
Abstract
Purpose
Growing evidence indicates that aberrant expression of microRNAs contributes to tumor development. However, the biological role of microRNA-4490 (miR-4490) in gastric cancer (GC) remains to be clarified.
Methods
To explore the function of miR-4490 in GC, we performed colony formation, EdU incorporation, qRT-PCR, Western blotting, in situ hybridization (ISH), immunohistochemistry (IHC), flow cytometry, ChIP and dual-luciferase reporter assays. In addition, the growth, migration and invasion capacities of GC cells were evaluated.
Results
We found that miR-4490 was significantly downregulated in primary GC samples and in GC-derived cell lines compared with normal controls, and that this expression level was negatively correlated with GC malignancy. Exogenous miR-4490 expression not only reduced cell cycle progression and proliferation, but also significantly inhibited GC cell migration, invasion and epithelial-mesenchymal transition (EMT) in vitro. Mechanistically, we found that miR-4490 directly targets USP22, which mediates inhibition of GC cell proliferation and EMT-induced metastasis in vitro and in vivo. Moreover, we found through luciferase and ChIP assays that transcription factor POU2F1 can directly bind to POU2F1 binding sites within the miR-4490 and USP22 promoters and, by doing so, modulate their transcription. Spearman’s correlation analysis revealed a positive correlation between USP22 and POU2F1 expression and negative correlations between miR-4490 and USP22 as well as miR-4490 and POU2F1 expression in primary GC tissues.
Conclusion
Based on our results we conclude that miR-4490 acts as a tumor suppressor, and that the POU2F1/miR-4490/USP22 axis plays an important role in the regulation of growth, invasion and EMT of GC cells.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
MicroRNAs (miRNAs) are small (21–25 nt) noncoding RNAs that can regulate gene expression through complementary base pairing with the 3′-untranslated regions (UTRs) of target messenger RNAs (mRNAs), resulting in mRNA degradation or suppression of mRNA translation [1]. As such, miRNAs may play major roles in the regulation of various cellular processes, including differentiation, proliferation, migration and apoptosis, and they may act as tumor suppressors or oncogenes [2,3,4]. Also, miRNAs are regulated by transcription factors [5]. MiR-646 has, for example, been reported to directly target FOXK1 and inhibit FOXK1-mediated proliferation and epithelial-mesenchymal transition (EMT)-induced metastasis [6]. In addition, transcription factor HMGA1 has been reported to bind to the miR-137 promoter and to activate its transcription [7]. Although it is one of the first miRNAs that has been discovered, the biological function of miR-4490 and the molecular mechanisms underlying its role in cancer initiation and progression are not yet understood.
The human ubiquitin-specific protease 22 (USP22) gene is located on chromosome 17p11.2. USP22 belongs to a family of more than 70 de-ubiquitinases in mammals [8] and has been identified as a member of an 11-gene “death-from-cancer” signature that can predict treatment resistance, tumor aggressiveness and the occurrence of metastasis [9]. Previous studies have shown that USP22 overexpression may promote cancer progression and may be associated with a poor prognosis in human gastric cancer (GC), breast cancer, and colorectal cancer patients [10,11,12]. A more recently published report indicates that USP22 may promote tumor progression and induce EMT in colorectal carcinoma patients [13]. Although we recently developed an algorithm to predict targeting of USP22 by miR-4490, it remains to be established whether this miRNA participates in the regulation of USP22 expression in GC.
In this study, we examined the relationship between miR-4490 and its target USP22 in GC, and provide evidence that the expression of miR-4490 is regulated by the upstream transcription factor POU2F1. Thus, we reveal a POU2F1-miR-4490-USP22 signaling axis that may play a regulatory role in GC development and progression and, as such, may serve as a therapeutic target.
2 Materials and methods
2.1 Cell culture and reagents
Human GC cell lines (AGS, BGC-803, BGC-823, MKN-45 and SGC-7901) were purchased from the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). The human normal gastric mucosal cell line GES-1 was acquired from Biowit Technologies Corporation (Shenzhen, China). All cell lines were cultured as described previously [14].
Mouse anti-cyclin B1(sc-245) and anti-Cyclin D1 (sc-8396) were purchased from Santa Cruz (Santa Cruz, CA, USA), whereas mouse anti-CDK4(#2906S)and anti-CDK6(#3136S)were purchased from Cell Signaling Technology (CST, MA, USA). Rabbit anti-USP22 (55110–1-AP), anti-OCT1 (POU2F1) (10387–1-AP), anti-E-cadherin (20874–1-AP), anti-γ-catenin (27872–1-AP), anti-Vimentin (Ag0489), and anti-MMP2 (10373–2-AP) were purchased from Proteintech (Wuhan, China). Rabbit anti-ERK1/2 (137F5), anti-p-ERK1/2 (Thr202/Tyr204), anti-AKT (C-terminal), and anti-p-AKT (Ser473) were purchased from CST (MA, USA). Rabbit anti-GAPDH and anti-Ki-67 (EPR3610) were purchased from Abcam (Abcam, Cambridge, UK).
2.2 Patients and specimens
Seventy GC tissues and 70 adjacent non-tumor tissues collected by the Department of Surgery of Nanfang Hospital, Southern Medical University, China between January 2017 and December 2017 were included. None of the patients had received radiation therapy or chemotherapy prior to surgery. GC diagnoses were made by endoscopic biopsy combined with histopathological examination after surgery. The diagnoses were confirmed by two pathologists in Nanfang hospital according to the American Joint Committee on Cancer (AJCC) TNM Staging Classification for Carcinoma of the Stomach. The Ethics Committee of the Southern Medical University approved our experimental protocols (authorization protocol number: NFEC-2017-062).
2.3 RNA ISH and IHC analyses
RNA ISH was performed on paraffin-embedded tissue sections. A double digoxigenin (DIG)-labeled mercury locked nucleic acid probe was used as miR-4490 probe [miRCURY LNA™ detection probes (Exiqon, Vedbaek, Denmark)]. The probe sequence was as follows: 5′- TATGC CCAAA TCTCT TACCA GA-3′. Sections (4 μm) of paraffin-embedded specimens were dewaxed in xylene and next rehydrated in ethanol and PBS. The sections were incubated with Pepsin for ISH (BOSTER, Wuhan, China) at 37 °C for 15 min and then washed 3 times for 15 min with 0.5 mM TBS. Next, the sections were incubated in prehybridization buffer at 50 °C for 2 h after which a DIG-labeled miR-4490 probe was added and hybridized overnight at 50 °C. Next, the sections were washed with a gradient diluted SSC solution at 50 °C for 30 min, followed by incubation with DIG blocking reagent (Roche) in maleic acid buffer at 30 °C for 15 min and alkaline phosphatase-conjugated anti-digoxigenin (diluted 1:500 in blocking reagent, Roche) at room temperature for 60 min. Finally, the hybridization signals were visualized using NBT/BCIP (Sigma). The reaction was stopped by washing with water for 5 min. The sections were counterstained with haematoxylin, fixed with an aqueous solution, and photographed. A scrambled probe was used as a negative control.
IHC was conducted as previously described using the following primary antibodies: anti-POU2F1, anti-USP22 and anti-E-cadherin (Abcam). The resulting staining patterns were evaluated by two independent reviewers using a semi-quantitative scoring system, as previously described [14,15,16].
2.4 DNA constructs and oligonucleotide transfection
pENTER-USP22, POU2F1 and empty vector (pENTER) plasmids encompassing a FLAG tag were purchased from Vigene (Rockville, MD, USA). A miR-4490 mimic (40 nmol/L/well, sense 5′- UCUGGUAAGAGAUUUGGGCA UA −3′; antisense 5′- UGCCCAAAUCUCUUACCAGAUU -3′), a miR-4490 inhibitor (100 nmol/L/well, 5′- UAUGCCCAAAUCUCUUACCAGA -3′, GenePharma Co, Ltd., Shanghai, China) or their corresponding controls (m-NC for mimics sense 5′-UUCUCCGAACGUGUCACGUTT-3′; antisense 5′-ACGUGACACGUUCGGA GAATT -3′and i-NC for inhibitor, 5’- CAGUACUUUUGUGUAGUACAA -3′) were transfected using Lipofectamine 2000 (Invitrogen, Foster City, CA, USA) according to the manufacturers’ protocols. Transfection efficiencies were evaluated using quantitative real-time PCR (qRT-PCR).
