
Abstract
Background
Chronic kidney disease (CKD) is characterized by sustained tissue damage and ongoing tubulo-interstitial inflammation and fibrosis. Pattern recognition receptors (PRRs) including Toll-like receptors (TLRs) and NOD-like receptors (NLRs) can sense endogenous ligands released upon tissue damage, leading to sterile inflammation and eventually irreversible kidney disease. It is known that NOD1 and NOD2 contribute to the pathogenesis of various inflammatory diseases, including acute kidney injury. However their role in chronic kidney disease is largely unknown. The aim of this study was therefore to investigate the contribution of NOD1 and NOD2 in renal interstitial fibrosis and obstructive nephropathy.
Methods
To do so, we performed unilateral ureteral obstruction (UUO) in wild type (WT) and NOD1/NOD2 double deficient (DKO) mice and analysed renal damage, fibrosis and inflammation. Data were analysed using the non-parametric Mann-Whitney U-test.
Results
Minor changes in inflammatory response were observed in NOD1/2 DKO mice, while no effects were observed on renal injury and the development of fibrosis.
Conclusion
No difference in renal injury and fibrosis between WT and NOD1/NOD2 DKO mice following obstructive nephropathy induced by ureteral obstruction.
Similar content being viewed by others


Background
NOD1 and NOD2 are members of the cytoplasmic PRR family of NLRs. PRRs are important in mediating a rapid response to pathogens via recognition of several highly conserved pathogen- associated molecular patterns (PAMP). In addition to PAMPs various (endogenous) damage-associated molecular patterns (DAMP) or stress signals have been identified that can initiate sterile inflammation [1]. Upon renal injury DAMPs are released such as biglycan, high-mobility group box 1 (HMGB1), and hyaluronic acid that can signal via TLRs and NLRs [2,3,4,5]. NOD1 and NOD2 detect specific substructures from bacterial peptidoglycan (PGN). NOD1 senses Gram−-derived PGN containing Tri-DAP [6, 7], while NOD2 senses Gram−- and Gram+-derived PGN containing MDP [8, 9]. In line, we found that NOD1/2 are involved in the development of acute renal disease during septic shock induced by bacterial components [10]. In addition to bacterial structures, Sabbah et al. reported the activation of NOD2 by single-stranded RNA viruses [11]. Recently, activation of NOD1 and NOD2 by the non-pathogenic derived cell permeable small molecule DMXAA was reported [12]. As far as we know, no endogenous DAMPs for NOD1 and NOD2 are described. However, based on their structural and functional similarities with other NLR family members and TLRs it could be speculated that NOD1 and NOD2 are also activated by currently unknown endogenous ligands. In line with this reasoning, Shigeoka et al. showed that NOD1 and NOD2 participate in acute renal ischemia reperfusion injury (IRI), suggesting that these receptors are able to respond to endogenous ligands released upon IRI [13]. NOD1 is widely expressed in many cell types and organs including the tubular epithelial cells (TEC) in human and mouse kidney [13,14,15,16]. NOD2 is expressed on murine TECs, mesangial cells, podocytes and on human TECs and glomerular endothelial cells [13, 17, 18]. Given the expression of NOD1 and NOD2 in the kidney and more specific in TEC and the fact that NOD1/2 play a deleterious role in acute kidney disease could suggest that these PRRs contribute to the pathogenesis of chronic renal damage as well. PRRs like NLRP3 and TLR4 have already been shown to play a role in obstructive nephropathy [19,20,21]. However, nothing is known about the role of NOD1 and NOD2 in inflammation and fibrosis during obstructive nephropathy. In the present study we therefore investigated the role of NOD1 and NOD2 in a model of obstructive nephropathy induced by ureteral obstruction.
