers’ to authors’ assessments. BMC Med Res Methodol. 2014;14:45.
Davies HT, Crombie IK, Tavakoli M. When can odds ratios mislead? BMJ. 1998;316(7136):989–91.
Janez S, Maucort-Boulch D. Odds ratio, hazard ratio and relative risk. Adv Methodol Stat. 2016;13(1):59–67.
Shor E, Roelfs D, Vang ZM. The “Hispanic mortality paradox” revisited: Meta-analysis and meta-regression of life-course differentials in Latin American and Caribbean immigrants’ mortality. Soc Sci Med. 2017;186:20–33.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–88.
Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ. 2003;327(7414):557–60.
Thompson MM, Garland C, Barrett-Connor E, Khaw KT, Friedlander NJ, Wingard DL. Heart disease risk factors, diabetes, and prostatic cancer in an adult community. Am J Epidemiol. 1989;129(3):511–7.
Severson RK, Nomura AM, Grove JS, Stemmermann GN. A prospective study of demographics, diet, and prostate cancer among men of Japanese ancestry in Hawaii. Cancer Res. 1989;49(7):1857–60.
Mills PK, Beeson WL, Phillips RL, Fraser GE. Cohort study of diet, lifestyle, and prostate cancer in Adventist men. Cancer. 1989;64(3):598–604.
Le Marchand L, Kolonel LN, Wilkens LR, Myers BC, Hirohata T. Animal fat consumption and prostate cancer: a prospective study in Hawaii. Epidemiology. 1994;5(3):276–82.
Hiatt RA, Armstrong MA, Klatsky AL, Sidney S. Alcohol consumption, smoking, and other risk factors and prostate cancer in a large health plan cohort in California (United States). Cancer Causes Control. 1994;5(1):66–72.
Adami HO, Bergström R, Engholm G, Nyrén O, Wolk A, Ekbom A, et al. A prospective study of smoking and risk of prostate cancer. Int J Cancer. 1996;67(6):764–8.
Engeland A, Andersen A, Haldorsen T, Tretli S. Smoking habits and risk of cancers other than lung cancer: 28 years’ follow-up of 26,000 Norwegian men and women. Cancer Causes Control. 1996;7(5):497–506.
Veierød MB, Laake P, Thelle DS. Dietary fat intake and risk of prostate cancer: a prospective study of 25,708 Norwegian men. Int J Cancer. 1997;73(5):634–8.
Cerhan JR, Torner JC, Lynch CF, Rubenstein LM, Lemke JH, Cohen MB, et al. Association of smoking, body mass, and physical activity with risk of prostate cancer in the Iowa 65+ Rural Health Study (United States). Cancer Causes Control. 1997;8(2):229–38.
Will JC, Vinicor F, Calle EE. Is diabetes mellitus associated with prostate cancer incidence and survival? Epidemiology. 1999;10(3):313–8.
Giovannucci E, Rimm EB, Ascherio A, Colditz GA, Spiegelman D, Stampfer HJ, et al. Smoking and risk of total and fatal prostate cancer in United States health professionals. Cancer Epidemiol Biomarkers Prev. 1999;8(4):277–82.
Putnam SD, Cerhan JR, Parker AS, Bianchi GD, Wallace RB, Cantor KP, et al. Lifestyle and anthropometric risk factors for prostate cancer in a cohort of Iowa men. Ann Epidemiol. 2000;10(6):361–9.
Lund Nilsen TL, Johnsen R, Vatten LJ. Socio-economic and lifestyle factors associated with the risk of prostate cancer. Br J Cancer. 2000;82(7):1358–63.
Lotufo PA, Lee IM, Ajani UA, Hennekens CH, Manson JE. Cigarette smoking and risk of prostate cancer in the physicians’ health study (United States). Int J Cancer. 2000;87(1):141–4.
Baglietto L, Severi G, English DR, Hopper JL, Giles GG. Alcohol consumption and prostate cancer risk: results from the Melbourne collaborative cohort study. Int J Cancer. 2006;119(6):1501–4.
Gonzalez A, Peters U, Lampe JW, White E. Boron intake and prostate cancer risk. Cancer Causes Control. 2007;18(10):1131–40.
Park SY, Murphy SP, Wilkens LR, Henderson BE, Kolonel LN. Fat and meat intake and prostate cancer risk: the multiethnic cohort study. Int J Cancer. 2007;121(6):1339–45.