2.5 Western blot analysis
Lysates were harvested from cells in RIPA containing a protease inhibitor cocktail. Next, equal amounts of protein were separated via sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). The resulting membranes were incubated with primary antibodies directed against target proteins diluted as recommended in 5% milk/TBST or 5% bovine serum albumin (BSA)/TBST overnight at 4 °C with agitation. Next, the membranes were washed in TBST for 4 × 10 min, and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (ZSGB-BIO, Beijing, CN) diluted 1:3500 in 5% milk/TBST for 1 h at room temperature. Subsequently, the membranes were washed again 4 × 10 min in TBST. Finally, detection was performed using an enhanced chemiluminescence kit (Millipore, MA, USA).
2.6 RNA isolation and qRT-PCR
Total RNA was extracted from fresh tissues and cells using TRIzol reagent (Invitrogen, San Diego, CA, USA) according to the manufacturer’s instructions. Complementary DNA was synthesized by reverse transcription using a PrimeScrip™ RT Master Mix (TaKaRa, Otsu, Shiga, Japan) or an All-in-One™ miRNA First-Strand cDNA Synthesis Kit (Gene Copoeia, Rockville, MD, USA). qPCR was performed using a Roche LightCycler 480® 96-well block PCR Machine (Roche, Mannheim, Germany). Relative mRNA and miRNA expression levels were calculated using the comparative 2-△△CT method with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and U6 small nuclear RNA levels as internal controls. The primers used are listed in Supplementary Table 1.
2.7 EdU incorporation and WST-1 assays
For EdU incorporation assays, cells were treated as described in the figure legends and subsequently incubated with 20 μM EdU (RiboBio, Guangzhou, China) according to the manufacturer’s instructions. Next, the cells were fixed with 4% paraformaldehyde/PBS (Santa Cruz Biotechnology, Dallas, TX, USA) for 10 min and permeabilized with 0.2% Triton-100 for 15 min at room temperature. Subsequently, the cells were conjugated to Apollo solution and Hoechst 33342. Finally, six random fields were selected and the numbers of proliferative cells were evaluated by fluorescence microscopy (Olympus, Tokyo, Japan).
For the WST-1 assays, treated cells were inoculated in 96-well plates (3000 cells /well). At 0, 24, 48, and 72 h, the culture media were removed and 100 μl fresh medium containing 10 μl WST-1 reagent was added to each well. After 2 h, absorbance was measured at 450 nm using a microplate reader (Molecular Decices, Sunnyvale, CA, USA).
2.8 Flow cytometry
For flow cytometry, cells were transfected with miR-4490 mimics and control m-NC. After 24 h the media were refreshed and the cells were harvested by trypsin. Next, the cells were fixed using Cytofix/Cytoperm buffer (BD Biosciences, CA, USA) and stored at 4 °C overnight. Subsequently, the cells were intracellularly stained using propidium oxide using a Cycle TEST PLUS DNA Reagent Kit (BD Biosciences, CA, USA) according to the manufacture’s protocol. Data were collected on a LSR II flow cytometer (BD Pharmingen) and analyzed using Flowjo software (Treestar).
2.9 Colony formation assay
Cells were transfected with m-NC and miR-4490 mimics, or i-NC and miR-4490 inhibitor. After 48 h, the cells were harvested by trypsin and counted. Next, the cells were seeded in 6-well plates at a density of 1 × 103 cells per well and cultured for 2–3 weeks. The resulting colonies were fixed in 70% ethanol, stained with 0.05% crystal violet (Sigma), and evaluated under a bright field microscope (Olympus, Tokyo, Japan) after being washed with PBS. Colonies containing more than 50 cells were counted and the data were analyzed using Image J software.
2.10 Immunofluorescence assay
Treated cells grown on 13-mm diameter coverslips were fixed with 4% paraformaldehyde and permeabilized in 0.2% Triton X-100. Next, the coverslips were washed with PBS and blocked with 1% bovine serum albumin for 30 min. Subsequently, the cells were incubated with primary antibodies in a humidified chamber overnight at 4 °C followed by incubation with Alexa Fluor 594-conjugated goat anti-rabbit/mouse IgG (1:200, Jackson lab) for 2–3 h at room temperature. Nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI; 1:1000; Sigma). After mounting, the cells were evaluated under an Olympus CKX 41 fluorescence microscope (Olympus Optical Co, Ltd., Tokyo, Japan).
2.11 Migration and invasion assays
After transfection, cells were cultured in 6-well plates for 48 h. For migration assays, the cells were grown to 90% confluence in complete medium, after which artificial wounds were prepared by scraping the monolayers with a 10-μl pipette tip and washing with PBS to remove floating cells. Next, the cells were grown in serum-free medium to inhibit cell proliferation at 37 °C with 5% CO2 for 24 and 48 h. Cell migration was assessed by microscopy and analyzed using ImageJ software (scratch wound healing assay).
For invasion assays, transwell chambers (Corning Incorporated, Corning, NY, USA) were used. Cell suspensions (1 × 105) in serum-free culture media were placed into chambers after which each insert was placed in a well of a 24-well plate filled with RPMI-1640 medium containing 20% FBS. After 24 h at 37 °C in an incubator at 5% CO2, noninvasive cells were removed from the upper chamber by wiping with cotton-tipped swabs, after which the filters were fixed with 4% paraformaldehyde for 30 min and subsequently stained with a dye solution containing 0.05% crystal violet for 30 min at room temperature. Five fields of adherent cells in each well were randomly photographed and counted under a bright field microscope (Olympus Corp, Tokyo, Japan) (Transwell invasion assay).
2.12 Bioinformatics analysis
Public web-based prediction sites TargetScan (http://www.targetscan.org), miRWalk (http://mirwalk.umm.uni-heidelberg.de/), miRanda (http://www.microrna.org/microrna/home.do) and miRmap (https://omictools.com/mirmap-tool) were used to predict potential miRNA-targeted gene transcripts.
2.13 Dual luciferase assay
For binding assessment of miR-4490 to the USP22 3’UTR, the 3’UTR segment was amplified by PCR and inserted into a vector. A mutant miR-4490 binding site in the USP22 3’UTR region was generated using a Quick Change Site-Directed Mutagenesis Kit (Agilent, Roseville City, CA, USA). Co-transfections of USP22 3’UTR or mutUSP22 3’UTR plasmids with miR-4490 lentivirus vector into cells were carried out using Lipofectamine 2000 (Invitrogen). The USP22-WT-3′-UTR and USP22-MUT-3′-UTR sequences are shown in Fig. 4b.
2.14 MiR-4490 promoter analysis
A 2-kb region directly upstream of the miR-4490 gene was predicted to serve as a promoter using UCSC software. Analysis of POU2F1 binding sites within this promoter was performed using the TF prediction programme promo (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3). Based on this information, a miR-4490 promoter luciferase reporter vector (miR-4490-p) containing POU2F1 binding site 1 (miR-4490-p-site 1: −1440 ~ −1430) was constructed. Next, a dual luciferase assay was performed using a Dual-luciferase Reporter Assay kit (Promega, Madison, WI, USA) as per the manufacturer’s instructions. The oligonucleotide primers used for the luciferase activity assays are listed in Supplementary Table 1.
2.15 Chromatin immunoprecipitation (ChIP) assay
ChIP was performed using a SimpleChIP Enzymatic Chromatin IP Kit (CST, #9003, MA, USA) according to the manufacturer’s recommended protocol. Chromatin was extracted from POU2F1-overexpressing GC cells using a sodium dodecyl sulfate lysis buffer and sheared to lengths ranging from 160 to 500 bp. Next, protein-chromatin complexes were immunoprecipitated using an anti-POU2F1 antibody (Abcam, Boston City, MA, USA) and control IgG. The purified DNAs were subjected to PCR using primers specific for the miR-4490 promoter. The sequences of the primers used are listed in Supplementary Table 1.