Methods
Mice
Pathogen-free 8- to 12-week old female C57Bl/6 WT mice were purchased from Janvier (Le Genest, France). NOD1/NOD2 DKO mice were generated from NOD1 and NOD2 knockout mice and backcrossed to C57Bl/6 background at least 10 generations as described before [22]. Previously, we have characterized the DKO mice phenotypically and this revealed that except for lower liver weight in NOD1/2 DKO mice, there were no differences in body/organ weight, leukocyte count/composition and plasma biochemical markers between both strains [10]. NOD1/2 DKO mice (Additional file 1) were bred in the animal facility of the Academic Medical Center in Amsterdam, The Netherlands. Age- and sex-matched mice were used in all experiments. The animal and Use Committee of the University of Amsterdam approved all experiments.
Unilateral ureter obstruction
Mice (N = 9/group) received a pre-operative dose of analgesia (0.15 mg/kg buprenorfine, subcutaneously) and were anesthetized by inhalation of 3% isoflurane, 0.2% N2O and 2% O2 during the surgical procedure. The right ureter was permanently ligated via a ventral approach using 6–0 silk (Tyco, Gosport, UK). The ureter was ligated at the height of the lower part of the kidney. Mice were sacrificed 3, 7 and 14 days after surgery via a heart puncture (blood collection) followed by cervical dislocation under general anaesthesia. Kidneys were snap frozen in liquid nitrogen and stored at -80 °C or fixed in 10% formalin o/n prior to further processing. Contralateral non-obstructed kidneys served as control.
Quantitative real-time RT-PCR
Total RNA was extracted from kidney using the TRIzol® reagent (Invitrogen, Breda, The Netherlands) and converted to cDNA. Quantitative real-time RT-PCR was performed on a LightCycler® 480 System (Roche, Mijdrecht, The Netherlands) using LightCycler® 480 SYBR Green I Master mix (Roche). Specific gene expression was normalized towards the reference gene TATA box binding protein (TBP). Primer sequences are as follows: NOD1 forward 5′-tcagactcagcgtcaaccag-3′ and reverse 5′-taaacccaggaacgtcacga-3′, NOD2 forward 5′-gggagatgttggagtggaac-3′ and reverse 5′-agcgaagagcacactcaacc-3′, and TBP forward 5′-ggagaatcatggaccagaaca-3′ and reverse 5′-gatgggaattccaggagtca-3′.
Histology and immunohistochemistry
Formalin-fixed tissue was embedded in paraffin using standard procedures. Four-μm thick sections were cut and used for all stainings. For examining renal histology, sections were stained with periodic acid-Schiff reagents after diastase digestion (PasD). Injury to tubules was assessed (blinded) by determining the percentage of affected tubules per 10 fields (magnification × 400) semi-quantitatively on a scale from 0 to 4 (0 = 0%, 1 = < 25%, 2 = 25–50%, 3 = 50–75%, and 4 = > 75%) according to the following criteria: tubular dilatation, epithelial simplification, and interstitial expansion in the cortex. For immunohistochemistry, sections were stained with FITC-labelled anti-mouse Ly-6G (Pharmingen, BD Biosciences, Alphen a/d Rijn, The Netherlands), rat anti-mouse F4/80 (Serotec, Oxford, UK), rabbit anti-mouse active caspase-3 (Cell Signaling Technology, Beverly, MA, USA), rabbit anti-human Ki67 (Neomarkers, Fremont, CA, USA), rabbit polyclonal to collagen type I (GeneTex, Irvine, CA, USA), or mouse anti-human αSMA (DAKO, Heverlee, Belgium) to detect granulocytes, macrophages, apoptosis, proliferation, collagen type I, and myofibroblasts respectively. The number of Ly6 positive cells and the number of caspase-3 and Ki67 positive TEC was counted in 10 non-overlapping fields (magnification × 400) in a blinded manner. The percentage of positive staining for F4/80, collagen type I, total collagen and αSMA in obstructed kidneys was analysed using a computer-assisted digital analysis program (Image Pro-plus®, Media Cybernetics). At least 15 visual fields were sampled from the cortex of each kidney (magnification × 20).
