
Abstract
Background
Transcobalamin deficiency is a rare inborn metabolic disorder, characterized by pancytopenia, megaloblastic anemia, failure to thrive, diarrhea, and psychomotor retardation.
Case presentation
We describe a patient who first presented at 3 months of age, with pancytopenia, hepatosplenomegaly, recurrent infection, metabolic acidosis, and acute hemolytic crisis. Extensive hematologic and immunologic investigations did not identify inherited bone marrow failure syndrome, acute leukemia or its related disorders. Whole exome sequencing identified a novel homozygous TCN2 mutation, c.428-2A > G and mRNA study confirmed an aberrant transcription of exon 4 skipping. The mutant protein is predicted to have an in-fame 51 amino acids deletion (NP_000346:p.Gly143_Val193del). The patient exhibited marked clinical improvement following hydroxocobalamin treatment.
Conclusions
Transcobalamin deficiency should be investigated in infants with unexplained pancytopenia and acute hemolytic crisis with or without typical evidence of vitamin B12 deficiency.
Similar content being viewed by others


Background
Transcobalamin II (TC), a cobalamin binding plasma protein, facilitates cellular uptake via receptor-mediated endocytosis [1,2,3]. Deficiency or defect of TC, caused by TCN2 mutations, results in intracellular cobalamin depletion that leads to a rare multisystemic disorder with autosomal recessive inheritance. Common clinical features include pancytopenia, megaloblastic anemia, failure to thrive, diarrhea, psychomotor retardation, and less frequently, immune deficiency [1]. The onset of symptoms is usually during infantile period, with the median age of 2 to 4 months [1].
Here, we reported an infant with overlapping clinical features of hematologic malignancy, inherited bone marrow failure syndrome (IBMFS), hemolytic crisis, primary immune deficiency (PID), and metabolic acidosis. The advance in next-generation sequencing had enabled the genetic diagnosis of the homozygous novel TCN2 mutation.
Case presentation
A 3-month-old male infant, a single child of a consanguineous Thai couple, was first presented to a local medical center with upper respiratory tract infection, anemia, petechiae, mild hepatosplenomegaly, and pancytopenia (Table 1). Acute leukemia and inherited bone marrow failure syndrome were suspected but not supported by the findings of bone marrow (BM) aspiration and biopsy, flow cytometry, and BM karyotype. At 4 to 5 months of age, the patient had two episodes of diarrhea. There was a 10% increase of circulating immature myeloid series and marked elevation of serum lactate dehydrogenase (LDH) levels, necessitating a re-evaluation for myeloid leukemia, but findings were negative.
At 5 months after arrival at our center, the patient appeared to have failure to thrive, hepatosplenomegaly, respiratory distress; and his chest X-ray indicated bilateral reticulonodular infiltration suggesting pneumonia (Fig. 1A). Bronchoalveolar lavage was performed and Pneumocystis jirovecii was confirmed (via PCR) as the causative pathogen. Persistent pancytopenia with monocytosis > 1000/mm3 was noted; thus, PID, IBMFS, myelodysplastic syndrome (MDS), juvenile myelomocytic leukemia (JMML), and autoimmune lymphoproliferative syndrome (ALPS) were investigated. The results did not support PID, including normal or slightly elevated number of absolute lymphocyte counts [CD3+, 7.3 K/μL (reference 2.2–6.4); CD4+, 4.2 K/μL (reference 1.6–4.6); CD8+, 3.1 K/μL (reference 0.7–2.4), CD19+, 0.6 K/μL (reference 0.5–1.5)] and normal or slightly increased immunoglobulin levels [IgA 1.19 mg/ml (reference 0.1–0.24), IgG 5.22 mg/ml (reference 4.57–8.83), and IgM 4.84 mg/ml (reference 0.27–0.93)]. Additional BM studies revealed normal karyotype, no canonical RAS pathway gene mutations, and normal double negative T-cell, excluding JMML and ALPS. The patient received aggressive supportive treatments including blood transfusion, parenteral nutrition and enteral tube feeding for 4 weeks plus antimicrobial agent for the pneumonia. This led to temporary improvement of hematologic profiles (Table 1). At 9 months, the patient developed mild diarrhea with inappropriate wide anion gap metabolic acidosis (serum TCO2 5; gap 20), prompting investigation for inborn metabolic disorders. Acylcarnitine profile, plasma amino acids, and urinary organic acids were within normal ranges. Whole exome sequencing was performed on Illumina HiSeq2500 (Macrogen, South Korea) and analyzed following previously published protocols [4], using clinical terms of ketoacidosis (HP:0001993) and pancytopenia (HP:0001876).

