
Abstract
Background
Intermittent theta burst stimulation (iTBS), a novel form of repetitive transcranial magnetic stimulation (rTMS), can be administered in 1/10th of the time of standard rTMS (~ 3 min vs. 37.5 min) yet achieves similar outcomes in depression. The brief nature of the iTBS protocol allows for the administration of multiple iTBS sessions per day, thus reducing the overall course length to days rather than weeks. This study aims to compare the efficacy and tolerability of active versus sham iTBS using an accelerated regimen in patients with treatment-resistant depression (TRD). As a secondary objective, we aim to assess the safety, tolerability, and treatment response to open-label low-frequency right-sided (1 Hz) stimulation using an accelerated regimen in those who do not respond to the initial week of treatment.
Methods
Over three years, approximately 230 outpatients at the Centre for Addiction and Mental Health and University of British Columbia Hospital, meeting diagnostic criteria for unipolar MDD, will be recruited and randomized to a triple blind sham-controlled trial. Patients will receive five consecutive days of active or sham iTBS, administered eight times daily at 1-hour intervals, with each session delivering 600 pulses of iTBS. Those who have not achieved response by the week four follow-up visit will be offered a second course of treatment, regardless of whether they initially received active or sham stimulation.
Discussion
Broader implementation of conventional iTBS is limited by the logistical demands of the current standard course consisting of 4–6 weeks of daily treatment. If our proposed accelerated iTBS protocol enables patients to achieve remission more rapidly, this would offer major benefits in terms of cost and capacity as well as the time required to achieve clinical response.
Trial registration
ClinicalTrials.gov Identifier: NCT04255784.
Similar content being viewed by others


Background and rationale
Major depressive disorder (MDD) is a highly prevalent and disabling disorder, in which one-third of patients are classified as treatment-resistant [1]. Electroconvulsive therapy (ECT) is the most effective treatment option for those with treatment-resistant depression (TRD); however, it is used in < 1% of patients with TRD [2] due in large part to the cognitive adverse effects [3,4,5], need for anesthesia and societal stigma [6], associated with ECT treatment. Thus, alternative treatment approaches are urgently needed.
Repetitive transcranial magnetic stimulation (rTMS) is a safe and effective treatment for TRD that uses powerful, focused magnetic field pulses, applied non-invasively, to induce lasting changes in the activity of brain regions involved in regulating thoughts, emotions, and behaviour [7,8,9]. Across several studies [10, 11] rTMS has demonstrated high response and remission rates of up to 50% and 35%, respectively. The current standard rTMS treatment protocol involves applying 10 Hz stimulation daily to the left dorsolateral prefrontal cortex (DLPFC), over 37.5 min. [12]. Additionally, average treatment courses last between 4 and 6 weeks, given that current studies suggest the effects of rTMS treatment are linearly cumulative [13,14,15], with maximal benefits after 25 to 28 treatments [14, 16, 17]. These long treatment sessions and courses result in two major drawbacks to rTMS: high cost and low capacity.
Intermittent theta burst stimulation (iTBS), a novel form of rTMS, addresses these key issues as treatment can be administered in 1/10th of the time of standard high frequency rTMS (~ 3 min vs. 37.5 min). Importantly, iTBS has resulted in similar or greater effects on neural plasticity [18] and clinical outcomes [19]. Major gains in rTMS efficiency and accessibility could be realized by administering multiple iTBS sessions per day, thus significantly reducing course length. Several recently published studies suggest that accelerated rTMS may be feasible, tolerable, and capable of achieving comparable remission rates to standard rTMS in shorter time-frames of 4 to 10 days [20,21,22,23,24,25]. However, the majority of these studies were small, open-label case series that did not control for the non-specific effects of multiple daily interventions.
Objectives
The primary objective of this study is to compare the efficacy and tolerability of active versus sham iTBS using an accelerated regimen of 8 daily sessions administered at 1-hour intervals, for five days, in patients with TRD. As a secondary objective, we aim to assess the safety, tolerability and treatment response to open-label low-frequency right-sided (1 Hz) stimulation using an accelerated regimen of 8 daily sessions for five days, in patients with TRD who do not respond to an initial week of blinded left DLPFC iTBS stimulation.
Methods
Trial design and setting
Over three years, approximately 230 outpatients at CAMH and the University of British Columbia Hospital, meeting diagnostic criteria for MDD, will be recruited and randomized to a triple-blind sham-controlled trial (patient, rater and technician blinding). Combined, these two centres receive 600–800 referrals annually for psychiatric brain stimulation from nurse practitioners, family physicians and psychiatrists. At the CAMH site, referrals are received from across the province of Ontario for patients from all genders and across the adult lifespan. A similarly diverse population are referred to the UBC site, from metro Vancouver areas. We believe that this recruitment approach will lead to the inclusion of a wide range of patients with diverse backgrounds.
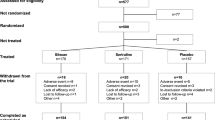
Patients will receive five consecutive days (Monday to Friday) of active or sham iTBS, administered eight times daily at 1-hour intervals, with each session delivering 600 pulses of iTBS over 3 min and 9 s. Sham treatments will be delivered using a shielded “sham coil” that reproduces auditory and tactile sensations of stimulation. Refer to Fig. 1 for a summary of the trial design.

