
Abstract
Background
Mitochondrial diseases present with variable multi-organ symptoms. Common disease-causing mutations in mitochondrial DNA (mtDNA) are regularly screened in diagnostic work-up, but novel mutations may remain unnoticed.
Methods
Patients (N = 66) with a clinical suspicion of mitochondrial disease were screened for their mtDNA coding region using conformation sensitive gel electrophoresis and sequencing. Long-PCR was used to detect deletions followed by POLG1 sequencing in patients with multiple deletions.
Results
We discovered three novel mtDNA variants that included m.8743G > C, m.11322A > G and m.15933G > A. The novel MTTT variant m.15933G > A is suggested to be pathogenic. Analysis revealed also multiple mtDNA deletions in two patients and five nonsynonymous variants that were putatively pathogenic according to in-silico prediction algorithms. In addition, a rare haplogroup H associated m.7585_7586insT variant was discovered.
Conclusions
Among patients with a suspected mitochondrial disease, a novel MTTT variant m.15933G > A was discovered and is suggested to be pathogenic. In addition, several putatively pathogenic nonsynonymous variants and rare variants were found. These findings highlight the importance of coding region mtDNA screening among patients with clinical features suggesting a mitochondrial disease, but who lack the common mitochondrial disease mutations.
Similar content being viewed by others

Background
More than 200 pathogenic point mutations and large-scale rearrangements have been described in mitochondrial DNA (mtDNA) [1]. Some of the most common mitochondrial diseases caused by mutations in mtDNA are Kearns-Sayre syndrome, Leber’s hereditary optic neuropathy, myoclonic epilepsy with ragged red fibers syndrome, MELAS syndrome (mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes) and the syndrome of neuropathy, ataxia and retinitis pigmentosa (NARP). The prevalence of mitochondrial diseases caused by mtDNA mutations has been calculated to be 1:5000 in the adult population [1, 2] and the total prevalence rises up to 1:4300, when disorders caused by mutations in nuclear-encoded genes contributing to mitochondrial functions are included [3].
Screening of established pathogenic mtDNA and nDNA mutations is straightforward, but detection of novel or rare mtDNA mutations is more tedious. Such mutations may remain unnoticed, unless the entire mtDNA coding region is sequenced after excluding known mitochondrial disease mutations. However, conventional sequencing methods do not detect large mtDNA deletions, nor are they good in recognizing extremes of mtDNA heteroplasmy. It is also imperative to be able to analyse the pathogenic potential of novel or rare mtDNA mutations and to determine their frequency in the population [4], as mtDNA is highly variable by nature. There is also emerging evidence suggesting that polymorphic variation can influence disease phenotype and raise the risk for developing a mitochondrial disorder [5–9]. Furthermore, mtDNA variants have been suggested to increase the risk of common neurodegenerative diseases, such as Alzheimer’s and Parkinson’s disease [10–13]. In this sense, the Finnish population is ideal for detecting underlying genetic risk factors, as it is considered to be a genetically isolated, homogenous population.
In the present study, we screened the mtDNA coding region of 66 patients with a clinical suspicion of a mitochondrial disease. Our aim was to determine mtDNA sequence for rare mtDNA variants and to assess if they were pathogenic mutations or polymorphic risk factors. Conformation sensitive gel electrophoresis (CSGE) and sequencing were used to determine the mtDNA coding region of the patients and long-PCR was used to detect multiple mtDNA deletions. Polymerase γ gene (POLG1) was sequenced in multiple deletions patients.
Methods
Patients and population controls
Finnish patients (N = 66) with a clinically suspected mitochondrial disease were examined. Muscle samples (N = 21), blood samples (N = 36) or both (N = 9) were obtained. Common symptoms among selected patients included exercise intolerance, myopathy, encephalomyopathy, cardiomyopathy, diabetes mellitus, sensorineural hearing impairment and progressive external ophthalmoplegia. The patients consisted of sporadic cases or cases with similar symptoms in maternal relatives. The ethics committee of the University of Oulu has approved the study protocol and all patients gave a written informed consent for participation in the study.