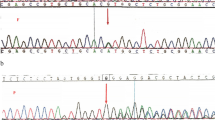
A Chest x-ray showing bilateral reticulonodular infiltration and hepatomegaly, B Peripheral blood smear (1000x) showing macro-ovalocytes (black arrow), variable-sized red blood cells as well as fragmented cell (red arrow), and hypersegmented neutrophils (green arrow). These findings give credence to megaloblastic anemia. C Bone marrow smear with Wright-Giemsa stain (1000x) showing megaloblastic change, large erythroblast (black arrow), nuclear and cytoplasmic maturational asynchrony of erythroid precursors (yellow arrow), dysplastic nuclei of erythroid precursors (green arrow), and giant band (red arrow). D Sanger sequencing, noted homozygous change from A to G at nucleotide c.428–2, and heterozygous A > G in the father (and mother: not shown). E mRNA (cDNA) bands showing only aberrant mRNA in the patient’s specimen (PT) and only wild type band in the normal control (NL). F mRNA sequencing demonstrating exon 4 skipping: coding sequence of exon 3 followed by the coding sequence of exon 5
At 12 months, macrocytic anemia with neutropenia was noted (Table 1 and Fig. 1B) leading to additional investigations. That revealed normal levels of serum folate (> 20; reference range 4.2–19.9 ng/ml), red-blood-cell folate (383; reference range 141–1038 ng/ml), and B12 (498; reference range 197–771 pg/ml). Serum holotranscobalamin which is an active form of vitamin B12 and an optimal marker for vitamin B12 deficiency was not measured in the present patient, owing to unavailability in Thailand. Another BM study was performed that revealed hypercellularity, megaloblastic and dysplastic changes (Fig. 1C). Targeted exome analysis of 25 genes associated with myeloid malignancy and MDS was carried out using QIAact Myeloid DNA UMI Panel (Qiagen, Hilden, Germany). Results were negative. At 14 months, the patient developed Bacillus cereus septicemia accompanying acute hemolytic crisis as evidenced by reticulocytosis at 22.7% (absolute count 679.6 × 103/mm3), reduction of serum haptoglobin (< 0.025; normal range 0.32–1.97 mg/ml) and marked elevation of LDH level whilst having normal G6PD enzyme activity, hemoglobin typing, and alpha-globin gene analysis.
At 15 months of age, the results of WES became available that revealed a homozygous mutation at the acceptor site of intron 3 of TCN2 gene, c.428-2A > G (IVS3-2A > G; NM_000355.3). Sanger sequencing confirmed the homozygous mutation in the patient and heterozygous alleles in both parents (Fig. 1D). mRNA study using previously published methods [5] demonstrated an aberrant transcript lacking exon 4 (Fig. 1E-F). Daily treatment with 1 mg of intramuscular hydroxocobalamin was initiated for 1 week, then twice a week. Serum LDH and plasma homocysteine levels were used to monitor the clinical and biochemical response, besides CBC (Table 1). The patient exhibited dramatic response to the cobalamin therapy by becoming energetic, exhibiting mouth feeding, achieving motor milestones, and requiring no further transfusion. Total plasma homocysteine rapidly declined while CBC revealed subsequent improvements in Hb, MCV, RDW and platelets. At the time of this report, the patient is 30 months-old and exhibits a mild autistic-spectrum disorder (repetitive behavior, delayed speech and poor social interaction) despite active motor development (Table 1).
Discussion and conclusion
We describe an unusual case with complex manifestations of TC deficiency, including hepatosplenomegaly, metabolic acidosis without methylmalonic aciduria, episodic hemolytic crisis, P.jirovecii infection, and high LDH levels. The hepatosplenomegaly, persistent elevation of LDH levels, hypercellularity and dysplastic changes of BM led to differential diagnosis of hematologic malignancies [6,7,8]. These complex features complicated and delayed the diagnosis which was eventually resolved by WES. Plausible mechanisms underlying the acute hemolytic episode in this patient are intramedullary hemolysis and premature lysis of erythroid precursor cells due to ineffective hematopoiesis [8]. Hemolytic crisis is one of the characteristics described in vitamin B12 deficiency [8]. The low-to-normal MCV in the present case could be explained by frequent blood transfusion and possible low MCV of the transfused blood due to high prevalence (40%) of thalassemia carrier among Thai people including blood donors [1, 9]. By retrospective examination of the peripheral blood and BM specimens, macro-ovalocytes and hypersegmented neutrophils were seen, and giant erythroblast with nuclear cytoplasmic asynchrony and giant band were noted (Fig. 1C). In fact, these findings could be diagnostic clues for TC deficiency. We experienced that serum homocysteine and LDH levels were good alternative biomarkers in monitoring the biochemical response to cobalamin treatment in the present patient (Table 1). Normal serum cobalamin level does not exclude cobalamin deficiency and other cobalamin-related disorders, since 80% of cobalamin binds to haptocorrin, which does not represent its intracellular utilization [2, 8, 10]. It should be mentioned that homocysteine is generally undetectable or barely detectable using quantitative plasma amino acid analysis; therefore, elevated homocysteine concentration was missed. Later, when transcobalamin defect and its associated increased homocysteine level was suspected, plasma homocysteine level was measured using a separate specific test.
