
Abstract
Background
Lung cancer is the most common malignant tumor, and it has a high mortality rate. However, the study of miRNA-mRNA regulatory networks in the plasma of patients with non-small cell lung cancer (NSCLC) is insufficient. Therefore, this study explored the differential expression of mRNA and miRNA in the plasma of NSCLC patients.
Methods
The Gene Expression Omnibus (GEO) database was used to download microarray datasets, and the differentially expressed miRNAs (DEMs) were analyzed. We predicted transcription factors and target genes of the DEMs by using FunRich software and the TargetScanHuman database, respectively. The Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used for GO annotation and KEGG enrichment analysis of downstream target genes. We constructed protein-protein interaction (PPI) and DEM-hub gene networks using the STRING database and Cytoscape software. The GSE20189 dataset was used to screen out the key hub gene. Using The Cancer Genome Atlas (TCGA) and UALCAN databases to analyze the expression and prognosis of the key hub gene and DEMs. Then, GSE17681 and GSE137140 datasets were used to validate DEMs expression. Finally, the receiver operating characteristic (ROC) curve was used to verify the ability of the DEMs to distinguish lung cancer patients from healthy patients.
Results
Four upregulated candidate DEMs (hsa-miR199a-5p, hsa-miR-186-5p, hsa-miR-328-3p, and hsa-let-7d-3p) were screened from 3 databases, and 6 upstream transcription factors and 2253 downstream target genes were predicted. These genes were mainly enriched in cancer pathways and PI3k-Akt pathways. Among the top 30 hub genes, the expression of KLHL3 was consistent with the GSE20189 dataset. Except for let-7d-3p, the expression of other DEMs and KLHL3 in tissues were consistent with those in plasma. LUSC patients with high let-7d-3p expression had poor overall survival rates (OS). External validation demonstrated that the expression of hsa-miR-199a-5p and hsa-miR-186-5p in peripheral blood of NSCLC patients was higher than the healthy controls. The ROC curve confirmed that the DEMs could better distinguish lung cancer patients from healthy people.
Conclusion
The results showed that miR-199a-5p and miR-186-5p may be noninvasive diagnostic biomarkers for NSCLC patients. MiR-199a-5p-KLHL3 may be involved in the occurrence and development of NSCLC.
Similar content being viewed by others


Background
Lung cancer is the most common malignant tumor, and it is the main cause of cancer-related death worldwide. In 2018, about 2.09 million people developed lung cancer, and nearly 1.8 million people died of lung cancer [1]. In China, lung cancer is the leading tumor in terms of male morbidity, male mortality, and female mortality, and it is the second leading tumor in terms of female morbidity [2]. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for about 85% of lung cancer cases [3]. The main subtypes of NSCLC are lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), and large cell carcinoma [3]. The pathogenesis of NSCLC is complex and involves genetic and immunologic changes. For instance, when tumor oncogenes are upregulated or when tumor suppressor genes are downregulated, the downstream signaling pathways of the genes are activated or inhibited, respectively. On the one hand, the migration, invasion, and proliferation abilities of the tumor cells are promoted; on the other hand, the tumor cells develop resistance to anti-tumor drugs [4]. Patients with early-stage NSCLC usually have no obvious symptoms; therefore, most patients have stage III or IV NSCLC at the time of diagnosis, which seriously affects their quality of life and subsequent treatment [4, 5]. With continuous medical technology advancements, new treatment methods, such as tumor immunotherapy and targeted therapy, have been widely developed in clinical settings. Moreover, precise and individualized treatment plans for tumors are gradually maturing. However, due to the lack of early diagnostic markers, NSCLC patients often miss the best opportunity for early treatment, and their 5-year survival rate is only 23% [6]. Therefore, identifying markers for the diagnosis and treatment of lung cancer has important clinical significance when it comes to improving the rate of early NSCLC diagnosis and selecting appropriate medical treatments.
MicroRNAs (miRNAs) are very short noncoding RNAs comprising about 20–24 nucleotides. MiRNAs were discovered in the research of Caenorhabditis elegans in 1993 [7]. There are obviously conserved miRNA sequences, which play vital roles in the regulatory pathways between mononuclear and multinuclear eukaryotes [7, 8]. MiRNA specifically binds to the 3′ untranslated region of target gene messenger RNA (mRNA) through base complementary pairing; it thus regulates the expression of target genes and participates in the regulation of cell migration, invasion, proliferation, apoptosis, and other biological processes [9]. A variety of miRNAs play important roles in the occurrence and development of lung cancer. By combining with corresponding target genes and regulating their expression levels, miRNAs can function as tumor suppressor genes or oncogenes to regulate the biological processes of lung cancer [10,11,12,13,14,15]. Previous studies have found that miRNAs can be stably expressed in blood, and different diseases have a distinct serum-miRNA profile [16]. Mitchell et al. found that many tumor-derived miRNAs existed in the peripheral blood of mice with prostate cancer xenograft models [17]. The expression of miRNAs in the blood may be related to miRNAs released by tumor cells into the surrounding environment [18]. Therefore, miRNAs in the circulation may provide new insight for the diagnosis and treatment of lung cancer. Leng et al. found that plasma miRNA biomarkers may be helpful for the early diagnosis of lung cancer and the classification of lung cancer subtypes [19]. They also found that some miRNAs (miR-126, miR-145, miR-210, and miR-205-5p) in plasma have high sensitivity and specificity when it comes to the early diagnosis of lung cancer and are independent of the stage and histological type of lung tumor as well as the age, sex, and race of the patient. Plasma miRNA signals may provide a blood-based detection method for the early diagnosis of lung cancer, thereby reducing related patient mortality and economic costs [19, 20]. Szczyrek et al. found that the expression levels of miR-27a-3p, miR-31, miR-182, and miR-195 in the plasma of lung cancer patients are different from those in the plasma of healthy people; and the detection of these miRNAs in the plasma can be useful in the noninvasive diagnosis of lung cancer [21]. Therefore, actively searching for miRNAs and mRNAs closely related to the occurrence of NSCLC can provide new targets for the diagnosis and treatment of the disease, which in turn may improve the prognosis of the disease.
Although many studies have reported on the expression levels and functions of miRNAs in lung cancer, there are still few studies regarding the miRNA-mRNA regulatory networks in the plasma of lung cancer patients. Therefore, the purpose of this study was to establish a potential miRNA-mRNA regulatory network in the plasma of NSCLC patients through corresponding analysis in order to provide new targets for the diagnosis and treatment of lung cancer.