Anonymous healthy blood donors (N = 480) were used as population controls. It was required that the donor and his or her mother were born in the same region, and that the donor did not report neurological ailments, diabetes or sensorineural hearing impairment in the family. MtDNA haplogroups have been determined in all of the 480 controls [14], the hypervariable segment I (HVSI) in the D-loop has been sequenced in 403 controls [15] and the entire mtDNA sequence has been determined in 192 controls [14]. The ethics committee of the Finnish Red Cross has approved the study protocol.
Molecular methods
Total DNA was extracted from blood by using the QIAmp Blood Kit (Qiagen, Hilden, Germany). Total DNA from muscle samples was extracted by using Wizard® Genomic DNA purification kit (Promega, Madison, WI, U.S.A.) and standard tissue DNA extraction method using phenol and chloroform. MtDNA haplogroups were determined by restriction fragment analysis [16].
Forty-one samples were analysed by using conformation sensitive gel electrophoresis (CSGE) as previously described [17, 18]. The mtDNA coding region, nucleotides m.577-m.16090, was amplified in 63 partially overlapping fragments. Each amplified fragment was mixed with the corresponding fragment amplified on a control template with a known sequence. Heteroduplex formation was also allowed to occur autogenously enabling the detection of heteroplasmic mutations. Heteroduplexes that differed in mobility in CSGE were sequenced (ABI PRISM ™ 377 Sequencer using DYEnamic ET Terminator Cycle Sequencing Kit; Amersham Pharmacia Biotech Inc., Buckinghamshire, U.K.) after purification with exonuclease I and shrimp alkaline phosphatase [19]. The primers used for sequencing were the same as those used for the amplification of the 63 CSGE fragments. Twenty-five samples were sequenced directly for their entire mtDNA. The mtDNA was first amplified in 11 fragments followed by sequencing using primers that were used for the amplification of mtDNA fragments.
Muscle DNA samples (N = 30) were examined for multiple mtDNA deletions by using long-PCR (Phusion expand XL-PCR, Boehringer Mannheim, Mannheim, Germany) as described previously [20]. Certain POLG1 mutations are fairly common in the Finnish population and, hence, the entire POLG1 gene (NCBI Reference Sequence: NM_002693) was sequenced in the two samples that were found to harbour multiple mtDNA deletions. Next-generation sequencing would have provided an effective tool for mtDNA analysis and heteroplasmy detection but, unfortunately, this method was beyond our resources at the time of investigation.
Analysis of mtDNA variants
Sequences were compared to the revised Cambridge reference sequence [GenBank: NC_012920] [21] and to mtDNA sequences available in the Mitomap database http://www.mitomap.org [22]. Variants were considered rare if they occurred ≤ 10 times in the Mitomap database among 32,059 mtDNA sequences. Novel substitutions were confirmed by RFLP or by sequencing in both directions at least twice from different PCR products. The possibility of m.15933G > A being an artefact caused by nuclear mitochondrial DNA was examined by amplifying total mtDNA of patient 41 with long-PCR (XL-PCR) Expand Long Template PCR system kit (Boehringer Mannheim) as described previously [20] and by subsequently sequencing the mutation site with a forward primer at m.15714 and a reverse primer at m.16090. Conservation of nucleotides among species was evaluated using Mamit tRNA: Compilation of Mammalian mitochondrial tRNA genes http://mamit-tRNA.u-strasbg.fr [23] in the case of tRNA genes and GiiB-JST mtSAP evaluation http://mtsnp.tmig.or.jp/cgi-bin/mtsnp/specAlign/ctrlSpecAlignE.cgi in the case of structural mtDNA genes [24]. The pathogenicity of m.15933G > A was assessed by means of a multifactorial probability-based classification method PON-mt-tRNA that predicts the pathogenicity of single nucleotide substitutions in human mitochondrial tRNAs [25].