2.16 Construction and transfection/transduction of lentivial vectors
A miR-4490 lentiviral expression vector (Ubi-MCS-SV40- EGFP-IRES) containing a green fluorescent protein gene (GeneChem Co, Ltd., Shanghai, China) was transfected into the lentiviral packaging cell line 293 T. Next, 1 ml viral supernatant containing 5 g polybrene was added to GC cells for stable transduction. After 14 days, puromycin-resistant cell pools were established. A Ubi-MCS-3FLAG- SV40- Cherry-IRES lentiviral expression vector was constructed containing the USP22 sequence without the 3’UTR region. Empty vectors were used as controls. To generate mir-4490/USP22-co-expressing cells, 3 ml of a concentrated USP22 lentiviral expression vector solution was added to miR-4490 overexpressing GC cells as described previously. Next, 5 g/ml polybrene was mixed with the cells, and after 72 h Western blotting was performed to detect USP22 expression.
2.17 Animal models
To generate an in vivo xenograft tumor model, fifteen 4–6 weeks old female BALB/c nu/nu mice were randomly divided into five groups and maintained under specific pathogen-free conditions at the Experiment Animal Center of South Medical University. Next, BGC-823 cells were transfected with m-NC, miR-4490, Vector, USP22 or miR-4490/USP22 lentivirus and resuspended (1 × 106) in 100 μl PBS, followed by subcutaneous inoculation into the flanks of the mice. After 35 days, the mice were sacrificed and the tumor tissues were resected and imaged using a whole-body GFP imaging system (Lighttools, Encinitas, CA, USA).
For in vivo metastasis assays, 4–6 weeks old female BALB/c nude mice were used. BGC-823 cells transfected with m-NC, miR-4490, Vector, USP22 or miR-4490/USP22 lentivirus were resuspended (5 × 106) in 100 μl PBS and injected into the tail vein of nude mice. After 35 days, the mice were sacrificed after which lung tissues were dissected and subjected to histological examination. The experiments were performed as previously described [6, 14]. The nude mice were provided by the Experimental Animal Center of the Southern Medical University. All animal studies complied with the Southern Medical University animal use guidelines and the protocols approved by the Southern Medical University Animal Care Committee.
2.18 Statistical analysis
SPSS 20.0 statistical software was used for all data analyses, and all data are expressed as the mean ± standard deviation (SD). Pearson’s χ2 test was used to analyse associations of miR-4490 expression with clinicopathologic features. USP22-miR-4490, USP22-POU2F1 and POU2F1-miR-4490 interaction tests were performed using linear regression models. Categorical data were analyzed using the Fisher’s exact test. The quantitative data obtained from experiments with biological replicates are presented as the mean ± SD. P < 0.05 was considered to indicate statistical significance.
3 Results
3.1 MiR-4490 expression is downregulated in GC cells and tissues
To determine the role of miR-4490 in GC development, we first examined its expression levels in the normal gastric epithelial cell line GES-1, in GC-derived cell lines (AGS, BGC-823, MKN-45, SGC-7901 and BGC-803) and in primary GC tissues. Using qRT-PCR we found that miR-4490 expression was significantly decreased in the GC cell lines compared with GES-1 cells (Fig. 1a). Moreover, we found that miR-4490 expression was decreased approximately three-fold in gastric cancer tissues compared with corresponding nontumor tissues (Fig. 1b) and to be downregulated in 74.3% (52 of 70) of GC tissues compared with nontumor tissues (Fig. 1c). We also observed a statistically significant decrease in the expression of this miRNA in advanced-stage GCs (stages III-IV, n = 41) compared with early-stage GCs (stages I-II, n = 29) (Fig. 1d). Furthermore, miR-4490 expression was found to be lower in tumors with a greater invasion depth (Fig. 1e, T3–4 vs. T1–2), suggesting that its deficiency may contribute to GC cell invasiveness (Fig. 1d, e). In addition, RNA in situ hybridization (ISH) revealed that miR-4490 was mainly localized in the nucleus of patient-derived GC cells and that, in accordance with the above results, its expression was lower in GC tissues than in adjacent normal gastric mucosa (Fig. 1f). Taken together, these results indicate that miR-4490 is downregulated in GC and may be negatively related to its metastasis.

MiR-4490 is downregulated in GC cells and tissues. (a) Expression of miR-4490 in human GC-derived cells compared with human normal gastric epithelial GES-1 cells. **, p < 0.05. (b) Relative expression of miR-4490 measured using qRT-PCR in 70 pairs of GC samples and corresponding non-cancerous samples; the results were normalized to endogenous U6 RNA. **, p < 0.05. (c) Expression of miR-4490 in paired GC tissues and matched normal tissues. (d) Expression of miR-4490 in different clinical GC stages. The patients were staged in accordance with the 7th Edition of the AJCC Cancer Tumor Node Metastasis Classification. **, p < 0.05. stages I-II vs. stages III-IV. (e) The miR-4490 expression level is lower in T3–4 than in T1–2 GC samples. **, p < 0.05. (f) ISH analysis of miR-4490 expression in normal gastric mucosa and GC tissues. Data are shown as the means ± SD from at least three independent experiments. Scale bars, 50 μm in F
3.2 miR-4490 suppresses the growth and cell cycle progression of GC cells
To assess putative correlations of miR-4490 with clinicopathologic features, we measured the relative expression levels of miR-4490 in 70 GC tissue samples. Next, the patients were divided into two groups, i.e., a high miR-4490 expression group and a low miR-4490 expression group, with a cut-off at the median expression value. After this, correlations between miR-4490 expression and clinicopathologic features of the patients were analyzed. We found that low miR-4490 expression closely correlated with tumor size (p = 0.029), differentiation (p = 0.001), invasion (T1–2 vs. T3–4, p = 0.001), lymph node metastasis (p = 0.0001) and TNM stage (I-II vs. III-IV, p = 0.008) (Supplementary Table 2). No significant correlations were observed between low miR-4490 expression and other clinicopathologic features, including sex (p = 0.799) and age (p = 0.473).
To assess the role of miR-4490 in GC development and progression, GC-derived cells were transfected with miR-4490 mimics, m-NC mimics, or the miR-4490 inhibitor i-NC, after which modulations in miR-4490 expression were confirmed by qRT-PCR (Supplementary Fig. 1). Next, we assessed the effects of miR-4490 expression modulation on GC cell growth characteristics using soft agar and WST-1 assays. We found that miR-4490 overexpression decreased the numbers and sizes of GC cell colonies, whereas miR-4490 downregulation increased the colony formation capacities (Fig. 2a). Also, there was a significant decrease in the growth rate of miR-4490 mimic-transfected cells at 24, 48, and 72 h compared with m-NC cells, whereas there was a significant increase in the growth rate of miR-4490 inhibitor-transfected cells compared with that of i-NC cells (Fig. 2b). An EdU incorporation assay confirmed that miR-4490 mimic-transfected cells exhibited a significantly decreased proliferation rate compared with control cells, and that miR-4490 inhibitors induced a decreased EdU incorporation (Fig. 2c). Cell cycle analysis revealed that miR-4490 caused a significant accumulation in GC cells in the G0/G1 phase (Fig. 2d). Cell cycle-related protein expression was subsequently assessed using Western blotting. We found that the cyclin D1, CDK and CDK6 levels were significantly decreased in miR-4490 mimic-treated GC cells compared with NC-treated cells, whereas the level of cyclin B1 remained unchanged (Fig. 2e). These results indicate that overexpression of miR-4490 inhibits the growth of GC cells.

MiR-4490 suppresses GC cell growth in vitro. (a) Representation (left) and quantification (right) of colony formation assay showing the growth of GC cells transfected with NC or miR-4490 mimics or inhibitors. ****, p < 0.001. (b) The proliferation of miR-4490 mimic-transfected GC cells is lower compared with the corresponding m-NC-transfected cells, and downregulation of miR-4490 reverses this effect. ***, p < 0.01; ****, p < 0.001. (c) The EdU-positive cell rate is decreased after miR-4490 mimic transfection, whereas miR-4490 inhibitor-treated GC cells show a higher proliferation rate. ****, p < 0.001. (d) Flow cytometric analysis of the effect of miR-4490 on GC cell cycle progression; the percentages in G0-G1, S and G2-M phase GC cells were quantified. The figures are representative of three independent experiments. (e) Protein expression detected by Western blotting. Scale bars, 100 μm in C
3.3 MiR-4490 inhibits migration, invasion and EMT of GC cells
To determine whether miR-4490 regulates human GC cell invasion and metastasis, we performed in vitro gain-of-function and loss-of-function assays using miRNA mimics and inhibitors. Using a scratch wound healing assay, we found that compared to the control, the miR-4490 mimics markedly inhibited GC cell migration. In contrast, the miR-4490 inhibitor significantly increased GC cell migration (Fig. 3a). Using Transwell invasion assays, we found that the invasiveness of GC cells transfected with miR-4490 mimics was significantly decreased, whereas it was increased after transfection with miR-4490 inhibitor (Fig. 3b). These data indicate that exogenous miR-4490 overexpression significantly suppresses GC cell migration and invasion.