Methods
Microarray data information
In the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), the screening of miRNA datasets related to lung cancer plasma was accomplished by using the terms “lung cancer” (research keyword), “non-coding RNA profile” (research type), and “Homo sapiens” (organism). Three miRNA datasets (GSE24709 [22], GSE31568 [23], and GSE61741 [24]) were selected for subsequent analysis. These datasets were all based on the GPL9040 platform (febit Homo Sapiens miRBase 13.0). The GSE24709 dataset included 47 samples (28 lung cancer samples and 19 healthy controls), the GSE31568 dataset included 102 samples (32 lung cancer samples and 70 healthy controls), and the GSE61741 dataset included 167 samples (73 lung cancer samples and 94 healthy controls). The details of the 3 datasets are shown in Table 1.
Screening of DEMs
The miRNA gene names within the datasets were obtained by using the R software (3.6.1) and relevant annotation packages. The limma package (3.40.6) was used to compare and screen differentially expressed miRNAs (DEMs) between lung cancer patients and healthy groups and to make corresponding thermograms and volcano maps. Adjusted P-values were obtained by using the Benjamini-Hochberg false discovery rate method. The adjusted P-values were used to correct the occurrence of false positives. Adjusted P-values < 0.05 and |log fold change| values > 1 were set as the threshold for identifying DEMs within the 3 datasets (GSE24709, GSE61741, and GSE31568; Tables S1, S2 and S3). A Venn diagram was used to analyze DEM overlap within the 3 datasets, and the overlapping miRNAs were selected as candidate DEMs. The research and design process is shown in Fig. 1.

Flow chart of the construction of the miRNA-mRNA network of lung cancer patients
Forecasting upstream transcription factors of DEMs
FunRich (http://www.funrich.org/) is an independent software tool that is mainly used to analyze the functional enrichment of genes and proteins and to perform interaction network analyses. It was used to predict the potential upstream transcription factors of the candidate DEMs. P-values < 0.05 were considered statistically significant.
Forecasting downstream target genes of DEMs
The online database TargetScanHuman Release 7.2 (http://www.targetscan.org/vert_72/) was used to predict the biological targets of the miRNAs (Table S4), including targets with conserved sites, as well as targets irrespective of conserved sites.
GO and KEGG analyses of target genes
To study the biological functions of the target genes of the DEMs, the online tool Database for Annotation, Visualization, and Integrated Discovery (DAVID; https://david.ncifcrf.gov/) was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses.
Building the PPI network and screening hub genes
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) online database (https://string-db.org/) was used to construct the protein-protein interaction (PPI) network of the target genes of the DEMs. The network was then visualized by using Cytoscape software (version 3.6.1). By using the cytoHubba plug-in, the nodes in the preloaded PPI network were sorted according to several topology algorithms and their features. The Maximal Clique Centrality (MCC) method was used to select the hub genes, which were considered to be the top 30 nodes of the PPI network, as this method has good performance when it comes to accurately predicting essential proteins [25].
Analysis of hub gene expression in the GSE20189 dataset
Since there is no other data on mRNA expression in the plasma of lung cancer patients, the GSE20189 dataset [26] was downloaded from the GEO database in order to analyze hub gene expression levels. This dataset, based on the GPL571 platform (Affymetrix Human Genome U133A 2.0 Array), contained 81 lung cancer patient plasma samples and 81 normal patient plasma samples. Student’s t-test was used to identify differentially expressed genes in the lung cancer and normal plasma samples. The key hub gene had to meet the following conditions: first, the upregulated target genes of the DEMs were downregulated or the downregulated target genes were upregulated; second, the P-value was < 0.05.
Verifying the expression levels of the key hub gene and DEMs in tissues and performing the survival analysis
The University of Alabama Cancer (UALCAN) database (http://ualcan.path.uab.edu) is a website for the online analysis and mining of datasets from The Cancer Genome Atlas (TCGA). It was used to analyze the expression levels of the key hub gene and DEMs in LUAD and LUSC tissues as well as the different stages of LUAD and LUSC, and to analyze the overall survival (OS) associated with the expression levels of these hub genes and DEMs in patients with lung cancer. A P-value < 0.05 was considered statistically significant.
The raw counts of the RNA sequencing data of the key hub gene and DEMs (Supplementary Tables S5, S6, S7, S8, S9, S10, S11, S12, S13, S14) and the clinical information of NSCLC patients (Supplementary Table S15) were obtained from the TCGA dataset (https://portal.gdc.cancer.gov/). The Kaplan-Meier survival analysis with the log-rank test was used to compare the 2 groups’ progression-free survival (PFS). All of the analytical methods described above were performed using R software. A P-values < 0.05 were considered statistically significant.
Subsequently, univariate and multivariate Cox analyses were performed to evaluate the proportional hazards model using the survival package of the R software. Variables included age, sex, and tumor stage. Nomogram and receiver operating characteristic (ROC) curves were used to evaluate the impact of DMEs on patient’s prognosis.
Verifying the translation levels of the key hub gene
The Human Protein Atlas (HPA) database (https://www.proteinatlas.org/) provides information regarding the distribution of human proteins in tissues and cells. The expression of target protein in tissues is characterized by the annotations Intensity and Quantity. According to the previous study, we transformed the four values (Strong, Moderate, Weak, and Negative) that used to describe Intensity into 3,2,1, and 0, respectively; and also transformed the five values (>75, 75–25%, <25%, Rare, and Negative) that used to describe Quantity into 75,50,25,5, and 0, respectively [27]. I and Q expressed the transformed Intensity and Quantity. The expression of the hub gene in NSCLC tissues and normal tissues was calculated using I × Q (Tables S16, S17, S18) [27]. Student’s t-test and Wilcoxon test were used to compare the expression of key genes in LUAD and normal tissues, and LUSC and normal tissues, respectively. A P-values < 0.05 were considered statistically significant.
External validation and efficacy evaluation
The GSE17681 [28] and GSE137140 [29] datasets were used for external validation data to verify DEMs expression in the peripheral blood of lung cancer patients and healthy controls. The GSE17681 dataset, based on the GPL9040 platform, included 17 lung cancer samples and 19 control samples, and the GSE137140 dataset, based on the GPL21263 platform, included 1746 lung cancer samples and 2178 control samples. GraphPad Prism 8.0 software was used to calculate ROC curves to assess the ability of the DEMs to distinguish between lung cancer patients and healthy people. The student’s t-test was used to compare the expression of 4 DEMs in blood of lung cancer patients and healthy groups. A P-values < 0.05 were considered statistically significant.