Analysis of nonsynonymous mtDNA mutations
Nonsynonymous mtDNA mutations were analysed for their pathogenic potential by using PolyPhen-2 version 2.2.2 [26] http://genetics.bwh.harvard.edu/pph2/, SIFT BLink [27] http://sift.jcvi.org/www/SIFT_BLink_submit.html, PMut [28, 29] http://mmb2.pcb.ub.es/PMut/ and SNAP [30] http://rostlab.org/services/snap/ algorithms. Pathogenicity classes were ascertained according to the guidelines suggested by Wallis et al. 2013 and Claustres et al. 2014 [31, 32].
PolyPhen-2 calculates a naïve Bayes posterior probability for each mutation to assess its damaging potential. Mutations are then classified as benign if the probability is less than 50%, possibly damaging if the probability is greater than 50% and probably damaging if the probability is greater than 90%.
SIFT BLink and PMut algorithms are based on sequence homology. These algorithms assume that functionally important protein sequences are conserved in evolution, whereas diverse positions are unimportant to protein function. SIFT scores for nonsynonymous mutations range between 0–1 and scores of ≤ 0.05 are considered to be deleterious. PMut also uses information on sequence conservation and neural networks to predict whether a variant is benign or pathogenic. PMut provides a reliability index ranging from 0 (most unreliable) to 9 (most reliable). SNAP predicts functional effect by using conservation and functional information of proteins, such as secondary structure and solvent accessibility. A variant is assigned either neutral or non-neutral. A reliability index (0 being the most reliable prediction and 7 being the most unreliable) and an accuracy percentage is also provided.
Exact test of population differentiation as implemented in Arlequin 3.5 was used to compare frequencies of mtDNA haplogroups between cases and controls [33]. Phylogenetic networks of mtDNA sequences were constructed on the basis of median algorithm [34].
Results
MtDNA sequence variation and novel mutations
The frequency of mtDNA haplogroups did not differ between the 66 patients with suspected mitochondrial disease and the controls. Three novel homoplasmic variants, m.8743G > C, m.11322A > G and m.15933G > A were discovered (Fig. 1). The m.11322A > G variant (p. N188S, MTND4) was found in two siblings, one (patient 21) with hypertrophic cardiomyopathy, chronic atrial fibrillation and sick sinus syndrome and the other (patient 22) with ptosis, restricted eye movements and exercise intolerance. The nucleotide position m.11322 is not notably conserved among 60 species which were investigated with GiiB-JST mtSAP evaluation http://mtsnp.tmig.or.jp/cgi-bin/mtsnp/specAlign/ctrlSpecAlignE.cgi [24]. The other two novel variants m.8743G > C (p.V73L, MTATP6) and m.15933G > A in MTTT were found in the muscle and blood of patient 41. The nucleotide at position m.8743 is not particularly conserved, whereas m.15933G is a highly conserved (Table 1, Fig. 2). The m.15933G > A mutation was predicted to be pathogenic with a probability score of 0.92 by PON-mt-tRNA method. Sequencing revealed that the m.15933G > A variant was present in an amplicon obtained in a long-range PCR amplification covering the whole mtDNA of patient 41.

Phylogenetic network of the mtDNA coding region in 66 patients with suspected mitochondrial disease. Patients are identified by numbers inside the nodes. Outgroup = an African sequence (GenBank: AF346980); CRS = the revised Cambridge Reference Sequence (GenBank: NC_012920). Unless marked otherwise, mtDNA variants are transitions. Superscript text indicates transversions. Insertions are marked with superscript (i), back mutations (@) and heteroplasmic mutations are marked with an asterisk (*). Underlined red font, variants predicted to be likely pathogenic, blue font novel mutations, green font rare mutations, purple font multiple mtDNA deletions and POLG1 mutations

The m.15933G > A mutation in MTTT. a Invariable nucleotides among 135 mammal species in MTTT according to sequence alignment in Mamit tRNA [24]. b Human polymorphisms and pathogenic mutations in MTTT. Polymorphisms are marked with white circles, black circles denote confirmed pathogenic mutations and undetermined variants are marked with a white square ([24, 42])
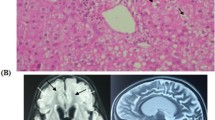
Patient 41 is a 51-year old woman. Her athletic achievements did not match those of the other girls at school. In adulthood she has had poor muscle fitness, and exercise-induced muscle weakness and myalgia. In addition, she has mild dysphagia and choking tendency. In her forties a nonsymmetrical ptosis developed and her right eye was operated at age 47 years. Psoriatic skin rash developed at age 49 years. Clinical neurological examination and blood lactate were normal. Muscle histology revealed ragged red fibers (RRF) and COX-negative fibers.