MiR-4490 suppresses GC cell migration/invasion via EMT in vitro. (a) Scratch wound healing assays were used to detect GC cell motility following transfection with NC and miR-4490 mimics or a miR-4490 inhibitor. Wound closure percentages are shown in the top panel. ****, p < 0.001. (b) Representation and quantification of matrigel assay indicating the invasive capacity of GC cells transfected with NC, miR-4490 mimics, or a miR-4490 inhibitor. ****, p < 0.001. Error bars represent the means ± SD from three independent experiments. (c) GC cell morphology observed under an inverted phase-contrast microscope. (d) Expression levels of E-cadherin, γ-cadherin, vimentin, ZEB1, N-cadherin and MMP2 in GC cells analyzed by Western blotting. GAPDH was used as a loading control. (e) Immunofluorescence analysis of E-cadherin and vimentin expression in human GC cells. Scale bars, 200 μm in C and 20 μm in E
Next, we investigated whether miR-4490 may regulate EMT. We found that i-NC-transfected cells displayed a uniform cobblestone morphology with intimate cell-cell contacts. After miR-4490 inhibitor treatment, however, we observed stick-like or long spindle-shaped mesenchymal cell populations, characteristic of EMT, using phase-contrast microscopy [17] (Fig. 3c). Furthermore, we found that miR-4490 mimics decreased MMP2, vimentin, N-cadherin and ZEB1 expression levels and increased E-cadherin and γ-catenin expression levels, whereas miR-4490 inhibition increased MMP2, vimentin, N-cadherin and ZEB1 expression levels and decreased E-cadherin and γ-catenin expression levels as determined by Western blot analysis in GC cells (Fig. 3d). Additionally, we found by immunofluorescence analysis that E-cadherin expression was decreased, but vimentin expression was increased in GC cells treated with miR-4490 inhibitor (Fig. 3e). Together, these results indicate that miR-4490 suppresses migration, invasion and EMT of GC cells.
3.4 MiR-4490 affects the proliferation, invasion and metastasis of GC cells by targeting USP22
To further investigate the mechanism underlying miR-4490-mediated inhibition of proliferation, migration and invasion of GC cells, we searched for potential target genes using prediction algorithms available through several online databases, including miRwalk (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2/), TargetScan (www. targetscan.org), miRanda (http://www.microrna.org/) and miRmap (https://mirmap.ezlab.org/). As a result of intersection of the four bioinformatics software programs (Fig. 4a), we identified eight genes including TRIM59, LIN28B TRAF6, SP1, WEE1, SP3, STMN1 and USP22. Subsequent qRT-PCR screening data suggested that the expression levels of TRIM59, LIN28B, TRAF6, SP1, WEE1, SP3 and STMN1 did not decrease significantly in the miR-4490 group, although only changes at the mRNA level were assessed [18,19,20,21]. We found, however, that the USP22 mRNA level was significantly reduced in the miR-4490 group compared with the m-NC group (p > 0.05) (Supplementary Fig. 2A).

USP22 is a direct downstream target of miR-4490 in GC cells. (a) miRmap, miRWalk, miRanda and TargetScan databases were used to predict miR-4490 targets; USP22 was identified as a potential target. (b) Putative miR-4490-binding sequence within the 3′UTR of USP22. Mutations in the complementary site for the seed region of miR-4490 in the 3′UTR of USP22 are indicated. (c) Reporter plasmids containing either the wild-type 3′UTR or a mutated 3′UTR of the USP22 gene were co-transfected into GC cells with a miR-4490-encoding plasmid, after which luciferase activity was measured. WT, wild type; MUT, mutated. *, p > 0.05; ****, p < 0.001. The data are presented as the means ± SD of three experiments. (d) USP22 expression in m-NC and miR-4490 mimic-transfected or i-NC and miR-4490 inhibitor-treated GC cells analyzed by Western blotting. GAPDH was used as a loading control. (e) Representative micrographs of crystal violet-stained cell colonies formed by the indicated GC cells at 12 days after seeding. (f) Assessment of the invasive activity of GC cells transfected with a USP22 expression plasmid and/or miR-4490 mimics. (g) USP22, ERK1/2, p-ERK1/2, Akt, and p-Akt protein expression in GC cells transfected with miR-4490 mimic, inhibitor or NC plasmid. (h) Expression levels of USP22, E-cadherin and vimentin detected by Western blotting in GC cells transfected with USP22 expression plasmid, miR-4490 mimic and/or its control plasmid. The figures are representative of three independent experiments
Next, we performed luciferase reporter assays to assess whether USP22 serves as a direct target of miR-4490 in GC cells. To this end, the target region sequence of the USP22 3’-UTR (WT 3’-UTR) and a mutant sequence containing a putative miR-4490 site (Mut 3’-UTR) were cloned into a luciferase reporter vector (Fig. 4b). Next, these constructed reporter vectors were co-transfected with miR-4490 mimics or m-NC into GC cells. By doing so, we found that miR-4490 inhibited the luciferase activity of the USP22 WT 3’-UTR construct, whereas mutation of the putative binding site abolished the miR-4490-mediated inhibition of luciferase activity. These findings indicate that miR-4490 targets a complementary sequence in the USP22 3’-UTR (Fig. 4c).
To further confirm that USP22 serves as a target of miR-4490 in GC cells, Western blot analysis was performed to assess USP22 expression. We found that exogenous overexpression of miR-4490 significantly suppressed USP22 expression in GC cells, and that miR-4490 silencing increased its expression in GC cells (Fig. 4d), indicating that miR-4490 regulates USP22 expression at the posttranscriptional level.
We also determined the effects of miR-4490-mediated USP22 downregulation on GC cell proliferation and metastasis by rescuing USP22 expression in miR-4490-overexpressing GC cells. Subsequent WST-1 (Supplementary Fig. 2B), colony formation (Fig. 4e and Supplementary Fig. 2C) and Transwell invasion (Fig. 4f and Supplementary Fig. 3D) assays indicated that USP22 overexpression partially reverses the inhibitory effects of miR-4490.
A recent report has stated that USP22 can sustain activation of multiple EGFR downstream signaling pathways, including the AKT/mTOR and MEK/ERK pathways [22]. We, therefore, set out to assess whether miR-4490 regulates the AKT and ERK signaling pathways in GC cells. We found that the levels of both phosphorylated Akt and phosphorylated ERK were decreased in miR-4490 mimic-transfected GC cells, but were elevated in miR-4490 inhibitor-transfected cells compared to control cells. In addition, we found that USP22 expression counteracted the downregulation of p-Akt and p-ERK induced by miR-4490 (Fig. 4g).
Previous studies have also shown that USP22 can promote tumor progression and induce EMT in various tumors [13, 23]. Here, we investigated whether miR-4490 may affect the EMT process by regulating USP22 expression in GC cells. We found that, compared with m-NC cells, GC cells transfected with miR-4490-mimics exhibited an increased expression of E-cadherin and a decreased expression of vimentin (Fig. 4h). USP22 transfection significantly reversed these changes by suppressing E-cadherin and promoting vimentin expression (Fig. 4h and Supplementary Fig. 2E). Together, these results indicate that miR-4490 may inhibit GC metastasis and EMT by targeting USP22.
