
Abstract
Background
Oguchi disease is a rare type of congenital stationary night blindness associated with an abnormal fundus appearance. It is inherited in an autosomal recessive manner where two types exist according to the gene affected; type 1 associated with S-antigen (SAG) gene mutations and type 2 associated with rhodopsin kinase (GRK1) gene mutations.
Purpose
The aim of this work was to describe the clinical and genetic findings of the first two reported families of Oguchi disease in Egypt and African region.
Methods
Four members of two consanguineous Egyptian families with history of night blindness since childhood underwent complete ophthalmological examination, standard automated static perimetry, fundus color photography, fundus autofluorescence (FAF), fundus fluorescein angiography (FFA) in light-adapted state and spectral-domain optical coherence tomography (SD-OCT) of both the macula and the optic nerve head as well as central corneal thickness with repeated fundus photography following prolonged dark adaptation. Mutation screening of 7 coding exons of GRK1 gene and 15 coding exons of SAG gene as well as some flanking regions were performed using Sanger sequencing technique. The variants were tested for pathogenicity using different in silico functional analysis tools.
Results
The clinical examination and investigations confirmed Oguchi disease phenotype. One patient showed p.R193* (c.577C > T) which is a previously reported SAG gene mutation in a homozygous form. The other three patients from a different family showed (c.649–1 G > C), a novel canonical splice site SAG gene mutation in a homozygous form.
Conclusion
The identification of the novel canonical splice site SAG gene variant in three members of the same family with clinically confirmed Oguchi disease reinforces its pathogenicity. A fourth patient from another family carried a previously reported mutation in the same gene. SAG gene variants may be the underlying genetic cause for Oguchi disease in Egypt. Our findings have expanded the spectrum of Oguchi disease-associated mutations in SAG gene and may serve as a basis for genetic diagnosis for Oguchi disease.
Similar content being viewed by others

Background
Oguchi disease is considered one of the rare forms of congenital stationary night blindness (CSNB) that is inherited in an autosomal recessive manner. It is classified into two types: type 1 and type 2, each related to separate genetic background. Mutations in arrestin gene (SAG, S-antigen; OMIM:181031) were first discovered in Japanese patients (Oguchi disease type 1) [1], later on, mutations involving the rhodopsin kinase gene (GRK1; OMIM:180381) were implicated in European patients (Oguchi disease type 2) [2]. Both genes encode proteins involved in recovery of rhodopsin after photoactivation [1].
GRK1 gene is mapped to chromosome 13. It is a specialized G-protein-coupled receptor kinase that eventually results in phototransduction cascade shutdown [3, 4]. Various mutations have been reported in this gene, which have been shown to result in a decline in the catalytic activity of the protein, leading to a delay in photoreceptor recovery [5, 6].
SAG gene is located on chromosome 2, consists of 16 exons of which 15 are coding, and it encodes for arrestin which is a 405 amino acid regulatory cytosolic protein. Arrestin forms a complex with photoactive rhodopsin, thereby uncoupling it from the G-protein transducin, and thus playing a critical role in recovery phase of phototransduction [3, 7]. Pathogenic variants detected in SAG gene can manifest as Oguchi inherited in autosomal recessive manner as well as retinitis pigmentosa (RP) inherited in autosomal dominant manner. To date, around 18 pathogenic variants have been detected in the gene including 1 gross insertion, 2 gross deletions, 2 small deletions and 13 missense/nonsense mutations [8].
Clinically, Oguchi disease is characterized by non-progressive retinal dysfunction as well as a unique golden-yellow fundus with a metallic sheen that reverts to normal fundus color after prolonged dark adaptation. It was first reported by Oguchi in 1907 as a variant of CSNB. Later, it was further phenotypically characterized by Mizuo in 1913 by demonstrating the Mizuo-Nakamura phenomenon where the fundus discoloration disappears, leaving normal fundus appearance after 3–4 h of dark adaptation, only to reappear again shortly after exposure to light [9].