In addition, three other variants were considered rare occurring ≤10 times in the Mitomap database of 32,059 sequences. The rare insertion m.7585_7586insT (N = 4 in Mitomap database) between MTTD and MTCO2 was found in patient 16. This insertion adds a thymidine between the last nucleotide of MTTD and the first nucleotide of MTCO2 (Fig. 2). Cloning revealed that the insertion was present in all 100 clones investigated suggesting that the mutation in muscle mtDNA is homoplasmic with high probability. The other rare variants were m.4924G > C (p. S152T, MTND2) and m.7706G > A (p. A41T, MTCO2). The frequency of the rare nonsynonymous variants among the 66 patients did not differ from that in 89 haplogroup-matched controls drawn from the population [14].
In-silico analysis of nonsynonymous variants
All the nonsynonymous mtDNA variants (N = 47) discovered in the 66 patients were analysed for their pathogenic potential using PolyPhen-2, SIFT BLink, PMut and SNAP algorithms. Five of the mutations were predicted to be deleterious by at least three of the algorithms (Table 2). The highest probability in favour of a pathogenic effect was obtained for m.8616G > T (p. L30F, MTATP6). With the exception of m.5095 T > C (p.I209T, MTND2) and m.14180 T > C (p. Y165C, MTND6), the three other variants predicted to be deleterious have previously been reported to be haplogroup-associated.
Multiple mtDNA deletions
Muscle samples from 30 patients were analysed for multiple mtDNA deletions. Patients 59 and 22 (Fig. 1) were found to harbour multiple deletions. Patient 59 suffered from bilateral progressive external ophthalmoplegia, diplopia, ptosis and mild exercise intolerance. His muscle histology revealed numerous RRFs and COX-negative muscle fibers. He harboured a heterozygous p.R722H allele in POLG1, but no potentially pathogenic mtDNA mutations.
Patient 22 harboured multiple mtDNA deletions and the novel m.11322A > G variant (p. N188S, MTND4), but not POLG1 mutations. Her daughter had increased lactate in blood. No mtDNA deletions were detected in her brother (patient 21).
Discussion
We discovered three previously unreported mtDNA variants, three rare variants and a total of 47 nonsynonymous variants among 66 patients with suspected mitochondrial disease. In addition, two patients harbored multiple mtDNA deletions. One of the novel variants was m.15933G > A in MTTT in a patient with ptosis, mild muscle weakness, myalgia, mild dysphagia, psoriasis and with RRF and COX-negative fibers in skeletal muscle. This variant is situated at the base of the T stem of tRNAThr in tRNA consensus position 49, which is highly conserved among mammals, including its pair nucleotide at position 65 [35]. Nuclear mitochondrial DNA as a source of m.15933G > A was excluded by whole mitochondrial genome amplification and subsequent sequencing. The insertion m.15933_15934insG in the same position has been reported in one subject (GenBank: JQ702629.1) [36]. This insertion is not considered to be deleterious, because it makes the preceding variable loop one base pair longer but it retains the conserved m.15933G. On the other hand, the m.15933G > A removes the highly conserved G in the beginning of the T stem and decreases the length of the T stem to four base pairs (Fig. 2). Another example of a similar pair of a pathogenic mutation and a polymorphic insertion is present in the T stem of MTTL2, where m.12311 T > C is pathogenic and m.12311_12312insA is polymorphic variant [35, 37, 38]. Altogether six confirmed pathogenic mutations have been reported in MTTT [22, 23]. The symptoms of the patient, the conserved nature of the nucleotide site and the predicted high probability in favour of pathogenicity suggest that m.15933G > A is pathogenic, but further functional studies will be needed to discern the biochemical consequences of the mutation.