Results
Identification of DEMs in the plasma of lung cancer patients
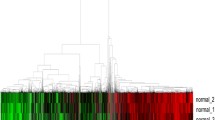
After screening with adjusted P-values < 0.05 and |log fold change| values > 1, a total of 72 DEMs were found in the GSE24709 dataset, of which 52 were upregulated and 20 were downregulated; 14 DEMs were found in the GSE31568 dataset, of which 11 were upregulated and 3 were downregulated; and 64 DEMs were found in the GSE61741 dataset, of which 37 were upregulated and 27 were downregulated. The heat and volcano maps of these data are shown in Fig. 2A-F. Through observation of Venn diagram intersections, 4 upregulated DEMs (hsa-miR-199a-5p, hsa-miR-328-3p, hsa-miR-186-5p, and hsa-let-7d-3p) were found to overlap between the 3 datasets (Fig. 2G, H).

Heat and volcano maps of DEMs in lung cancer and normal plasma samples. A, B. GSE24709 dataset. C, D. GSE31568 dataset. E, F. GSE61741 dataset. Red indicates higher expression, and green indicates lower expression. G. Venn diagram of the expression levels of the DEMs in the 3 datasets. H. Log fold change heat map of the candidate DEMs
Prediction of upstream transcription factors and downstream target genes of the DEMs
The FunRich software was used to predict the upstream transcription factors of the 4 candidate upregulated DEMs. Specificity protein 1 (SP1), early growth response protein 1 (EGR1), POU domain class 2 transcription factor 1 (POU2F1), RXR-alpha (RXRA), ROR-alpha (RORA), and E74-like factor1 (ELF1) were considered to potentially be involved in regulating the expression of the candidate DEMs (Fig. 3A, B). Further, a total of 2253 potential downstream target genes of the DEMs were predicted by using the TargetScanHuman 7.2 database (Fig. 3C-F). The number of target genes corresponding to each DEM is listed in Table 2.

The upstream transcription factors and downstream target genes of the DEMs. A-B. FunRich prediction of potential upstream transcription factors of the candidate DEMs. C-F. miRNA-target genes network diagram of the 4 DEMs. C. hsa-let-7d-3p. D. hsa-miR-186-5p. E. hsa-miR-199a-5p. F. hsa-miR-328-3p
GO and KEGG enrichment analyses of the downstream target genes of the DEMs
The DAVID database was used to perform GO and KEGG enrichment analyses of the 2253 target genes of the DEMs. As shown in Fig. 4A-F, biological process analysis results showed that the target genes were mainly concentrated in transcription, positive regulation of transcription, positive regulation of transcription from RNA polymerase II promoter, and negative regulation of transcription from RNA polymerase II promoter. Additionally, cell component analysis results showed that the target genes were mainly enriched in the nucleus, cytoplasm, and nucleoplasm. Furthermore, molecular function analysis results showed that the target genes were enriched in protein binding, metal ion binding, DNA binding, poly(A) RNA binding, and transcription factor activity sequence-specific DNA binding. KEGG pathway analysis results showed that the target genes were significantly enriched in cancer pathways, PI3K-Akt signaling pathways, proteoglycans, focal adhesions, endocytosis, Ras signaling pathways, actin cytoskeleton regulation, HTLV-I infection, and MAPK signaling pathways (Fig. 5A, B).

GO annotation analysis of the DEMs target genes in biological processes, cell components, and molecular functions. A, C, E. Bar plots. B, D, F. Bubble charts

KEGG pathway enrichment analysis of the DEMs target genes. A. Bar plots. B. Bubble charts
PPI and DEM-hub gene networks
The STRING database was used to establish a PPI network for the target genes of the DEMs, then the cytoHubba plug-in was used to screen the top 30 hub genes (Fig. 6A, Table 3). To better study the molecular mechanisms of these DEMs in the plasma of lung cancer patients, Cytoscape was used to construct a DEM-hub gene network (Fig. 6B). The interactions between the miRNAs and genes were as follows: miR-199a-5p interacted with 8 hub genes (KLHL3, FBXO9, BTRC, ARIH2, ITCH, UBE2Q1, FBXO30, and RLIM); let-7d-3p interacted with 4 hub genes (FBXL5, ATG7, HACE1, and SH3RF1); miR-328-3p interacted with 2 hub genes (TRIM71 and KLHL42); and miR-186-5p interacted with 20 hub genes (NEDD4, SKP1, CUL3, KLHL11, BTRC, CCNF, ATG7, SMURF2, KBTBD7, SPSB1, ASB7, UBE2K, TRAF7, ARIH2, UBE2R2, UBE2Q2, SH3RF1, UBAC1, UBE2B, and UBR2).

PPI and DEM-hub gene networks. A. PPI network of the top 30 hub genes of the DEMs. B. miRNA-hub gene regulatory network
Verification of hub gene expression
The GSE20189 dataset was used to identify the expression levels of the first 30 hub genes in the plasma samples. Some of these genes (FBXO30, RLIM, HACE1, SH3RF1, KLHL42, KBTBD7, SPSB1, TRAF7, UBE2R2, UBE2Q2, and SH3RF1) were not checked in the dataset. Compared with the healthy people, in the plasma of lung cancer patients, only the expression of KLHL3 was continuously downregulated, while NEDD4 and UBAC1 were increased. (Fig. 7A-S). Therefore, miR-199a-5p-KLHL3 may be a potential regulatory pathway in lung cancer plasma.

Identification of the mRNA expression levels of the top 30 hub genes in the GSE20189 dataset. A. NEDD4. B. SKP1. C. FBXO9. D. CUL3. E. KLHL11. F. FBXL5. G. BTRC. H. CCNF. I. ATG7. J. SMURF2. K. ITCH. L. KLHL3. M. ASB7. N. UBE2B. O. ARIH2. P. UBE2K. Q. UBE2Q1. R. UBAC1. S. UBR2. **p<0.01, ****p<0.0001
Expression levels of the DEMs and KLHL3 in tissues
To further explore the function of DEMs and KLHL3 in lung cancer, we analyze the expression of the DEMs and KLHL3 in lung cancerous tissues by using UALCAN database. As a result, an elevated expression of miR-199a-5p, miR-186-5p, and miR-328-3p were observed in LUAD and LUSC tissues compared to normal tissues (Fig. 8A, C, E, G, I, K). Also, the miR-199a-5p, miR-328-3p, and miR-186-5p expression were increased in diverse stages in LUAD patients (Fig. 8B, F, J). Nevertheless, in LUSC patients with stage 4, the expression of miR-199a-5p, miR-328-3p, miR-186-5p were not significantly increased (Fig. 8D, H, L). The expression of let-7d-3p in LUAD samples and diverse stages of LUAD patients were lower than the normal controls (Fig. 8M, N). In LUSC samples, the let-7d-3p expression was increased compared to the normal samples, but the expression of let-7d-3p was not obvious in different cancer stages with LUSC patients (Fig. 8O, P). In addition, the expression of KIHL3 was decreased in LUAD and LUSC patients, but the phenomenon was unconspicuous in different cancer stages (Fig. 8Q, R, S, T). Next, using HPA database, we found that a lower expression of KLHL3 protein was explored in LUAD and LUSC tissue versus normal tissues (Fig. 8U). As shown in Fig. 8V, the expression of KLHL3 was reduced in LUSC tissues.