Oguchi disease has been demonstrated to be more prevalent among the Japanese population as compared to other populations [1]. To the authors’ knowledge, around 50 cases of Oguchi disease have been reported worldwide and those have been primarily concentrated in Japan with few reported cases from Europe [10,11,12,13], Turkey [14], China [15], India [16], Pakistan [17, 18] and Iran [19].
Oguchi disease has not been described so far in the Egyptian population or in any African population, and the mutation spectrum remains unknown whether SAG or GRK1 mutations will prevail. Thus, we set out this study to report and characterize the first cases encountered both clinically and genetically.
Methods
Four members of two indigenous Egyptian families with history of night blindness were examined at a general comprehensive clinic of Al Mouneer Diabetic Eye Center, Cairo, Egypt.
The enrolled patients underwent a complete ophthalmological examination that included recording of medical, ocular and family histories, unaided distance visual acuity (UDVA) and corrected distance visual acuity (CDVA) testing using the Snellen chart, subjective refraction, color vision testing by Ishihara chart (38-plate edition), ocular motility examination, intraocular pressure measurements using Goldmann applanation tonometry, as well as slit-lamp biomicroscopy and a dilated fundus examination. All patients refrained from undergoing electrophysiological studies as they were unavailable in our center and had to be performed elsewhere.
Standard automated static perimetry (Vision Monitor, Metrovision, Pérenchies, France) was done for both eyes of each patient pre-dilation of the pupils. Fundus color photography, fundus autofluorescence (FAF) and fundus fluorescein angiograms (FFA) were done in light-adapted state post-dilation of the pupils. In order to confirm the diagnosis, patients were dark adapted for 3–4 h following which the fundi were re-imaged, and spectral-domain optical coherence tomography (SD-OCT) of both the macula and the optic nerve head as well as central corneal thickness were performed with Topcon 3D OCT-2000FA plus (Oakland, NJ, USA).
This study adhered to the tenets of the Declaration of Helsinki and the study protocol was approved by the International Review Board of Watany Eye Hospitals, Cairo, Egypt (registration code RET-2020–002), and informed consents were obtained from all patients after providing an explanation of the purpose of this study.
Mutation screening
Genomic DNA was extracted from peripheral blood leukocytes using the salting out protocol [20]. The 7 coding exons of GRK1 gene (NM_002929.3), the 15 coding exons of SAG gene (NM_000541.5) as well as a flanking intronic sequences of at least 50 bp have been amplified to detect any splice site variant using specially designed primers by primer3 software. Table 1 shows the primers for coding exons of SAG gene. Successfully amplified fragments were purified using Exo-SAP PCR kit (Fermentas, Germany) followed by sequencing in both directions using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and analyzed on the ABI Prism 3500 Genetic Analyzer (Applied Biosystems) according to instructions supplied by manufacturer. Obtained sequencing data was aligned against reference genomic and complementary DNA of each gene. Detected variants were checked in publicly available control databases followed by check for pathogenicity using in silico pathogenicity prediction tools as MutationTaster (http://www.mutationtaster.org), NNSplice (http://www.fruitfly.org/seq_tools/splice.html), NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/), GENSCAN (http://genes.mit.edu/GENSCAN.html), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), and Spliceman (http://fairbrother.biomed.brown.edu/spliceman/index.cgi).
Results
We examined four patients of two unrelated families, where both families gave history of consanguinity. Table 2 shows the results of our collected data.
Clinical findings
Our first patient (patient 1) was a 55-year-old female patient who was coming for a baseline checkup for diabetic retinopathy following her new diagnosis of type 2 Diabetes mellitus (DM) a month earlier. She was aware of night blindness dating since childhood which was diagnosed as vitamin A deficiency for which she received medical treatment with no improvement. Otherwise, her ocular history was unremarkable. There was no history of night blindness in either the mother or the father. The patient was unsure about her grandparents ever complaining of night blindness.