Study flow chart
Following randomization, patients will undergo a series of assessments and motor threshold testing to determine the appropriate strength of stimulation. Depressive symptoms will be assessed using the Hamilton Depression Rating Scale (HRSD-17) [26]. The primary outcome analysis will include the change in HRSD scores from baseline to the end of the acute treatment. The HRSD-17 was selected to facilitate the comparison of outcomes in this trial to that of previous trials, since the HRSD has been standard in both rTMS and pharmacotherapy trials of patients with depression for decades [27,28,29,30,31]. The HRSD-6 [32], derived from the HRSD-17, will also be included as a secondary outcome measure. The Scale for Suicide Ideation (SSI) and self-report scales of depression and anxiety (Beck Depression Inventory, BDI-II and the General Anxiety Disorder, GAD-7), will also be completed. Side effects will be recorded at every visit and after every treatment session. Final assessments will be completed immediately after the last treatment. The one-week and four-week follow up assessments will be allowed within 1–2 days of the scheduled time points. Refer to Table 1 for a detailed schedule of assessments.
All treatments will be conducted with minimized personal contact (i.e., verbal communication) between the technician and patient to reduce the impact of nonspecific therapeutic contact on outcomes. The relevance of alliance extends beyond psychotherapy and has been shown to impact pharmacological interventions [33]. As a way to quantify the nonspecific active component of the treatment, a modified Human Connections Scale (HCS) will be administered following each treatment course (blinded and open label) to determine the impact of the patient’s sense of alliance with the technician on the therapeutic outcome [34]. This scale will be used as a secondary measure and explored with respect to its role in the treatment outcome.
Open Label: To ensure that all patients can receive a form of active treatment, those who have not achieved the response criterion (i.e., a 50% improvement from baseline on the HRSD-17) at the week four follow-up visit will be offered a second course of treatment, regardless of whether they initially received active or sham treatment.
The blind will be maintained, and no further assessment contributing to the primary hypotheses will occur after the 4-week time point. A different technician will administer the open-label second course of treatment so as not to unblind the technician to the original treatment course. To ensure that patients do not simply receive the same treatment pattern that previously failed to exert an adequate effect, the second course of treatment will apply active rTMS using low-frequency (1 Hz) stimulation for 600 pulses (10 min), 8 times daily at 1-hour intervals for five days. 1 Hz right-sided rTMS has been shown in large-scale trials to achieve outcomes similar to left high-frequency stimulation [35], which has equivalent outcomes to iTBS [19]. All patients completing the second course of treatment will undergo the same schedule of clinical assessments during and after the course of treatment. Should the participant be available to start the second course of treatment immediately, the four-week follow-up assessment from the first course of treatment will serve as the baseline for the unblinded 1 Hz treatment. Those who go on to receive the second treatment course but cannot attend treatment immediately, will have a separate baseline assessment. Participants will be allowed to start the open-label course of treatment up to 3 months after the 4-week follow-up of the first course. Female participants will complete an abbreviated reproductive history immediately before the second course of treatment.
Eligibility criteria
Inclusion Criteria: Patients will be included if they: (1) are outpatients between the ages of 18 and 65; (2) are voluntary and competent to consent to treatment; (3) have a Mini-International Neuropsychiatric Interview (MINI) confirmed diagnosis of MDD, single or recurrent; (4) have failed to achieve a clinical response to an adequate dose of an antidepressant based on an Antidepressant Treatment History Form (ATHF) score for that antidepressant trial of ≥ 3 in the current episode [36, 37] OR have been unable to tolerate at least 2 separate trials of antidepressants of inadequate dose and duration (ATHF score of 1 or 2 on those 2 separate antidepressants) in the current episode; (5) have a minimum of moderate depression as indicated by a score of > 9 on the PHQ-9, in order to separate inclusion criteria from the primary outcome assessment; (6) have had no increase or initiation of any antidepressant or augmentation medication in the 4 weeks prior to screening; (7) able to adhere to the treatment schedule; (8) Pass the TMS adult safety screening (TASS) questionnaire; (9) have normal thyroid functioning based on pre-study blood work.
Exclusion Criteria: Patients are excluded if they: (1) have a MINI confirmed diagnosis of substance dependence or abuse within the last 3 months; (2) have a concomitant major unstable medical illness, cardiac pacemaker or implanted medication pump; (3) have active suicidal intent; (4) are pregnant; (5) have a lifetime MINI diagnosis of bipolar I or II disorder, schizophrenia, schizoaffective disorder, schizophreniform disorder, delusional disorder, or current psychotic symptoms; (6) have a MINI diagnosis of obsessive compulsive disorder, post-traumatic stress disorder (current or within the last year), anxiety disorder (generalized anxiety disorder, social anxiety disorder, panic disorder), or dysthymia, that is assessed by a study investigator to be primary and causing greater impairment than MDD; (7) have a diagnosis of any personality disorder, assessed by a study investigator to be primary and causing greater impairment than MDD; (8) have failed a course of ECT in the current episode or previous episode; (9) have received rTMS for any previous indication due to the potential compromise of subject blinding; (10) have any significant neurological disorder or insult including, (e.g., any condition likely associated with increased intracranial pressure, space occupying lesion, history of seizure except those therapeutically induced by ECT or a febrile seizure of infancy, cerebral aneurysm, Parkinson’s disease, Huntington’s chorea, multiple sclerosis, significant head trauma with clear radiological evidence of cerebrovascular injury on imaging); (11) have an intracranial implant (e.g., aneurysm clips, shunts, stimulators, cochlear implants, or electrodes) or any other metal object within or near the head, excluding the mouth, that cannot be safely removed; (12) if participating in psychotherapy, must have been in stable treatment for at least 3 months prior to entry into the study, with no anticipation of change in the frequency of therapeutic sessions, or the therapeutic focus over the duration of the study; (13) clinically significant laboratory abnormality, in the opinion of the one of the principal investigators or study physicians; (14) currently take more than lorazepam 2 mg daily (or equivalent) or any dose of an anticonvulsant due to the potential to limit rTMS efficacy.