Expression levels of the DEMs and KLHL3 in NSCLC tissues. A-D. hsa-miR-199a-5p. E-H. hsa-miR-186-5p. I-L. hsa-miR-328-3p. M-P. hsa-let-7d-3p. Q-T. KLHL3. U. KLHL3 protein expression in lung cancer tissue and normal tissue was verified by using the Human Protein Atlas database. Sample size (8 LUAD, 4 LUSC, 3 normal). *p<0.05, ***p<0.001, ****p<0.0001
Survival analysis and prognostic roles of the DEMs and KLHL3
Subsequently, we explored the prognostic value of the DEMs and KLHL3 in lung cancer patients. The expression levels of miR-199a-5p, miR-186-5p, miR-328-3p, and KLHL3 in the LUAD and LUSC patients did not affect OS (Fig. 9A-F, I-J). For LUSC, patients with high let-7d-3p expression had poor OS, but not in the LUAD (Fig. 9G, H). Therefore, let-7d-3p may be a potential prognostic marker of LUSC. The expression levels of the DEMs and KLHL3 did not affect PFS in NSCLC patients (Fig. 10).

Overall survival analysis of the DEMs and KLHL3 in NSCLC patients. A-B. hsa-miR-199a-5p. C-D. hsa-miR-186-5p. E-F. hsa-miR-328-3p. G-H. hsa-let-7d-3p. I-J. KLHL3

Progression free survival analysis of the DEMs and KLHL3 in NSCLC patients. A-B. hsa-let-7d-3p. C-D. hsa-miR-186-5p. E-F. hsa-miR-199a-5p. G-H. hsa-miR-328-3p. I-J. KLHL3
Kaplan-Meier curves showed that high hsa-let-7d-3p expression was closely associated with low OS in LUSC patients. Univariate and multivariate Cox regression analyses, nomograms, and ROC curves are required to evaluate the accuracy of differential expression genes for patient prognosis [30, 31]. Therefore, we researched whether hsa-let-7d-3p and variables such as age, sex, and tumor stage were risk factors for survival in LUSC patients.
Using univariate Cox regression, we know that hsa-let-7d-3p was not a risk factor in LUSC patients. The multivariate Cox regression revealed that let-7d-3p was not an independent prognostic factor in LUSC patients (Table 4). We also constructed a nomogram and ROC curve (Fig. 11A, B).

Nomogram and ROC curve for the prognostic value of hsa-let-7D-3p. A. Nomogram. B. ROC curve
External validation and effectiveness assessment
To further verify the reliability of our results, we used GSE17681 and GSE137140 datasets to verify the expression levels of the 4 DEMs in the peripheral blood of lung cancer patients and healthy controls. In both GSE datasets, we found that the expression of hsa-miR-199a-5p and hsa-miR-186-5p in lung cancer patients were higher than those in the healthy controls, while the expression of hsa-let-7d-3p and hsa-328-3p was contrary to previous prediction results (Fig. 12A-H). In addition, ROC curves were used to assess the ability of the 4 DEMs to distinguish between lung cancer patients and healthy people. As shown in Fig. 12I-L, the 4 DEMs had good accuracy in the diagnosis of lung cancer, with the following results: hsa-miR-328-3p (AUC = 0.7416, 95%CI [0.6856–0.7977], P < 0.0001); hsa-let-7d-3p (AUC = 0.8021, 95%CI [0.7513–0.8529], P < 0.0001); hsa-miR-186-5p (AUC = 0.6970, 95%CI [0.6375–0.7565], P < 0.0001); and hsa-miR-199a-5p (AUC = 0.8013, 95%CI [0.7520–0.8506], P < 0.0001).

External validation and ROC analysis of the 4 DEMs. A-H. External validation of the 4 DEMs. A. hsa-miR-199a-5p (GSE17681). B. hsa-miR-186a-5p (GSE17681). C. hsa-miR-199a-5p (GSE137140). D. hsa-186-5p (GSE137140). E. hsa-let-7d-3p (GSE176181). F. hsa-miR-328-3p (GSE176181). G. hsa-let-7d-3p (GSE137140). H. hsa-328-3p (GSE137140). I-J. ROC analysis of the 4 DEMs. I. hsa-miR-328-3p. J. hsa-let-7d-3p. K. hsa-miR-186-5p. L. hsa-miR-199a-5p. *p<0.05, ****p<0.0001
Discussion
Although many studies regarding the diagnosis and treatment of lung cancer have been performed in recent years, the 5-year survival rate of lung cancer patients remains low due to the lack of biomarkers for early diagnosis and insufficient research on lung cancer pathogenesis. The number of studies involving microarrays is gradually increasing, which will help to reveal the genetic changes involved in disease progression. However, most micro-matrix studies on lung cancer are focused on tumors and adjacent tissues. Invasive operations can cause complications, such as hemothorax, pneumothorax, and infection; therefore, it is extremely important to actively seek effective noninvasive operation inspection strategies. Leidinger et al. found that miRNA in the blood can be used as a diagnostic marker for lung cancer, and it can also distinguish lung cancer from chronic obstructive pulmonary disease [32]. In this study, we found that 4 upregulated DEMs (hsa-miR-328-3p, hsa-miR-199a-5p, hsa-miR-186-5p, and let-7d-3p) had overlap in the 3 datasets, so they were used as candidate DEMs for further research. Recent studies have found that these DEMs play important roles in the diagnosis, treatment, and progression of different tumors. Ma et al. found that overexpressed miR-328-3p can improve the sensitivity of cancer cells to radiotherapy by changing the DNA damage and repair signaling pathway in NSCLC patients [33]. Yang et al. found that hsa-miR-574-5p, hsa-miR-328-3p, and hsa-miR-423-3p are involved in the Wnt/β-catenin signaling pathway to promote bone metastasis in lung cancer and may be biomarkers for bone metastasis in NSCLC patients [34]. Zheng et al. showed that plasma exosomal miR-30d-5p and let-7d-3p can be used as valuable biomarkers for the noninvasive screening of cervical cancer and its precancerous lesions [35]. Furthermore, the upregulation of miR-186-5p expression can increase the sensitivity of NSCLC to cisplatin by targeting the expression of the SIX1 protein; on the other hand, targeting the expression of the PTEN protein can promote LUAD migration, invasion, and proliferation [36, 37]. Some studies have found that miR-199a-5p dysregulation is related to the progression and pathogenesis of cancer. However, miR-199a-5p has conflicting roles in tumor progression and carcinogenesis. In thyroid cancer, oral squamous cell carcinoma, liver cancer, and ovarian cancer, miR-199a-5p can inhibit tumor growth and metastasis [38,39,40,41]. Conversely, miR-199a-5p can promote tumor growth and invasion in osteosarcoma, cervical cancer, and skin squamous cell carcinoma [42,43,44]. MiR-199a-5p can also inhibit the growth of lung cancer cells by targeting the MAP3K11 protein, as shown through in vivo and in vitro experiments of lung cancer [45]. It is believed that miR-199a-5p and miR-495 can be used in NSCLC as diagnostic biomarkers for activated unfolded protein response. Ahmadi et al. found that miR-199a-5p and miR-495 can regulate the endoplasmic reticulum stress response by regulating the expression of the GRP78 protein, thereby regulating the progression of lung cancer. In addition, the manipulation of the expression of these miRNAs may have potential therapeutic applications when it comes to lung cancer [46]. A recent study found that miR-199a-5p expression was significantly reduced in doxorubicin-resistant A549 and H469 cells. Increasing the expression of miR-199a-5p leads to the resistance of NSCLC to doxorubicin via the regulation of the expression of the ABCC1 and HIF1A proteins [47]. Therefore, the roles of miRNA-mRNA regulatory networks in the occurrence and development of lung cancer deserve further research.