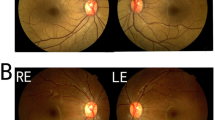
Her medical history was unremarkable, except for the recent diagnosis of type 2 DM. CDVA was 20/20 bilaterally with a hyperopic refraction of + 2.25 D sphere, -1.5 D cylinder in the right eye, and + 1.75 D sphere, -0.25 D cylinder in the left eye. The Mizuo-Nakamura phenomenon was demonstrated after three hours of dark adaptation. No changes were identified on both FAF and FFA. Central corneal thickness was 575 µm in the right eye, and 579 µm in the left eye. SD-OCT was unable to detect the hyporeflective band corresponding to outer segment of photoreceptors between the hyper-reflective layers associated with the ellipsoid zone (EZ), the interdigitation zone (IZ) and a densely packed retinal pigment epithelium (RPE)/Bruch's membrane complex in the parafoveal region as shown in Fig. 1.

Clinical and genetic data of the 55-year-old patient from the first family (patient 1). A Color fundus photograph of the right and left eye showing the characteristic golden-yellow reflex involving the macula. B Mizuo-Nakamura phenomenon is seen as the fundus color of both eyes reverts back to normal following prolonged dark adaptation. C Spectral domain optical coherence tomography of the right and left eyes showing indiscernible hyporeflective band of outer segments of photoreceptors between the hyper-reflective layers associated with the ellipsoid zone, interdigitation zone and the densely packed RPE/Bruch’s membrane complex in the parafoveal region. Enlarged images of boxed regions (1, patient, 2, control) shows the outer retina in detail. D Four-generation family pedigree highlighting the patient
The other highly consanguineous family had three members; a 65-year-old male patient (patient 2) along with his two sons (patient 3 and 4), who presented for a routine checkup. The father was medically-free, with past ocular history significant for bilateral phacoemulsification with posterior chamber intraocular implants done 2 years prior to the current date. When questioned about night blindness, he assented that he had it all his life. Moreover, he confirmed that his late mother as well as both his sons suffered from the same condition.
CDVA was 20/25 bilaterally. Anterior segment examination showed well-centered posterior chamber intraocular implants. Upon presentation, his IOP was found to be elevated with pressures of 36 mmHg bilaterally. His fundus examination revealed the characteristic golden-yellow reflex in all areas of the fundus. The Mizuo-Nakamura phenomenon was demonstrated and documented. FAF and FFA imaging were normal. Central corneal thickness was 599 µm in the right eye, and 613 µm in the left eye.
SD-OCT of the macula showed the same finding of an indiscernible hyporeflective layer of outer segment of photoreceptors between the hyper-reflective layers associated with the EZ, IZ and a densely packed RPE/Bruch's membrane complex in the parafoveal region as demonstrated in the first patient as shown in Fig. 2.

Clinical and genetic data of the 65-year-old patient from the second family (patient 2). A Color fundus photograph of the right and left eye showing the characteristic golden-yellow reflex involving the macula. B Mizuo-Nakamura phenomenon is seen as the fundus color reverts back to normal following prolonged dark adaptation. C Spectral domain optical coherence tomography of macula of right and left eyes showing indiscernible outer segments of photoreceptors. D Three-generation family pedigree highlighting the patient and other affected family members; note the consanguinity in the two generations on both sides of the family
Both of his sons were recruited for complete ophthalmological examination, the elder was a 31-year-old (patient 3) and the younger (patient 4) was a 28-year old. They both suffered from stationary night blindness since childhood and consented to giving blood samples for genetic testing. UDVA was 20/20 bilaterally. Anterior segment examination was within normal limits. Examination revealed within normal intraocular pressures. Fundus examination revealed the characteristic golden-metallic sheen. The Mizuo-Nakamura phenomenon was ascertained and documented in the younger son, while the elder patient refrained from undergoing prolonged dark adaptation. All the investigations were within normal limits except for the same finding of indiscernible outer segment of photoreceptors on SD-OCT as shown in Figs. 3 and 4 respectively.