Informed consent procedure
The informed consent process is initiated before the patient agrees to participate in the study and will be obtained according to REB and GCP guidelines. All patients referred by a general physician or psychiatrist will undergo an extensive consultation with a brain stimulation psychiatrist at each study site, who will determine suitability for referral to this trial. Patients referred to the trial will then be assessed for eligibility by qualified research personnel who will obtain consent. Patients will be informed that they can withdraw participation at any point during the study, and the rights and welfare of the patients will be protected.
Randomization and blinding
Participants will be randomized into the study, stratified by site and by medication resistance (2 or fewer adequate trial failures versus more than 2 adequate trial failures; an adequate trial of antidepressant is defined as an antidepressant trial ranked with an ATHF (score ≥ 3)). Prior rTMS trials have demonstrated that the degree of treatment resistance is a key predictor of response, and therefore, it is essential to ensure that the groups are balanced concerning this variable [12, 38, 39].
Patients will be randomized using random permuted blocks of varying sizes, with study personnel blinded to the block sizes. An independent assistant external to the study will manage the randomization using a computer generator. The participants’ number and treatment code will be assigned after their details (i.e., initials and number of failed adequate antidepressant trials) have been obtained. A unique treatment code, designated to either active or sham stimulation, allows for the technicians to remain blinded. Once the code is entered into the machine, it indicates which side of the A/P coil to place over the stimulation site and will only deliver pulses when the designated coil is placed correctly.
A specially designed rTMS coil containing both an active and a sham inductor head and identical external appearance will be used to ensure blinding during treatment. A computerized sensor instructs the technician to rotate the coil so that one side is in contact with the scalp. A pair of electrodes will also be placed close to the treatment spot, directly on the forehead, 1 cm apart from each other and secured with medical tape, in both active and sham groups. In the sham group, electrical pulses will be delivered through the scalp electrodes on each TMS pulse to further mimic the scalp sensations and muscle contractions associated with verum TMS. This approach is most likely to achieve both patient and technician blinding and is seen as the best possible sham for blinded rTMS treatment trials [24, 40].
An independent, blinded rater at each site will administer the clinical assessment scales the week before and after the final treatment and at four weeks post-treatment. At baseline, participants will be asked about their expectancy of improving following the treatment course using the Stanford Expectancy Scale [41]. In addition, to assess the integrity of blinding, after their first treatment day and at the 4-week follow-up (for participants)/1-week follow-up (for technicians), all patients and technicians will be asked whether or not they believe the active or sham treatment was delivered along with their degree of certainty (0 guess – 10 certain). Collecting this information will help determine the extent of underlying placebo or nocebo effects concerning their expectations and therapeutic outcomes [42, 43].
Intervention
rTMS treatment parameters
rTMS will employ the MagPro X100/R30 stimulator (MagVenture, Farum, Denmark) equipped with the B70 A/P fluid-cooled coil, which has both an active and a placebo head actuated under computer control to maintain blinding. The modified BeamF3 scalp heuristic will be used to localize the treatment site over the left DLPFC [44].
Before the first treatment, each participant’s motor threshold will be determined according to published methods [45, 46]. This location and the stimulation target site will be marked at the first session on the scalp, and standard methods will be used to target this site during treatment. All patients will undergo 8 treatment sessions per day for 5 consecutive days, with the start of each session timed to be at least 50 min from the previous session. While the optimal time interval between treatment sessions remains unknown, longer intersession intervals (ISI), of approximately 1 h and above, are suggested to induce cumulative effect on synaptic strengthening compared to shorter ISIs [47,48,49,50]. Additionally, this intersession interval was chosen to be line with previously published high dose aiTBS trials [25, 51]. Each session will deliver 600 pulses of iTBS (bursts of 3 pulses at 50 Hz, bursts repeated at 5 Hz, with a duty cycle of 2 s on, 8 s off, over 60 cycles / ~3 min) at a target of 120% of the participant’s resting motor threshold. Based on their tolerability, intensity can be decreased to a minimum of 90%. We selected 600 pulses for each session, given recent research suggesting that 600 vs. 1800 pulses of TBS may have similar neurophysiological [52] and clinical [53] effects.
For those undergoing the second course of treatment, rTMS will be delivered with an active, B70 fluid-cooled coil over the right DLPFC located using the scalp heuristic above for the F4 electrode. The schedule of treatments will remain the same: 600 pulses of 1 Hz stimulation over 10 min and 8 sessions per day for 5 consecutive days, with the start of each session timed to be at least 50 min from the previous session.
rTMS treatment side effects/risks
Many thousands of people have received rTMS treatment over the last 20 years. rTMS has certain risks; while some of these risks are known, there is a possibility of risks that we do not know about and have not been seen in study participants to date.
rTMS is recognized in the most recent consensus safety guidelines as a safe and well-tolerated treatment for the vast majority of individuals [54], with an all-causes dropout rate several-fold lower than for antidepressant medications [55]. Common and rare but serious risks are described as below (the numbers in brackets show how often the side-effect happened.):
-
1.
Common: Headache (30%), discomfort or pain at the stimulation site (20%), lightheadedness or dizziness after the treatment (20%), facial muscle twitching (30%). These side effects are mild, generally diminish over treatment, and can usually be managed with rest or over-the-counter pain medications such as acetaminophen or ibuprofen.
-
2.
Less common: (1–7%) fatigue, headache persisting after treatment, dizziness or fainting during the initial sessions of rTMS treatment.
-
3.
Rare but serious: Onset of suicidal thinking (< 1%); hypomanic episode (< 1%).
-
4.
Very rare but serious: There are rare cases of an epileptic seizure resulting from rTMS (less than 0.1%) [56]. Safety guidelines have been in place since 1997 to minimize the risk of seizures from rTMS, and this study follows those guidelines.