Transcription factors can regulate the expression of miRNAs, so the transcription factors that might regulate the candidate DEMs in this study were predicted. We found that SP1, EGR1, and POU2F1 accounted for a high proportion of the transcription factors of the candidate DEMs. These transcription factors have been widely reported in studies of lung cancer and other tumors. Hu et al. found that high SP1 expression can inhibit the expression of the prenyl diphosphate synthase subunit 2 (PDSS2) promoter in lung cancer cells [48]. Higher SP1 expression and lower PDSS2 expression have been found to be significantly associated with poor prognosis in lung cancer patients [48]. SP1 can be combined with long noncoding RNA (lncRNA) promoters to promote the proliferation, migration, and invasion of lung cancer cells by regulating lncRNA expression [49, 50]. When hypoxia occurs, the level of EGR1 expression decreases, but SP1 and HIF1A expression levels increase, which can induce erythropoietin secretion and promote NSCLC growth [51]. POU2F1, which is also known as OCT1, promotes tumor growth and metastasis by activating downstream signaling pathways in liver cancer, colon cancer, ovarian cancer, and gastric cancer [52,53,54,55]. Xiao et al. found that ELF1/CASC2/miR-18a axis-mediated IRF2 expression is significantly related to the proliferation, migration, and invasion of cisplatin-resistant NSCLC, and they believe that this regulatory axis may be a new therapeutic target for NSCLC [56]. In general, many transcription factors have been reported in lung cancer, which supports the importance of the candidate DEMs in the pathogenesis of lung cancer.
The KEGG pathway enrichment analysis results of this study showed that the target genes of the DEMs were mainly enriched in cancer pathways, PI3K-Akt signaling pathways, proteoglycans, focal adhesions, endocytosis, Ras signaling pathways, actin cytoskeleton regulation in cancer, HTLV-I infection, and MAPK signaling pathways. In addition, cancer pathways included many pathways of growth factors, such as insulin-like growth factor I, fibroblast growth factor, recombinant human epidermal growth factor, platelet-derived growth factor, and human hepatocyte growth factor. These factors play important roles in the development and resistance of lung cancer [57,58,59,60,61,62,63,64]. Previous studies have shown that PI3k/Akt signaling pathway activation is closely related to lung cancer progression. Li et al. found that overexpressed miR-133b inhibits the proliferation of cisplatin-induced NSCLC cells through the regulation of the PI3K/Akt and JAK2/STAT3 signaling pathways by targeting epidermal growth factor receptor (EGFR) [65]. Wu et al. showed that the PAX6 protein can directly bind to the promoter region of the ZEB2 gene and mediate the downregulation of E-cadherin through the PI3K/AKT signaling pathway, thereby mediating cell migration, stem cell transformation, and cisplatin resistance, and ultimately affecting the survival of NSCLC patients [66]. In addition, some studies have found that the Ras and MAPK signaling pathways are involved in lung cancer progression. Wei et al. showed that miR-330-3p promotes NSCLC brain metastasis by activating MAPK/ERK signaling pathways and enhancing cell proliferation, migration, invasion, and angiogenesis [67]. A recent study found that miR-148a-3p, which is a tumor suppressor, is often downregulated in NSCLC cells and can inhibit the proliferation and epithelial-mesenchymal transition processes of NSCLC by regulating SOS2/MAPK/ERK signal transduction, thereby providing new insight into the pathogenesis of NSCLC [68]. These studies suggest that the downstream target genes of DEMs are involved in many biological processes of lung cancer. They further suggest that the 4 upregulated DEMs in this study play key roles in the pathogenesis of lung cancer.
A DEM-hub gene network was constructed, and it was found that most of the hub genes may be targeted by miR-328-3p, miR-199a-5p, miR-186-5p, and let-7d-3p. Due to different samples, among the top 30 hub genes, only the expression of KLHL3 was consistent with the expression in the GSE20189 database. Further, it was verified that the expression levels of KLHL3 in tissues were consistent with those in plasma. The KLHL3 (kelch-like 3) and CUL3 proteins are components of the Cullin-RING E3 ubiquitin ligase complex, and they belong to the ubiquitin proteasome system. Their function is to degrade proteins, and they have important roles in maintaining cell function. The complex interacts with WNK1 and WNK4, which are part of the WNK kinase family, to induce WNK1 and WNK4 ubiquitination and regulate the levels of these proteins through proteasome degradation [69]. The secreted protein acidic and rich in cysteine (SPARC) secreted by extracellular matrix components can activate the expression of the WNK1 protein to promote the migration and invasion of lung cancer cells [70]. Hsu et al. found that activated lung fibroblasts produce the tryptophan metabolite kynurenine, which promotes the migration and growth of lung cancer cells by activating AKT and WNK1 [71, 72]. KLHL3 can degrade the expression of the WNK1 protein through ubiquitination, so it may have an important role in the progression of lung cancer.