3.5 MiR-4490 regulates USP22-mediated GC growth and metastasis in vivo
To further explore the effects of miR-4490 on tumor growth in vivo, BGC-823 cells stably expressing LV-m-NC, LV-miR-4490, LV-vector, LV-USP22, or LV-miR-4490/USP22 were inoculated subcutaneously into nude mice, after which the growth of resultant primary tumors was monitored (Fig. 5a). We found that the mice injected with miR-4490 cells developed smaller tumors than those injected with m-NC cells. The mice injected with USP22 cells developed larger tumors than those injected with vector or miR-4490 cells, whereas mice injected with miR-4490/USP22 cells showed a partially rescued growth (Fig. 5a, b). We also explored expression of the cell proliferation marker Ki-67 in xenograft tumors in the five groups. Representative images of the tumors after IHC staining are shown in Fig. 5c. The miR-4490 group showed significantly decreased expression of Ki-67 compared with the m-NC group. The USP22 group showed increased expression of Ki-67 compared with that of the vector group and the miR-4490 cell group, whereas miR-4490/USP22 showed decreased expression of Ki-67 compared with that of the USP22 group (Fig. 5d).

MiR-4490 suppresses USP22-mediated GC cell growth and invasion in vivo. (a) Representative images of tumors in nude mice after inoculation of BGC-823 cells stably expressing LV-m-NC, LV-miR-4490, LV-vector, LV-USP22 and LV-4490/USP22 (n = 3). (b) Tumor volumes measured after GC cell inoculation in each group. ****, p < 0.001, m-NC vs. miR-4490; ****, p < 0.001, miR-4490 vs. miR-4490/USP22. (c) and (d) Immunohistochemical (IHC) detection and quantification of Ki-67 expression in subcutaneous tumors from mice injected with GC cells. Student’s t test. ****, p < 0.001, m-NC vs. miR-4490; ***, p < 0.01, miR-4490 vs. miR-4490/USP22 and Ki-67. (e) Lungs of mice orthotopically transplanted with GC cells (n = 3 in each group). (f) Quantification of the number of metastatic loci in the lungs. ***, p < 0.01, m-NC vs. miR-4490; **, p < 0.05, miR-4490 vs. miR-4490/USP22. (g) E-cadherin expression in tumors derived from GC cells measured by qRT-PCR; **, p < 0.05, m-NC vs. miR-4490 and miR-4490 vs. miR-4490/USP22. (h) Immunohistochemical (IHC) detection of E-cadherin and vimentin expression in lung metastases. Scale bars, 200 μm in C and H
To assess the effects of miR-4490 on GC metastasis, LV-m-NC, LV-miR-4490, LV-vector, LV-USP22 and LV-miR-4490/USP22 cells were injected into the tail vein of nude mice. We found that miR-4490 cells formed only a few small metastatic nodules in the lungs compared with the m-NC cells. Large lung metastatic nodules were detected in the USP22 groups, whereas only a few small nodules were detected in the miR-4490/USP22 and vector groups (Fig. 5e). The number of metastatic nodules was clearly reduced in mice injected with miR-4490 cells compared to those injected with m-NC cells, whereas USP22 cells formed more metastatic nodules than USP22/miR-4490 co-expressing cells and empty vector transfected cells (Fig. 5f).
We next set out to investigate whether miR-4490 may affected the EMT process by regulating USP22 expression. Using qRT-PCR, we found that miR-4490 cells exhibited an increased E-cadherin expression compared to that in m-NC cells, whereas USP22 cells exhibited a decreased E-cadherin expression compared to that in USP22/miR-4490 co-expressing cells and empty vector transfected cells (Fig. 5g). Additionally, the presence of GC metastases in the lungs was confirmed by IHC using anti-E-cadherin and anti-vimentin antibodies (Fig. 5h). Based on these results, it is reasonable to conclude that miR-4490 suppresses the proliferation, invasion and EMT of GC through USP22 downregulation.
3.6 POU2F1 regulates EMT through transactivation of miR-4490 expression
To further understand the upstream regulatory factors controlling miRNA expression, we analyzed the miR-4490 promoter using Promo software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB = TF_8.3) and found several putative binding sequences for transcription factors, including c-JUN, YY1 and POU2F1 [14, 16, 17, 24]. Since c-JUN and YY1 have previously been implicated in the pathogenesis of gastrointestinal cancer [14, 16], we next questioned whether these factors regulate miR-4490 transcription in GC cells. We found that the miR-4490 level remained unchanged after exogenous expression of YY1 and c-JUN (Fig. 6a). More recent data have shown that POU2F1 is upregulated in GC patients and is associated with a poor survival [22]. Thus, we next investigated whether miR-4490 may be regulated by POU2F1. We found that POU2F1 upregulation significantly decreased miR-4490 expression in GC cells (Fig. 6b). Based on this observation, we next set out to survey the miR-4490 promoter (2000 bp, miR-4490-p) and identified a likely POU2F1 binding motif within the −1430 bp to −1440 bp region (Fig. 6c). We subsequently subcloned the miR-4490 promoter into a pGL3-basic vector, after which a dual-luciferase reporter assay was performed to assess interaction between POU2F1 and miR-4490. By doing so, we found that the activity of the miR-4490 site in POU2F1-expressing cells decreased more than 4-fold compared with vector-transfected GC cells (Fig. 6d). Subsequent ChIP assays showed that POU2F1 directly binds to the −1430 bp to −1440 bp region within the miR-4490 promoter in GC cells (Fig. 6e). Together, these data indicate that POU2F1 can bind to the miR-4490 promoter to suppress its expression in GC cells.

MiR-4490 is directly regulated by POU2F1. (a) and (b) Expression of miR-4490 in GC cells overexpressing of YY1, c-JUN or POU2F1 determined by qRT-PCR. *, p > 0.05; **, p < 0.05. (c) Luciferase (Luc) reporter constructs containing a miR-4490 promoter fragment with a potential POU2F1 binding site upstream of a luciferase gene. (d) Luciferase activity of the miR-4490 promoter construct after transfection of POU2F1 or vector plasmid in GC cells. ****, p < 0.001. (e) ChIP assay showing direct binding of POU2F1 to the miR-4490 promoter in GC cells. (f) and (h) Effects of POU2F1 and POU2F1/miR-4490, or POU2F1 siRNA and POU2F1 siRNA/miR-4490 on GC cell proliferation using a colony formation assay. (g) and (i) Effects of POU2F1 and POU2F1/miR-4490, or POU2F1 siRNA and POU2F1 siRNA/miR-4490 on GC cell invasion using a transwell assay. (j) POU2F1, E-cadherin, γ-catenin, Vimentin, ZEB1, p-Akt, Akt, p-ERK1/2 and ERK1/2 protein expression levels in GC cells transfected with POU2F1, POU2F1 plus miR-4490 or m-NC plasmids. The figures are representative of three independent experiments. (k) and (m) Mice were injected subcutaneously or systemically via the tail vein with BGC-823/Vector/m-NC, BGC-823/POU2F1/m-NC and BGC-823/POU2F1/miR-4490 cells, respectively. (l) Representative IHC images of Ki-67 expression in mouse tumor tissues. Scale bars, 100 μm. (n) Quantification of the numbers of metastatic loci in the lungs. ****, p < 0.001, vector vs. POU2F1; **, p < 0.01, POU2F1 vs. POU2F1/miR-4490 respectively
To further investigate whether miR-4490 affects the function of POU2F1 in mediating the growth and progression of GC in vitro, GC cells were co-transfected with a POU2F1 expression plasmid or a POU2F1 siRNA and/or miR-4490 mimic. The GC cells were co-transfected with empty vector, scrambled siRNA or mimic NC as controls. Next, we performed in vitro soft agar and invasion assays. Compared with the vector/mimic-NC group, the POU2F1 overexpression group exhibited increased colony formation and invasion capacities, whereas miR-4490 reintroduction attenuated these effects (Fig. 6f and Supplementary Fig. 3A and Fig. 6g and Fig. 3b). In addition, we found that POU2F1 knockdown inhibited the colony formation and invasion capacities compared with those in the vector group, while POU2F1 downregulation in miR-4490-overexpressing cells reduced their colony formation and invasion capacities compared to those in cells transfected with miR-4490 mimic (Fig. 6h and Supplementary Fig. 3C and Fig. 6I and Supplementary Fig. 3D).
Previous studies have shown that POU2F1 overexpression may promote EMT in different carcinomas [25, 26]. Therefore, we set out to investigate whether a correlation may exist between POU2F1 and miR-4490 expression that regulates EMT in GC cells. We found that exogenous expression of POU2F1 led to decreased E-cadherin and γ-catenin expression and increased vimentin and ZEB1 expression, whereas POU2F1 overexpression combined with miR-4490 mimic transfection led to increased E-cadherin and γ-catenin expression and decreased vimentin and ZEB1 expression, and vice versa (Fig. 6j and Supplementary Fig. 3E).