Clinical images of the 31-year-old patient 3. A Color fundus photograph of the right and left eye showing the characteristic metallic reflex involving the macula. B Spectral domain optical coherence tomography of macula of right and left eyes showing indiscernible outer segment of photoreceptors

Clinical images of the 28-year-old patient 4. A Color fundus photograph of the right and left eye showing the characteristic metallic reflex involving the macula, extending beyond the vascular arcades. B Mizuo-Nakamura phenomenon is seen as the fundus color reverts back to normal following prolonged dark adaptation. C Spectral domain optical coherence tomography of macula of right and left eyes showing indiscernible outer segment of photoreceptors
Molecular genetic study
Sequencing of the seven exons of the GRK1 gene disclosed no sequence changes for any of the four patients. On the other hand, Sanger sequencing for the 15 coding exons of SAG gene showed two pathogenic variants. Patient 1 had the previously reported nonsense mutation p.R193* (c.577C > T) in a homozygous form, whereas patients 2, 3 and 4 from the same family had a novel canonical splice site mutation in a homozygous form (Chr2:g.234238138G > C (hg19); NM_000541.5; c.649–1 G > C) with accession assigned by ClinVar (SCV001451934.1). The novel variant wasn’t previously present in publicly available control databases as 1000 genomes, NHLBI exome sequencing project or gnomAD. Performed in silico functional analysis favored a pathogenic effect for the novel splice site mutation as shown in Table 3.
Discussion
Oguchi disease is an exceedingly rare autosomal recessive disease. It was first described in Japanese subjects and is characterized by Mizuo-Nakamura phenomenon as well as abnormally slow dark adaptation manifesting as night blindness. Other visual functions, such as visual acuity, visual field, color vision and daytime vision are usually within normal [1]. The disease has two forms: Oguchi type 1 (OMIM 258100) caused by mutations in the SAG gene (OMIM 181031) and Oguchi type 2 (OMIM 613411) caused by mutations in the GRK1 gene (OMIM 181031) [21].
It has been suggested that Oguchi type 1 and 2 diseases are liable to ethnic-specific or geographical distribution [14]. Indeed, SAG mutations are concentrated in Japanese patients of Oguchi disease while GRK1 mutations are very rarely reported in Japanese patients [22]. Compound heterozygous variations in the SAG gene were detected in a Chinese family clinically diagnosed as Oguchi disease [15], whereas another showed homozygous variation [21]. On the other hand, GRK1 gene mutations are mostly found in southern Asians and Europeans patients with Oguchi disease, with very few reports of families harboring SAG gene mutations [16, 17, 23]. No studies reported any patients with Oguchi disease in Egyptian or African population, and it remained unclear whether they harbor SAG gene or GRK1 gene mutations.
Moreover, there were reports of slowly progressive visual dysfunction in SAG-associated Oguchi disease (type 1) secondary to minimally progressive retinal degeneration, which resulted in some cases of the disease being re-classified as RP many years after initial diagnosis with Oguchi disease [24, 25]. Hence, the importance of identifying the exact genotype as it may imply life-long follow up of patients for fear of possibility of RP development.
Normally, in the fovea, three hyper-reflective bands representing the EZ, the IZ, and the RPE/Bruch’s membrane complex are visible. However, several studies [3, 7, 26] reported that only two distinct lines were discernible in the parafoveal area on SD-OCT findings in Oguchi disease suggesting that microarchitectural changes may occur in the photoreceptors. The densely packed structure in the parafovea was observed in all of our four patients which may suggest that it is a rather constant feature of Oguchi disease.
Among the mutations related to Oguchi disease in SAG gene are; a gross deletion of 3224 bp including exon 2 [15], a small deletion of one nucleotide in codon 309 [1], and around 5 missense/nonsense mutations. These include one mutation in exon 5 p.96Lfs*28 [21], two mutations in exon 8 p.Arg175* [27], p.Arg193* [16], and two mutations in exon 11 p.Arg292* and p.Glu306* [17].
In our study, molecular screening of the 7 coding exons of the GRK1 gene revealed normal sequence excluding Oguchi disease type 2. On carrying molecular screening of the 15 coding exons of the SAG gene, patient 1 showed the previously reported nonsense mutation p.R193* (c.577C > T) in a homozygous form. Production of a truncated arrestin protein of 193 amino acid instead of the 405 amino acid would lead to configuration problems.