Our research found that the expression levels of miR-199a-5p and miR-186-5p were increased in both blood and tissues. In this study, only GSE24709, GSE137140, and GSE20189 datasets provide the clinical information of patients, who were mainly concentrated in TNM stages I and II. Therefore, we speculated that the identified DEMs might be useful for diagnosing early stage lung cancer, while more experiments are needed to validate our hypothesis. Survival analysis showed that let-7d-3p expression was negatively correlated with the prognosis of patients with LUSC. However, the univariate Cox regression, the multivariate Cox regression and ROC curves showed that hsa-let-7d-3p might not be predicting the outcomes of LUSC patients. Through the above assessments, a potential miRNA-mRNA regulatory network in the plasma of NSCLC patients was constructed and analyzed for the first time.
There are some limitations in our research. First, we only focused on the expression levels of miRNAs and mRNAs in the plasma of lung cancer patients and healthy subjects. However, some miRNA and mRNA expression levels may differ in the peripheral blood of lung cancer patients with different tumor stages, tissue types, sexes, ages, and smoking statuses. Second, our data are mainly from public databases, some of which do not provide clinical information of patients, and due to the limited sample sizes of these datasets, the results may be biased. Third, we did not perform external experimental verification. The molecular mechanisms and roles of the miRNAs and target genes in regulating lung cancer’s migration, invasion, proliferation, and immune microenvironment need to be further studied. Therefore, in subsequent research, we will collect more clinical samples to verify the ability of the DEMs and target genes on the diagnosis and prognosis of NSCLC patients. At the same time, we need to further study the effects of DEMs and target genes on lung cancer biological characteristics and the immune microenvironment by in vitro and in vivo experiments.
Conclusion
In conclusion, we found that miR-199a-5p and miR-186-5p may be noninvasive diagnostic biomarkers for NSCLC patients. MiR-199a-5p-KLHL3 may be involved in the development of NSCLC. We hope that more deep studies can provide new targets for the noninvasive diagnosis and treatment of lung cancer.
Availability of data and materials
In the study, the Gene Expression Omnibus database was used to download microarray datasets (GSE24709, GSE61741, GSE31568, GSE20189, GSE17681, GSE137140) about miRNA and mRNA microarray datasets (https://www.ncbi.nlm.nih.gov/geo/).The datasets generated and analyzed during this study are available in the TCGA database (https://portal.gdc.cancer.gov).
Abbreviations
- NSCLC:
-
Non-small cell lung cancer
- LUAD:
-
Lung Adenocarcinoma
- LUSC:
-
Lung Squamous Cell Carcinoma
- miRNAs:
-
microRNAs
- DEGs:
-
Differentially expressed genes
- DEM:
-
Differentially expressed miRNA
- GO:
-
Gene Ontology
- PPI:
-
Protein-protein interaction
- GEO:
-
Gene Expression Omnibus
- FC:
-
Fold change
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- UALCAN:
-
University of Alabama Cancer
- HPA:
-
Human Protein Atlas
- TCGA:
-
The Cancer Genome Atlas
- OS:
-
Overall survival
- PFS:
-
Progression free survival.
- HR:
-
Hazard ratio
- CI:
-
Confifidence interval
References
Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–53.
Feng RM, Zong YN, Cao SM, et al. Current cancer situation in China: good or bad news from the 2018 global Cancer statistics? Cancer Commun (Lond). 2019;39(1):22.
Woodman C, Vundu G, George A, et al. Applications and strategies in nanodiagnosis and nanotherapy in lung cancer. Semin Cancer Biol. 2020;S1044-579X(20):30042.
Misra P, Singh S. Role of cytokines in combinatorial immunotherapeutics of non-small cell lung cancer through systems perspective. Cancer Med. 2019;8(5):1976–95.
Miller KD, Nogueira L, Mariotto AB, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. 2019;69(5):363–85.
Ettinger DS, Wood DE, Aggarwal C, et al. NCCN guidelines insights: non-small cell lung Cancer, version 1.2020. J Natl Compr Cancer Netw. 2019;17(12):1464–72.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):43–54.
Aghanoori MR, Mirzaei B, Tavallaei M. MiRNA molecular profiles in human medical conditions: connecting lung cancer and lung development phenomena. Asian Pac J Cancer Prev. 2014;15(22):9557–65.
Iqbal MA, Arora S, Prakasam G, et al. MicroRNA in lung cancer: role, mechanisms, pathways and therapeutic relevance. Mol Asp Med. 2019;70:3–20.
Demes M, Aszyk C, Bartsch H, et al. Differential miRNA-expression as an adjunctive diagnostic tool in neuroendocrine tumors of the lung. Cancers (Basel). 2016;8(4):38.
Inamura K. Diagnostic and therapeutic potential of MicroRNAs in lung Cancer. Cancers (Basel). 2017;9(5):49.
He L, He X, Lim LP, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447(7148):1130–4.
Du L, Schageman JJ, Subauste MC, et al. miR-93, miR-98, and miR-197 regulate expression of tumor suppressor gene FUS1. Mol Cancer Res. 2009;7(8):1234–43.
Prudkin L, Behrens C, Liu DD, et al. Loss and reduction of FUS1 protein expression is a frequent phenomenon in the pathogenesis of lung cancer. Clin Cancer Res. 2008;14(1):41–7.
Crawford M, Brawner E, Batte K, et al. MicroRNA-126 inhibits invasion in non-small cell lung carcinoma cell lines. Biochem Biophys Res Commun. 2008;373(4):607–12.
Chen X, Ba Y, Ma L, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006.
Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513–8.
Chin LJ, Slack FJ. A truth serum for cancer--microRNAs have major potential as cancer biomarkers. Cell Res. 2008;18(10):983–4.
Leng Q, Wang Y, Jiang F. A direct plasma miRNA assay for early detection and histological classification of lung Cancer. Transl Oncol. 2018;11(4):883–9.
Leng Q, Lin Y, Jiang F, et al. A plasma miRNA signature for lung cancer early detection. Oncotarget. 2017;8(67):111902–11.
Szczyrek M, Kuźnar-Kamińska B, Grenda A, et al. Diagnostic value of plasma expression of microRNAs complementary to Drosha and dicer in lung cancer patients. Eur Rev Med Pharmacol Sci. 2019;23(9):3857–66.
Leidinger P, Keller A, Borries A, et al. Specific peripheral miRNA profiles for distinguishing lung cancer from COPD. Lung Cancer. 2011;74(1):41–7.
Keller A, Leidinger P, Bauer A, et al. Toward the blood-borne miRNome of human diseases. Nat Methods. 2011;8(10):841–3.
Keller A, Leidinger P, Vogel B, et al. miRNAs can be generally associated with human pathologies as exemplified for miR-144. BMC Med. 2014;12:224.
Chin CH, Chen SH, Wu HH, et al. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Rotunno M, Hu N, Su H, et al. A gene expression signature from peripheral whole blood for stage I lung adenocarcinoma. Cancer Prev Res (Phila). 2011;4(10):1599–608.