Next, we performed Western blot analysis to assess phosphorylation of AKT and ERK, known to be involved in EMT signaling. We found that exogenous POU2F1 expression increased Akt and ERK phosphorylation compared with that in vector transfected cells, whereas the increase in AKT and ERK1/2 phosphorylation levels was attenuated after co-transfection with miR-4490 mimic. The total AKT and ERK1/2 protein levels were unaltered (Fig. 6j).
To validate the association of miR-4490 expression with POU2F1-mediated GC development and progression in vivo, vector/m-NC, POU2F1/mimic-NC and POU2F1/miR-4490 transfected cells were divided into 3 groups. After subcutaneous inoculation of the respective BGC-823 cells into nude mice (Fig. 6k), we found that the tumor volumes of the POU2F1-overexpressing group were markedly larger than those of the vector group. In contrast, we found that tumors derived from miR-4490/POU2F1-overexpressing cells were markedly smaller than those derived from POU2F1 cells (Supplementary Fig. 3F). Next, we employed IHC for Ki-67 to assess cell proliferation. The Ki-67 levels in the POU2F1-overexpressing group were found to be increased compared to those in the vector group, whereas the POU2F1/miR-4490 group exhibited a decreased proliferation rate (i.e., a lower Ki-67 expression) compared to those of the POU2F1-overexpressing group (Fig. 6l).
In addition, the respective BGC-823 cells were injected via the tail vein to generate lung metastases in nude mice (Fig. 6m). By doing so, we found that POU2F1 upregulation led to increased numbers of metastatic lung nodules in the vector group. In contrast, the miR-4490/POU2F1-overexpressing group exhibited a decreased number of metastatic lung nodules compared to that in the POU2F1 group (Fig. 6n). IHC analysis revealed that the expression of the epithelial marker E-cadherin was downregulated, whereas the expression of the mesenchymal marker vimentin was upregulated in cancer tissues compared to that in adjacent normal lung tissues (Supplementary Fig. 3G). These data suggest that POU2F1 negatively regulates transactivation of miR-4490 expression in GC cells.
3.7 MiR-4490 expression negatively correlates with USP22 and POU2F1 expression in GC tissues
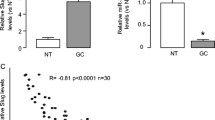
To validate the expression relationship between miR-4490, USP22 and POU2F1 in primary GC tissues, we assessed their expression in 12 paired human GC clinical specimens. Using Western blot and/or qRT-PCR analyses we found that the expression of both USP22 and POU2F1 were significantly upregulated (Fig. 7a, b), while that of miR-4490 was downregulated in the 12 samples examined (Fig. 7b) relative to the paired adjacent noncancerous tissues from the same patients. Spearman’s correlation analyses revealed a negative correlation between miR-4490 and USP22 (Fig. 7C-a) and between POU2F1 and miR-4490 (Fig. 7C-b), and positive correlations between POU2F1 and USP22 (Fig. 7C-c) expression in the twelve GC tissues, respectively (Fig. 7c).

Identification of a POU2F1/miR-4490/USP22 axis in primary human GC samples. (a) USP22 and POU2F1 protein levels detected by Western blotting in 12 paired GC tissues; GAPDH was used for normalization. (b) Average tumor/adjacent normal gastric epithelium (N/T) ratios of USP22 (a) POU2F1 and (b), miR-4490 expression quantified by qRT-PCR and normalized against GAPDH or U6 (n = 12). **, p < 0.05; ***, p < 0.01. (c) Correlation between miR-4490 and USP22 expression, between miR-4490 and POU2F1 expression, or between USP22 and POU2F1 expression in 12 GC tissues. (d) MiR-4490, USP22, POU2F1, E-cadherin, and Vimentin expression patterns in human GC tissues and normal adjacent epithelial tissues determined by in situ hybridization (ISH) or IHC staining. Scale bars, 50 μm
Additionally, we performed ISH and IHC analyses and again found that the miR-4490 expression levels were negatively correlated with those of the POU2F1 and USP22 proteins (Fig. 7d). We also assessed the expression of EMT markers and observed upregulation of E-cadherin and downregulation of vimentin in the noncancerous tissues. In contrast, decreased E-cadherin and increased vimentin expression was observed in the GC tissues. Taken together, these data suggest that the POU2F1-miR-4490-USP22 signaling axis may serve as an important regulator of GC development and progression.
4 Discussion
MiRNAs have been reported to play important roles in the initiation and progression of GC, and aberrant miRNA expression levels have been associated with GC metastasis [25, 27]. However, the role of and molecular mechanisms underlying the mode of action of miR-4490 in GC development and invasion have not been reported. Here, we found that miR-4490 was downregulated in most GC cancer cell lines and primary GC tissues tested. We also found that exogenous miR-4490 expression inhibited the proliferation, migration and invasion of GC cells. MiR-4490 targets USP22, which is known to play an important role in GC progression. Furthermore, we found that the miR-4490 promoter is negatively regulated by the transcription factor POU2F1. These results suggest that the POU2F1/miR-4490/USP22 axis may regulate the proliferation and invasion of GC cells.
The miR-4490 gene is located at chromosome region 11q14.3. Previous studies have suggested an involvement of genes in this region in cancer development [28, 29]. For example, the NOXIN gene, located at 11q14.1, has been identified as an oncogene in hepatocellular carcinoma (HCC). NOXIN overexpression has been found to promote cellular proliferation, colony formation, migration and in vivo tumorigenicity [28]. However, the role of miR-4490 in GC is not well characterized. We found that that the expression of miR-4490 is downregulated in GC, and that exogenous miR-4490 expression caused a significant G1/G0 arrest and a significant inhibition of GC cell growth. More interestingly, we found that miR-4490 deficiency contributes to GC cell migration and invasion.
Several studies have shown that miRNAs can regulate cancer invasion and metastasis through EMT-related mechanisms [30, 31]. The EMT process, through which cells lose epithelial features and acquire mesenchymal features, is activated during cancer invasion and metastasis [32, 33]. Since we found that miR-4490 can regulate GC cell migration and invasion, we hypothesized that it may affect the EMT process. We indeed found that miR-4490 inhibition induced EMT-like phenotypic features in GC cells. Furthermore, we found that miR-4490 expression was closely associated with the expression of EMT markers. Both in vitro and in vivo studies showed that exogenous miR-4490 expression inhibited GC cell invasiveness. These results suggest that miR-4490 deficiency affects GC cell migration and invasion by enhancing the EMT process.
MiRNA binding to the 3′-UTRs of target mRNAs and its association with translational repression and/or mRNA degradation has amply been studied in recent years, and this may provide an important approach to investigate their roles in cancer [3, 5, 6, 34]. By exploring the molecular mechanisms underlying the inhibition of GC cell motility and invasion induced by miR-4490, we identified putative targets using the online software TargetScan, miRanda, miRWalk and miRmap. Among the predicted targets, we found that USP22 acted as a critical effector of miR-4490. Several reports previously showed that USP22 may be tightly linked to the progression of GC [35, 36]. In the present study, using a luciferase reporter assay, we showed that miR-4490 can bind to a sequence within the 3′-UTR of USP22. Moreover, we found that miR-4490 overexpression inhibited the expression of USP22 in GC cells, and that exogenous overexpression of USP22 partially counteracted the effects of miR-4490 on the suppression of GC cell proliferation, migration and invasion. In addition, it has been reported that USP22 is a critical oncogene that is overexpressed and plays critical roles in the EMT process in various tumors by affecting E-cadherin expression [37, 38]. We found that miR-4490 inhibited vimentin expression, upregulated E-cadherin expression and regulated cell invasion and EMT by targeting USP22 in GC cells. Thus, these results imply that miR-4490 modulates GC cell proliferation and metastasis through EMT induction by targeting USP22.