Less than 60 patients and 50 TCN2 variants have been reported worldwide in which many cases were reported from regions with a high rate of consanguinity, including Turkey [1, 2, 11,12,13,14]. To our knowledge, the present patient is the first case report from a Southeast Asian country with a novel TCN2 variant, c.428-2A > G. The variant was subsequently submitted to ClinVar database and now is accessible using a number SCV001981507 (https://www.ncbi.nlm.nih.gov/clinvar/). This mutation leads to a mutant TCN2 transcript with deletion of 153 nucleotides (NM_000355.3:r.428_580del) and results in an in-frame deletion of 51 amino acids (NP_000346:p.Gly143_Val193del). The mutant protein is predicted to damage cobalamin transport due to loss of two of the three cobalamin binding sites, codon 152–156, 190–194, and 395–397 (www://uniprot.org) [15]. Moreover, the loss of cysteine at codon 165 could disrupt one (Cys165/Cys205) of the four disulfide bonds which are essential for maintaining the secondary structure of the TC protein [15, 16].
Cobalamin plays an important role in initiating and maintaining myelination of the central nervous system for almost one-third of patients with TC deficiency demonstrated developmental delay [1, 17]. Early diagnosis and treatment could alleviate and prevent permanent neurologic sequelae [1, 17]. A case report of a TC deficiency patient who exhibited urinary methylmalonic acid excretion at birth may indicate intrauterine onset of the metabolic derangement and neurologic damage of this intriguing disorder [18]. This may explain neurocognitive impairment in some patients despite receiving prompt diagnosis and treatment at birth [14].
In summary, TC deficiency should be suspected in infant with chronic pancytopenia, hepatosplenomegaly, metabolic acidosis of unclear cause, and recurrent infection with or without overt megaloblastic change.
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the ClinVar repository (accession number SCV001981507, https://www.ncbi.nlm.nih.gov/clinvar/).
Abbreviations
- ALPS:
-
Autoimmune lymphoproliferative syndrome
- ASD:
-
Autistic spectrum disorder
- BM:
-
Bone marrow
- CBC:
-
Complete blood count
- EN + PN:
-
Enteral and parenteral nutrition
- G6PD:
-
Glucose 6 phosphate dehydrogenase
- Hb:
-
Hemoglobin
- IBMFS:
-
Inherited bone marrow failure syndrome
- IM:
-
Intramuscular
- JMML:
-
Juvenile myelomocytic leukemia
- JP:
-
Just palpable
- LDH:
-
Lactate dehydrogenase
- MCV:
-
Mean corpuscular volume
- MDS:
-
Myelodysplastic syndrome
- ND:
-
No data
- NG:
-
Nasogastric tube
- PCR:
-
Polymerase chain reaction
- PEM:
-
Protein energy malnutrition
- PID:
-
Primary immune deficiency
- PJP:
-
Pneumocystis jirovecii
- RDW:
-
Red blood cell distribution width
- TC:
-
Transcobalamin II
- URI:
-
Upper respiratory tract infection
- WES:
-
Whole exome sequencing
References
Trakadis Y, Alfares A, Bodamer O, Buyukavci M, Christodoulou J, Connor P, et al. Update on transcobalamin deficiency: clinical presentation, treatment and outcome. J Inherit Metab Dis. 2014;37(3):461–73.
Huemer M, Baumgartner MR. The clinical presentation of cobalamin-related disorders: from acquired deficiencies to inborn errors of absorption and intracellular pathways. J Inherit Metab Dis. 2019;42(4):686–705.
Quadros EV. Advances in the understanding of cobalamin assimilation and metabolism. Br J Haematol. 2010;148(2):195–204.