Common side effects of rTMS treatment are expected and will be recorded separately from adverse events. Participants will be asked to defer any changes to their antidepressant medications for four weeks before and during the course of rTMS to avoid confounding effects. A numeric rating scale (0 no pain – 10 worst pain they have experienced) will be administered at every visit and after every treatment session, to rate the severity of pain from side effects along with one open-ended question asking about other side effects. The technician will complete these questions so that side effect severity and resolution can be assessed and verified appropriately over time.
Schedule of events
Screening evaluation
Patients will be assessed using the MINI to assess current and lifetime depression and other psychiatric disorders, and will be used to verify psychiatric inclusion. The screening information based on the current episode, will be determined from the patient report and the records of a pre-study clinical assessment, conducted by a psychiatrist trained in TRD. The ATHF is a commonly used and reliable method of assessing the adequacy of prior antidepressant treatment [36, 37] and will be used to confirm eligibility based on inclusion criteria for treatment resistance. To address the potential for rater bias, whereby baseline scores are inflated to ensure patient eligibility [57], we will employ two separate depression rating scales, one to determine eligibility and a different scale to serve as the primary outcome measure [58]. As such, we will require a minimum severity of moderate depression on a separate measure, the patient health questionnaire (PHQ-9 > 9) [59], as an inclusion criterion. The Transcranial Magnetic Stimulation Adult Safety Screen (TASS) will be used to assess potential rTMS risk factors. A pre-study blood test that includes electrolytes, complete blood count, and thyroid stimulating hormone will be required to rule out any underlying medical causes of the depression. These results will be accepted if completed within the lesser of the current depressive episode or six months.
Clinical assessments during and after treatment
Clinical measures will be assessed the week before treatment and immediately after treatment completion (Friday, Visit 5), as well as at 1 and 4 weeks after treatment completion. The schedule of assessments is located in Table 1.
To compare results to other depression studies, we will examine depressive symptoms using clinician-rated and self-report scales. Secondary outcome measures will include the HRSD-6, SSI, the self-rated BDI-II, and the GAD-7. Female participants will also complete a complete reproductive history the week before treatment since the effects of high-frequency rTMS have been shown to vary based on circulating hormones [60].
Attendance and withdrawal criteria
Patients will be encouraged to attend all scheduled treatments. Those that meet the following criteria will be excluded from the per protocol analysis if they:
-
1)
Miss / fail to attend any one of the five treatment days in the course overall.
-
2)
Miss / fail to attend more than five treatment sessions over the five days.
-
3)
Cannot tolerate stimulation of at least 90% RMT for the entire session on more than five treatment sessions.
-
4)
Develop active suicidal ideation with intent during the course of stimulation, or in the opinion of the site PI, that participation is not clinically indicated.
-
5)
Are admitted to hospital during the course of treatment.
-
6)
Withdraw consent to participate.
Patients will be discontinued if they experience worsening in depression, defined as an increase in HRSD-17 from a baseline of more than 25% at the post-treatment assessment or development of active suicidal intent or attempted suicide.
Sample size
We will consider the 8 × 5 protocol successful if the active stimulation group achieves superior improvement (i.e., a larger reduction in the mean HRSD-17 score) versus sham on the HRSD-17. To detect the minimally clinically significant difference of three points [61] on the HRSD-17 with 80% power (based on post-treatment HRSD-17 standard deviation of 8 and pre-post correlation of 0.27 in the iTBS group in the THREE-D trial [19]), 105 patients per group are needed. We expect a low attrition of 10% given that the treatment course is only five days, increasing the number required to be randomized to 116 per group (232 in total).
Statistical methods
Clinical outcomes analysis
Primary Outcome Analysis: The primary outcome analysis on the change in HRSD-17 between baseline and end of treatment, will be conducted on an intention-to-treat basis. We plan to use multiple imputation methods developed by Schafer [62] to account for potential bias that may be incurred by missing data. Before the main effects analysis, descriptive statistics will be generated to summarize the data on all randomized participants to confirm that there are no group differences between the two conditions concerning baseline demographics and clinical characteristics. To assess for significant differences in treatment effects over time, a mixed-effects model with predictor variables for group (active vs. sham), time (baseline vs. end of treatment), and group-by-time interaction will be fitted to the data from the primary outcome measure (HRSD-17). The mixed-effects model will also be used to estimate the treatment effect over the full course of treatment.
Secondary Outcome Analyses: We will repeat the primary and secondary analyses in the same manner for the one-week post-treatment and four-week post-treatment time points. The same approach will be employed to assess treatment effects on the HRSD-6, SSI, BDI-II and GAD-7. Response and remission will be included as secondary clinical outcomes. Response will be defined as ≥ 50% reduction in symptoms on the primary outcome measure (HRSD-17) and remission will be defined as a post-treatment HRSD-17 < 8 [1]. We will use a conservative imputation that assumes non-remission or non-response if data is missing at the post-treatment assessment. A two-tailed Chi-squared test will assess the significance of any observed differences in the proportion of responders and remitters between groups based on pre- to post-treatment HRSD-17. Finally, we will summarize and compare the rates of adverse events, severe adverse events and dropouts in both groups.
Exploratory Outcome Analyses: A multivariable regression model will be used to explore whether or not treatment resistance, patient expectancy, patient experience, or HCS modify the baseline adjusted treatment effect on the HRSD-17 (at day 5). The same analytic approach used for the primary outcome will be employed to assess for treatment effects on the HRSD-6, BDI-II, SSI and GAD-7. We will descriptively report the change from baseline to 5 days, 1-week, and 4-week post-treatment in the open-label 1 Hz right DLPFC phase and compare change, response, and remission in those who received active compared to sham stimulation in the blinded phase. Additionally, we will build a multiple regression model to explore how prior treatment condition, expectancy, and HCS score impact the baseline adjusted treatment effect in the open-label 1 Hz right DLPFC treatment group.