Statistical analyses
All statistical analyses were performed using GraphPad Prism 5 software (San Diego, CA, USA). Data were analysed using the non-parametric Mann-Whitney U-test. Results are expressed as mean ± standard error of the mean (SEM). P < 0.05 was considered statistically significant.
Results
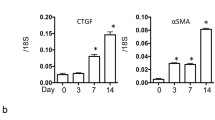
The role of NOD1 and NOD2 in CKD was investigated using the mouse model UUO. First we analysed the expression of NOD1 and NOD2 mRNA in WT kidney at several time points after UUO. We found expression of both transcripts in the kidney (Fig. 1), which were not altered during the development of obstructive nephropathy. Tubular injury, as assessed by scoring PAS-D-stained kidney sections, increased markedly after UUO with a similar degree of damage in WT and NOD1/2 DKO at all time points examined (Fig. 2a, c). In line with the injury score, there were no differences in KIM-1 expression between the WT and the KO mice at all time points (Fig. 2b). KIM-1 was declined at day 14 of UUO possibly as an adaptation to prevent or slow down KIM-1 mediated chronic inflammation and renal fibrosis, as described previously [23]. Tubulointerstitial injury in obstructed kidneys can result in an imbalance between TEC apoptosis and proliferation. Apoptosis and proliferation of TEC was increased at all investigated time points after obstruction (Fig. 3). However, no difference between WT and NOD1/2 DKO mice was observed. Fibrosis was determined by collagen type I (Fig. 4a) and total collagen deposition (Additional file 2: Figure S2). In both WT and NOD1/2 DKO obstructed kidneys fibrosis increased progressively, however no difference between the WT and NOD1/2 DKO mice was observed. Next we analysed the amount of myofibroblasts by αSMA immunohistochemistry (Fig. 4b). In line with tubular injury and fibrosis, the amount of myofibroblasts increased after UUO. Although myofibroblast accumulation was lower in NOD1/2 DKO mice 3 days following ureteral obstruction compared to WT mice, no differences were found after 7 and 14 days (Fig. 4b). One of the early events in progressive renal injury is the induction of chemokines and the subsequent recruitment of inflammatory cells. The granulocyte chemoattractant KC (Fig. 5a) and the monocyte chemoattractant MCP-1 (Fig. 5b) increased significantly following obstruction in both WT and NOD1/2 DKO kidneys. Except for a slight but significant higher MCP-1 level in kidneys from NOD1/2 DKO mice compared with WT 7 days following obstruction, no difference in KC and MCP-1 levels were observed between the WT and NOD1/2 DKO mice. The influx of granulocytes (Fig. 5c, e) and accumulation of macrophages (Fig. 5d, f) increased in the obstructed WT and NOD1/2 DKO kidneys, yet there were no differences in these parameters between the WT and NOD1/2 DKO mice, 3 and 14 days post UUO. Seven days following obstruction there was a slight but significant decrease in granulocyte influx and a significant increase in macrophage accumulation in NOD1/2 DKO kidneys compared with WT kidneys. To get more insight in the activation of the NF-kB signaling pathway, we measured the downstream cytokines IL-1b and TNF-α in total kidney homogenates. This revealed no significant differences between the WT and NOD1/2 DKO mice, expect for IL-1b day 3 (Additional file 3: Figure S3). Together these results reveal that there are no or minor changes in the inflammatory response, renal damage and fibrosis following CKD induced by ureteral obstruction in NOD1/2 DKO mice compared with WT mice.