Chiang SC, Han CL, Yu KH, Chen YJ, Wu KP. Prioritization of cancer marker candidates based on the immunohistochemistry staining images deposited in the human protein atlas. PLoS One. 2013;8(11):e81079.
Keller A, Leidinger P, Borries A, et al. miRNAs in lung cancer - studying complex fingerprints in patient's blood cells by microarray experiments. BMC Cancer. 2009;6(9):353.
Urabe F, Matsuzaki J, Yamamoto Y, et al. Large-scale circulating microRNA profiling for the liquid biopsy of prostate Cancer. Clin Cancer Res. 2019;25(10):3016–25.
Dang HH, Ta HDK, Nguyen TTT, et al. Identifying GPSM family members as potential biomarkers in breast Cancer: a comprehensive bioinformatics analysis. Biomedicines. 2021;9(9):1144.
Le NQK, Yapp EKY, Nagasundaram N, et al. Computational identification of vesicular transport proteins from sequences using deep gated recurrent units architecture. Comput Struct Biotechnol J. 2019;25(17):1245–54.
Leidinger P, Brefort T, Backes C, et al. High-throughput qRT-PCR validation of blood microRNAs in non-small cell lung cancer. Oncotarget. 2016;7(4):4611–23.
Ma W, Ma CN, Zhou NN, et al. Up- regulation of miR-328-3p sensitizes non-small cell lung cancer to radiotherapy. Sci Rep. 2016;6:31651.
Yang XR, Pi C, Yu R, et al. Correlation of exosomal microRNA clusters with bone metastasis in non-small cell lung cancer. Clin Exp Metastasis. 2021;38(1):109–117.
Zheng M, Hou L, Ma Y, et al. Exosomal let-7d-3p and miR-30d-5p as diagnostic biomarkers for non-invasive screening of cervical cancer and its precursors. Mol Cancer. 2019;18(1):76.
Liu X, Zhou X, Chen Y, et al. miR-186-5p targeting SIX1 inhibits cisplatin resistance in non-small-cell lung cancer cells (NSCLCs). Neoplasma. 2020;67(1):147–57.
Feng H, Zhang Z, Qing X, et al. miR-186-5p promotes cell growth, migration and invasion of lung adenocarcinoma by targeting PTEN. Exp Mol Pathol. 2019;108:105–13.
Hao F, Bi YN, Wang L, et al. miR-199a-5p suppresses epithelial mesenchymal transition in anaplastic thyroid carcinoma cells via targeting snail signals. Cancer Biomark. 2020;29(3):317–26.
Wei D, Wang W, Shen B, et al. MicroRNA-199a-5p suppresses migration and invasion in oral squamous cell carcinoma through inhibiting the EMT-related transcription factor SOX4. Int J Mol Med. 2019;44(1):185–95.
Shen Q, Cicinnati VR, Zhang X, et al. Role of microRNA-199a-5p and discoidin domain receptor 1 in human hepatocellular carcinoma invasion. Mol Cancer. 2010;9:227.
Lian XY, Zhang H, Liu Q, et al. Ovarian cancer-excreted exosomal miR-199a-5p suppresses tumor metastasis by targeting hypoxia-inducible factor-2α in hypoxia microenvironment. Cancer Commun (Lond). 2020;40(8):380–5.
Wang C, Ba X, Guo Y, et al. MicroRNA-199a-5p promotes tumour growth by dual-targeting PIAS3 and p27 in human osteosarcoma. Sci Rep. 2017;7:41456.
Qu D, Yang Y, Huang X. miR-199a-5p promotes proliferation and metastasis and epithelial-mesenchymal transition through targeting PIAS3 in cervical carcinoma. J Cell Biochem. 2019;120(8):13562–72.
Wang S, Cao KE, He Q, et al. miR-199a-5p induces cell invasion by suppressing E-cadherin expression in cutaneous squamous cell carcinoma. Oncol Lett. 2016;12(1):97–101.
Li Y, Wang D, Li X, et al. MiR-199a-5p suppresses non-small cell lung cancer via targeting MAP3K11. J Cancer. 2019;10(11):2472–9.
Ahmadi A, Khansarinejad B, Hosseinkhani S, et al. miR-199a-5p and miR-495 target GRP78 within UPR pathway of lung cancer. Gene. 2017;620:15–22.
Jin Y, Wang H, Zhu Y, et al. miR-199a-5p is involved in doxorubicin resistance of non-small cell lung cancer (NSCLC) cells. Eur J Pharmacol. 2020;878:173105.
Hu L, Chen Q, Wang Y, et al. Sp1 mediates the constitutive expression and repression of the PDSS2 gene in lung Cancer cells. Genes (Basel). 2019;10(12):977.
Xie JJ, Guo QY, Jin JY, et al. SP1-mediated overexpression of lncRNA LINC01234 as a ceRNA facilitates non-small-cell lung cancer progression via regulating OTUB1. J Cell Physiol. 2019;234(12):22845–56.
Zhang R, Niu Z, Pei H, et al. Long noncoding RNA LINC00657 induced by SP1 contributes to the non-small cell lung cancer progression through targeting miR-26b-5p/COMMD8 axis. J Cell Physiol. 2020;235(4):3340–9.
Su T, Liu P, Ti X, et al. ΗΙF1α, EGR1 and SP1 co-regulate the erythropoietin receptor expression under hypoxia: an essential role in the growth of non-small cell lung cancer cells. Cell Commun Signal. 2019;17(1):152.
Zhu HY, Cao GY, Wang SP, et al. POU2F1 promotes growth and metastasis of hepatocellular carcinoma through the FAT1 signaling pathway. Am J Cancer Res. 2017;7(8):1665–79.
Vázquez-Arreguín K, Bensard C, Schell JC, et al. Oct1/Pou2f1 is selectively required for colon regeneration and regulates colon malignancy. PLoS Genet. 2019;15(5):e1007687.
Zhang R, Lu H, Lyu YY, et al. E6/E7-P53-POU2F1-CTHRC1 axis promotes cervical cancer metastasis and activates Wnt/PCP pathway. Sci Rep. 2017;7:44744.
Xiao Y, Liu S, Li J, et al. The POU2F1/miR-4490/USP22 axis regulates cell proliferation and metastasis in gastric cancer. Cell Oncol (Dordr). 2020;43(6):1017–33.
Xiao XH, He SY. ELF1 activated long non-coding RNA CASC2 inhibits cisplatin resistance of non-small cell lung cancer via the miR-18a/IRF-2 signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24(6):3130–42.
Liu P, Zhang R, Yu W, et al. FGF1 and IGF1-conditioned 3D culture system promoted the amplification and cancer stemness of lung cancer cells. Biomaterials. 2017;149:63–76.