Discussion
Broader implementation of rTMS is limited by logistical demands associated with the current standard 4–6-week course of daily treatment. This can be a significant barrier for patients who live farther from a treatment centre or those who can work or want to reduce the length of their medical leave. If the treatment regimen can be accelerated by administering multiple sessions per day, this would allow outpatients to undergo treatment with less disruption of daily activities and ultimately improve access to rTMS. This may also position rTMS as an option for patients with a need for rapid treatment effects, potentially allowing for remission in days rather than weeks. As such, there is a strong rationale to study the efficacy, safety, and tolerability of accelerated iTBS in patients with TRD.
Importantly, if our accelerated iTBS protocol demonstrates superiority over sham stimulation, it could be rapidly integrated into clinical practice with current equipment and minor modifications to clinic scheduling. Additionally, through strict adherence to blinding procedures, an advanced sham rTMS technique, and an assessment of patient expectancy and patient-technician relationship, this proposed study has the potential to provide significant insights into the role of non-specific TMS effects on clinical outcomes [43]. This large-scale, multi-site randomized control trial may broaden the use of neurostimulation by demonstrating the efficacy of a new rapid-acting treatment for this challenging, common, and burdensome illness.
Trial status
The study is currently recruiting participants. This trial began recruitment in February 2020.
Data availability
The final dataset generated from the current protocol will be available from the corresponding author upon reasonable request.
Abbreviations
- aiTBS:
-
accelerated intermittent theta burst stimulation
- ATHF:
-
Antidepressant Treatment History Form
- BDI-II:
-
Beck’s Depression Inventory
- CAMH:
-
Centre for Addiction and mental Health
- DLPFC:
-
dorsolateral prefrontal cortex
- eCRF:
-
electronic case report forms
- ECT:
-
Electroconvulsive therapy
- GAD-7:
-
Generalized Anxiety Disorder 7-Item
- HRSD:
-
Hamilton Rating Scale for Depression
- ITT:
-
intention to treat
- MDD:
-
Major depressive disorder
- MINI:
-
Mini-International Neuropsychiatric Interview
- PHQ-9:
-
Patient Health Questionnaire
- rTMS:
-
repetitive transcranial magnetic stimulation
- SSI:
-
Scale of Suicidal Ideation
- TASS:
-
Transcranial Magnetic Stimulation Adult Safety Screen
- TRD:
-
treatment-resistant depression
References
Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, Niederehe G, Thase ME, Lavori PW, Lebowitz BD, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163(11):1905–17.
The media’s damaging impact on public perception of electroconvulsive therapy. [http://healthydebate.ca/opinions/the-medias-damaging-impact-on-public-perception-of-electroconvulsive-therapy].
Sackeim HA, Prudic J, Fuller R, Keilp J, Lavori PW, Olfson M. The cognitive effects of electroconvulsive therapy in community settings. Neuropsychopharmacology. 2007;32(1):244–54.
Ingram A, Saling MM, Schweitzer I. Cognitive side effects of brief pulse electroconvulsive therapy: a review. J ECT. 2008;24(1):3–9.
Brakemeier EL, Berman R, Prudic J, Zwillenberg K, Sackeim HA. Self-evaluation of the cognitive effects of electroconvulsive therapy. J ECT. 2011;27(1):59–66.
Bustin J, Rapoport MJ, Krishna M, Matusevich D, Finkelsztein C, Strejilevich S, Anderson D. Are patients’ attitudes towards and knowledge of electroconvulsive therapy transcultural? A multi-national pilot study. Int J Geriatr Psychiatry. 2008;23(5):497–503.
Rossi S, Hallett M, Rossini PM, Pascual-Leone A. Safety of TMSCG: Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol. 2009;120(12):2008–39.
Lipsman N, Sankar T, Downar J, Kennedy SH, Lozano AM, Giacobbe P. Neuromodulation for treatment-refractory major depressive disorder. CMAJ. 2014;186(1):33–9.
Downar J, Geraci J, Salomons TV, Dunlop K, Wheeler S, McAndrews MP, Bakker N, Blumberger DM, Daskalakis ZJ, Kennedy SH, et al. Anhedonia and reward-circuit connectivity distinguish nonresponders from responders to dorsomedial prefrontal repetitive transcranial magnetic stimulation in major depression. Biol Psychiatry. 2014;76(3):176–85.
Mutz J, Vipulananthan V, Carter B, Hurlemann R, Fu CHY, Young AH. Comparative efficacy and acceptability of non-surgical brain stimulation for the acute treatment of major depressive episodes in adults: systematic review and network meta-analysis. BMJ. 2019;364:l1079.
Brunoni AR, Chaimani A, Moffa AH, Razza LB, Gattaz WF, Daskalakis ZJ, Carvalho AF. Repetitive Transcranial Magnetic Stimulation for the Acute treatment of major depressive episodes: a systematic review with Network Meta-analysis. JAMA Psychiatry. 2017;74(2):143–52.
O’Reardon JP, Solvason HB, Janicak PG, Sampson S, Isenberg KE, Nahas Z, McDonald WM, Avery D, Fitzgerald PB, Loo C, et al. Efficacy and safety of transcranial magnetic stimulation in the acute treatment of major depression: a multisite randomized controlled trial. Biol Psychiatry. 2007;62(11):1208–16.
Fitzgerald PB, Hoy K, Gunewardene R, Slack C, Ibrahim S, Bailey M, Daskalakis ZJ. A randomized trial of unilateral and bilateral prefrontal cortex transcranial magnetic stimulation in treatment-resistant major depression. Psychol Med. 2011;41(6):1187–96.