Renal expression of NOD1 (a) and NOD2 (b) after 0, 3, 7, and 14 days following obstruction. Gene expression was normalized towards the reference gene TBP. Data are expressed as mean ± SEM. N = 9/group

Renal injury in WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. Renal damage was evaluated by blinded scoring of the necrotic tubules in PAS-D-stained sections (a, c) and by measuring the mRNA expression of KIM-1 (b) in total kidney homogenates. Data are expressed as mean ± SEM. Results were analysed with the non-parametric two-tailed Mann-Whitney U-test. *P < 0.05. N = 9/group

Apoptosis and proliferation of TECs in WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. The amount of apoptotic TEC was determined by scoring caspase-3+ TEC (a, c) and the amount of proliferating TEC (b, d) was determined by scoring Ki67+ TEC. Data are expressed as mean ± SEM. N = 9/group

Renal fibrosis in WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. Collagen type I deposition in kidneys was determined by Collagen type I staining and digitally analysed (a, d). Myofibroblast accumulation in kidneys was assessed by αSMA staining and digitally analysed (b, c). Data are expressed as mean ± SEM. Results were analysed with the non-parametric two-tailed Mann-Whitney U-test. *P < 0.05. N = 9/group

Renal inflammation in WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. KC (a) and MCP-1 (b) were measured in total kidney homogenates with specific ELISAs. Influx of granulocytes was determined by scoring Ly6+ cells (c, e). Influx of macrophages was assessed by F4/80 staining which was digitally analysed (d, f). Data are expressed as mean ± SEM. Results were analysed with the non-parametric two-tailed Mann-Whitney U-test. *P < 0.05. N = 9/group
Discussion
To investigate the role of NOD1 and NOD2 in chronic renal inflammation, injury and fibrosis we subjected NOD1/2 DKO and WT mice at various time points to UUO. UUO initiates a sequence of events in the obstructed kidney, including interstitial inflammation and TEC death, ultimately leading to renal fibrosis which is the final common pathway for numerous forms of progressive renal disease. Recently, the role of the PRRs TLR2, TLR9, TLR4 and NLRP3 in progressive renal injury was investigated. Although TLR2 initiates the inflammatory response during obstructive nephropathy, it does not play a significant role in the development of renal progressive injury and fibrosis [3, 24]. Similarly, TLR9 was not involved in the pathogenesis of UUO [24]. On the other hand, TLR4 attenuates tubular damage and does contribute to renal fibrosis during obstructive nephropathy as demonstrated by an increased injury score and decreased collagen deposition in TLR4-deficient mice [20, 25]. Other work implied a central role for NLRP3 in renal inflammation, fibrosis and tubular damage at different phases of UUO [19, 21]. Apparently, different PRR members have unique response during obstructive nephropathy that lead to a profoundly different outcome of local injury and tubulointerstitial inflammation and fibrosis.
From our study we conclude that NOD1 and NOD2 do not play a significant role in the development of tubulointerstitial fibrosis and inflammation nor in the progression of renal damage after UUO-induced injury. No differences were observed between WT and NOD1/2 DKO obstructed kidneys regarding tubular injury score, apoptosis, proliferation and myofibroblast accumulation. A marginal effect of NOD1/2 deficiency could be detected in the inflammatory response during obstructive nephropathy. Slightly more MCP-1 and concomitant increased macrophage accumulation was observed in the NOD1/2 DKO kidney 7 days following obstruction, while granulocyte influx was lower at this time point. The majority of infiltrating leukocytes into the UUO-damaged kidney are macrophages, which produce cytokines responsible for tubular apoptosis and fibroblast proliferation and activation. However, enhanced macrophage accumulation did not affect the progression of renal fibrosis in NOD1/2 DKO mice. In another study on progressive kidney disease, namely diabetes, it was shown that NOD2 is upregulated and promoted the transcription of extracellular matrix genes and renal injury by inducing inflammation and podocyte insulin resistance [18]. Moreover, in sepsis- and ischemia-induced acute kidney disease models, NOD1/2 DKO mice were demonstrated to be protected against renal disease [10, 13]. Apparently, as it was the case for TLR2 [3, 26], NOD1 and NOD2 are involved in the initiation of inflammation but do not necessarily contribute to further renal damage and fibrosis. Considering that we found no difference between WT and NOD1/2 DKO mice in renal pathology, one could speculate that despite their structural and functional similarities with other PRRs, NOD1 and NOD2 are not activated by DAMPs that are released after UUO. This would be in line with current literature in which solely bacterial ligands and not DAMPs are described to activate NOD1 and NOD2. We anticipate that the difference in renal pathology between WT and NOD1/2DKO mice in the renal ischemia reperfusion injury model might be due to translocation of bacterial products across the leaky intestinal barrier that activate NOD1/2 resulting in inflammation-associated nephropathy. The phenomenon of intestinal barrier disruption is known to occur after renal ischemia reperfusion injury [27] but is not described for UUO.