Many studies have shown that transcription factors (TFs) can positively or negatively regulate miRNA expression [35, 36, 38, 39]. To explore the mechanisms of miR-4490 regulation in GC cells, we subjected the miR-4490 promoter region to further detailed analysis. Bioinformatics analysis predicted that the miR-4490 promoter contains putative POU2F1 binding sites and that a highly conserved binding element is composed of a consensus sequence (5′- ATGCAAATCA -3′). A previous study has shown that 74% of the GC samples tested displayed increased POU2F1 protein levels [27]. More recently, POU2F1 was reported to be upregulated in hepatocellular carcinoma and to modulate EMT [25, 26]. Therefore, we hypothesized that POU2F1 may bind to the promoter region of miR-4490 to affect its expression. Our results revealed that POU2F1 indeed directly binds to the miR-4490 promoter and decreases its activity using ChIP and luciferase reporter assays, respectively. POU2F1 also markedly decreased the expression of miR-4490. Thus, we conclude that POU2F1 binds to the miR-4490 promoter to suppress miR-4490 expression in GC cells.
In conclusion, we found that a novel miRNA, miR-4490, is downregulated in GC cell lines and tissues and functions as a tumor suppressor that inhibits GC cell proliferation, invasion and EMT in vitro and in vivo. Exogenous miR-4490 expression at least partially caused downregulation of USP22 expression. Moreover, we found that POU2F1 directly binds to the miR-4490 promoter to regulate its transcription in GC cells. We conclude that the POU2F1/miR-4490/USP22 signaling axis may serve as a promising target for the treatment of GC.
Change history
09 February 2021
Editor's Note:: Readers are alerted that concerns have been raised regarding Figures 3a and 4f. We will update readers once we have further information and all parties have been given an opportunity to respond in full.
29 December 2022
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1007/s13402-022-00768-4
Abbreviations
- GC:
-
Gastric cancer
- USP22:
-
ubiquitin-specific protease 22
- UTR, 3′:
-
untranslated region
- EMT:
-
epithelial-mesenchymal transition
- ChIP :
-
chromatin immunoprecipitation
- RLU:
-
relative luciferase unit
References
S. Volinia, G.A. Calin, C.G. Liu, S. Ambs, A. Cimmino, F. Petrocca, R. Visone, M. Iorio, C. Roldo, M. Ferracin, R.L. Prueitt, N. Yanaihara, G. Lanza, A. Scarpa, A. Vecchione, M. Negrini, C.C. Harris, C.M. Croce, A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103, 2257–2261 (2006)
J. Lu, G. Getz, E.A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B.L. Ebert, R.H. Mak, A.A. Ferrando, J.R. Downing, T. Jacks, H.R. Horvitz, T.R. Golub, MicroRNA expression profiles classify human cancers. Nature 435, 834–838 (2005)
G.A. Calin, C.M. Croce, MicroRNA-cancer connection: The beginning of a new tale. Cancer Res 66, 7390–7394 (2006)
G.F. Zhu, Y.W. Xu, J. Li, H.L. Niu, W.X. Ma, J. Xu, P.R. Zhou, X. Liu, D.L. Ye, X.R. Liu, T. Yan, W.K. Zhai, Z.J. Xu, C. Liu, L. Wang, H. Wang, J.M. Luo, L. Liu, X.Q. Li, S. Guo, H.P. Jiang, P. Shen, H.K. Lin, D.H. Yu, Y.Q. Ding, Q.L. Zhang, Mir20a/106a-WTX axis regulates RhoGDIa/CDC42 signaling and colon cancer progression. Nat Commun 10, 112 (2019)
N. Zhang, X. Li, C.W. Wu, Y. Dong, M. Cai, M.T. Mok, H. Wang, J. Chen, S.S. Ng, M. Chen, J.J. Sung, J. Yu, microRNA-7 is a novel inhibitor of YY1 contributing to colorectal tumorigenesis. Oncogene 32, 5078–5088 (2013)
P. Zhang, W.M. Tang, H. Zhang, Y.Q. Li, Y. Peng, J. Wang, G.N. Liu, X.T. Huang, J.J. Zhao, G. Li, A.M. Li, Y. Bai, Y. Chen, Y.X. Ren, G.X. Li, Y.D. Wang, S.D. Liu, J.D. Wang, MiR-646 inhibited cell proliferation and EMT-induced metastasis by targeting FOXK1 in gastric cancer. Brit J Cancer 117, 525–534 (2017)
L. Liang, X. Li, X. Zhang, Z. Lv, G. He, W. Zhao, X. Ren, Y. Li, X. Bian, W. Liao, W. Liu, G. Yang, Y. Ding, MicroRNA-137, an HMGA1 target, suppresses colorectal cancer cell invasion and metastasis in mice by directly targeting FMNL2. Gastroenterol 144, 624–635 e624 (2013)
H.J. Lee, M.S. Kim, J.M. Shin, T.J. Park, H.M. Chung, K.H. Baek, The expression patterns of deubiquitinating enzymes, USP22 and Usp22. GEP 6, 277–284 (2006)
G.V. Glinsky, Genomic models of metastatic cancer: Functional analysis of death-from-cancer signature genes reveals aneuploid, anoikis-resistant, metastasis-enabling phenotype with altered cell cycle control and activated Polycomb group (PcG) protein chromatin silencing pathway. Cell Cycle 5, 1208–1216 (2006)
Y. He, Y.J. Jin, Y.H. Zhang, H.X. Meng, B.S. Zhao, Y. Jiang, J.W. Zhu, G.Y. Liang, D. Kong, X.M. Jin, Ubiquitin-specific peptidase 22 overexpression may promote cancer progression and poor prognosis in human gastric carcinoma. J Lab Clin Med 165, 407–416 (2015)
J. Hu, Y.L. Liu, S.L. Piao, D.D. Yang, Y.M. Yang, L. Cai, Expression patterns of USP22 and potential targets BMI-1, PTEN, p-AKT in non-small-cell lung cancer. Lung Cancer 77, 593–599 (2012)
Y.L. Liu, Y.M. Yang, H. Xu, X.S. Dong, Increased expression of ubiquitin-specific protease 22 can promote cancer progression and predict therapy failure in human colorectal cancer. J Gastroenterol Hepatol 25, 1800–1805 (2010)
Y. Li, Y. Yang, J. Li, H. Liu, F. Chen, B. Li, B. Cui, Y. Liu, USP22 drives colorectal cancer invasion and metastasis via epithelial-mesenchymal transition by activating AP4. Oncotarget 8, 32683–32695 (2017)
Y. Peng, P. Zhang, X. Huang, Q. Yan, M. Wu, R. Xie, Y. Wu, M. Zhang, Q. Nan, J. Zhao, A. Li, J. Xiong, Y. Ren, Y. Bai, Y. Chen, S. Liu, J. Wang, Direct regulation of FOXK1 by C-Jun promotes proliferation, invasion and metastasis in gastric cancer cells. Cell Death Dis 7, e2480 (2016)
S.B. Edge, C.C. Compton, The American joint committee on Cancer: The 7th edition of the AJCC cancer staging manual and the future of TNM. Ann Surg Oncol 17, 1471–1474 (2010)
W. Tang, W. Zhou, L. Xiang, X. Wu, P. Zhang, J. Wang, G. Liu, W. Zhang, Y. Peng, X. Huang, J. Cai, Y. Bai, L. Bai, W. Zhu, H. Gu, J. Xiong, C. Ye, A. Li, S. Liu, J. Wang, The p300/YY1/miR-500a-5p/HDAC2 signalling axis regulates cell proliferation in human colorectal cancer. Nat Commun 10, 663 (2019)
T. Yu, J. Li, M. Yan, L. Liu, H. Lin, F. Zhao, L. Sun, Y. Zhang, Y. Cui, F. Zhang, J. Li, X. He, M. Yao, MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene 34, 413–423 (2015)
S. Bagga, J. Bracht, S. Hunter, K. Massirer, J. Holtz, R. Eachus, A.E. Pasquinelli, Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 122, 553–563 (2005)
L. Wu, J. Fan, J.G. Belasco, MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A 103, 4034–4039 (2006)
G. Ye, K. Huang, J. Yu, L. Zhao, X. Zhu, Q. Yang, W. Li, Y. Jiang, B. Zhuang, H. Liu, Z. Shen, D. Wang, L. Yan, L. Zhang, H. Zhou, Y. Hu, H. Deng, H. Liu, G. Li, X. Qi, MicroRNA-647 targets SRF-MYH9 axis to suppress invasion and metastasis of gastric cancer. Theranostics 7, 3338–3353 (2017)