McDonald WM, Durkalski V, Ball ER, Holtzheimer PE, Pavlicova M, Lisanby SH, Avery D, Anderson BS, Nahas Z, Zarkowski P, et al. Improving the antidepressant efficacy of transcranial magnetic stimulation: maximizing the number of stimulations and treatment location in treatment-resistant depression. Depress Anxiety. 2011;28(11):973–80.
Downar J, Geraci J, Salomons TV, Dunlop K, Wheeler S, McAndrews MP, Bakker N, Blumberger DM, Daskalakis ZJ, Kennedy SH et al. Anhedonia and reward-circuit connectivity distinguish nonresponders from responders to Dorsomedial Prefrontal Repetitive Transcranial Magnetic Stimulation in Major Depression. Biol Psychiatry 2013.
Carpenter LL, Janicak PG, Aaronson ST, Boyadjis T, Brock DG, Cook IA, Dunner DL, Lanocha K, Solvason HB, Demitrack MA. Transcranial magnetic stimulation (TMS) for major depression: a multisite, naturalistic, observational study of acute treatment outcomes in clinical practice. Depress Anxiety. 2012;29(7):587–96.
Connolly KR, Helmer A, Cristancho MA, Cristancho P, O’Reardon JP. Effectiveness of transcranial magnetic stimulation in clinical practice post-FDA approval in the United States: results observed with the first 100 consecutive cases of depression at an academic medical center. J Clin Psychiatry. 2012;73(4):e567–573.
Di Lazzaro V, Dileone M, Pilato F, Capone F, Musumeci G, Ranieri F, Ricci V, Bria P, Di Iorio R, de Waure C, et al. Modulation of motor cortex neuronal networks by rTMS: comparison of local and remote effects of six different protocols of stimulation. J Neurophysiol. 2011;105(5):2150–6.
Blumberger DM, Vila-Rodriguez F, Thorpe KE, Feffer K, Noda Y, Giacobbe P, Knyahnytska Y, Kennedy SH, Lam RW, Daskalakis ZJ, et al. Effectiveness of theta burst versus high-frequency repetitive transcranial magnetic stimulation in patients with depression (THREE-D): a randomised non-inferiority trial. Lancet. 2018;391(10131):1683–92.
Holtzheimer PE 3rd, McDonald WM, Mufti M, Kelley ME, Quinn S, Corso G, Epstein CM. Accelerated repetitive transcranial magnetic stimulation for treatment-resistant depression. Depress Anxiety. 2010;27(10):960–3.
Hadley D, Anderson BS, Borckardt JJ, Arana A, Li X, Nahas Z, George MS. Safety, tolerability, and effectiveness of high doses of adjunctive daily left prefrontal repetitive transcranial magnetic stimulation for treatment-resistant depression in a clinical setting. J ECT. 2011;27(1):18–25.
McGirr A, Van den Eynde F, Tovar-Perdomo S, Fleck MP, Berlim MT. Effectiveness and acceptability of accelerated repetitive transcranial magnetic stimulation (rTMS) for treatment-resistant major depressive disorder: an open label trial. J Affect Disord. 2015;173:216–20.
Baeken C, Marinazzo D, Wu GR, Van Schuerbeek P, De Mey J, Marchetti I, Vanderhasselt MA, Remue J, Luypaert R, De Raedt R. Accelerated HF-rTMS in treatment-resistant unipolar depression: insights from subgenual anterior cingulate functional connectivity. World J Biol Psychiatry. 2014;15(4):286–97.
Cole EJ, Phillips AL, Bentzley BS, Stimpson KH, Nejad R, Barmak F, Veerapal C, Khan N, Cherian K, Felber E, et al. Stanford Neuromodulation Therapy (SNT): a double-blind randomized controlled trial. Am J Psychiatry. 2022;179(2):132–41.
Cole EJ, Stimpson KH, Bentzley BS, Gulser M, Cherian K, Tischler C, Nejad R, Pankow H, Choi E, Aaron H, et al. Stanford Accelerated Intelligent Neuromodulation Therapy for Treatment-Resistant Depression. Am J Psychiatry. 2020;177(8):716–26.
Hamilton M. Development of a rating scale for primary depressive Illness. Br J Soc Clin Psychol. 1967;6(4):278–96.
Kupfer DJ. Achieving adequate outcomes in geriatric depression: standardized criteria for remission. J Clin Psychopharmacol. 2005;25(4 Suppl 1):24–8.
Reynolds CF 3rd, Frank E, Kupfer DJ, Thase ME, Perel JM, Mazumdar S, Houck PR. Treatment outcome in recurrent major depression: a post hoc comparison of elderly (young old) and midlife patients. Am J Psychiatry. 1996;153(10):1288–92.
Petrides G, Fink M, Husain MM, Knapp RG, Rush AJ, Mueller M, Rummans TA, O’Connor KM, Rasmussen KG Jr., Bernstein HJ, et al. ECT remission rates in psychotic versus nonpsychotic depressed patients: a report from CORE. J ECT. 2001;17(4):244–53.
Sackeim HA, Dillingham EM, Prudic J, Cooper T, McCall WV, Rosenquist P, Isenberg K, Garcia K, Mulsant BH, Haskett RF. Effect of concomitant pharmacotherapy on electroconvulsive therapy outcomes: short-term efficacy and adverse effects. Arch Gen Psychiatry. 2009;66(7):729–37.
Sackeim HA, Prudic J, Devanand DP, Nobler MS, Lisanby SH, Peyser S, Fitzsimons L, Moody BJ, Clark J. A prospective, randomized, double-blind comparison of bilateral and right unilateral electroconvulsive therapy at different stimulus intensities. Arch Gen Psychiatry. 2000;57(5):425–34.