This study however, has some limitations which have to be pointed out. By using double knockout mice we cannot rule out the possibility that either NOD1 or NOD2 play a different role in UUO and compensate each other. Alternatively, the function of NOD1 or NOD2 might be masked in our knockout model by redundancy or compensatory mechanism. Our experiments tested moreover only unilateral ureteral obstruction with its own advantages and limitations [28] and no other animal models of chronic kidney disease and fibrosis. Other possible roles of NOD1/NOD2 in these disorders that may be activated under different circumstances remain therefore to be tested.
Taken together, our data do not show a functional role for NOD1/2 in kidney injury and fibrosis following chronic kidney disease induced by ureteral obstruction and suggest that similar to infection, different forms of sterile kidney disease will be sensed by different PRRs triggering different signalling pathways which culminate in different kidney disease outcomes.
Conclusion
Together these results reveal that there are no or minor changes in the inflammatory response, renal damage and fibrosis following obstructive nephropathy induced by ureteral obstruction in NOD1/2 DKO mice compared with WT mice.
Abbreviations
- CKD:
-
Chronic kidney disease
- DAMP:
-
Danger associated molecular patterns
- DKO:
-
Double deficient
- HMGB1:
-
High-mobility group box 1
- IRI:
-
Ischemia reperfusion injury
- NLRs:
-
NOD-like receptors
- PAMP:
-
Pathogen associated molecular patterns
- PGN:
-
Peptidoglycan
- PRRs:
-
Pattern recognition receptors
- SEM:
-
Standard error of the mean
- TEC:
-
Tubular epithelial cell
- TLRs:
-
Toll-like receptors
- UUO:
-
Unilateral ureteral obstruction
- WT:
-
Wild type
References
Leemans JC, et al. Pattern recognition receptors and the inflammasome in kidney disease. Nat Rev Nephrol. 2014;10(7):398–414.
Iyer SS, et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci U S A. 2009;106(48):20388–93.
Leemans JC, et al. The role of toll-like receptor 2 in inflammation and fibrosis during progressive renal injury. PLoS One. 2009;4(5):e5704.
Schaefer L, et al. Absence of decorin adversely influences tubulointerstitial fibrosis of the obstructed kidney by enhanced apoptosis and increased inflammatory reaction. Am J Pathol. 2002;160(3):1181–91.
Wu H, et al. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117(10):2847–59.
Chamaillard M, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702–7.
Girardin SE, et al. Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science. 2003;300(5625):1584–7.
Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869–72.
Inohara N, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn's disease. J Biol Chem. 2003;278(8):5509–12.
Stroo I, et al. Phenotyping of Nod1/2 double deficient mice and characterization of Nod1/2 in systemic inflammation and associated renal disease. Biol Open. 2012;1(12):1239–47.
Sabbah A, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073–80.
Cheng G, et al. Activation of the nucleotide oligomerization domain signaling pathway by the non-bacterially derived xanthone drug 5′6-dimethylxanthenone-4-acetic acid (Vadimezan). J Biol Chem. 2010;285(14):10553–62.
Shigeoka AA, et al. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J Immunol. 2010;184(5):2297–304.
Dharancy S, et al. Neutrophil migration during liver injury is under nucleotide-binding oligomerization domain 1 control. Gastroenterology. 2010;138(4):1546–56. 1556.e1–5
Inohara N, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem. 1999;274(21):14560–7.
Uehara A, et al. Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44(12):3100–11.
Anders HJ, Muruve DA. The inflammasomes in kidney disease. J Am Soc Nephrol. 2011;22(6):1007–18.
Du P, et al. NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int. 2013;84(2):265–76.