Rohrmann S, Genkinger JM, Burke A, Helzlsouer KJ, Comstock GW, Alberg AJ, et al. Smoking and risk of fatal prostate cancer in a prospective U.S. study. Urology. 2007;69(4):721–5.
Butler LM, Wang R, Wong AS, Koh WP, Yu MC. Cigarette smoking and risk of prostate cancer among Singapore Chinese. Cancer Causes Control. 2009;20(10):1967–74.
Watters JL, Park Y, Hollenbeck A, Schatzkin A, Albanes D. Cigarette smoking and prostate cancer in a prospective US cohort study. Cancer Epidemiol Biomarkers Prev. 2009;18(9):2427–35.
Grundmark B, Zethelius B, Garmo H, Holmberg L. Serum levels of selenium and smoking habits at age 50 influence long term prostate cancer risk; a 34 year ULSAM follow-up. BMC Cancer. 2011;11:431.
Li Q, Kuriyama S, Kakizaki M, Yan H, Nagai M, Sugawara Y, et al. History of cholelithiasis and the risk of prostate cancer: the Ohsaki Cohort Study. Int J Cancer. 2011;128(1):185–91.
Geybels MS, Verhage BA, van Schooten FJ, van den Brandt PA. Measures of combined antioxidant and pro-oxidant exposures and risk of overall and advanced stage prostate cancer. Ann Epidemiol. 2012;22(11):814–20.
Karppi J, Kurl S, Laukkanen JA, Kauhanen J. Serum β-carotene in relation to risk of prostate cancer: the Kuopio Ischaemic Heart Disease Risk Factor study. Nutr Cancer. 2012;64(3):361–7.
Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS. Coffee consumption and prostate cancer risk: further evidence for inverse relationship. Nutr J. 2012;11:42.
Bae JM, Li ZM, Shin MH, Kim DH, Lee MS, Ahn YO. Cigarette smoking and prostate cancer risk: negative results of the Seoul Male Cancer Cohort Study. Asian Pac J Cancer Prev. 2013;14(8):4667–9.
Lemogne C, Consoli SM, Geoffroy-Perez B, Coeuret-Pellicer M, Nabi H, Melchior M, et al. Personality and the risk of cancer: a 16-year follow-up study of the GAZEL cohort. Psychosom Med. 2013;75(3):262–71.
Rohrmann S, Linseisen J, Allen N, Bueno-de-Mesquita HB, Johnsen NF, Tjønneland A, et al. Smoking and the risk of prostate cancer in the European Prospective Investigation into Cancer and Nutrition. Br J Cancer. 2013;108(3):708–14.
Sawada N, Inoue M, Iwasaki M, Sasazuki S, Yamaji T, Shimazu T, et al. Alcohol and smoking and subsequent risk of prostate cancer in Japanese men: the Japan Public Health Center-based prospective study. Int J Cancer. 2014;134(4):971–8.
Everatt R, Kuzmickienė I, Virvičiūtė D, Tamošiūnas A. Cigarette smoking, educational level and total and site-specific cancer: a cohort study in men in Lithuania. Eur J Cancer Prev. 2014;23(6):579–86.
Ho T, Howard L, Vidal A, Gerber L, Moreira D, McKeever M, et al. Smoking and risk of low- and high-grade prostate cancer: results from the REDUCE study. Clin Cancer Res. 2014;20(20):5331–8.
Jacob L, Freyn M, Kalder M, Dinas K, Kostev K. Impact of tobacco smoking on the risk of developing 25 different cancers in the UK: a retrospective study of 422,010 patients followed for up to 30 years. Oncotarget. 2018;9(25):17420–9.
Viner B, Barberio AM, Haig TR, Friedenreich CM, Brenner DR. The individual and combined effects of alcohol consumption and cigarette smoking on site-specific cancer risk in a prospective cohort of 26,607 adults: results from Alberta’s Tomorrow Project. Cancer Causes Control. 2019;30(12):1313–26.
Weber MF, Sarich PEA, Vaneckova P, Wade S, Egger S, Ngo P, et al. Cancer incidence and cancer death in relation to tobacco smoking in a population-based Australian cohort study. Int J Cancer. 2021;149(5):1076–88.
Hippisley-Cox J, Coupland C. Predicting the risk of prostate cancer in asymptomatic men: a cohort study to develop and validate a novel algorithm. Br J Gen Pract. 2021;71(706):e364–71.
Allen NE, Sauvaget C, Roddam AW, Appleby P, Nagano J, Suzuki G, et al. A prospective study of diet and prostate cancer in Japanese men. Cancer Causes Control. 2004;15(9):911–20.