Bech P, Allerup P, Gram LF, Reisby N, Rosenberg R, Jacobsen O, Nagy A. The Hamilton depression scale. Evaluation of objectivity using logistic models. Acta Psychiatr Scand. 1981;63(3):290–9.
Totura CMW, Fields SA, Karver MS. The role of the therapeutic relationship in Psychopharmacological Treatment outcomes: a Meta-analytic review. Psychiatric Serv. 2018;69(1):41–7.
Mack JW, Block SD, Nilsson M, Wright A, Trice E, Friedlander R, Paulk E, Prigerson HG. Measuring Therapeutic Alliance between oncologists and patients with Advanced Cancer: the human connections scale. Cancer. 2009;115(14):3302–11.
Fitzgerald PB, Hoy KE, Reynolds J, Singh A, Gunewardene R, Slack C, Ibrahim S, Daskalakis ZJ. A pragmatic randomized controlled trial exploring the relationship between pulse number and response to repetitive transcranial magnetic stimulation treatment in depression. Brain Stimul 2019.
Oquendo MA, Baca-Garcia E, Kartachov A, Khait V, Campbell CE, Richards M, Sackeim HA, Prudic J, Mann JJ. A computer algorithm for calculating the adequacy of antidepressant treatment in unipolar and bipolar depression. J Clin Psychiatry. 2003;64(7):825–33.
Sackeim HA, Prudic J, Devanand DP, Decina P, Kerr B, Malitz S. The impact of medication resistance and continuation pharmacotherapy on relapse following response to electroconvulsive therapy in major depression. J Clin Psychopharmacol. 1990;10(2):96–104.
George MS, Lisanby SH, Avery D, McDonald WM, Durkalski V, Pavlicova M, Anderson B, Nahas Z, Bulow P, Zarkowski P, et al. Daily left prefrontal transcranial magnetic stimulation therapy for major depressive disorder: a sham-controlled randomized trial. Arch Gen Psychiatry. 2010;67(5):507–16.
Hsu JH, Downar J, Vila-Rodriguez F, Daskalakis ZJ, Blumberger DM. Impact of prior treatment on remission with intermittent theta burst versus high-frequency repetitive transcranial magnetic stimulation in treatment resistant depression. Brain Stimul. 2019;12(6):1553–5.
Borckardt JJ, Nahas ZH, Teal J, Lisanby SH, McDonald WM, Avery D, Durkalski V, Pavlicova M, Long JM, Sackeim HA, et al. The painfulness of active, but not sham, transcranial magnetic stimulation decreases rapidly over time: results from the double-blind phase of the OPT-TMS trial. Brain Stimul. 2013;6(6):925–8.
Younger J, Gandhi V, Hubbard E, Mackey S. Development of the Stanford Expectations of Treatment Scale (SETS): a tool for measuring patient outcome expectancy in clinical trials. Clin Trails. 2012;9:767–76.
Colloca L, Barsky AJ. Placebo and Nocebo effects. N Engl J Med. 2020;382(6):554–61.
Burke MJ. A fundamental change is needed for appraising placebo responses in psychiatry. Lancet Psychiatry. 2023;10(5):316–7.
Mir-Moghtadaei A, Caballero R, Fried P, Fox MD, Lee K, Giacobbe P, Daskalakis ZJ, Blumberger DM, Downar J. Concordance between BeamF3 and MRI-neuronavigated Target sites for Repetitive Transcranial Magnetic Stimulation of the Left Dorsolateral Prefrontal Cortex. Brain Stimul. 2015;8(5):965–73.
Schutter DJ, van Honk J. A standardized motor threshold estimation procedure for transcranial magnetic stimulation research. J ECT. 2006;22(3):176–8.
Julkunen P, Saisanen L, Sarasti M, Kononen M. Effect of electrode cap on measured cortical motor threshold. J Neurosci Methods. 2009;176(2):225–9.
Kramar EA, Babayan AH, Gavin CF, Cox CD, Jafari M, Gall CM, Rumbaugh G, Lynch G. Synaptic evidence for the efficacy of spaced learning. Proc Natl Acad Sci U S A. 2012;109(13):5121–6.
Lynch G, Kramar EA, Babayan AH, Rumbaugh G, Gall CM. Differences between synaptic plasticity thresholds result in new timing rules for maximizing long-term potentiation. Neuropharmacology. 2013;64:27–36.
Smolen P, Zhang Y, Byrne JH. The right time to learn: mechanisms and optimization of spaced learning. Nat Rev Neurosci. 2016;17(2):77–88.
Rogasch NC, Daskalakis ZJ, Fitzgerald PB. Mechanisms underlying long-interval cortical inhibition in the human motor cortex: a TMS-EEG study. J Neurophysiol. 2013;109(1):89–98.
Williams NR, Sudheimer KD, Bentzley BS, Pannu J, Stimpson KH, Duvio D, Cherian K, Hawkins J, Scherrer KH, Vyssoki B, et al. High-dose spaced theta-burst TMS as a rapid-acting antidepressant in highly refractory depression. Brain. 2018;141(3):e18.
McCalley DM, Lench DH, Doolittle JD, Imperatore JP, Hoffman M, Hanlon CA. Determining the optimal pulse number for theta burst induced change in cortical excitability. Sci Rep. 2021;11(1):8726.
Konstantinou GN, Downar J, Daskalakis ZJ, Blumberger DM. Accelerated intermittent Theta Burst Stimulation in Late-Life Depression: a possible option for older depressed adults in need of ECT during the COVID-19 pandemic. Am J Geriatr Psychiatry. 2020;28(10):1025–9.
Rossi S, Antal A, Bestmann S, Bikson M, Brewer C, Brockmoller J, Carpenter LL, Cincotta M, Chen R, Daskalakis JD, et al. Safety and recommendations for TMS use in healthy subjects and patient populations, with updates on training, ethical and regulatory issues: Expert guidelines. Clin Neurophysiol. 2021;132(1):269–306.