A. Abdeyrim, X. Cheng, M. Lian, Y. Tan, miR4905p regulates the proliferation, migration, invasion and epithelialmesenchymal transition of pharyngolaryngeal cancer cells by targeting mitogenactivated protein kinase kinasekinase 9. Int J Mol Med 44, 240–252 (2019)
J. Hu, D. Yang, H. Zhang, W. Liu, Y. Zhao, H. Lu, Q. Meng, H. Pang, X. Chen, Y. Liu, L. Cai, USP22 promotes tumor progression and induces epithelial-mesenchymal transition in lung adenocarcinoma. Lung Cancer 88, 239–245 (2015)
J. Qian, X. Kong, N. Deng, P. Tan, H. Chen, J. Wang, Z. Li, Y. Hu, W. Zou, J. Xu, J.Y. Fang, OCT1 is a determinant of synbindin-related ERK signalling with independent prognostic significance in gastric cancer. Gut 64, 37–48 (2015)
S.H. Xu, J.Z. Huang, M.L. Xu, G. Yu, X.F. Yin, D. Chen, G.R. Yan, ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol 236, 175–185 (2015)
W.W. Hwang-Verslues, P.H. Chang, Y.M. Jeng, W.H. Kuo, P.H. Chiang, Y.C. Chang, T.H. Hsieh, F.Y. Su, L.C. Lin, S. Abbondante, C.Y. Yang, H.M. Hsu, J.C. Yu, K.J. Chang, J.Y. Shew, E.Y. Lee, W.H. Lee, Loss of corepressor PER2 under hypoxia up-regulates OCT1-mediated EMT gene expression and enhances tumor malignancy. Proc Natl Acad Sci U S A 110, 12331–12336 (2013)
Z.Z. Zhang, J. Huang, Y.P. Wang, B. Cai, Z.G. Han, NOXIN as a cofactor of DNA polymerase-primase complex could promote hepatocellular carcinoma. Int J Cancer 137, 765–775 (2015)
Y. Zhong, H. Huang, M. Chen, J. Huang, Q. Wu, G.R. Yan, Chen, POU2F1 over-expression correlates with poor prognoses and promotes cell growth and epithelial-to-mesenchymal transition in hepatocellular carcinoma. Oncotarget 8, 44082–44095 (2017)
S. Deneberg, M. Kanduri, D. Ali, S. Bengtzen, M. Karimi, Y. Qu, E. Kimby, L. Mansouri, R. Rosenquist, A. Lennartsson, S. Lehmann, microRNA-34b/c on chromosome 11q23 is aberrantly methylated in chronic lymphocytic leukemia. Epigenetics 9, 910–917 (2014)
W.K. Wu, C.W. Lee, C.H. Cho, D. Fan, K. Wu, J. Yu, J.J. Sung, MicroRNA dysregulation in gastric cancer: A new player enters the game. Oncogene 29, 5761–5771 (2010)
Y. Xu, F. Zhao, Z. Wang, Y. Song, Y. Luo, X. Zhang, L. Jiang, Z. Sun, Z. Miao, H. Xu, MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene 31, 1398–1407 (2012)
J. Yu, F. Xie, X. Bao, W. Chen, Q. Xu, miR-300 inhibits epithelial to mesenchymal transition and metastasis by targeting Twist in human epithelial cancer. Mol Cancer 13, 121 (2014)
C.W. Lin, S.H. Kao, P.C. Yang, The miRNAs and epithelial-mesenchymal transition in cancers. Curr Pharmaceut Design 20, 5309–5318 (2014)
J. Tang, L. Li, W. Huang, C. Sui, Y. Yang, X. Lin, G. Hou, X. Chen, J. Fu, S. Yuan, S. Li, W. Wen, S. Tang, D. Cao, M. Wu, L. Chen, H. Wang, MiR-429 increases the metastatic capability of HCC via regulating classic Wnt pathway rather than epithelial-mesenchymal transition. Cancer Lett 364, 33–43 (2015)
D. Kim, A. Hong, H.I. Park, W.H. Shin, L. Yoo, S.J. Jeon, K.C. Chung, Deubiquitinating enzyme USP22 positively regulates c-Myc stability and tumorigenic activity in mammalian and breast cancer cells. J Cell Physiol 232, 3664–3676 (2017)
X. Yang, H. Zang, Y. Luo, J. Wu, Z. Fang, W. Zhu, Y. Li, High expression of USP22 predicts poor prognosis and advanced clinicopathological features in solid tumors: A meta-analysis. OncoTargets Ther 11, 3035–3046 (2018)
H. Liu, N. Liu, Y. Zhao, X. Zhu, C. Wang, Q. Liu, C. Gao, X. Zhao, J. Li, Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging 11, 9643–9660 (2019)
H.Y. Zhu, G.Y. Cao, S.P. Wang, Y. Chen, G.D. Liu, Y.J. Gao, J.P. Hu, POU2F1 promotes growth and metastasis of hepatocellular carcinoma through the FAT1 signaling pathway. Am J Cancer Res 7, 1665–1679 (2017)
D.J. Sharpe, K.S. Orr, M. Moran, S.J. White, S. McQuaid, T.R. Lappin, A. Thompson, J.A. James, POU2F1 activity regulates HOXD10 and HOXD11 promoting a proliferative and invasive phenotype in head and neck cancer. Oncotarget 5, 8803–8815 (2014)
R. Zhang, H. Lu, Y.Y. Lyu, X.M. Yang, L.Y. Zhu, G.D. Yang, P.C. Jiang, Y. Re, W.W. Song, J.H. Wang, C.C. Zhang, F. Gu, T.J. Luo, Z.Y. Wu, C.J. Xu, E6/E7-P53-POU2F1-CTHRC1 axis promotes cervical cancer metastasis and activates Wnt/PCP pathway. Sci Rep 7, 44744 (2017)
Acknowledgments
We acknowledge the generous support of the Guangdong Provincial Key Laboratory of Gastroenterology, Department of Gastroenterology, Nanfang Hospital, Southern Medical University, China. This study was supported by grants from the National Natural Science Funds of China (Nos. 81974448, 81672875, 81700489 and 81772964), the Science and Technology Project of Guangzhou (Grant NO. 201707010276), the Science and Technology Planning Project of Guangdong Province (Nos. 2017B020209003 and 2017B030314037) and the National Science and Technology Major Project of China (No. 2020ZX09201017). We acknowledge the generous support of the Department of Gastroenterology, Longgang District People’s Hospital, Shenzhen, China, the Special Foundation for Economic and Technological Development of Longgang District, Shenzhen (Grant/Award Number: LGKCYLWS2018000141), the Special Scientific Research Fund of Public Welfare Profession of National Health and Family Planning Commission (Grant/Award Number: 201502026) and the Shenzhen Science and Technology Innovation Commission (Grant No: JCYJ20180306170328854).
Availability of data and materials
All data are available within the article and supplementary files, or from the authors upon request.
Author information
Authors and Affiliations
Contributions
This study was designed and conceived by Jide Wang, Aimin Li and Side Liu. Experiments in vitro were performed by Yizhi Xiao, Weiyu Dai and Jiaying Li. Experiments in vivo were performed by Yizhi Xiao, Li Xiang, Weimei Tang, Wenjing Zhang, Jianjiao Lin, Jing Wang. Yizhi Xiao, Xiaosheng Wu, Guangnan Liu and Yuyang Liu. Yaying Chen helped with data analysis. Huiqiong Zhu and Qiong Yang were responsible for the collection of patient specimens. Yusi Wang, Zhizhao Lin, Tianming Chen and Yong Sun provided technical support. Aimin Li and Side Liu supervised the project and Jide Wang wrote the manuscript. Jing Xiong revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was reviewed and approved by the Medical Ethics Committee of Nanfang Hosital (NFEC-2017-062), Southern Medical University, Guangzhou, China. All animal studies were approved by the Institutional Animal Care and Use Committee of Committee of Nanfang Hosital.
Consent for publication
All authors have agreed to publish this manuscript.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail: https://doi.org/10.1007/s13402-022-00768-4
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xiao, Y., Liu, S., Li, J. et al. RETRACTED ARTICLE: The POU2F1/miR-4490/USP22 axis regulates cell proliferation and metastasis in gastric cancer. Cell Oncol. 43, 1017–1033 (2020). https://doi.org/10.1007/s13402-020-00553-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-020-00553-1