Pulskens WP, et al. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS One. 2014;9(1):e85775.
Pulskens WP, et al. TLR4 promotes fibrosis but attenuates tubular damage in progressive renal injury. J Am Soc Nephrol. 2010;21(8):1299–308.
Vilaysane A, et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J Am Soc Nephrol. 2010;21(10):1732–44.
Geddes K, et al. Nod1 and Nod2 regulation of inflammation in the Salmonella colitis model. Infect Immun. 2010;78(12):5107–15.
Humphreys BD, et al. Chronic epithelial kidney injury molecule-1 expression causes murine kidney fibrosis. J Clin Invest. 2013;123(9):4023–35.
Skuginna V, et al. Toll-like receptor signaling and SIGIRR in renal fibrosis upon unilateral ureteral obstruction. PLoS One. 2011;6(4):e19204.
Campbell MT, et al. Toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. J Surg Res. 2011;168(1):e61–9.
Leemans JC, et al. Renal-associated TLR2 mediates ischemia/reperfusion injury in the kidney. J Clin Invest. 2005;115(10):2894–903.
Kim M, et al. Isoflurane activates intestinal sphingosine kinase to protect against renal ischemia-reperfusion-induced liver and intestine injury. Anesthesiology. 2011;114(2):363–73.
Eddy AA, et al. Investigating mechanisms of chronic kidney disease in mouse models. Pediatr Nephrol. 2012;27(8):1233–47.
Funding
This study is financially supported by the Dutch Kidney Foundation (to IS and LMB) and ZonMW (MKMD) 114024013 (to JCL). SEG is supported by funding from the Canadian Institutes of Health Research (CIHR). Funding bodies had no role in the design of the study, collection, analysis and interpretations of data and in writing the manuscript.
Availability of data and materials
All data underlying the findings are incorporated within the paper.
Author information
Authors and Affiliations
Contributions
JCL was responsible for conception and design of the study, interpretation of results and revising of the paper. IS participated in experimental setup, performing experiments, interpretation of data, analysis of data and writing the manuscript. DE was involved in interpretation of data, analysis of data, drafting, revising and writing of the manuscript. LMB, and NC participated in laboratory analysis of samples. GJT performed all animal experiments. SF and MCD participated in understanding data and revising the manuscript. SEG provided the NOD1/NOD2 DKO mice. All authors read and agreed on the final version of the paper.
Corresponding author
Ethics declarations
Ethics approval
All animal experiments were approved by the Animal and Use Committee of the University of Amsterdam. C57Bl/6 WT mice were purchased from Janvier (Le Genest, France). NOD1/NOD2 DKO were provided by Girardin’s lab via a verbal consent between JCL and SEG.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1. The genotype of NOD1/2 DKO mice. Genomic DNA from mice was amplified by PCR with specific primers to detect the disrupted sequences on a 1% agarose gel with a 100 bp marker. First 5 bands are the KO mice and the last bands are the WT mice in A and B. N = 5/3 per group. (TIFF 12868 kb)
Additional file 2:
FigureS2. Total collagen in kidneys of WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. Total collagen was assessed by Picro Sirius Red staining which was digitally analysed (A, B). Data are expressed as mean ± SEM. N = 9/group. (TIFF 1068 kb)
Additional file 3:
Figure S3. Renal inflammation in WT (white bars) and NOD1/2 DKO (black bars) mice after 0, 3, 7, and 14 days following obstruction. IL-1b (A) and TNF-α (B) were measured in total kidney homogenates with specific ELISAs. Data are expressed as mean ± SEM. Results were analysed with the non-parametric two-tailed Mann-Whitney U-test. *P < 0.05. N = 9/group. (TIFF 2728 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Stroo, I., Emal, D., Butter, L.M. et al. No difference in renal injury and fibrosis between wild-type and NOD1/NOD2 double knockout mice with chronic kidney disease induced by ureteral obstruction. BMC Nephrol 19, 78 (2018). https://doi.org/10.1186/s12882-018-0867-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-018-0867-8