Pradier MF, McCoy TH Jr., Hughes M, Perlis RH, Doshi-Velez F. Predicting treatment dropout after antidepressant initiation. Transl Psychiatry. 2020;10(1):60.
Taylor JJ, Newberger NG, Stern AP, Phillips A, Feifel D, Betensky RA, Press DZ. Seizure risk with repetitive TMS: Survey results from over a half-million treatment sessions. Brain Stimul. 2021;14(4):965–73.
Kobak KA, Kane JM, Thase ME, Nierenberg AA. Why do clinical trials fail? The problem of measurement error in clinical trials: time to test new paradigms? J Clin Psychopharmacol. 2007;27(1):1–5.
Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170(7):723–33.
Kroenke K, Spitzer RL, Williams JB. The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med. 2001;16(9):606–13.
Chung SW, Thomson CJ, Lee S, Worsley RN, Rogasch NC, Kulkarni J, Thomson RH, Fitzgerald PB, Segrave RA. The influence of endogenous estrogen on high-frequency prefrontal transcranial magnetic stimulation. Brain Stimul. 2019;12(5):1271–9.
National Collaborating Centre for Mental Health, National Institute for Health & Clinical Excellence. Depression in adults with a chronic physical health problem: treatment and management. National clinical practice guideline 91. edn. London: British Psychological Society, The Royal College of Psychiatrists; 2010.
Schafer JL. Analysis of incomplete Multivariate Data. New York: Chapman and Hall; 1997.
Funding
This work is supported by Brain Canada.
Author information
Authors and Affiliations
Contributions
DMB obtained funding and developed the manuscript, with important intellectual input from all co-authors.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval was obtained by the Centre for Addiction and Mental Health Research Ethics Board (reference number 109–2021) and the University of British Columbia and the Vancouver Coastal Health Research Institute Research Ethics Boards, (reference number H22-00151). The informed consent process is initiated before the patient agrees to participate in the study and will be obtained according to REB and GCP guidelines. All patients referred by a general physician or psychiatrist will undergo an extensive consultation with a brain stimulation psychiatrist at each study site, who will determine suitability for referral to this trial. Patients referred to the trial will then be assessed for eligibility by qualified research personnel who will obtain consent. Patients will be informed that they can withdraw participation at any point during the study, and the rights and welfare of the patients will be protected.
Consent for publication
Not applicable.
Competing interests
FVR receives research support from the Canadian Institutes of Health Research, Brian Canada, Michael Smith Foundation for Health Research, Vancouver Coastal Health Research Institute, and in-kind equipment support for an investigator-initiated trial from MagVenture. He has participated in an advisory board for Janssen. MB receives salary support from the SickKids Research Institute, and unrelated implementation research funding from CIHR, NIH, and the Helmsley Trust. MJB has received research salary support from the Liu Fu Yu Charity Foundation, Sunnybrook Foundation and University of Toronto Department of Psychiatry. MJB reports no conflicts of interest. JD has received research support from NIH, CIHR, Brain Canada, Ontario Brain Institute, the Klarman Family Foundation, the Krembil Foundation, Arrell Family Foundation, and the Buchan Family Foundation, in-kind equipment support for investigator-initiated trials from MagVenture, is an advisor for BrainCheck, Arc Health Partners and Salience Neuro Health, and is a co-founder of Ampa Health. JH has received research support from CIHR, NIH, SUAP, the University Health Network-Mount Sinai Alternative Funding Program, Physicians Services Incorporated Foundation, Canadian Breast Cancer Research Alliance, and Science of Caring, He receives research and salary support from the Pencer Family Chair in Applied General Psychiatry at Sinai Health, funded by Sinai Health and the University of Toronto. TSK has received research support from CIHR and the AFP Innovation Fund. RM has received research support from CIHR, University Health Network-Mount Sinai Alternative Funding Program, and Heart & Stroke Foundation (Canada). He receives research and salary support from the Chair in Health and Behaviour at Sinai Health, funded by Sinai Health and the University of Toronto. DV holds the Labatt Family Professorship in Depression Biology, a University Named Professorship at the University of Toronto. She receives research support from CIHR, NIMH, the Centre for Addiction and Mental Health (CAMH), The Centre for Mental Health at University Health Network and the Department of Psychiatry at the University of Toronto. DV declares no biomedical interests or conflicts. WZ acknowledges the support of the Michael Smith Health Research Scholar Award. Her research has also been funded by CIHR, UBC Health Ministry of Health Research Seed Grant Program, BC Lung Foundation, BC SUPPORT Unit, Canadian Glycomics Network, and Pfizer Canada. Neither of the above research funding is in relation to this submitted work and related subjects. WZ declares that she has no competing interests. DMB receives research support from the Canadian Institutes of Health Research (CIHR), National Institutes of Health – US (NIH), Brain Canada Foundation and the Temerty Family through the CAMH Foundation and the Campbell Family Research Institute. He received research support and in-kind equipment support for an investigator-initiated study from Brainsway Ltd. and he was the site principal investigator for three sponsor-initiated studies for Brainsway Ltd. He received in-kind equipment support from Magventure for investigator-initiated studies. He received medication supplies for an investigator-initiated trial from Indivior. He has participated in an advisory board for Janssen. He has participated in an advisory board for Welcony Inc. MSG, YK, KT, PK, and APT report no completing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Goodman, M.S., Vila-Rodriguez, F., Barwick, M. et al. A randomized sham-controlled trial of high-dosage accelerated intermittent theta burst rTMS in major depression: study protocol. BMC Psychiatry 24, 28 (2024). https://doi.org/10.1186/s12888-023-05470-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-023-05470-9