Upon carrying the mutation analysis of patients 2, 3 and 4 (the father and his two sons), all patients carried a novel canonical splice site mutation at the acceptor region of intron 9. The mutation was one base before exon 9 (c.649–1 G > C) in a highly conserved region. We couldn’t obtain a blood sample from a healthy member of the family for segregation analysis as the mother was deceased and there were no grandchildren to date. On performing in silico functional analysis using different tools, they all favored the pathogenic effect of the novel canonical splice site mutation affecting the intronic AG region as shown in Table 2. The mutation variations that affect intronic GT‐AG dinucleotides compromise pre‐mRNA splicing [28]. Functional protein production requires accurate splicing of pre‐mRNA otherwise hereditary disorders might be a consequence [29]. Correct splicing is affected by the process of recognition and selection of the proper donor and acceptor splice sites which define the introns’ borders [30].
In our study, the novel splice site mutation was encountered in the 3’ AG acceptor region of intron 9 just before exon 9 which lies within the C-terminal domain of arrestin [31]. C-domain is responsible for membrane engagement and stabilization of the formed arrestin-receptor complex through its way to tight binding. It is also involved in the binding specificity since the activation of the C-domain doesn’t occur unless arrestin is activated by the phosphorylated receptor C terminus [32]. c.649–1 G > C splice site mutation is predicted to hinder the proper transcription of the gene and hence the proper production of normal mRNA which might be subjected to nonsense mediated mRNA decay or end up in an aberrant nonfunctioning protein. This splice site variant is the third to be found in SAG gene as the first splice site mutation in SAG gene was described by Liu et al. in a Chinese Oguchi patient in a homozygous form [21] and the second one was described in another Chinese Oguchi patient in a compound heterozygous form [33].
Mutations in the non-coding region of SAG gene may play a role in the pathogenesis of Oguchi disease, so should be considered in genetic testing of Oguchi disease patients. Furthermore, identification of additional families with the same mutation would enable better understanding of genotype–phenotype correlation.
Conclusion
To the best of our knowledge, this is the first study documenting cases of Oguchi disease in Egypt and in whole Africa. SAG gene mutations were confirmed in all four of our enrolled patients diagnosed with Oguchi disease. The novel splice site mutation (c.649–1 G > C) is the third splice site mutation to be detected in the SAG gene so far. Our findings have expanded the spectrum of Oguchi disease associated mutations in SAG gene and may serve as a basis for genetic diagnosis for Oguchi disease.
Availability of data and materials
All data generated or analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
Abbreviations
- SAG:
-
S-antigen
- GRK1:
-
Rhodopsin kinase
- FAF:
-
Fundus autofluorescence
- FFA:
-
Fundus fluorescein angiography
- SD-OCT:
-
Spectral-domain optical coherence tomography
- CSNB:
-
Congenital stationary night blindness
- RP:
-
Retinitis pigmentosa
- UDVA:
-
Unaided distance visual acuity
- CDVA:
-
Corrected distance visual acuity
- PCR:
-
Polymerase chain reaction
- DM:
-
Diabetes mellitus
- EZ:
-
Ellipsoid zone
- IZ:
-
Interdigitation zone
- RP:
-
Retinitis pigmentosa
- RPE:
-
Retinal pigment epithelium
References
Fuchs S, Nakazawa M, Maw M, Tamai M, Oguchi Y, Gal A. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10(3):360–2.
Yamamoto S, Sippel KC, Berson EL, Dryja TP. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet. 1997;15(2):175–8.
Khani SC, Abitbol M, Yamamoto S, Maravic-Magovcevic I, Dryja TP. Characterization and chromosomal localization of the gene for human rhodopsin kinase. Genomics. 1996;35(3):571–6.
Ohguro H, Van Hooser JP, Milam AH, Palczewski K. Rhodopsin phosphorylation and dephosphorylation in vivo. J Biol Chem. 1995;270(24):14259–62.
Hayashi T, Gekka T, Takeuchi T, Goto-Omoto S, Kitahara K. A novel homozygous GRK1 mutation (P391H) in 2 siblings with Oguchi disease with markedly reduced cone responses. Ophthalmology. 2007;114(1):134–41.