The patient with the novel m.15933G > A harboured also the novel variant m.8743G > C (p.V73L, MTATP6). This position holds a known polymorphism of m.8743G > A (p.V73M, MTATP6), but the transversion m.8743G > C has not been previously reported in databases. This nucleotide position is variable and the amino acid at this position is not evolutionarily conserved. We consider this novel variant unlikely to be pathogenic.
The three rare variants included the insertion m.7585_7586insT that was discovered in a patient with a multi-organ phenotype. The m.7585_7586insT variant is interesting, as it adds a nucleotide in between the contiguous MTTD and MTCO2 genes and may interfere with translation by one of three mechanisms. First, the MTTD transcript could be one nucleotide longer thus interrupting its proper function. Alternatively, the m.7585_7586insT insertion may affect the translation of MTCO2. The third possible mechanism entails that the RNAase cleavage site in between MTTD and MTCO2 is disrupted. However, m.7585_7586insT has previously been discovered in four subjects belonging to haplotype H35, three of whom are Finnish. Our patient belonged to haplotype H1 suggesting that m.7585_7586insT has arisen at least twice in this population. Additional studies are needed to verify, if m.7585_7586insT indeed affects the function of mitochondria. Unfortunately, only a few blood samples were available from this family and no autopsy had been performed on the proband after his death.
The frequency of rare nonsynonymous variants among the 66 patients did not differ from that in 89 haplogroup-matched controls drawn from the population [14]. Five out of the 47 nonsynonymous variants were predicted to be pathogenic in effect. Four algorithms were used including SNAP, SIFT BLink, PolyPhen-2 and PMut. These algorithms have been reported to accurately predict 76% of non-neutral variants, and SNAP is among the most accurate methods for predicting damaging variants [39]. We assumed the best candidates for pathogenic mutations to be those that three out of four algorithms predicted to be pathogenic. Three out of five predicted pathogenic variants were haplogroup-associated and, indeed, we found these variants in the same haplogroups as previously reported, suggesting that they are haplogroup-associated polymorphisms. On the other hand, m.5095 T > C (p.I209T, MTND2) has previously been reported in multiple haplogroups (B, C, T, A, U5a, R) [23], whereas we discovered it in haplogroup H. This patient presented with proximal muscle weakness and atrophy, orbicularis oculi muscle weakness, myalgia and a large psoriasis-type rash. Inflammatory changes were detected in muscle histology. The true pathogenicity of m.5095 T > C remains, however, unknown despite the high pathogenicity prediction score, as this mutation has been discovered in healthy individuals. Multiple mtDNA deletions were discovered in two patients aged 33 and 42 years with suspected mitochondrial disease. Multiple mtDNA deletions do occur normally in the course of healthy aging, but up to 48% of younger mitochondrial disease patients with multiple deletions harbour POLG1 mutations [40]. We found a heterozygous p.R722H allele in one patient, while the other patient with multiple deletions did not harbour any POLG1 mutations. The clinical features of the patient with the heterozygous p.R722H allele included progressive external ophthalmoplegia, diplopia, ptosis, mild exercise intolerance, and numerous RRFs and COX negative fibers in skeletal muscle. The p.R722H POLG1 allele has not been associated with disease in a heterozygous state, whereas in the homozygous state it leads to progressive external ophthalmoplegia, sensorineural hearing impairment, diabetes mellitus, dysphagia, a limb myopathy and dementia [41]. Patient 22 with multiple deletions, but without POLG1 mutations, also harboured a novel nonsynonymous mtDNA mutation m.11322A > G (p. N188S, MTND4) that was predicted to be neutral.