Onitilo AA, Berg RL, Engel JM, Stankowski RV, Glurich I, Williams GM, et al. Prostate cancer risk in pre-diabetic men: a matched cohort study. Clin Med Res. 2013;11(4):201–9.
Thune I, Lund E. Physical activity and the risk of prostate and testicular cancer: a cohort study of 53,000 Norwegian men. Cancer Causes Control. 1994;5(6):549–56.
Tulinius H, Sigfússon N, Sigvaldason H, Bjarnadóttir K, Tryggvadóttir L. Risk factors for malignant diseases: a cohort study on a population of 22,946 Icelanders. Cancer Epidemiol Biomarkers Prev. 1997;6(11):863–73.
Chamie K, DeVere White RW, Lee D, Ok JH, Ellison LM. Agent Orange exposure, Vietnam War veterans, and the risk of prostate cancer. Cancer. 2008;113(9):2464–70.
Laukkanen JA, Pukkala E, Rauramaa R, Makikallio TH, Toriola AT, Kurl S. Cardiorespiratory fitness, lifestyle factors and cancer risk and mortality in Finnish men. Eur J Cancer. 2010;46(2):355–63.
Karlsen RV, Bidstrup PE, Christensen J, Larsen SB, Tjønneland A, Dalton SO, et al. Men with cancer change their health behaviour: a prospective study from the Danish diet, cancer and health study. Br J Cancer. 2012;107(1):201–6.
Rolison JJ, Hanoch Y, Miron-Shatz T. Smokers: at risk for prostate cancer but unlikely to screen. Addict Behav. 2012;37(6):736–8.
Littlejohns TJ, Travis RC, Key TJ, Allen NE. Lifestyle factors and prostate-specific antigen (PSA) testing in UK Biobank: Implications for epidemiological research. Cancer Epidemiol. 2016;45:40–6.
Tangen C, Goodman P, Till C, Schenk J, Lucia M, Thompson I. Biases in recommendations for and acceptance of prostate biopsy significantly affect assessment of prostate cancer risk factors: results from two large randomized clinical trials. J Clin Oncol. 2016;34(36):4338–44.
Johnson JA, Moser RP, Ellison GL, Martin DN. Associations of prostate-specific antigen (PSA) testing in the US population: results from a national cross-sectional survey. J Community Health. 2021;46(2):389–98.
Moses KA, Zhao Z, Bi Y, Acquaye J, Holmes A, Blot WJ, et al. The impact of sociodemographic factors and PSA screening among low-income Black and White men: data from the Southern Community Cohort Study. Prostate Cancer Prostatic Dis. 2017;20(4):424–9.
Pickles K, Scherer LD, Cvejic E, Hersch J, Barratt A, McCaffery KJ. Preferences for more or less health care and association with health literacy of men eligible for prostate-specific antigen screening in Australia. JAMA Netw Open. 2021;4(10): e2128380.
de Nunzio C, Tema G, Trucchi A, Cicione A, Sica A, Lombardo R, et al. Smoking reduces PSA accuracy for detection of prostate cancer: Results from an Italian cross-sectional study. Minerva Urol Nefrol. 2019;71(6):583–9.
GBD 2019 Tobacco Collaborators. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990–2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet. 2021;397(10292):2337-2360.
Sturgis EM, Wei Q, Spitz MR. Descriptive epidemiology and risk factors for head and neck cancer. Semin Oncol. 2004;31(6):726–33.
Burger M, Catto JW, Dalbagni G, Grossman HB, Herr H, Karakiewicz P, et al. Epidemiology and risk factors of urothelial bladder cancer. Eur Urol. 2013;63(2):234–41.
Cook MB, Kamangar F, Whiteman DC, Freedman ND, Gammon MD, Bernstein L, et al. Cigarette smoking and adenocarcinomas of the esophagus and esophagogastric junction: a pooled analysis from the international BEACON consortium. J Natl Cancer Inst. 2010;102(17):1344–53.
Duffy SA, Ronis DL, McLean S, Fowler KE, Gruber SB, Wolf GT, et al. Pretreatment health behaviors predict survival among patients with head and neck squamous cell carcinoma. J Clin Oncol. 2009;27(12):1969–75.
Centers for Disease Control and Prevention (CDC). Smoking-attributable mortality, years of potential life lost, and productivity losses--United States, 2000-2004. MMWR Morb Mortal Wkly Rep. 2008;57(45):1226–8.