Xu H, Lee MS, Tsai PY, et al. Ablation of insulin receptor substrates 1 and 2 suppresses Kras-driven lung tumorigenesis. Proc Natl Acad Sci U S A. 2018;115(16):4228–33.
Hegab AE, Ozaki M, Kameyama N, et al. Effect of FGF/FGFR pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J Pathol. 2019;249(2):193–205.
Schoenfeld AJ, Chan JM, Kubota D, et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line Osimertinib in EGFR-mutant lung Cancer. Clin Cancer Res. 2020;26(11):2654–63.
Lin C, Qin Y, Zhang H, et al. EGF upregulates RFPL3 and hTERT via the MEK signaling pathway in non-small cell lung cancer cells. Oncol Rep. 2018;40(1):29–38.
He L, Wang X, Liu K, et al. Integrative PDGF/PDGFR and focal adhesion pathways are downregulated in ERCC1-defective non-small cell lung cancer undergoing sodium glycididazole-sensitized cisplatin treatment. Gene. 2019;691:70–6.
Naidu S, Shi L, Magee P, et al. PDGFR-modulated miR-23b cluster and miR-125a-5p suppress lung tumorigenesis by targeting multiple components of KRAS and NF-kB pathways. Sci Rep. 2017;7(1):15441.
Jiao D, Chen J, Li Y, et al. miR-1-3p and miR-206 sensitizes HGF-induced gefitinib-resistant human lung cancer cells through inhibition of c-met signalling and EMT. J Cell Mol Med. 2018;22(7):3526–36.
Li B, Ding CM, Li YX, et al. Over-regulation of microRNA-133b inhibits cell proliferation of cisplatin-induced non-small cell lung cancer cells through PI3K/Akt and JAK2/STAT3 signaling pathway by targeting EGFR. Oncol Rep. 2018;39(3):1227–34.
Wu DM, Zhang T, Liu YB, et al. The PAX6-ZEB2 axis promotes metastasis and cisplatin resistance in non-small cell lung cancer through PI3K/AKT signaling. Cell Death Dis. 2019;10(5):349.
Wei CH, Wu G, Cai Q, et al. MicroRNA-330-3p promotes cell invasion and metastasis in non-small cell lung cancer through GRIA3 by activating MAPK/ERK signaling pathway. J Hematol Oncol. 2017;10(1):125.
Xie Q, Yu Z, Lu Y, et al. microRNA-148a-3p inhibited the proliferation and epithelial-mesenchymal transition progression of non-small-cell lung cancer via modulating Ras/MAPK/Erk signaling. J Cell Physiol. 2019;234(8):12786–99.
Louis-Dit-Picard H, Kouranti I, Rafael C, et al. Mutation affecting the conserved acidic WNK1 motif causes inherited hyperkalemic hyperchloremic acidosis. J Clin Invest. 2020;130(12):6379–94.
Hung JY, Yen MC, Jian SF, et al. Secreted protein acidic and rich in cysteine (SPARC) induces cell migration and epithelial mesenchymal transition through WNK1/snail in non-small cell lung cancer. Oncotarget. 2017;8(38):63691–702.
Hsu YL, Hung JY, Chiang SY, et al. Lung cancer-derived galectin-1 contributes to cancer associated fibroblast-mediated cancer progression and immune suppression through TDO2/kynurenine axis. Oncotarget. 2016;7(19):27584–98.
Gallolu Kankanamalage S, Karra AS, Cobb MH. WNK pathways in cancer signaling networks. Cell Commun Signal. 2018;16(1):72.
Acknowledgments
Not applicable.
Funding
This study was supported by grants from Natural Science Foundation of China (No.82060010), Guizhou Provincial Natural Science Foundation Project ([2018]5623), Guizhou Provincial Respiratory Critical Disease Clinical Research and Prevention and Treatment Talent Base Project ([2020]8), Zunyi Respiratory Medicine Talent Base Project ([2019]69).
Author information
Authors and Affiliations
Contributions
WZ designed the study and drafted the manuscript. QZ, LC collected of data. ZX, XC analysis and interpretation of data, LG, ZL assisted in manuscript revision and figure organization. As the corresponding author, SL and DL are responsible for critical revision of the manuscript. All authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Ethics approval was exempted by the Ethics Committee of the First Affiliated Hospital of Jinan University (Guangzhou, China). The data used in this paper come from GEO and TCGA databases, which are publicly available and need not ethical review. All of our methods in this study are carried out in accordance with the relevant guidelines and regulations in the section “Ethical Approval and Consent to participate.”
Consent for publication
Not applicable.
Competing interests
All authors agreed to publish the paper and there was no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
DEMs between Lung cancer and normal plasma from GSE24709 dataset. Table S2. DEMs between Luan cancer and normal plasma from GSE31568 dataset. Table S3. DEMs between Luan cancer and normal plasma from GSE61741 dataset. TargetScanHuman database. Table S4. Target genes of DEMs predicted by TargetScanHuman. Table S5. Raw counts of RNA-sequencing data of hsa-let-7d-3p in LUAD from the TCGA. Table S6. Raw counts of RNA-sequencing data of hsa-miR-186-5p in LUAD from the TCGA. Table S7. Raw counts of RNA-sequencing data of hsa-miR-199a-5p in LUAD from the TCGA. Table S8. Raw counts of RNA-sequencing data of hsa-miR-328-3p in LUAD from the TCGA. Table S9. Raw counts of RNA-sequencing data of KLHL3 in LUAD from the TCGA. Table S10. Raw counts of RNA-sequencing data of hsa-let-7d-3p in LUSC from the TCGA. Table S11. Raw counts of RNA-sequencing data of hsa-miR-186-5p in LUSC from the TCGA. Table S12. Raw counts of RNA-sequencing data of hsa-miR-199a-5p in LUSC from the TCGA. Table S13. Raw counts of RNA-sequencing data of hsa-miR-328-3p in LUSC from the TCGA. Table S14. Raw counts of RNA-sequencing data of KLHL3 in LUSC from the TCGA. Table S14. Raw counts of RNA-sequencing data of KLHL3 in LUSC from the TCGA. Table S16. The immunohistochemical score of KLHL3 in LUAD patients. Table S17. The immunohistochemical score of KLHL3 in LUSC patients. Table S18. The immunohistochemical score of KLHL3 in normal group.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, W., Zhang, Q., Che, L. et al. Using biological information to analyze potential miRNA-mRNA regulatory networks in the plasma of patients with non-small cell lung cancer. BMC Cancer 22, 299 (2022). https://doi.org/10.1186/s12885-022-09281-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-022-09281-1