Thongpradit S, Jinawath N, Javed A, Jensen LT, Chunsuwan I, Rojnueangnit K, et al. Novel SOX10 mutations in Waardenburg syndrome: functional characterization and genotype-phenotype analysis. Front Genet. 2020;11. https://doi.org/10.3389/fgene.2020.589784.
Suwannarat P, Keeratichamroen S, Wattanasirichaigoon D, Ngiwsara L, Cairns J, Svasti J, et al. Molecular characterization of type 3 (neuronopathic) Gaucher disease in Thai patients. Blood Cells Mol Dis. 2007;39(3):348–52.
Parmentier S, Meinel J, Oelschlaegel U, Mohr B, Ehninger G, Schaich M, et al. Severe pernicious anemia with distinct cytogenetic and flow cytometric aberrations mimicking myelodysplastic syndrome. Ann Hematol. 2012;91(12):1979–81.
Rana SR, Colman N, Goh KO, Herbert V, Klemperer MR. Transcobalamin II deficiency associated with unusual bone marrow findings and chromosomal abnormalities. Am J Hematol. 1983;14(1):89–96.
Stabler SP. Vitamin B12 deficiency. N Engl J Med. 2013;368(2):149–60.
Panich V, Pornpatkul M, Sriroongrueng W. The problem of thalassemia in Thailand. Southeast Asian J Trop Med Public Health. 1992;23:1–6.
Snow CF. Laboratory diagnosis of vitamin B12 and folate deficiency: a guide for the primary care physician. Arch Intern Med. 1999;159(12):1289–98.
Zhan S, Cheng F, He H, Hu S, Feng X. Identification of transcobalamin deficiency with two novel mutations in the TCN2 gene in a Chinese girl with abnormal immunity: a case report. BMC Pediatr. 2020;20(1):460.
Ünal S, Karahan F, Arıkoğlu T, Akar A, Kuyucu S. Different presentations of patients with transcobalamin II deficiency: a single-center experience from Turkey. Turk J Hematol. 2019;36(1):37.
Nashabat M, Maegawa G, Nissen PH, Nexo E, Al-Shamrani H, Al-Owain M, et al. Long-term outcome of 4 patients with transcobalamin deficiency caused by 2 novel TCN2 mutations. J Pediatr Hematol Oncol. 2017;39(8):e430–6.
Kose E, Besci O, Gudeloglu E, Suncak S, Oymak Y, Ozen S, et al. Transcobalamin II deficiency in twins with a novel variant in the TCN2 gene: case report and review of literature. J Pediatr Endocrinol Metab. 2020;33(11):1487–99.
Wuerges J, Garau G, Geremia S, Fedosov SN, Petersen TE, Randaccio L. Structural basis for mammalian vitamin B12 transport by transcobalamin. Proc Natl Acad Sci U S A. 2006;103(12):4386–91.
Wuerges J, Geremia S, Fedosov SN, Randaccio L. Vitamin B12 transport proteins: crystallographic analysis of β-axial ligand substitutions in cobalamin bound to Transcobalamin. IUBMB Life. 2007;59(11):722–9.
Hall CA. The neurologic aspects of transcobalamin II deficiency. Br J Haematol. 1992;80(1):117–20.
Ratschmann R, Minkov M, Kis A, Hung C, Rupar T, Mühl A, et al. Transcobalamin II deficiency at birth. Mol Genet Metab. 2009;98(3):285–8.
Acknowledgements
The authors are thankful of the family for participating in this study, the referring physicians, the Ramathibodi Foundation for financial support for the cost of whole exome sequencing, and the Faculty of Medicine Ramathibodi Hospital for giving Research Career Development Awards to DW, TT, and NS.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
PP and DW designed and wrote the paper. DW, TT, and AK interpreted the WES and mRNA study. PP, NS, and DS prepared and described peripheral blood smear and bone marrow smear. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by Human Research Ethics Committee, Faculty of Medicine Ramathibodi Hospital, Mahidol University (COA. MURA2021/632). Written informed consent for publication was obtained from parents.
Consent for publication
Written informed consent for publication was obtained from parents.
Competing interests
The corresponding author, DW, is a member of the editorial board (Associate Editor) of this journal.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visithttp://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Pongphitcha, P., Sirachainan, N., Khongkraparn, A. et al. A novel TCN2 mutation with unusual clinical manifestations of hemolytic crisis and unexplained metabolic acidosis: expanding the genotype and phenotype of transcobalamin II deficiency. BMC Pediatr 22, 233 (2022). https://doi.org/10.1186/s12887-022-03291-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-022-03291-5