Poulter JA, Gravett MSC, Taylor RL, Fujinami K, De Zaeytijd J, Bellingham J, Rehman AU, Hayashi T, Kondo M, Rehman A, Ansar M, Donelly D, Toomes C, Ali M, UK Inherited Retinal Disease Consortium, Genomics England Research Consortium, De Baere E, Leroy BP, Davies NP, Henderson RH, Webster AR, Rivolta C, Zeitz C, Mahroo OA, Arno G, Black GCM, McKibbin M, Harris SA, Khan KN, Inglehearn CF. New variants and in silico analyses in GRK1 associated Oguchi disease. Hum Mutat. 2021;42(2):164–76.
Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol. 2000;130(5):547–63.
Stenson PD, Mort M, Ball EV, Shaw K, Philips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): 2003 Update. Hum Mutat. 2003;21:577–81.
Usui T, Ichibe M, Ueki S, Takagi M, Hasegawa S, Abe H, Sekiya K, Nakazawa M. Mizuo phenomenon observed by scanning laser ophthalmoscopy in a patient with Oguchi disease. Am J Ophthalmol. 2000;130(3):359–61.
Boissonnot M, Robert MF, Gilbert-Dussardier B, Dighiero P. Syndrome d’Oguchi ou cécité nocturne congénitale stationnaire: à propos d’un cas [Oguchi disease or stationary congenital night blindness: a case report]. J Fr Ophtalmol. 2007;30(1):e2.
Colombo L, Abeshi A, Maltese PE, Frecer V, Miertuš J, Cerra D, Bertelli M, Rossetti L. Oguchi type I caused by a homozygous missense variation in the SAG gene. Eur J Med Genet. 2019;62(9):103548.
Mucciolo DP, Sodi A, Murro V, Passerini I, Palchetti S, Pelo E, Virgili G, Rizzo S. A novel GRK1 mutation in an Italian patient with Oguchi disease. Ophthalmic Genet. 2018;39(1):137–8.
Skorczyk-Werner A, Kociecki J, Wawrocka A, Wicher K, Krawczyniski MR. The first case of Oguchi disease, type 2 in a Polish patient with confirmed GRK1 gene mutation. Klin Oczna. 2015;117(1):27–30.
Teke MY, Citirik M, Kabacam S, Demircan S, Alikasifoglu M. A novel missense mutation of the GRK1 gene in Oguchi disease. Mol Med Rep. 2016;14(4):3129–33.
Huang L, Li W, Tang W, Zhu X, Ou-Yang P, Lu G. A Chinese family with Oguchi’s disease due to compound heterozygosity including a novel deletion in the arrestin gene. Mol Vis. 2012;18:528–36.
Maw M, Kumaramanickavel G, Kar B, John S, Bridges R, Denton M. Two Indian siblings with Oguchi disease are homozygous for an arrestin mutation encoding premature termination. Hum Mutat. 1998;Suppl 1:S317-9.
Waheed NK, Qavi AH, Malik SN, Maria M, Riaz M, Cremers FPM, Azam M, Qamar R. A nonsense mutation in S-antigen (p.Glu306*) causes Oguchi disease. Mol Vis. 2012;18:1253–9.
Azam M, Collin RW, Khan MI, Shah STA, Qureshi N, Ajmal M, den Hollander AI, Qamar R, Cremers FPM. A novel mutation in GRK1 causes Oguchi disease in a consanguineous Pakistani family. Mol Vis. 2009;15:1788–93.
Aryan H, Bahadori A, Farhud DD, Zarif Yeganeh M, Pourkalhor H. A homozygote mutation in S-antigen visual arrestin SAG gene in an Iranian patient with Oguchi type one: a case report. Iran J Public Health. 2020;49(5):995–1000.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Liu X, Gao L, Wang G, Long Y, Ren J, Fujinami K, Meng X, Li S. Oguchi disease caused by a homozygous novel SAG splicing alteration associated with the multiple evanescent white dot syndrome: a 15-month follow-up. Doc Ophthalmol. 2020;141(3):217–26.