Foerster B, Pozo C, Abufaraj M, Mari A, Kimura S, D’Andrea D, et al. Association of smoking status with recurrence, metastasis, and mortality among patients with localized prostate cancer undergoing prostatectomy or radiotherapy: a systematic review and meta-analysis. JAMA Oncol. 2018;4(7):953–61.
Darcey E, Boyle T. Tobacco smoking and survival after a prostate cancer diagnosis: a systematic review and meta-analysis. Cancer Treat Rev. 2018;70:30–40.
Prueitt RL, Wallace TA, Glynn SA, Yi M, Tang W, Luo J, et al. An immune-inflammation gene expression signature in prostate tumors of smokers. Cancer Res. 2016;76(5):1055–65.
Enokida H, Shiina H, Urakami S, Terashima M, Ogishima T, Li LC, et al. Smoking influences aberrant CpG hypermethylation of multiple genes in human prostate carcinoma. Cancer. 2006;106(1):79–86.
Creamer MR, Wang TW, Babb S, Cullen KA, Day H, Willis G, et al. Tobacco product use and cessation indicators among adults - United States, 2018. MMWR Morb Mortal Wkly Rep. 2019;68(45):1013–9.
Rigotti NA, Kruse GR, Livingstone-Banks J, Hartmann-Boyce J. Treatment of tobacco smoking: a review. JAMA. 2022;327(6):566–77.
Warren GW, Marshall JR, Cummings KM, Toll BA, Gritz ER, Hutson A, et al. Addressing tobacco use in patients with cancer: a survey of American Society of Clinical Oncology members. J Oncol Pract. 2013;9(5):258–62.
Li WHC, Ho KY, Wang MP, Cheung DYT, Lam KKW, Xia W, et al. Effectiveness of a brief self-determination theory-based smoking cessation intervention for smokers at Emergency Departments in Hong Kong: a randomized clinical trial. JAMA Intern Med. 2020;180(2):206–14.
Stead LF, Hartmann-Boyce J, Perera R, Lancaster T. Telephone counselling for smoking cessation. Cochrane Database Syst Rev. 2013;(8):CD002850.
Lancaster T, Stead LF. Self-help interventions for smoking cessation. Cochrane Database Syst Rev. 2005;(3):CD001118.
Civljak M, Sheikh A, Stead LF, Car J. Internet-based interventions for smoking cessation. Cochrane Database Syst Rev. 2010;(9):CD007078.
Kim AS, Ko HJ, Kwon JH, Lee JM. Exposure to secondhand smoke and risk of cancer in never smokers: a meta-analysis of epidemiologic studies. Int J Environ Res Public Health. 2018;15(9):1981.
Andreotti G, Freedman ND, Silverman DT, Lerro CC, Koutros S, Hartge P, et al. Tobacco use and cancer risk in the agricultural health study. Cancer Epidemiol Biomarkers Prev. 2017;26(5):769–78.
Zhao J, Stockwell T, Roemer A, Chikritzhs T. Is alcohol consumption a risk factor for prostate cancer? A systematic review and meta-analysis. BMC Cancer. 2016;16(1):845.
Sharma R, Lodhi S, Sahota P, Thakkar MM. Nicotine administration in the wake-promoting basal forebrain attenuates sleep-promoting effects of alcohol. J Neurochem. 2015;135(2):323–31.
Acknowledgements
Not applicable.
Funding
This work was supported by the Sanming Project of Medicine in Shenzhen (grant number: SZSM202011011). The funder had no role in the study design, data collection, analysis and interpretation, or writing of the report.
Author information
Authors and Affiliations
Contributions
XWY, HC and JP conceptualized the study and developed the registered protocol for the review. XWY and HC conducted the literature search, quality assessment, data extraction, and drafted the manuscript. SQZ, XJC and YYS performed statistical analyses. JP revised the manuscript, obtained funding and supervised the project. JP is responsible for the overall content and serves as the guarantor. All authors helped refine the final version of the manuscript and approve with its submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
A. Characteristics of the 39 studies included in the meta-analysis. B. Characteristics of the 5 studies not included in the meta-analysis due to lack of information.
Additional file 2.
Results of quality assessment using the Newcastle-Ottawa Scale for cohort studies.
Additional file 3.
Sensitivity analyses of association between smoking status and risk of prostate cancer.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, X., Chen, H., Zhang, S. et al. Association of cigarette smoking habits with the risk of prostate cancer: a systematic review and meta-analysis. BMC Public Health 23, 1150 (2023). https://doi.org/10.1186/s12889-023-16085-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12889-023-16085-w