Conclusions
We determined the coding region mtDNA sequence of 66 Finnish patients with suspected mitochondrial disease. Among the 47 nonsynonymous variants, five were predicted to be deleterious in effect by using four algorithms in parallel, but three of them were considered haplogroup-associated polymorphisms. A likely pathogenic novel mutation m.15933G > A (MTTT) was discovered in patient with a suspected mitochondrial disease. Sequencing of the mtDNA coding region is recommended for patients presenting with symptoms of a mitochondrial disorder but lacking the common disease-causing mutations in mtDNA.
Abbreviations
- COX:
-
Cytochrome oxidase
- CRS:
-
The Cambridge reference sequence
- CSGE:
-
Conformation sensitive gel electrophoresis
- D-loop:
-
Displacement loop, non-coding mtDNA segment
- HVSI:
-
Hypervariable segment I
- MELAS:
-
Mitochondrial encephalomyopathy, lactic acidosis with stroke-like episodes
- MtDNA:
-
Mitochondrial DNA
- MTTT :
-
Mitochondrial tRNA Threonine gene
- NARP:
-
Neuropathy, ataxia and retinitis pigmentosa
- nDNA:
-
Nuclear DNA
- POLG1 :
-
Polymerase γ gene
- RFLP:
-
Restriction fragment length polymorphism
- RRF:
-
Ragged red fiber
References
Chinnery PF, Elliott HR, Hudson G, Samuels DC, Relton CL. Epigenetics, epidemiology and mitochondrial DNA diseases. Int J Epidemiol. 2012;41(1):177–87.
Gorman GS, Schaefer AM, Ng Y, Gomez N, Blakely EL, Alston CL, Feeney C, Horvath R, Yu-Wai-Man P, Chinnery PF, Taylor RW, Turnbull DM, Mcfarland R. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77(5):753–9.
Ylikallio E, Suomalainen A. Mechanisms of mitochondrial diseases. Ann Med. 2012;44(1):41–59.
Breen MS, Kondrashov FA. Mitochondrial pathogenic mutations are population-specific. Biol Direct. 2010;5:68.
Zhou X, Qian Y, Zhang J, Tong Y, Jiang P, Liang M, Dai X, Zhou H, Zhao F, Ji Y, Mo JQ, Qu J, Guan MX. Leber’s hereditary optic neuropathy is associated with the T3866C mutation in mitochondrial ND1 gene in three Han Chinese families. Invest Ophthalmol Vis Sci. 2012;53(8):4586–94.
Cai W, Fu Q, Zhou X, Qu J, Tong Y, Guan MX. Mitochondrial variants may influence the phenotypic manifestation of Leber’s hereditary optic neuropathy-associated ND4 G11778A mutation. J Genet Genomics. 2008;35(11):649–55.
Shu L, Zhang YM, Huang XX, Chen CY, Zhang XN. Complete mitochondrial DNA sequence analysis in two southern Chinese pedigrees with Leber hereditary optic neuropathy revealed secondary mutations along with the primary mutation. Int J Ophthalmol. 2012;5(1):28–31.
Achilli A, Olivieri A, Pala M, Hooshiar Kashani B, Carossa V, Perego UA, Gandini F, Santoro A, Battaglia V, Grugni V, Lancioni H, Sirolla C, Bonfigli AR, Cormio A, Boemi M, Testa I, Semino O, Ceriello A, Spazzafumo L, Gadaleta MN, Marra M, Testa R, Franceschi C, Torroni A. Mitochondrial DNA backgrounds might modulate diabetes complications rather than T2DM as a whole. Plos One. 2011;6(6):e21029.
Soini HK, Moilanen JS, Finnila S, Majamaa K. Mitochondrial DNA sequence variation in Finnish patients with matrilineal diabetes mellitus. BMC Res Notes. 2012;5(1):350.