Oishi A, Akimoto M, Kawagoe N, Mandai M, Takahashi M, Yoshimura N. Novel mutations in the GRK1 gene in Japanese patients With Oguchi disease. Am J Ophthalmol. 2007;144(3):475–7.
Sergouniotis PI, Davidson AE, Sehmi K, Webster AR, Robson AG, Moore AT. Mizuo-Nakamura phenomenon in Oguchi disease due to a homozygous nonsense mutation in the SAG gene. Eye. 2011;25(8):1098–101.
Nishiguchi KM, Ikeda Y, Fujita K, Kunikata H, Akiho M, Hashimoto K, Hosono K, Kurata K, Koyanagi Y, Akiyama M, Suzuki T, Kawasaki R, Wada Y, Hotta Y, Sonoda KH, Murakami A, Nakazawa M, Nakazawa T, Abe T. Phenotypic features of Oguchi disease and retinitis pigmentosa in patients with s-antigen mutations: a long-term follow-up study. Ophthalmology. 2019;126(11):1557–66.
Nishiguchi KM, Oguchi Y, Nakazawa T. Progression from classical Oguchi disease to retinitis pigmentosa after 50 years. Ophthalmology. 2020;127(1):51.
Cideciyan AV, Zhao X, Nielsen L, Khani SC, Jacobson SG, Palczewski K. Null mutation in the rhodopsin kinase gene slows recovery kinetics of rod and cone phototransduction in man. Proc Natl Acad Sci U S A. 1998;95(1):328–33.
Nakamura M, Yamamoto S, Okada M, Ito S, Tano Y, Miyake Y. Novel mutations in the arrestin gene and associated clinical features in Japanese patients with Oguchi’s disease. Ophthalmology. 2004;111(7):1410–4.
Ohno K, Takeda JI, Masuda A. Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdiscip Rev RNA. 2018;9(1):e1451.
Baralle D, Buratti E. RNA splicing in human disease and in the clinic. Clin Sci. 2017;131(5):355–68.
Wimmer K, Schamschula E, Wernstedt A, Traunfellner P, Amberger A, Zschocke J, Kroisel P, Chen Y, Callens T, Messiaen L. AG-exclusion zone revisited: Lessons to learn from 91 intronic NF1 3’ splice site mutations outside the canonical AG-dinucleotides. Hum Mutat. 2020;41(6):1145–56.
Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97(2):257–69.
Lally CC, Bauer B, Selent J, Sommer ME. C-edge loops of arrestin function as a membrane anchor. Nat Commun. 2017;8:14258.
Deng Z, Fan F, Tang D, Wu Y, Shu Y, Wu K. A compound heterozygous mutation in the S-Antigen Visual Arrestin SAG gene in a Chinese patient with Oguchi type one: a case report. BMC Ophthalmol. 2022;22(1):99.
Acknowledgements
Not applicable.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB). No funding or sponsorship was received for this study.
Author information
Authors and Affiliations
Contributions
C.A.T., N.M.E., N.I.K. and M.L.E. all contributed to the study conception and design. Material preparation, data collection and analysis were done by C.A.T. and N.M.E. The first draft of the manuscript was written by C.A.T. and N.M.E., and C.A.T., N.M.E., N.I.K. and M.L.E. all commented on previous versions of the manuscript. C.A.T., N.M.E., N.I.K. and M.L.E. all read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study adhered to the tenets of the Declaration of Helsinki, and the study protocol was reviewed and approved by International Review Board of Watany Eye Hospitals, Cairo, Egypt (RET-2020–002), approval number (RET-2020–002). A written informed consent was obtained from participants before enrolling in the study.
Consent for publication
Consent for publication has been obtained from the patients enrolled in this study.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Tawfik, C.A., Elbagoury, N.M., Khater, N.I. et al. Mutation analysis reveals novel and known mutations in SAG gene in first two Egyptian families with Oguchi disease. BMC Ophthalmol 22, 217 (2022). https://doi.org/10.1186/s12886-022-02444-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12886-022-02444-5