Santoro A, Balbi V, Balducci E, Pirazzini C, Rosini F, Tavano F, Achilli A, Siviero P, Minicuci N, Bellavista E, Mishto M, Salvioli S, Marchegiani F, Cardelli M, Olivieri F, Nacmias B, Chiamenti AM, Benussi L, Ghidoni R, Rose G, Gabelli C, Binetti G, Sorbi S, Crepaldi G, Passarino G, Torroni A, Franceschi C. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. Plos One. 2010;5(8):e12037.
Coskun P, Wyrembak J, Schriner SE, Chen HW, Marciniack C, Laferla F, Wallace DC. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820(5):553–64.
Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322(1–2):254–62.
Khusnutdinova E, Gilyazova I, Ruiz-Pesini E, Derbeneva O, Khusainova R, Khidiyatova I, Magzhanov R, Wallace DC. A mitochondrial etiology of neurodegenerative diseases: evidence from Parkinson’s disease. Ann N Y Acad Sci. 2008;1147:1–20.
Finnilä S, Lehtonen MS, Majamaa K. Phylogenetic network for European mtDNA. Am J Hum Genet. 2001;68(6):1475–84.
Meinilä M, Finnilä S, Majamaa K. Evidence for mtDNA admixture between the Finns and the Saami. Hum Hered. 2001;52(3):160–70.
Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus ML, Wallace DC. Classification of European mtDNAs from an analysis of three European populations. Genetics. 1996;144(4):1835–50.
Körkkö J, Annunen S, Pihlajamaa T, Prockop DJ, Ala-Kokko L. Conformation sensitive gel electrophoresis for simple and accurate detection of mutations: comparison with denaturing gradient gel electrophoresis and nucleotide sequencing. Proc Natl Acad Sci U S A. 1998;95(4):1681–5.
Finnilä S, Hassinen IE, Ala-Kokko L, Majamaa K. Phylogenetic network of the mtDNA haplogroup U in Northern Finland based on sequence analysis of the complete coding region by conformation-sensitive gel electrophoresis. Am J Hum Genet. 2000;66(3):1017–26.
Werle E, Schneider C, Renner M, Volker M, Fiehn W. Convenient single-step, one tube purification of PCR products for direct sequencing. Nucleic Acids Res. 1994;22(20):4354–5.
Remes AM, Majamaa-Voltti K, Kärppä M, Moilanen JS, Uimonen S, Helander H, Rusanen H, Salmela PI, Sorri M, Hassinen IE, Majamaa K. Prevalence of large-scale mitochondrial DNA deletions in an adult Finnish population. Neurology. 2005;64(6):976–81.
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. 1999;23(2):147.
Lott MT, Leipzig JN, Derbeneva O, Xie HM, Chalkia D, Sarmady M, Procaccio V, Wallace DC. mtDNA Variation and Analysis Using MITOMAP and MITOMASTER. Curr Protoc Bioinformatics. 2013;1(23):1–26.
Putz J, Dupuis B, Sissler M, Florentz C. Mamit-tRNA, a database of mammalian mitochondrial tRNA primary and secondary structures. RNA. 2007;13(8):1184–90.
Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M, Shinoda K, Umetsu K, Yamada Y, Oshida Y, Sato Y, Hattori N, Mizuno Y, Arai Y, Hirose N, Ohta S, Ogawa O, Tanaka Y, Kawamori R, Shamoto-Nagai M, Maruyama W, Shimokata H, Suzuki R, Shimodaira H. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;14(10A):1832–50.
Niroula A, Vihinen M. PON-mt-tRNA: a multifactorial probability-based method for classification of mitochondrial tRNA variations. Nucleic Acids Res. 2016;44(5):2020–7.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74.
Ferrer-Costa C, Orozco M, de la Cruz X. Sequence-based prediction of pathological mutations. Proteins. 2004;57(4):811–9.
Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics. 2005;21(14):3176–8.
Bromberg Y, Yachdav G, Rost B. SNAP predicts effect of mutations on protein function. Bioinformatics. 2008;24(20):2397–8.
Claustres M, Kozich V, Dequeker E, Fowler B, Hehir-Kwa JY, Miller K, Oosterwijk C, Peterlin B, van Ravenswaaij-Arts C, Zimmermann U, Zuffardi O, Hastings RJ, Barton DE, European Society of Human Genetics. Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular Genetic). Eur J Hum Genet. 2014;22(2):160–70.
Wallis Y, Payne S, Mcanulty C, Bodmer D, Sistermans E, Robertson K, Moore D, Abbs S, Deans Z, Devereau A. Practice guidelines for the evaluation of pathogenicity and the reporting of sequence variants in clinical molecular genetics. Birmingham: Association for Clinical Genetic Science (ACGS); 2013. p. 1–16.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2007;1:47–50.
Bandelt HJ, Forster P, Rohl A. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol. 1999;16(1):37–48.
Vilmi T, Moilanen JS, Finnilä S, Majamaa K. Sequence variation in the tRNA genes of human mitochondrial DNA. J Mol Evol. 2005;60(5):587–97.
Behar DM, van Oven M, Rosset S, Metspalu M, Loogvali EL, Silva NM, Kivisild T, Torroni A, Villems R. A “Copernican” reassessment of the human mitochondrial DNA tree from its root. Am J Hum Genet. 2012;90(4):675–84.
Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N. Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups. Am J Hum Genet. 2002;70(5):1152–71.
van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat. 2009;30(2):E386–94.
da Hao C, Feng Y, Xiao R, Xiao PG. Non-neutral nonsynonymous single nucleotide polymorphisms in human ABC transporters: the first comparison of six prediction methods. Pharmacol Rep. 2011;63(4):924–34.
Ferreira M, Evangelista T, Almeida LS, Martins J, Macario MC, Martins E, Moleirinho A, Azevedo L, Vilarinho L, Santorelli FM. Relative frequency of known causes of multiple mtDNA deletions: two novel POLG mutations. Neuromuscul Disord. 2011;21(7):483–8.
Komulainen T, Hinttala R, Kärppä M, Pajunen L, Finnilä S, Tuominen H, Rantala H, Hassinen I, Majamaa K, Uusimaa J. POLG1 p.R722H mutation associated with multiple mtDNA deletions and a neurological phenotype. BMC Neurol. 2010;10:29.
Ruiz-Pesini E, Lott MT, Procaccio V, Poole JC, Brandon MC, Mishmar D, Yi C, Kreuziger J, Baldi P, Wallace DC. An enhanced MITOMAP with a global mtDNA mutational phylogeny. Nucleic Acids Res. 2007;35(Database issue):D823–8.
Acknowledgements
We greatly appreciate the technical assistance of Ms. Anja Heikkinen and Ms. Pirjo Keränen.
Funding
This study was supported by grants from Academy of Finland (project numbers 107174, 127764 and 266498), the Sigrid Juselius Foundation, the Finnish Graduate School for Population Genetics, the Päivikki and Sakari Sohlberg Foundation, The Alma and K.A. Snellman Foundation and the University of Oulu, Finland.
Availability of data and materials
Patient mtDNA sequences are available in GenBank: KY496857-KY496890.
Authors’ contributions
RH, JU and KM designed the study. HKS, AV, LK and RH generated and analysed the data. MK and JSM collected and analysed patient data. HKS and KM wrote the paper with contributions from other authors. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The ethics committee of the University of Oulu has approved the study protocol. All procedures performed in studies involving human participants were in accordance with the ethical standards with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Soini, H.K., Väisänen, A., Kärppä, M. et al. A novel MTTT mutation m.15933G > A revealed in analysis of mitochondrial DNA in patients with suspected mitochondrial disease. BMC Med Genet 18, 14 (2017). https://doi.org/10.1186/s12881-017-0377-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-017-0377-8